

Abstract
Computational screening is a method to prioritize small-molecule compounds based on the structural and biochemical attributes built from ligand and target information. Previously, we have developed a scalable virtual screening workflow to identify novel multitarget kinase/bromodomain inhibitors. In the current study, we identified several novel N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide derivatives that scored highly in our ensemble docking protocol. We quantified the binding affinity of these compounds for BRD4(BD1) biochemically and generated cocrystal structures, which were deposited in the Protein Data Bank. As the docking poses obtained in the virtual screening pipeline did not align with the experimental cocrystal structures, we evaluated the predictions of their precise binding modes by performing molecular dynamics (MD) simulations. The MD simulations closely reproduced the experimentally observed protein–ligand cocrystal binding conformations and interactions for all compounds. These results suggest a computational workflow to generate experimental-quality protein–ligand binding models, overcoming limitations of docking results due to receptor flexibility and incomplete sampling, as a useful starting point for the structure-based lead optimization of novel BRD4(BD1) inhibitors.
Introduction
Epigenetics refers to changes in gene expression that are not due to DNA sequence modifications.1 Epigenetic modifications occur due to the coordinated activities of three classes of epigenetic proteins, namely, writers, readers, and erasers.2 Epigenetic readers recognize specific modifications, such as histone acetylation and nucleotide methylation, which promote nucleosome remodeling. Importantly, regulatory posttranslational modifications of proteins such as lysine acetylation have emerged as essential marks for regulating transcription, metabolism, and cell signaling.3 Bromodomain proteins are a family of epigenetic reader domain proteins that bind acetylated lysine residues, particularly in the N-terminal tails of histones, facilitating decondensation of chromatin and regulation of gene expression. Of the 61 bromodomain modules found in 42 diverse proteins of the human proteome, bromodomain and extra-terminal (BET) proteins include family members BRD2, BRD3, BRD4, and BRDT.4 These proteins contain two bromodomains termed bromodomain 1 (BD1) and bromodomain 2 (BD2) and exhibit a high level of sequence conservation in the amino terminal; however, more divergence has been characterized in the carboxy terminal of this protein family.5 Bromodomain-containing protein 4 (BRD4) has been linked to the expression of oncogenes such as MYC and is a promising target for cancer therapy.6 Specifically, researchers observe BRD4 overexpression in various cancers, including glioblastoma and squamous cell carcinoma, among others.7 Recently, the discovery and development of BET protein inhibitors allow for the treatment of cancer cells in vitro and in vivo, with many studies demonstrating promising results.
Additionally, the structural topology of BRD4 consists of four α helices (αz, αA, αB, and αC) linked by ZA and BC loops that have been shown to be responsible for substrate specificity.8 Previous structure determination of BRD4 with acetylated histone peptides demonstrates acetyl-lysine binding in the central hydrophobic cavity, where a hydrogen bond forms with N140. This conserved binding motif was also observed in previously characterized BRD4 inhibitors.8
Recent advances in computational methods and power facilitate high-throughput approximation of binding free energies and dynamics with increasing accuracy.9 The classical “lock-and-key” theory of protein–ligand binding, in which a rigid receptor is thought to accommodate a small molecule, has given way to dynamic molecular models that aim to incorporate induced changes of small-molecule ligand and receptor conformation upon binding. Accordingly, such models can lead to better results and more accurate predictions.10,11 Structure-based virtual screening methods, such as docking, typically search for energetically favorable conformations and atomic interactions of a small-molecule binding to a protein pocket and often use empirical scoring functions to estimate binding free energy to various degrees of accuracy but minimally to enrich actives out of a pool of mostly inactive compounds. In contrast to ligand-based models, which do not use a protein structure and rely on correlations between chemical topology and binding affinity, structure-based models can predict the atomic interactions through which a new small molecule binds to a protein of interest and can estimate how energetically favorable that interaction is. If correct, these models are useful for the development and optimization of lead compounds. Although structure-based methods can be highly predictive, for flexible proteins it can be challenging to find the best “induced-fit” binding pose and accurately predict (or rank order) their binding affinity. Molecular dynamics (MD) simulation can help solve that problem, and advances in computer hardware and user-friendly software have enabled much higher throughput simulation of multicomponent dynamic molecular systems, including those with protein and ligand components.12−14 Several examples have demonstrated that MD can improve structure-based virtual screening by predicting stable low-energy ligand–receptor binding conformations.15−19 Although computationally intensive, this approach has the potential to produce an accurate binding model even if no cocrystal structure is available, which is often a limiting factor with many druggable targets.20 Furthermore, alchemical free energy calculations have recently been shown to provide robust and accurate protein–ligand binding energies. However, these are among the most computationally intensive methods and require extensive sampling to converge accurately.21−24 In this study, we were primarily interested in accurate pose prediction and activity ranking and therefore did not computationally investigate more expensive free-energy methods.
