

Abstract
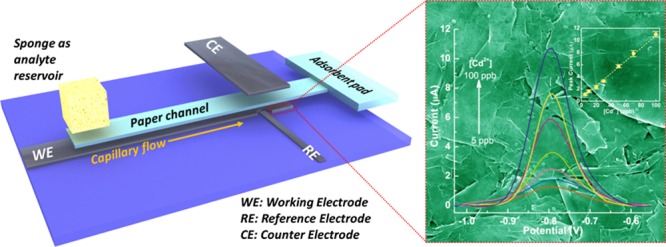
Heavy-metal pollution poses severe threat to ecological systems and presents a great challenge for global sustainability. Portable point-of-care sensing platform for detection/monitoring of heavy-metal pollution in the environment is urgently demanded. Herein, a highly sensitive, robust, and low-cost microfluidic electrochemical carbon-based sensor (μCS) for detection of trace heavy metals is presented. The miniaturized μCS devices are based on a microfluidic paper channel combined with a novel three-dimensional layout with working and counter electrodes facing each other and analyte flowing along the microfluidic channel between these two electrodes. Pristine graphite foil free of any surface modifier is not only used as the electronically conductive pad but also directly employed as the working electrode for fabricating the μCS. The resulting simple and portable device was applied in Cd2+ and Pb2+ detection using square-wave anodic stripping voltammetry. Detection limits down to 1.2 μg/L for Cd2+ and 1.8 μg/L for Pb2+ can be achieved over the μCS. The μCS devices are also found to be highly robust, and 10 repetitive measurements with a single μCS device resulted to be highly reproducible.
Introduction
Heavy metals are widely used in the manufacture of batteries, pigments, alloys, electroplating, coating, and so forth.1,2 However, mining, pouring, casting, processing, and inappropriate disposal of heavy metals have made them hazardous pollutants to the environment.3−6 Their toxic and nonbiodegradable nature imposes severe risks to human health.6−9 For example, increasing amounts of lead (Pb) in human body, as indicated by blood Pb levels, can impair neurobehavioral development in children, increase blood pressure, and cause kidney injury and anemia.2,5,9,10 Cadmium (Cd) has been proved to be a carcinogenic agent and may cause lung cancer, osteomalacia, and proteinuria, even at low doses.1,9,11,12 Conventional methods for heavy-metal detection are mainly based on atomic adsorption spectroscopy, X-ray fluorescence, inductively coupled plasma atomic emission spectroscopy, and inductively coupled plasma mass spectroscopy.5,12−14 Nevertheless, these ponderous, sophisticated, and expensive instruments are not suitable for fast and point-of-care analysis.5,13 Hence, researchers have been striving to develop simple, cost-effective, and portable sensing devices for fast and point-of-care analysis of heavy-metal pollution in the environment, especially in developing countries and areas with insufficient infrastructures.
Numerous detection platforms, including colorimetric, fluorescent, and electrochemical methods, have already been adopted to fabricate miniaturized portable devices for heavy-metal detections.15 Among them, the electrochemical method has attracted intensive attention due to its capability for achieving better quantitative results, more rapid analysis, and higher sensitivity.13,15,16 However, for the electrochemical detection of even trace amounts of heavy metals, it is usually mandatory to modify the surface of the working electrode with active electrocatalysts (receptors), which are supposed to have high collection capacity of target metal ions. This surface modification is especially of importance for the miniaturized devices because of the small amount of analyte (typically less than 100 μL) to be used for the analysis. Following this line of reasoning, a wide range of organic and inorganic materials have been used to modify the working electrode for the electrochemical detection of trace heavy metals. In early times, hanging mercury drop electrodes and electrodes modified with a mercury film were commonly used for heavy-metal detection because of the capability of mercury to form amalgam with heavy-metal ions, which results in its high sensitivity and good reproducibility.15,17−19 However, the well-known toxicity of mercury limits the development of mercury electrode sensor.20 Later on, bismuth has been introduced as an alternative surface modifier due to its unique behavior to form multicomponent Bi alloys with numerous heavy metals and less toxicity compared to mercury.21−27 Detection by bismuth-modified electrodes is usually carried out by simultaneous electroreduction of Bi ions and heavy-metal ions onto carbon-based solid electrodes (e.g., glassy carbon,28−30 carbon nanofibers (CNFs)/nanotubes,21,28,31−33 graphite,7,34−36 graphene,13,37,38 and carbon paste/ink23,25,39,40). The prereduced Bi could promote the deposition of heavy-metal ions from analytes by forming “fuse alloys”, which are analogous to the amalgam mercury forms.21,26,27 However, bismuth-modified electrode usually needs to be activated using a tedious pretreatment to improve the sensing reproducibility and is not applicable for some in situ and online trace metal measurements in natural or bioenvironment because it introduces Bi ions additionally.9,41 Besides, Bi ions are easy to hydrolyze to form insoluble compound in aqueous solution.21,42 For this reason, pH of the sample media should be controlled below 5 to avoid hydrolysis of Bi ions, which makes this method unpractical for biological and clinical analyses.9,21 In recent years, a great variety of innovative nanomaterials, such as metal (Au, Ag, Pd) nanoparticles,5,43,44 carbonaceous materials (carbon nanotubes, graphene, carbon spheres),20,35,37 and biological materials (DNA, protein/enzyme),45,46 have been tested as surface modifier for electrochemical detection of heavy-metal ions. Considering the tedious synthesis procedure and relatively high cost of these materials, using these innovative nanomaterials could be a big burden in mass production of electrochemical sensors and prohibit cost effectiveness.
Carbon-based materials, including bare glassy carbon electrode (GCE), in principle can also be directly used for heavy-metal detection. Hashemi et al. also demonstrated that surface modifier-free carbon-fiber microelectrode in combination with an high-performance liquid chromatography unit can be employed for copper-ion detection.47,48 In the current work, we are intending to directly use the inexpensive graphite foil without any surface modifier for electrochemical detection of heavy-metal ions in water, and graphite foils were chosen because of their excellence in both chemical stability and electrical conductivity. However, the challenge lies in two aspects: (1) how to achieve low limit of detection (LOD) (e.g., drinking-water standards established by World Health Organization (WHO) or United States Environmental Protection Agency (US-EPA)) by directly using graphite foils without any surface modifier and (2) how to transfer the sensing performance to a miniaturized device. Herein, these challenges are overcome by integrating a microfluidic paper channel into the miniaturized carbon-based sensor, where the pristine graphite foils were used as working, pseudoreference, and counter electrodes. The microfluidic channel continuously and efficiently delivers the aqueous analyte to the detection sites to enable a shortening of the overall current response time. Moreover, the μCS possesses a novel three-dimensional (3D) structure with working and reference electrodes directly facing each other but separated by the microfluidic paper channel, as shown schematically in Figure 1. This configuration brings the working electrode in close proximity to the counter electrode, and the resulting more homogeneous and efficient electric field is supposed to facilitate the reductive deposition of metal ion on the graphite-foil working electrode.49 The sensing performance of the μCS built solely from paper and graphite foil as benign and inexpensive materials is evaluated by detecting heavy-metal Cd2+ and Pb2+, which are common pollutants in the environment. It is found that impressive low detection limits of 1.2 μg/L for Cd2+ and 1.8 μg/L for Pb2+ can be achieved on the μCS. The μCS also exhibits stable sensing performance up to 10 repetitive measurements, demonstrating the unprecedented robustness of a sensing device for heavy-metal detections. We also successfully demonstrate that even for a miniaturized device, a proper design in the device configuration can completely eliminate the necessity to modify the working electrode by using additional surface modifier, such as mercury or bismuth. We believe these findings would have significant implications in developing other fast and cost-effective detection platforms, such as clinical diagnosis and security inspection.