Here, we report the identification of six novel N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide derivatives that scored high in an ensemble docking virtual screening workflow. We determined their BRD4(BD1) binding affinity biochemically and resolved the cocrystal structures for these compounds, which were deposited in the Protein Data Bank (PDB).25 Because the docking poses obtained in the virtual screening pipeline did not align with the later determined experimental cocrystal structures, we ran MD simulations on the original docking poses and were able to closely reproduce the experimental cocrystal structure binding poses for all compounds. These results suggest that binding conformation and atomic protein–ligand interactions of a novel class of BRD4 inhibitors can be accurately predicted using published cocrystal structures and MD simulations. Docking studies based on the MD-optimized binding models suggest improved performance in terms of enriching known actives and to rank order activity of a novel chemotype, which we extensively characterized. Our results provide a useful starting point for the structure-based lead optimization of BRD4(BD1) inhibitors.
Results
We had previously reported a scalable virtual screening workflow using kinase machine learning classifiers and parallel ensemble docking to identify a novel dual kinase BET inhibitor.26 Briefly, we first prioritized 908 compounds using kinase machine learning models and then docked these compounds into several structures of BRD4(BD1) obtained from the PDB that were selected and processed to represent diverse cocrystal ligands as well as to include and exclude conserved cocrystal water. Here, we report the biochemical AlphaScreen, an AlphaScreen counter assay, and differential scanning fluorimetry (DSF) to confirm the activity of six BRD4 inhibitors identified via the virtual screening pipeline (see Experimental Section). Several of these compounds had IC50 values in the range of 11–44 μM as determined via a BRD4(BD1)-acetylated histone interaction assay (Table 1, Supplementary Table 1). All six compounds are N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamides, a previously unreported druglike scaffold for BRD4. As they were novel compounds, we produced cocrystals and resolved their structures (Figure 1). Analysis of the cocrystal structures displayed some interactions that were predicted by our ensemble docking model (Supplementary Figures 1 and 2). The cocrystal structures are also very useful to interpret the structure–activity relationships (SARs) observed for that series (see Discussion and Conclusions). However, when comparing the best docking poses with the experimentally determined structures, we observed differences in ligand binding conformation with ligand root-mean-square deviation (RMSD) ranging from 1.57 to 2.35 Å (Supplementary Table 2). We were interested in improving the computational pose predictions, and specifically, whether MD simulations would converge to the experimental cocrystal binding pose. Our motivation was to reliably predict the correct binding pose of a new chemotype of BRD4(BD1) in the absence of a cocrystal structure, which is much more time-consuming and expensive to obtain than running MD simulations.
Table 1. Biochemical Activity and Docking Scores of N-[3-(2-Oxo-pyrrolidinyl)phenyl]-benzenesulfonamide Derivatives.

Values expressed as mean ± standard error of the mean of three experiments in triplicate.
Figure 1.

Cocrystal structures of N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide BRD4 binders. Conserved interactions are observed between all compounds and the BRD4 N140 acetyl-lysine binding motif. Interactions with P82, V87, P82, D145, I146, and M149 are also observed across binding interactions with this scaffold.
A 50 ns all-atom explicit water MD simulation was performed for all active N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide derivatives starting from the docking results. Each MD system was created from the docked protein–ligand complex that corresponded to the best docking score (i.e., the BRD4(BD1) PDB structure that resulted in the highest absolute docking score with its corresponding docked ligand). Simulation trajectory analysis showed ligand binding pose stabilization after 20 to 30 ns as quantified by ligand RMSD from starting coordinates. Over that time frame, ligand RMSD relative to the starting (docked) conformation changed by approximately 2 Å and was then stable (Supplementary Table 2). Interactions throughout the trajectories showed conserved interactions with N140, which is a well-described residue necessary for BRD4 inhibitor binding (Figure 2).8 To assess whether the ligand conformation approaches the experimental binding pose, we transformed the default trajectory ligand RMSD relative to frame zero (the starting docked pose) into a representation relative to the cocrystal ligand pose using E1. The definition of RMSD is found in E2. The transformed trajectories are depicted in Figure 3 and illustrate how the docking poses, which are quite different from the experimental cocrystal structure, converge to RMSD within 0.08–0.46 Å, corresponding to the experimentally determined reference pose.
Figure 2.

Binding interactions of compound 8302 with BRD4 throughout the molecular simulation. Primary receptor ligand interactions (hydrogen bonds, hydrophobic, ionic, water bridges) of 8302 for the duration of the MD simulation with BRD4(1). The top panel shows the total number of specific contacts the protein makes with the ligand over the course of the trajectory. The bottom panel shows which residues interact with the ligand over the simulation time. Some residues make more than one specific contact with the ligand, which is represented by a darker shade of orange, according to the scale to the right of the plot. The visualizations were generated using the Desmond simulation interactions diagram component.
Figure 3.

N-[3-(2-Oxo-pyrrolidinyl)phenyl]-benzenesulfonamide ligand RMSD relative to the cocrystal pose over the duration of the MD simulation. Each panel illustrates how the ligand pose RMSD (Å) converged during the MD simulation from the original docking pose (obtained using a published cocrystal structure of a different chemotype) toward the experimentally determined cocrystal structure.
For further illustration, we extracted the final frame from each simulation and overlaid it onto the experimental cocrystal structure. Figure 4 shows that these poses are essentially identical. For the most active compound (8302), we calculated the heavy-atom RMSD of the protein binding site (5 Å around the ligand) before and after the simulations and observed a 1.84 Å change in the binding site geometry during the simulation.
Figure 4.
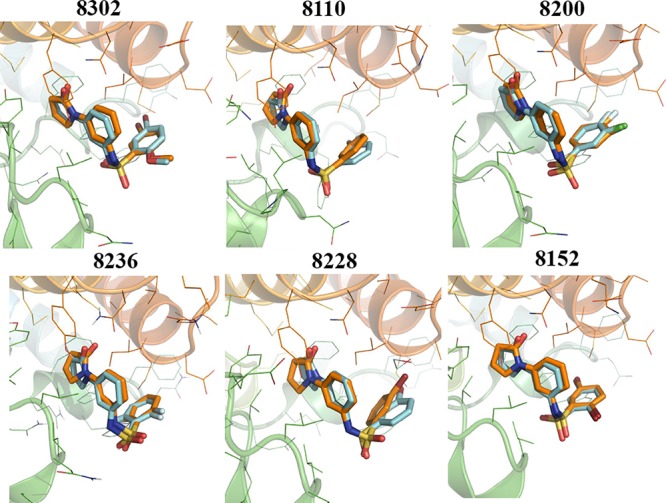
Overlay of ligand poses obtained by docking after MD optimization and experimental cocrystal structure. Bound ligand after MD shown in orange, and cocrystal ligand shown in teal. Ligand RMSD < 0.5 Å in all cases. Ligand poses after MD corresponded much more closely to the experimentally determined conformations compared to the initial docking poses.
To demonstrate that the protein structure and the protein–ligand pose remain stable after the 50 ns MD simulation and that the simulation time of 50 ns is appropriate, we ran a 500 ns simulation of the best compound (8302) in BRD4(BD1) starting with the same docking pose as the initial simulations. Supplementary Figure 10 shows the protein and ligand heavy-atom RMSD of the 8302–BRD4(BD1) complex over the duration of 500 ns and clearly illustrates that both the protein and the ligand RMSD remain stable. The protein RMSD levels out at between 3 and 3.5 Å, which suggests that the binding site adjustment throughout the simulation (RMSD 1.8 Å) is significant.