Figure 1.
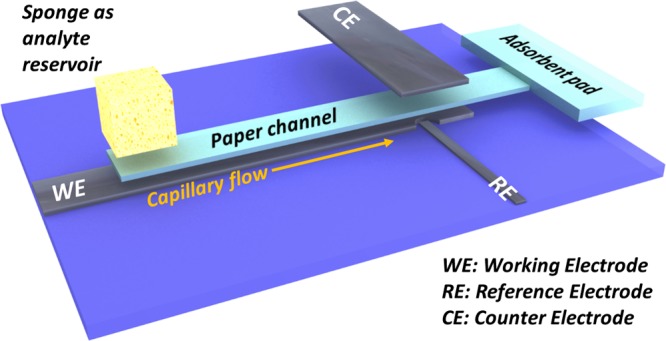
Scheme for the μCS device based on paper (light blue) and graphite foil (black). The dimensions are detailed in Figure S1.
Results and Discussion
Optimization of Square-Wave Voltammetry Parameters
To evaluate the sensing performance of the as-prepared μCS devices for heavy-metal detections, square-wave anodic stripping voltammetry (SWASV) measurements, which is well-known for its high sensitivity, were carried out for detections of Cd2+ and Pb2+. To realize a sensitive measurement, experimental parameters, including electrodeposition potential, deposition time, and electrolyte pH, which are considered as the most important influential factors in SWASV, were optimized, and the results are shown in Figure 2. The effect of electrodeposition potential was investigated by studying the anodic stripping peak current of 100 μg/L Cd2+ and 100 μg/L Pb2+ in 0.1 M acetate buffer solution (pH = 4.6) while varying the electrode potentials (Figure 2a). First, it can be observed that Cd2+ requires a more negative potential (−1.1 V) to be deposited on the graphite-foil electrode than Pb2+ (−0.9 V), which is not surprising considering their difference in standard reduction potentials. In principle, lower deposition potential could be more efficient to promote the reductive deposition of metal ions on the working electrodes (WEs). Interestingly, the peak currents for both metals exhibit a volcano dependence on the applied potentials with the maximum current obtained at potentials of −1.2 and −1.1 V for Cd2+ and Pb2+, respectively, which were later chosen as the deposition potentials for SWASV measurements. The decrease of the peak current at relatively lower deposition potential is caused by the competing hydrogen evolution reaction (H+ + e– → H2),50 where hydrogen gas bubbles can be clearly observed on the surface of the WE at low deposition potential. The effect of deposition time was investigated by varying the deposition time in the range of 1–10 min for Cd2+ and 1–15 min for Pb2+ (Figure 2b). Although longer deposition time always leads to higher current intensity, it could lower the upper range of heavy-metal detection due to the fast surface saturation in higher ion concentration. Herein, the deposition time was fixed at 1 and 3 min for detecting Cd2+ and Pb2+, respectively, as the trade-off between fast analysis and significant current response. A shorter deposition time was chosen for Cd2+ simply because a good linear calibration plot has already been obtained with a shorter time. As shown in Figure 2b, the proportional increase in the peak current with increasing the deposition time from 7 to 10 min indicates that the electrode surface is still far from being saturated. It is therefore possible to further improve the detection sensitivity by increasing the deposition time. The effect of electrolyte pH was studied by varying the pH values of acetate buffer solution from 3.0 to 6.0 (Figure 2c). It turns out that the square-wave stripping peak current was increasing rapidly with the increase of the pH values from 3.0 to 4.6, whereas further increasing pH to 6.0 has imposed minor influence on the peak currents for Cd2+ and Pb2+ detection. In contrast, it is well documented that the stripping current on Bi-modified electrode would decrease with increasing pH values.50,51 These results demonstrate that the detection performance of the μCS would be compromised for the sample with low pH values (≤4.0), which is also the case for the modified electrodes.50−53 Therefore, for analyte with low pH values (e.g., acidified water samples with pH < 2), we cannot expect the modifier-free μCS to exhibit superior sensing performance with respect to the conventional modified electrodes. However, for the samples with relatively high pH values, the μCS does show better tolerance on the pH values of the water samples. As the maximum stripping current can be obtained at pH = 4.6, the acetate buffer solution with pH 4.6 was used in subsequent experiments.
Figure 2.

Effect of deposition time (a), deposition potential (b), and electrolyte pH (c) on Cd2+ and Pb2+ detection. The concentration of both Cd2+ and Pb2+ is 100 μg/L.
As the sensing performance of a microfluidic device is also sensitive to the flow rate of an analyte to the detection sites, herein we also made attempts to evaluate the average flow rate of analytes within the paper channel. A typical capillary flow follows the Lucas–Washburn equation, which predicts that the flow rate decreases with increasing time. It has been experimentally demonstrated that the capillary flow behavior would significantly deviate from the Lucas–Washburn equation in the presence of an absorbent pad, and in this case, the capillary flow is sustained over time (quasi-stationary flow with time) because the liquid in the microfluidic channel would encounter a continuous increase in unwetted pore volume as it advanced in the porous absorbent pad. The constant cross section assumed by the Lucas–Washburn equation does not apply anymore.54 Similarly, in the current work, due to the presence of an absorbent pad at the end of the paper channel, we found that the stripping peak current was rather constant over a 15 min measurement time. This implies that the flow rate of analyte within the paper channel is not diminishing significantly over time and thus allows us to estimate the average flow rate of analyte by quantifying the mass (volume) of solution within the sampling sponge before and after the measurement over 15 min. The average flow rate or consumption rate of an aqueous analyte is estimated to be 130 μL/min. The sampling volumes for a single detection are ca. 130 and 390 μL for Cd2+ and Pb2+, respectively, which appear to be sufficient to get well-resolved stripping signals.