Our results demonstrate that via MD simulations (starting from the best docking poses) we can reliably reproduce the experimentally observed cocrystal protein–ligand complexes much more accurately than docking alone (Supplementary Table 2). Although not unexpected, this is an important and encouraging result from the perspective of virtual screening, rational drug design, and lead optimization.
We next determined whether the protein–ligand complexes optimized by MD (but not using our experimental cocrystal structures) would improve the ability to identify and rank order active compounds compared to the original models based on the previously published PDB cocrystal structures. We asked whether optimizing ligand docking poses using MD could improve computational predictions without the need to obtain cocrystal structures. Although all-atom explicit solvent MD simulations can be computationally expensive, methods such as minimization with implicit solvent neglect important water contributions to BRD4(BD1) binding and do not sample the protein–ligand interface sufficiently to escape static local minima and reliably predict the correct pose. To test this, we extracted three frames from the MD trajectory (after stabilization of the ligand RMSD) of the three most active ligands (8302, 8110, and 8200) and prepared docking models. Known active and decoy compounds were redocked using the three models, and docking scores were aggregated as before (see Methods). Using the MD-optimized model, docking predictions for the experimentally determined active compounds better reflected their BRD4 activity; the most active compounds received better docking scores. The receiver operating characteristic area under the curve (ROC) score using previously known BRD4 actives was slightly improved, suggesting that the MD-optimized docking models do not result in decreased predictive performance for other BRD4 scaffolds but tend to globally improve predictive performance (Supplementary Figure 4).
Using the optimized models, we then redocked the 908 compounds from which our lead compounds had originally been identified. All N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide derivatives scored consistently better compared to the original models, and they better correlated quantitatively to their experimentally determined BRD4 activity (Figure 5, Supplementary Figure 5). In addition to correlating experimental pIC50 values with docking scores (Supplementary Figure 5a), we also computed molecular mechanics-generalized Born surface area (MM-GBSA) binding free energies and observed the same trend in which the computed estimated binding free energy tracked the experimental activity much better postoptimization, compared to preoptimization (Supplementary Figure 5b).
Figure 5.
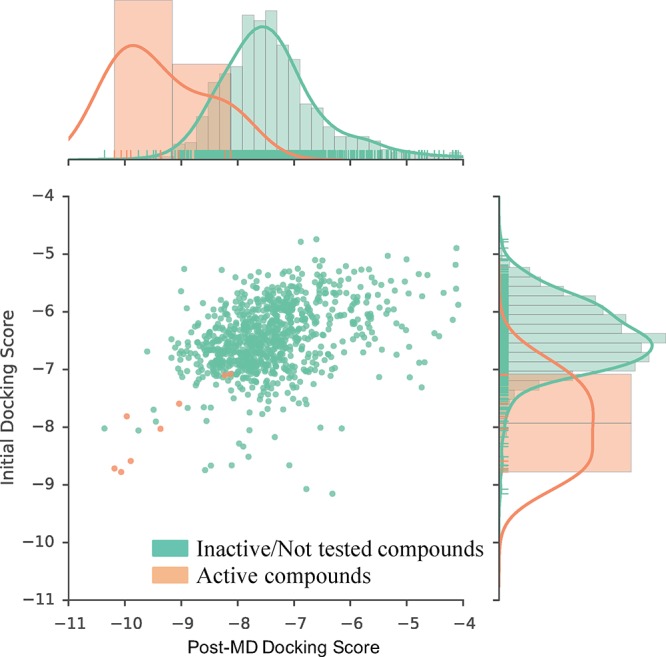
Ensemble docking score distribution of the initial and the MD-optimized docking models. The plot shows the distribution of active compounds (peach) and inactive compounds (teal). The vertical (y) axis and histogram on the right correspond to the distribution of the original docking scores (before MD optimization). The horizontal (x) axis and histogram on the top of the plot correspond to the docking scores after MD optimization. The MD-optimized docking scores show much better separation between active and inactive compounds.