Individual Detection of Cd2+ and Pb2+
After optimizing the square-wave voltammetry parameters, the μCS was applied in individual detection of Cd2+ and Pb2+ in acetate buffer solution. Typical square-wave voltammograms in the presence of Cd2+ and Pb2+ with different concentrations are displayed in Figure 3. Well-defined anodic stripping peaks centering at −0.78 and −0.52 V (vs carbon pseudoreference electrode) were obtained for Cd2+ and Pb2+, respectively. The peak currents of both Cd2+ and Pb2+ increase linearly with concentration from 5 to 100 μg/L (Figure 3b,d). However, peak broadening occurs at analyte concentration below 10 μg/L for both Cd2+ and Pb2+, which might stem from the heterogeneity of surface-active sites on the surface of graphite foils and associated varied interactions with the deposited metal particles. This might also lead to the loss of linear response of SWASV signals at lower analyte concentrations (<5 μg/L) by using the μCS. For instance, it is difficult for us to discriminate the analytes with Cd2+ concentrations between 1 and 2 ppb by comparing their SWASV signals, although their stripping peaks are still well resolved. Attempts were also made to study the upper limit of the linear range for both metals. As shown in Figure S2, the linear range for Cd2+ can be extended up to at least 500 μg/L. In contrast, we found that although the peak current response for Pb2+ would still get increased with the concentration, however, it deviates from the linear calibration plot obtained at the concentration range of 5–100 μg/L, which would stem from saturation of surface sites for metal ions with higher concentrations. The calibration plots and correlation coefficients are shown in Figure 3b,d (x: concentration/μg/L, y: current/μA). The limits of detection for Cd2+ and Pb2+ are 1.2 and 1.8 μg/L based on 3σ method (detailed in the Supporting Information), respectively, which are below the allowable limits in drinking water proposed by US-EPA (Cd: 5 μg/L, Pb: 15 μg/L) and WHO (Cd: 3 μg/L, Pb: 10 μg/L).55,56 Specifically, in the case of Cd2+ detection, the μCS device is capable of not only quantifying Cd2+ in water samples with ion concentration above the US-EPA allowable limits (5 μg/L) but also raising an alert for the water sample that contains ions with concentration below the allowable limit. Moreover, the impressive low detection limits and wide linear detection range achieved by using the μCS for both analytes also indicate that the potential selective absorption of Cd2+ or Pb2+ by the sensor components, that is, paper, graphite foil, or sampling sponge, plays a minor role in the electrochemical sensing on the μCS, at least for analytes with concentration above the lower limit of linear detection range. This is consistent with the finding reported by Pickering that the presence of competing electrolyte could significantly reduce the absorption/uptake of heavy-metal ions (e.g., Pb2+, Cd2+) by filter paper.57 It is proposed that absorption of metal ions (Pb2+, Cd2+, etc.) by filter paper may be attributed to an ion-exchange process, whereas abundant cations in the electrolyte would suppress this exchange process due to the preferential retention of cations in the electrolyte.57 The capability and performance of μCS are also compared to those of some modified electrodes, screen-printing electrodes (SPEs), and miniaturized sensors in recent literatures in Table 1. Apart from using miniaturized sensors, electrochemical sensing measurement can also be conducted using the bulk electrochemical cell configuration, where a surface-modified glassy carbon electrode, as the WE, is placed in excess amount of electrolyte with analyte (e.g., 100 mL). The bulk electrochemical cell configuration usually gives lower detection limit compared to that of miniaturized sensors because of the significantly much larger amount of analyte being used during the sensing process. It can be seen that the μCS devices exhibits almost comparable sensing performance to some state-of-the-art surface-modified electrodes in bulk electrochemical cell configuration and is also among the best miniaturized on-chip sensing devices in terms of detection limit and sensitivity. To be noted, for practical application, usually an analyte solution with large collection volume (e.g., 100 mL) is provided, and the small analyte volume required by using these μCS devices for a single analysis would enable strict replicate analysis and allow to achieve even lower detection limit by concentrating the analyte solution.
Figure 3.

Square-wave voltammograms for Cd2+ (a) and Pb2+ (c) in 0.1 M acetate buffer (pH = 4.6) on μCS. Linear correction from 5 to 100 μg/L for Cd2+ (b) and Pb2+ (d).
Table 1. Performance of Various Sensing Devices for Heavy-Metal Detectiona.
| linear
range (μg/L) |
detection of limit (μg/L) |
sensitivityb (μA (μg/L)−1) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| sensor or electrode | modifier | method | deposition time (s) | Cd | Pb | Cd | Pb | Cd | Pb | refs |
| electrospun CNF-modified GCE | CNF | SWV | 300 | 46–182 | 27–145 | 0.17 | 0.19 | 0.107 | 0.25 | (20) |
| CNF array-modified GCE | CNF array | SWV | 120 | 10–100 | 0.4 | 0.04 | (58) | |||
| Pd NPs incorporated porous activated carbon-modified GCE | Pd/C | DPV | not mentioned | 56–1440 | 104–4640 | 4.6 | 10.4 | (43) | ||
| porous bismuth–carbon nanocomposite electrode | Bi | SWV | 300 | 1–100 | 1–100 | 0.6 | 0.6 | 0.2 | 0.15 | (59) |
| bismuth bulk electrode | Bi | SWV | 180 | 10–100 | 10–100 | 0.54 | 0.93 | 0.112 | 0.125 | (60) |
| bismuth citrate-modified SPE | Bi | DPV | 120 | 5–40 | 10–80 | 1.1 | 0.9 | 0.028 | 0.04 | (61) |
| bismuth-coated microband SPE | Bi | SWV | 120 | 5–45 | 1.3 | 0.01 | (62) | |||
| Bi nanoparticle porous carbon composite SPE | Bi | SWV | 120 | 5–100 | 5–100 | 2.1 | 3.9 | 0.024 | 0.025 | (63) |
| disposable Bi2O3 SPE | Bi2O3 | SWV | 120 | 10–150 | 10–150 | 5 | 10 | 0.013 | 0.0085 | (64) |
| on-chip sensor with silver electrode | Ag | SWV | 300 | 1–1000 | 0.55 | 0.028 | (65) | |||
| bismuth screen-printing microfluidic sensor | Bi | SWV | 120 | 5–100 | 1 | 0.17 | (16) | |||
| MWCNT and graphite powder-modified screen-printing sensor | MWCNTs | SWV | 240 | 5–150 | 5–150 | 1 | 1 | 0.0066 | 0.0068 | (66) |
| Bi2O3-modified screen-printing sensor | Bi2O3 | SWV | 300 | 20–100 | 20–100 | 1.5 | 2.3 | 0.065 | 0.0022 | (67) |
| screen-printing (graphite ink) microfluidic sensor | graphite ink | SWV | 120 | 10–100 | 10–100 | 11 | 7 | 0.015 | 0.0025 | (8) |
| on-chip microfluidic channel sensor using Bi electrode | Bi | SWV | Cd: 90 | 28–280 | 25–400 | 9.3 | 8 | 0.065 | 0.0022 | (9) |
| Pb: 60 | ||||||||||
| microfluidic carbon-based sensor (μCS) | none | SWV | Cd: 60 | 5–500 | 5–100 | 1.2 (6)c | 1.8 (6)c | 0.101 | 0.101 | this work |
| Pb: 180 | ||||||||||
CNF: carbon nanofiber; GCE: glassy carbon electrode; SPE: screen-printing electrode; MWCNT: multiwalled carbon nanotube; SWV: square-wave voltammetry; DPV: differential pulse voltammetry.
Sensing sensitivity is expressed as the slope of the calibration plot.
Detection limit in the simultaneous detection of Cd2+ and Pb2+.
Compared to these sensing platforms, where relatively expensive (Pd, Ag, CNF) or other heavy-metal (Bi/Bi2O3) surface modifiers have to be used, in the current work, only inexpensive and environmental friendly materials (cellulose paper, graphite foil) were used to fabricate the μCS devices. The low cost and environmental friendly nature make these μCS devices entirely disposable without causing additional pollution to the environment, thus holding great application prospect in future point-of-care analysis of heavy-metal ions.
Simultaneous Detection of Cd2+ and Pb2+
The performance of the μCS for simultaneous detection of Cd2+ and Pb2+ was also evaluated. The measurements were carried out in 0.1 M acetate buffer solution (pH = 4.6) containing Cd2+ and Pb2+ by first holding the deposition potential at −1.2 V and then square-wave anodic stripping. The reductive deposition time is fixed at 3 min for simultaneous detection of both metals, to ensure that SWASV signal for Pb is strong enough for quantification analysis. As shown in Figure 4a, stripping peaks for Cd and Pb slightly overlap with each other and detection limits for both ions are worse than their corresponding individual detection. A similar peak intensity is obtained for Pb by comparing with the individual Pb detection. However, the peak intensity of Cd decreased in comparison with the individual analysis. This phenomenon may arise from the competitive adsorption of Cd2+ and Pb2+ on the surface of graphite foil, which leads to a reduction in the stripping peak intensity of Cd. Due to the overlap between these two stripping peaks, peak deconvolution was applied by using a Gaussian line shape, in a hope to obtain the “real” current contributed from individual metal ion. We found that calibration plots obtained on the basis of apparent or deconvoluted peak current gave quite comparable slopes (0.11 for Pb and 0.05 for Cd), and only minor differences in interception (<0.1 μA for both metals) can be observed. Therefore, in principle, both apparent and deconvoluted peak currents can be used to quantify the heavy-metal concentration, and for simplicity, we chose to use the apparent peak current later on. The slope changes in calibration plots, shown in Figure 4b, indicate that there are some interactions between Cd2+ and Pb2+ deposition processes. Nevertheless, these two ions could still be simultaneously detected within the linear range of 20–100 μg/L using μCS devices, with the detection limit of 6 μg/L for both metals. It should be noted that the μCS device actually still holds the potential to exhibit further improved sensing sensitivity for simultaneous detection by simply increasing the deposition time, given that recommended deposition time is as long as 10 min for some commercially available screen-printed electrodes for heavy-metal detection.68
Figure 4.