To demonstrate that the MD-optimized docking model improves the enrichment of commercially available compounds determined to be active against BRD4, a log–enrichment curve was calculated. The MD-optimized docking model better ranked active compounds compared to the initial docking model before MD (Supplementary Figure 6). Enrichment factors (EFs) for the top 1% best scoring compounds were 51 and 38 for the MD-optimized and initial docking models, respectively (Supplementary Table 3). The 908 compounds from which the N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide derivatives were identified included four additional sulfonamide compounds that were not prioritized in our original docking study and therefore not ordered or tested. Although the optimized docking scores of these compounds also improved (scores initially ranging from −6.56 to −6.83 and improving to the range −7.09 to −7.54), their optimized score would not have placed them among the top compounds for experimental testing, suggesting that our method does not indiscriminately result in better docking scores for the chemotype it was optimized with but better separates actives from inactives. In addition to better separating the active sulfonamide compounds from the remaining compounds and thus more confidently predicting their activity, the optimized model generally better separated active compounds from their presumed inactive decoys. Compounds that were purchased, but found to be inactive, scored more poorly (Figure 5). Together, these results suggest that the predictive performance of a docking model can be improved upon structural optimization using MD simulations. As expected, this improvement is particularly effective for compounds of the same scaffold as the optimized bound ligand, but an overall improvement of enrichment and predictive performance for known active BRD4 compounds is also evident. Most importantly however, we were able to reproduce the experimental binding pose via MD starting from a docking pose of a receptor of a different cocrystal ligand. This approach therefore appears useful for structure-guided lead optimization without the need for a cocrystal structure.
To investigate the novelty of the sulfonamide compound series, Tanimoto similarities against all known BRD4 compounds using ECFP4 chemical fingerprints were computed (Supplementary Figure 7a). Nine of the most similar (0.3–0.4) reported BRD4 inhibitors are shown in Supplementary Figure 7b. Although this cutoff would not be significant enough to consider them likely active based on the similarity alone, we further investigated the compounds, which included PFI-1 (compound 4 in Supporting Figure 7) and derivatives as well as phenyl isoxazole sulfonamide compounds (compounds 1–3 in Supporting Figure 7) from GSK.27,28 These compounds are structurally distinct from our N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamides, particularly the 2-oxo-pyrrolidinyl moiety that forms the characteristic hydrogen bonding interaction with BRD4 N140. We also computed the 3D shape similarity of our novel sulfonamide BRD4 binders to all known inhibitors using the ROCS package from OpenEye29 (see Methods). There was no single shape or color ROCS similarity score greater than 0.5 and the Tanimoto Combo score, which includes both shape and color scoring, varied between 0.2 and 0.7 for all of the known BRD4 compounds (Supplementary Figure 8a). Tanimoto Combo is a combination of both shape Tanimoto and color Tanimoto scoring functions (the score ranges from 0 to 2), and many perceive it as the best global 3D similarity scoring metric in the ROCS package.29Supplementary Figure 8b shows the top six most shape-similar compounds, including phenyl isoxazole sulfonamide compounds also enriched in the ECFP4 similarity study, and isoxazole azepine compounds, which share similar binding interactions in their amino-isoxazole moiety.28,30
To investigate perceived pharmacophoric similarity of the well-characterized PFI-1 to our compounds, we aligned the cocrystal structure of the most active compound in our series, 8302, with PFI-1. Supplementary Figure 9 illustrates similarity in binding orientation; we determined RMSD to be 1.1 Å. The sulfonamide and the aromatic substituents are well-aligned, and both compounds exhibit the key hydrogen bond interaction with N140 via their 2-oxo-pyrrolidinyl (8302) and tetrahydroquinazoline-2-one moieties. However, this interaction is to be expected and is the characteristic binding motif of most BRD4(BD1) inhibitors. Additionally, 8302 maintains hydrophobic interactions with Y81, W83, and I146, which is distinct from PFI-1 (see Discussion). This characterization suggested that our compounds are pharmacophorically somewhat similar to PFI-1 but topologically and electrostatically distinct.