Square-wave voltammograms for simultaneous detection of Cd2+ and Pb2+ in 0.1 M acetate buffer on μCS (a). Linear correction from 20 to 100 μg/L for Cd2+ and Pb2+ (b).
Detection of Cd2+ and Pb2+ in Commercial Mineral Water
To test the sensing performance of these μCS devices under more complex conditions, attempts herein were made to use commercial mineral water, which contains Na+ (109 mg/L), K+ (13.3 mg/L), Mg2+ (28.6 mg/L), Ca2+ (175 mg/L), Cl– (128 mg/L), sulfate (31 mg/L), and bicarbonate (706 mg/L) according to its specification, as a practical sample for the detection of Cd2+ and Pb2+. Prior to the measurement, bottled mineral water was used to prepare acetate buffer solution (0.1 M, pH = 4.6). It can be seen that no stripping peak for either Cd2+ or Pb2+ can be detected for the pristine mineral water sample (Figure S3). However, clear stripping peaks can be seen on the mineral water samples with intentionally added Cd2+ or Pb2+. The peak intensities were increased proportionally by increasing the concentration of both metal ions from 10 to 50 μg/L, and the calculated concentrations using the calibration plots (Figure 3b,d) are consistent with their nominal values (i.e., 10 or 50 μg/L), as listed in Table 2. On the basis of these results, we can draw two conclusions: (1) there is no or little amount (below the limit of detection) of these heavy metals in this commercial mineral water and (2) the presence of various mineral ions in the commercial mineral water imposes little effect on the sensing performance of the μCS, again demonstrating the great prospect of the μCS to be used in practical sample analysis.
Table 2. Determination of Cd2+ and Pb2+ in Commercial Mineral Water by Using the μCS.
Influence of Interfering Ions on Cd2+ and Pb2+ Detection
To test the selectivity behavior of μCS toward Cd2+ and Pb2+ detection, Ba2+, Mn2+, Zn2+, Fe2+, Co2+, Ni2+, Fe3+, and Cu2+ were chosen as interfering ions. The standard reduction potentials of these ions are shown in Table 3. Acetate buffer (pH = 4.6) containing 100 μg/L Cd2+ or Pb2+ together with 5 mg/L interfering ions was tested using the μCS. In principle, ions with higher reduction potential could affect the detection of ions with lower reduction potential. For example, the existence of Co2+, Ni2+, Fe3+, and Cu2+ may have influence on the electroreduction process of Cd2+. To investigate the influence of interfering ions on Cd2+ detection using μCS, the eight interfering ions were divided into four groups: (1) Ba2+, Mn2+, Zn2+, and Fe2+, which have lower reduction potential than Cd2+; (2) Co2+ and Ni2+, which have similar reduction potentials; (3) Fe3+; and (4) Cu2+. Figure 5a shows the influence of interfering ions on Cd2+ detection, which clearly shows that Ba2+, Mn2+, Zn2+, Fe2+, Co2+, Ni2+, and Fe3+ do not have much influence on Cd2+ detection, even though their concentration is 50-fold higher than that of Cd2+. The impact of Cu2+ on Cd2+ detection may be due to its oxidation property. For real applications, it thus becomes necessary to choose the appropriate calibration plot according to the absence/presence of the stripping peak characteristic of the interfering ion as Cu2+. Similar results could be obtained on Pd2+ detection against interfering ions, as shown in Figure 5b.
Table 3. Standard Reduction Potentials of Cd2+, Pb2+, and Interfering Ions69.
| half-reaction | standard reduction potential (V) | |
|---|---|---|
| 0.337 | ||
| –0.04 | ||
| –0.13 | ||
| –0.25 | ||
| –0.28 | ||
| –0.40 | ||
| –0.44 | ||
| –0.76 | ||
| –1.18 | ||
| –2.91 |
Figure 5.

Influence of interfering ions on Cd2+ (a) and Pb2+ (b) detection. The concentration of both Cd2+ and Pb2+ was 100 μg/L. The concentration of each interfering ions was controlled at 5 mg/L.
Influence of 3D Configuration and Paper Channel
To study the influence of the newly introduced 3D sandwich configuration, a sensor with a two-dimensional (2D) configuration was prepared. Similar to the design of some commercial screen-printed electrochemical sensors, three electrodes are parallel to each other and lie in the same horizontal plane, whereas the microfluidic channel is placed across the three electrodes (as illustrated in Figure S4). However, in contrast to the 3D configuration, the 2D one always lead to rather small current signal and can hardly be used to detect any trace amount of heavy metals (Figure S4). The poor performance of the 2D-structured μCS would originate from the small amount of analyte solution being used for the analysis and the relatively long distance between electrodes, which would cause significant resistance between WE and pseudoreference electrode (RE) (solution resistance) and the inefficient electric field between the WE and counter electrode (CE) for metal-ion migration and deposition. This drawback seems to be overcome by the 3D configuration, where the WE and CE face each other and the electrolyte is along the microfluidic channel between these two electrodes.
We also made attempts to study the exact role of the paper channel in the μCS device. To achieve this, a drop of analyte (200 μL) was added to the gap between the working electrode and reference electrode. This droplet stays well between these two electrodes because of the large surface tension of water on the graphite foil. However, detection of 100 μg/L Cd2+ over this stagnant analyte design sensor gives weak stripping peak intensity (4 μA), which is much lower than the value obtained in microfluidic configuration (Figure S5). After introducing the paper channel into the sensor, which changes the stagnant analyte into fluidic one, the square-wave voltammetry (SWV) signal increased from 4.0 to 11.5 μA, as shown in Figure S5. Therefore, besides the 3D structure, the paper channel is another key point in the sensor design, which could efficiently facilitate the mass transfer of analyte to detection sites.
Stability and Reproducibility of the μCS
The reusability is a highly desirable feature for a sensing device, which might result in significant cost and waste reduction during the field analysis. For this reason, the electrochemical stability/reusability of the μCS was studied by subjecting a single μCS device to 10 repetitive SWASV measurements for detecting Cd2+ and Pb2+ with concentrations of 50 and 100 μg/L, and the current response is plotted against the cycle number, as shown in Figure 6. Three sensors are studied in each stability test. It can be seen that the anodic stripping current shows negligible drop after 10 times repetitive measurements regardless of the type/concentration of metal ions, demonstrating the great robustness of these μCS devices for heavy-metal detections. Specifically, the signal drops by 6 and 9% for 50 and 100 μg/L Cd detection, respectively. The signal drops by 6 and 13% for 50 and 100 μg/L Pb detection, respectively. After 10 repetitive measurements, the stripping current would gradually decrease by further increasing the cycle numbers, which stems from the depletion of the analyte within the sampling sponge. Considering that for practical application, it is less likely to frequently reuse these portable devices due to their low cost and possible cross-contamination from different samples, and no further attempt was made to increase the cycle number by refilling the sampling sponge. In addition to the superior stability of a single device, the good reproducibility of the μCS has already been reflected by the small relative standard deviation (<5%) over at least four independent measurements (Figure 3). Attempt was also made to investigate the electrochemical sensing capability of a Bi-modified electrode (denoted as Bi-WE, WE modified with a mixture ink of Bi2O3 powder and carbon black). First, we investigated the effect of loading amount of Bi2O3 within the electrocatalysts on the electrochemical sensing performance by varying the loading from 2 to 50 wt % and found that the highest stripping peak current can be obtained at the loading around 25 wt %, which was later used for further investigation. Surprisingly, it was found that the current response differs significantly from one electrode to another and is highly sensitive to the exposure time of the Bi-WE in air (Figure S7). First, it can be seen from Figure 6c that the stripping peak current on fresh Bi-WE is actually ca. 25% higher than that on the modifier-free μCS, demonstrating that in principle Bi-modified electrode could exhibit higher sensitivity for Cd detection. Second, the SWASV signal decreases by 43% relative to its initial value after exposing the Bi-WE to air for only 5 min, and it is further decreased by over 67% when the air exposure time is increased to 30 min. Although the deactivation mechanism of the Bi-WE is still not fully understood, it can be seen that the ill-defined initial status of the Bi-WE would lead to scattered current response in electrochemical sensing measurement. In contrast, the sensing performance is rather consistent over different graphite-foil WEs free from any surface modifiers (Figure 6c). We believe that these results echo the importance of elimination of surface modifier in developing high-performing sensing platform with superior stability and reproducibility.