Discussion and Conclusions
We discovered a new class of BRD4 inhibitors, characterized the compounds biochemically, produced cocrystal structures, and studied the binding dynamics of these novel compounds as a starting point for future optimization.
Our virtual screening protocol used a high-throughput ensemble docking protocol employing various BRD4 protein structures that were prepared to incorporate or exclude conserved cocrystal water. The rationale of our approach was to sample a number of representative and feasible receptor conformations and binding modes (e.g., with or without water bridges). However, despite using an ensemble method, significant deviations of predicted versus experimental cocrystal ligand binding conformations were observed on the order of ∼2 Å heavy-atom RMSD per ligand. Receptor flexibility and specifically induced receptor conformational changes upon small-molecule binding as well as water molecules participating in binding are highly complex processes and challenging to predict accurately, in particular via high-throughput virtual screening such as docking, including ensemble docking with data fusion. Novel chemotypes with no ligand-similar experimental cocrystal structures are therefore especially difficult to model. We identified several novel active compounds, and we extensively characterized these inhibitors biochemically and determined the cocrystal structure of all six compounds. With that information, we explored the binding dynamics of these ligands in the BRD4(BD1) structure with the goal to predict the correct, experimentally determined, cocrystal binding pose starting from previously published cocrystal structures, such that it would not be required to obtain a cocrystal structure for the new chemotype. Secondarily, we were interested to improve the relative binding affinity predictions and enrichment of active compounds. The consideration of binding dynamics is particularly relevant for flexible protein domains and for targets where water frequently participates in binding such as with BET proteins.
Addressing these issues, we demonstrated that we can generate accurate binding models of N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide derivatives in BRD4 using molecular docking followed by 50 ns all-atom explicit water MD simulations. The obtained binding poses were essentially equivalent to experimental cocrystal structures. With the availability of advanced force fields and algorithms implemented in user-friendly software and increased computational power, relatively short (50 ns) MD simulations can now be employed routinely. The time frame of 50 ns appears sufficient to generate a stable ligand receptor conformation almost exactly reproducing the experimentally determined binding pose, if the starting docking conformation can assist in separating active from inactive compounds. This was further supported by a simulation with a 10-fold increased simulation time (500 ns) of the most active compound in which the protein and ligand RMSD remain stable (Supplementary Figure 10). We showed that these optimized models can better prioritize compounds and result in improved enrichment of known actives and in particular the sulfonamide compounds. This improvement is likely due to the increased sampling of ligand and protein states and convergence toward more energetically favorable conformations, which are likely different from the initial crystal structure conformation bound to a different chemotype. RMSD of just the BRD4(BD1) binding site heavy atoms during the simulation illustrated the adjustment of the binding site geometry compared to other chemotypes. Our results are significant in that N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide represents a novel scaffold of BRD4 binders, quantified by 2D structure and 3D shape-based similarity comparisons to previously reported actives.
The first potent inhibitors of BET proteins, (+)-JQ131 and I-BET762,32,33 were developed based on compounds found to increase the apolipoprotein A-1 (ApoA1) activity in anti-inflammatory phenotypic screens. Both compounds are triazolodiazepines and share a common binding mode in the acetyl-lysine binding site of BET proteins. The structure of the BRD4-1 and (+)-JQ1 complex (PDB ID: 3MXF) shows that the triazole ring functions as an acetyl-lysine mimic by occupying the same position as the acetyl group in histone tail peptides. The two adjacent nitrogen atoms of the triazole ring replicate the carbonyl group of acetyl-lysine with one hydrogen bond interaction with the amide side chain of the conserved asparagine residue (N140 in BRD4-1) and another to the hydroxyl side chain of Y97, courtesy of a bridging hydrogen bond through a water molecule of the structurally conserved ZA channel. (+)-JQ1 binding is further stabilized through extensive hydrophobic interactions: the pendant 4-chlorophenyl substituent of the diazepine ring occupies the hydrophobic WPF (tryptophan, proline, and phenylalanine) shelf (W81-P82-F83) present in all BET family members and the dimethyl-substituted thieno ring packs between the same WP motif and L92 of the ZA loop. Notably, the natural bend of the thienodiazepine core arcs (+)-JQ1 is effectively over the WPF shelf and is critical to attaining the near-perfect shape complementarity of this chemotype. Like (+)-JQ1, the pyrrolidinone moiety of our N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide chemotype functions as an acetyl-lysine mimic with the oxygen atom, establishing both a direct hydrogen bond to N140 and a ZA channel-mediated bridging hydrogen bond to Y97. In contrast to (+)-JQ1, these compounds form an additional bridging water-mediated hydrogen bond through the sulfonamide nitrogen atom to the main chain carbonyl of L92. Although our compounds also contain substituted phenyl rings which occupy a similar position to the 4-chlorophenyl substituent of (+)-JQ1, they lack many of the stabilizing hydrophobic interactions afforded to (+)-JQ1 by the larger thienodiazepine-based core, such as π–π stacking and van der Waals interactions between the dimethyl-substituted thieno ring and P82 and L92, respectively. Given the importance of extensive hydrophobic interactions to the positioning of small-molecule inhibitors in the BRD4 acetyl-lysine binding site, the reduced availability of these in our chemotype likely explains their reduced affinity compared to (+)-JQ1 (IC50:BRD4(BD1) = 77 nM).