Figure 6.

Stability measurement under 50 and 100 μg/L concentrations of Cd2+ (a) and Pb2+ (b). Sensing performance of graphite-foil WEs with and without Bi2O3 modification for Cd2+ detection after being exposed to air for a certain time (c).
Structural Analysis of the Graphite Foil
To probe the possible structural change of the graphite foil, Raman spectra were captured on the graphite foils before and after the electrochemical measurements. As shown in Figure 7, three characteristic peaks at 1350, 1580, and 2716 cm can be clearly resolved, which refer to disordered carbon structure (D-band), graphite structure (G-band), and second-order double-resonant scattering in graphite (G′-band), respectively. The identical Raman spectra recorded on these two samples indicate that graphite foil is rather robust during the electrochemistry measurement. Specifically, the intensity ratio of D-band to G-band (ID/IR), which is usually taken as an indicator of the graphitization degree for a carbon sample, is also comparable (0.04) on the graphite foils with and without experiencing the electrochemical measurements. These results confirm that the graphite foil has a high graphitization degree and is chemically stable during the electrochemistry measurement, which could rationalize the aforementioned well-reproducible performance of the μCS device in the repetitive heavy-metal sensing.
Figure 7.
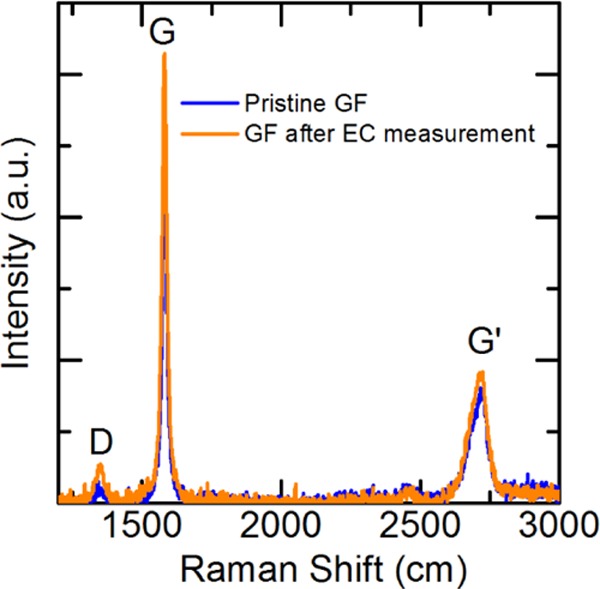
Raman spectrum for graphite foil before and after electrochemical measurements.
We have demonstrated that the modifier-free graphite foil is capable of detecting heavy metals with high sensitivity, low detection limit, and wide linear calibration region. However, it is still not well understood why the inherently less active pristine carbon, that is, graphite foil, could provide sufficient sites for the preconcentration of metal ions. To explore the nature of active sites for electrodeposition of metal ions on the graphite foil, we conducted scanning electron microscopy (SEM) analysis on the graphite foils before and after electrodeposition of Cd2+. As shown in Figure 8a, first, it can be seen that the surface of graphite foils consists of interconnected flakes. After electrodeposition of Cd2+, sphere-like particles (Cd or CdO) can be clearly seen on the surface of graphite flakes (Figure 8b). Interestingly, it can be found that the majority of the particles are selectively located at the edge position of the graphite flakes, as illustrated by the marked squares in Figure 8b. These results demonstrate that the edge surface positions of graphite flakes, where the carbon atoms have lower coordination number, could be more active to catalyze the electrodeposition of metal ions and provide the basis for graphite foils to be directly used as WE for heavy-metal detection. This phenomenon is actually not surprising, considering that it has been well documented that electrochemical reaction at graphite edge sites is preferential as the kinetics of bond formation at basal plane sites is much slower than that at edge/defect sites.70 Nevertheless, as shown previously, the properly designed sensor configuration is also a necessity for achieving high sensing performance of these μCS devices.
Figure 8.

Representative SEM images of pristine graphite foil (a) and graphite foil after electroreductive deposition of Cd2+ (b). The square areas which are magnified in (b-1), (b-2), (b-3), and (b-4) indicate that the electrodeposited Cd species are selectively located at the edge positions of graphite flakes.
Conclusions
A heavy-metal electrochemical sensor assembled using graphite foil and paper was developed, which is capable of detecting heavy-metal ions (Cd2+ and Pb2+) with wide linear range and low LODs. The sensor is modifier free, less expensive, reusable, and easy to fabricate, which would benefit from the combined microfluidic configuration and novel 3D electrode layout. We also disclose that the graphite foil comprising graphite flakes with abundant edge sites could favor the metal deposition, thus providing a simple block material to be used in electrochemical sensors. This easy-handling method could provide some new ideas for other portable electroanalytical/sensing systems.
Experimental Section
Materials and Chemicals
Cadmium chloride (Sigma-Aldrich), lead nitrate (Sigma-Aldrich), sodium acetate (Sigma-Aldrich), acetate acid glacial (VWR), and ethanol (VWR) were all analytical-grade regents and used as received. Graphite foil was purchased from Sigraflex. Filter paper (pore size: 12–15 μm; thickness: 305 μm) was purchased from VWR. All of the solutions were prepared using double-distilled water.
Device Fabrication
Three-electrode configuration was employed to construct the sensing devices. A piece of graphite foil with thickness of 0.5 mm was first cleaned by ethanol and deionized water under ultrasonication (300 W), followed by being cut into certain shapes as working electrode (WE), counter electrode (CE), and pseudoreference electrode (RE). Cleaning protocols for all of the other components, that is, poly(methyl methacrylate) (PMMA) substrate and sampling sponge, were also critically applied using ethanol and deionized water under ultrasonication before fabricating each μCS device to prevent any contaminations. The design and dimensions are detailed in Figure S1. A piece of microfluidic paper was sandwiched between the CE and WE, whereas the RE will be placed adjacent to the RE to reduce the ohmic resistance. To complete the device, the electrodes were taped onto a PMMA substrate for easy operation, as shown in Figure 1. A piece of sponge, placed on one end of the paper channel, was used as analyte reservoir, which contains 2 mL (initial volume) of analyte solution and can constantly feed the analyte solution in 15 min. During the measurement, a glass vial was used to cover the sampling sponge to prevent the solvent evaporation or concentration change of an analyte. An absorbent pad cut in pieces is located at the other end of the paper channel. The function of the absorbent pad is to wick the fluid through the paper channel, create a quasi-steady flow of analyte within the paper channel, and accumulate the amount of analyte flowing through the detection sites. For preparation of the Bi-modified WE, the catalyst suspension ink was first prepared by dispersing 5 mg of Bi2O3 and 15 mg of carbon black (Vulcan XC-72) together with 0.5 mL of 20 wt % Nafion in 1.5 mL of ethanol solution. The modified electrode was then prepared by applying 5 μL of the above ink solution onto graphite foils, covering an area of 0.5 × 0.5 mm2, which is comparable to the effective surface area of WE in a modifier-free μCS device (as detailed in the Supporting Information). The function of carbon black is to better disperse the Bi species and also improve the electrical conductivity within the catalyst layer.
Electrochemical Measurements
Electrochemical measurements were carried out on PARSTAT Multichannel-1000 (AMETEK) controlled by the VersaStudio software. The SWASV measurements were conducted at room temperature, and no deaeration was performed to the supporting electrolyte (0.1 M acetate buffer solution). Heavy-metal concentrations were evaluated using square-wave anodic stripping voltammetry (SWASV). The SWASV experiments comprise an electrochemical deposition step and a square-wave anodic stripping voltammetry scan. The SWASV was performed under the following conditions: pulse height, 25 mV; step height, 10 mV; frequency, 25 Hz. The calibration plots were generated by conducting four independent measurements using four μCS devices. On each device, the measurements were conducted with one standard solution after the other, starting from the lowest to highest concentration. In total, four sets of data were obtained, which are later used to generate the error bars (defined as the relative standard deviation) for each concentration.
Structural Analysis
Raman spectra were recorded with a Bruker Senterra Raman microscope spectrometer with 532 nm excitation (laser power: 0.5 mW) and a 3 cm–1 resolution and were captured from three different points for each sample. The morphology of graphite foils was investigated using a scanning electron microscope (Philips XL30 FEG) operated at an accelerating voltage of 30 kV.
Acknowledgments
The authors thank Dominik Ohlig and Hendryk Steldinger for their kind help in the Raman and SEM analyses. L.-L.S. acknowledges the financial support from China Scholarship Council (CSC, No. 201506210077). This work was supported by the ERC Consolidator Grant IL-E-CAT (No. 681719).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00611.
Additional information about determination of detection limits and about 2D sensor layout, optimization of SWASV parameters, limit of detection linearity, sensing performance of μCS in commercial mineral water, sensing performance of Bi-modified electrode and WE with varied surface area, and stability of graphite foils (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Llewellyn T. O.Cadmium (Materials Flow); Department of the Interior, Bureau of Mines: Washington, DC, 1994. [Google Scholar]
- Gidlow D. A. Lead toxicity. Occup. Med. 2004, 54, 76–81. 10.1093/occmed/kqh019. [DOI] [PubMed] [Google Scholar]
- Mejáre M.; Bülow L. Metal-binding proteins and peptides in bioremediation and phytoremediation of heavy metals. Trends Biotechnol. 2001, 19, 67–73. 10.1016/S0167-7799(00)01534-1. [DOI] [PubMed] [Google Scholar]
- Lodeiro P.; Barriada J.; Herrero R.; De Vicente M. S. The marine macroalga Cystoseira baccata as biosorbent for cadmium (II) and lead (II) removal: kinetic and equilibrium studies. Environ. Pollut. 2006, 142, 264–273. 10.1016/j.envpol.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Noh M. F. M.; Tothill I. E. Development and characterisation of disposable gold electrodes, and their use for lead (II) analysis. Anal. Bioanal. Chem. 2006, 386, 2095–2106. 10.1007/s00216-006-0904-5. [DOI] [PubMed] [Google Scholar]
- Apostoli P. Elements in environmental and occupational medicine. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2002, 778, 63–97. 10.1016/S0378-4347(01)00442-X. [DOI] [PubMed] [Google Scholar]
- Kachoosangi R. T.; Banks C. E.; Ji X.; Compton R. G. Electroanalytical determination of cadmium (II) and lead (II) using an in-situ bismuth film modified edge plane pyrolytic graphite electrode. Anal. Sci. 2007, 23, 283–289. 10.2116/analsci.23.283. [DOI] [PubMed] [Google Scholar]
- Medina-Sánchez M.; Cadevall M.; Ros J.; Merkoçi A. Eco-friendly electrochemical lab-on-paper for heavy metal detection. Anal. Bioanal. Chem. 2015, 407, 8445–8449. 10.1007/s00216-015-9022-6. [DOI] [PubMed] [Google Scholar]
- Zou Z.; Jang A.; MacKnight E.; Wu P.-M.; Do J.; Bishop P. L.; Ahn C. H. Environmentally friendly disposable sensors with microfabricated on-chip planar bismuth electrode for in situ heavy metal ions measurement. Sens. Actuators, B 2008, 134, 18–24. 10.1016/j.snb.2008.04.005. [DOI] [Google Scholar]
- Lo W.; Chua H.; Lam K.-H.; Bi S.-P. A comparative investigation on the biosorption of lead by filamentous fungal biomass. Chemosphere 1999, 39, 2723–2736. 10.1016/S0045-6535(99)00206-4. [DOI] [PubMed] [Google Scholar]
- Bostwick D. G.; Burke H. B.; Djakiew D.; Euling S.; Ho S.-m.; Landolph J.; Morrison H.; Sonawane B.; Shifflett T.; Waters D. J.; Timms B. Human prostate cancer risk factors. Cancer 2004, 101, 2371–2490. 10.1002/cncr.20408. [DOI] [PubMed] [Google Scholar]
- Silva E. L.; dos Santos Roldan P. Simultaneous flow injection preconcentration of lead and cadmium using cloud point extraction and determination by atomic absorption spectrometry. J. Hazard. Mater. 2009, 161, 142–147. 10.1016/j.jhazmat.2008.03.100. [DOI] [PubMed] [Google Scholar]
- Li J.; Guo S.; Zhai Y.; Wang E. High-sensitivity determination of lead and cadmium based on the Nafion-graphene composite film. Anal. Chim. Acta 2009, 649, 196–201. 10.1016/j.aca.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Wanekaya A. K. Applications of nanoscale carbon-based materials in heavy metal sensing and detection. Analyst 2011, 136, 4383–4391. 10.1039/c1an15574a. [DOI] [PubMed] [Google Scholar]
- Aragay G.; Pons J.; Merkoçi A. Recent trends in macro-, micro-, and nanomaterial-based tools and strategies for heavy-metal detection. Chem. Rev. 2011, 111, 3433–3458. 10.1021/cr100383r. [DOI] [PubMed] [Google Scholar]
- Nie Z.; Nijhuis C. A.; Gong J.; Chen X.; Kumachev A.; Martinez A. W.; Narovlyansky M.; Whitesides G. M. Electrochemical sensing in paper-based microfluidic devices. Lab Chip 2010, 10, 477–483. 10.1039/B917150A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J. W.; DeMars R. D.; Shain I. Analytical Applications of Hanging Mercury Drop Electrode. Anal. Chem. 1956, 28, 1768–1771. 10.1021/ac60119a039. [DOI] [Google Scholar]
- Florence T. Anodic stripping voltammetry with a glassy carbon electrode mercury-plated in situ. J. Electroanal. Chem. Interfacial Electrochem. 1970, 27, 273–281. 10.1016/S0022-0728(70)80189-9. [DOI] [Google Scholar]
- Wu H. P. Dynamics and performance of fast linear scan anodic stripping voltammetry of Cd, Pb, and Cu using in situ-generated ultrathin mercury films. Anal. Chem. 1996, 68, 1639–1645. 10.1021/ac950879e. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Wang T.; Han D.; Rusinek C.; Steckl A. J.; Heineman W. R. Electrospun Carbon Nanofiber Modified Electrodes for Stripping Voltammetry. Anal. Chem. 2015, 87, 9315–9321. 10.1021/acs.analchem.5b02017. [DOI] [PubMed] [Google Scholar]
- Hočevar S. B.; Ogorevc B.; Wang J.; Pihlar B. A study on operational parameters for advanced use of bismuth film electrode in anodic stripping voltammetry. Electroanalysis 2002, 14, 1707–1712. 10.1002/elan.200290014. [DOI] [Google Scholar]
- Kadara R. O.; Tothill I. E. Stripping chronopotentiometric measurements of lead (II) and cadmium (II) in soils extracts and wastewaters using a bismuth film screen-printed electrode assembly. Anal. Bioanal. Chem. 2004, 378, 770–775. 10.1007/s00216-003-2351-x. [DOI] [PubMed] [Google Scholar]
- Baldrianova L.; Svancara I.; Sotiropoulos S. Anodic stripping voltammetry at a new type of disposable bismuth-plated carbon paste mini-electrodes. Anal. Chim. Acta 2007, 599, 249–255. 10.1016/j.aca.2007.07.067. [DOI] [PubMed] [Google Scholar]
- Kokkinos C.; Economou A.; Raptis I.; Efstathiou C. E.; Speliotis T. Novel disposable bismuth-sputtered electrodes for the determination of trace metals by stripping voltammetry. Electrochem. Commun. 2007, 9, 2795–2800. 10.1016/j.elecom.2007.09.022. [DOI] [Google Scholar]
- Švancara I.; Baldrianová L.; Tesařová E.; Hočevar S. B.; Elsuccary S. A.; Economou A.; Sotiropoulos S.; Ogorevc B.; Vytřas K. Recent advances in anodic stripping voltammetry with bismuth-modified carbon paste electrodes. Electroanalysis 2006, 18, 177–185. 10.1002/elan.200503391. [DOI] [Google Scholar]
- Economou A. Bismuth-film electrodes: recent developments and potentialities for electroanalysis. TrAC, Trends Anal. Chem. 2005, 24, 334–340. 10.1016/j.trac.2004.11.006. [DOI] [Google Scholar]
- Wang J. Stripping analysis at bismuth electrodes: a review. Electroanalysis 2005, 17, 1341–1346. 10.1002/elan.200403270. [DOI] [Google Scholar]
- Wang J.; Lu J.; Hocevar S. B.; Farias P. A.; Ogorevc B. Bismuth-coated carbon electrodes for anodic stripping voltammetry. Anal. Chem. 2000, 72, 3218–3222. 10.1021/ac000108x. [DOI] [PubMed] [Google Scholar]
- Wang J.; Lu J.; Kirgöz Ü. A.; Hocevar S. B.; Ogorevc B. Insights into the anodic stripping voltammetric behavior of bismuth film electrodes. Anal. Chim. Acta 2001, 434, 29–34. 10.1016/S0003-2670(01)00818-2. [DOI] [Google Scholar]
- Kefala G.; Economou A.; Voulgaropoulos A.; Sofoniou M. A study of bismuth-film electrodes for the detection of trace metals by anodic stripping voltammetry and their application to the determination of Pb and Zn in tapwater and human hair. Talanta 2003, 61, 603–610. 10.1016/S0039-9140(03)00350-3. [DOI] [PubMed] [Google Scholar]
- Liu G.; Lin Y.; Tu Y.; Ren Z. Ultrasensitive voltammetric detection of trace heavy metal ions using carbon nanotube nanoelectrode array. Analyst 2005, 130, 1098–1101. 10.1039/b419447k. [DOI] [PubMed] [Google Scholar]
- Hwang G. H.; Han W. K.; Park J. S.; Kang S. G. Determination of trace metals by anodic stripping voltammetry using a bismuth-modified carbon nanotube electrode. Talanta 2008, 76, 301–308. 10.1016/j.talanta.2008.02.039. [DOI] [PubMed] [Google Scholar]
- Xu H.; Zeng L.; Xing S.; Xian Y.; Shi G.; Jin L. Ultrasensitive voltammetric detection of trace lead (II) and cadmium (II) using MWCNTs-nafion/bismuth composite electrodes. Electroanalysis 2008, 20, 2655–2662. 10.1002/elan.200804367. [DOI] [Google Scholar]
- Kırgöz Ü. A.; Marín S.; Pumera M.; Merkoçi A.; Alegret S. Stripping voltammetry with bismuth modified graphite-epoxy composite electrodes. Electroanalysis 2005, 17, 881–886. 10.1002/elan.200403167. [DOI] [Google Scholar]
- Aragay G.; Pons J.; Merkoçi A. Enhanced electrochemical detection of heavy metals at heated graphite nanoparticle-based screen-printed electrodes. J. Mater. Chem. 2011, 21, 4326–4331. 10.1039/c0jm03751f. [DOI] [Google Scholar]
- Nasraoui R.; Floner D.; Geneste F. Analytical performances of a flow electrochemical sensor for preconcentration and stripping voltammetry of metal ions. J. Electroanal. Chem. 2009, 629, 30–34. 10.1016/j.jelechem.2009.01.024. [DOI] [Google Scholar]
- Li J.; Guo S.; Zhai Y.; Wang E. Nafion–graphene nanocomposite film as enhanced sensing platform for ultrasensitive determination of cadmium. Electrochem. Commun. 2009, 11, 1085–1088. 10.1016/j.elecom.2009.03.025. [DOI] [Google Scholar]
- Teng Y.; Chen T.; Xu F.; Zhao W.; Liu W. Screen-printed Carbon Electrode Modified with Commercial Multilayer Graphene for Lead Detection in Soybean Sauces by Differential Pulse Stripping Voltammetry. Int. J. Electrochem. Sci. 2016, 11, 1907–1917. [Google Scholar]
- Wang J.; Lu J.; Hocevar S. B.; Ogorevc B. Bismuth-coated screen-printed electrodes for stripping voltammetric measurements of trace lead. Electroanalysis 2001, 13, 13–16. . [DOI] [Google Scholar]
- Królicka A. Bismuth-film-plated carbon paste electrodes. Electrochem. Commun. 2002, 4, 193–196. 10.1016/S1388-2481(01)00301-0. [DOI] [Google Scholar]
- Pauliukaitė R.; Hočevar S. B.; Ogorevc B.; Wang J. Characterization and applications of a bismuth bulk electrode. Electroanalysis 2004, 16, 719–723. 10.1002/elan.200302783. [DOI] [Google Scholar]
- Bučková M.; Gründler P.; Flechsig G. U. Adsorptive stripping voltammetric detection of daunomycin at a bismuth bulk electrode. Electroanalysis 2005, 17, 440–444. 10.1002/elan.200403178. [DOI] [Google Scholar]
- Veerakumar P.; Veeramani V.; Chen S.-M.; Madhu R.; Liu S.-B. Palladium Nanoparticle Incorporated Porous Activated Carbon: Electrochemical Detection of Toxic Metal Ions. ACS Appl. Mater. Interfaces 2016, 8, 1319–1326. 10.1021/acsami.5b10050. [DOI] [PubMed] [Google Scholar]
- Prakash S.; Shahi V. K. Improved sensitive detection of Pb2+ and Cd2+ in water samples at electrodeposited silver nanonuts on a glassy carbon electrode. Anal. Methods 2011, 3, 2134–2139. 10.1039/c1ay05265a. [DOI] [Google Scholar]
- Shen L.; Chen Z.; Li Y.; He S.; Xie S.; Xu X.; Liang Z.; Meng X.; Li Q.; Zhu Z.; Li M.; Chris Le X.; Shao Y. Electrochemical DNAzyme sensor for lead based on amplification of DNA–Au Bio-Bar codes. Anal. Chem. 2008, 80, 6323–6328. 10.1021/ac800601y. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Rowe A. A.; Plaxco K. W. Electrochemical detection of parts-per-billion lead via an electrode-bound DNAzyme assembly. J. Am. Chem. Soc. 2007, 129, 262–263. 10.1021/ja067278x. [DOI] [PubMed] [Google Scholar]
- Pathirathna P.; Yang Y.; Forzley K.; McElmurry S. P.; Hashemi P. Fast-scan deposition-stripping voltammetry at carbon-fiber microelectrodes: real-time, subsecond, mercury free measurements of copper. Anal. Chem. 2012, 84, 6298–6302. 10.1021/ac301358r. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Pathirathna P.; Siriwardhane T.; McElmurry S. P.; Hashemi P. Real-time subsecond voltammetric analysis of Pb in aqueous environmental samples. Anal. Chem. 2013, 85, 7535–7541. 10.1021/ac401539f. [DOI] [PubMed] [Google Scholar]
- Winkelmann C.; Lang W. Influence of the electrode distance and metal ion concentration on the resulting structure in electrochemical micromachining with structured counter electrodes. Int. J. Mach. Tools Manuf. 2013, 72, 25–31. 10.1016/j.ijmachtools.2013.05.004. [DOI] [Google Scholar]
- Xuan X.; Hossain M. F.; Park J. Y. A Fully Integrated and Miniaturized Heavy-metal-detection Sensor Based on Micro-patterned Reduced Graphene Oxide. Sci. Rep. 2016, 6, 33125 10.1038/srep33125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barón-Jaimez J. A.; Marulanda-Arévalo J. L.; Barba-Ortega J. J. Electrodes friendly with the environment for detect heavy metal. DYNA 2014, 81, 122–128. 10.15446/dyna.v81n187.40758. [DOI] [Google Scholar]
- Xiong S.; Yang B.; Cai D.; Qiu G.; Wu Z. Individual and Simultaneous Stripping Voltammetric and Mutual Interference Analysis of Cd2+, Pb2+ and Hg2+ with Reduced Graphene Oxide-Fe3O4 Nanocomposites. Electrochim. Acta 2015, 185, 52–61. 10.1016/j.electacta.2015.10.114. [DOI] [Google Scholar]
- Zhao G.; Wang H.; Liu G. Electrochemical Determination of Trace Cadmium in Soil by a Bismuth Film/Graphene-beta-cyclodextrin-Nafion Composite Modified Electrode. Int. J. Electrochem. Sci. 2016, 11, 1840–1851. [Google Scholar]
- Mendez S.; Fenton E. M.; Gallegos G. R.; Petsev D. N.; Sibbett S. S.; Stone H. A.; Zhang Y.; López G. P. Imbibition in porous membranes of complex shape: quasi-stationary flow in thin rectangular segments. Langmuir 2010, 26, 1380–1385. 10.1021/la902470b. [DOI] [PubMed] [Google Scholar]
- Table of Regulated Drinking Water Contaminants. https://www.epa.gov/ground-water-and-drinking-water/table-regulated-drinking-water-contaminants (accessed Feb 28, 2017).
- World Health Organization. Guidelines for Drinking Water Quality, 4th ed.; World Health Organization: Geneva, 2011; pp 327–383. [Google Scholar]
- Pickering W. Inorganic adsorption paper chromatography: III. The adsorption of divalent metal ions by filter paper. J. Chromatogr. A 1960, 4, 481–484. 10.1016/S0021-9673(01)98437-7. [DOI] [Google Scholar]
- Robinson J. E.; Heineman W. R.; Sagle L. B.; Meyyappan M.; Koehne J. E. Carbon nanofiber electrode array for the detection of lead. Electrochem. Commun. 2016, 73, 89–93. 10.1016/j.elecom.2016.11.002. [DOI] [Google Scholar]
- Gich M.; Fernández-Sánchez C.; Cotet L. C.; Niu P.; Roig A. Facile synthesis of porous bismuth–carbon nanocomposites for the sensitive detection of heavy metals. J. Mater. Chem. A 2013, 1, 11410–11418. 10.1039/c3ta12190a. [DOI] [Google Scholar]
- Armstrong K. C.; Tatum C. E.; Dansby-Sparks R. N.; Chambers J. Q.; Xue Z.-L. Individual and simultaneous determination of lead, cadmium, and zinc by anodic stripping voltammetry at a bismuth bulk electrode. Talanta 2010, 82, 675–680. 10.1016/j.talanta.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezi N.; Economou A.; Dimovasilis P. A.; Trikalitis P. N.; Prodromidis M. I. Disposable screen-printed sensors modified with bismuth precursor compounds for the rapid voltammetric screening of trace Pb (II) and Cd (II). Anal. Chim. Acta 2012, 728, 1–8. 10.1016/j.aca.2012.03.036. [DOI] [PubMed] [Google Scholar]
- Zaouak O.; Authier L.; Cugnet C.; Castetbon A.; Potin-Gautier M. Bismuth-Coated Screen-Printed Microband Electrodes for On-Field Labile Cadmium Determination. Electroanalysis 2009, 21, 689–695. 10.1002/elan.200804465. [DOI] [Google Scholar]
- Niu P.; Fernández-Sánchez C.; Gich M.; Navarro-Hernández C.; Fanjul-Bolado P.; Roig A. Screen-printed electrodes made of a bismuth nanoparticle porous carbon nanocomposite applied to the determination of heavy metal ions. Microchim. Acta 2016, 183, 617–623. 10.1007/s00604-015-1684-4. [DOI] [Google Scholar]
- Kadara R. O.; Jenkinson N.; Banks C. E. Disposable bismuth oxide screen printed electrodes for the high throughput screening of heavy metals. Electroanalysis 2009, 21, 2410–2414. 10.1002/elan.200900266. [DOI] [Google Scholar]
- Jung W.; Jang A.; Bishop P. L.; Ahn C. H. A polymer lab chip sensor with microfabricated planar silver electrode for continuous and on-site heavy metal measurement. Sens. Actuators, B 2011, 155, 145–153. 10.1016/j.snb.2010.11.039. [DOI] [Google Scholar]
- Rattanarat P.; Dungchai W.; Cate D.; Volckens J.; Chailapakul O.; Henry C. S. Multilayer paper-based device for colorimetric and electrochemical quantification of metals. Anal. Chem. 2014, 86, 3555–3562. 10.1021/ac5000224. [DOI] [PubMed] [Google Scholar]
- Hwang G.-H.; Han W.-K.; Park J.-S.; Kang S.-G. An electrochemical sensor based on the reduction of screen-printed bismuth oxide for the determination of trace lead and cadmium. Sens. Actuators, B 2008, 135, 309–316. 10.1016/j.snb.2008.08.039. [DOI] [Google Scholar]
- Bismuth Oxide Carbon Screen-Printed Carbon Electrode; Parque Tecnológico de Asturias: Spain. http://dropsens.com/en/pdfs_productos/new_brochures/110bi.pdf (accessed Feb 28, 2017). [Google Scholar]
- Haynes W. M.CRC Handbook of Chemistry and Physics, 95th ed.; CRC Press, 2014, pp 80–89. [Google Scholar]
- Banks C. E.; Davies T. J.; Wildgoose G. G.; Compton R. G. Electrocatalysis at graphite and carbon nanotube modified electrodes: edge-plane sites and tube ends are the reactive sites. Chem. Commun. 2005, 829–841. 10.1039/b413177k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.