Following the discovery that triazolodiazepines are potent inhibitors of BET proteins, further research aimed to identify new chemotypes which mimicked the acetyl-lysine moiety using fragment-based screening approaches. Lead optimization of one such hit led to the development of the dihydroquinazolinone PFI-1 (IC50:BRD4(BD1) = 220 nM).34 An analysis of the BRD4(BD1) and PFI-1 complex (PMID: 4E96) reveals that the carbonyl and NH of the cyclic urea function as a hydrogen bond donor/acceptor pair with the side chain of N140. Like the acetyl-lysine carbonyl, the quinaolinone carbonyl of PFI-1 also establishes a ZA channel-mediated bridging hydrogen bond to Y97. Furthermore, PFI-1 forms an additional bridging water-mediated hydrogen bond through the sulfonamide nitrogen atom to the backbone carbonyl of L92 in the ZA loop. Similarly to (+)-JQ1, the 2-methoxyphenyl substituent is directed into the hydrophobic WPF shelf. Superimposition of our cocrystal structures with that of the BRD4-PFI-1 complex reveals a remarkably conserved binding mechanism. The pyrrolidinone and urea carbonyls of our N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamides and PFI-1, respectively, overlap with one another and serve as the principal acetyl-lysine mimicking moieties, establishing direct hydrogen bonds to N140 and water-mediated hydrogen bonds to Y97. The central phenyl ring of our compounds occupies the same space as the dihydroquinazolinone phenyl of PFI-1. Noticeably, both chemotypes contain a sulfonamide linker that introduces a sharp ∼120° turn in their planar core structures, thereby directing the sulfonamide phenyl rings toward the hydrophobic WPF shelf. Likewise, the sulfonamide nitrogen in both chemotypes establishes an additional bridging water-mediated hydrogen bond to the main chain carbonyl of L92.
Another widely explored chemotype is the dimethylisoxazoles, as represented by the prototypical member I-BET151/GSK1210151A (IC50:BRD4(BD1) = 790 nM).35 The BRD4-1 and I-BET-151 cocrystal structures (PDB ID: 3ZYU) demonstrate that the isoxazole moiety serves as an acetyl-lysine mimic binding deep within the pocket, forming a direct hydrogen bond to the side chain of N140 and a water-mediated hydrogen bond to the side chain of Y97 through the isoxazole oxygen and nitrogen atoms, respectively. Additionally, the quinolone nitrogen atom forms a water-mediated bridging hydrogen bond to the main chain carbonyls of P82 and Q85 and to the amide side chain of Q85. Like the thienodiazepine core of (+)-JQ1, the quinolone core of I-BET151 packs between the WP motif and Leu92, and the pyridyl group interacts with the hydrophobic WPF shelf (albeit in a different orientation due to the stereochemistry of the preceding carbon). Comparison of the I-BET151 and N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide binding modes shows that the carbonyl oxygens of the respective isoxazole and pyrrolidinone moieties overlap in agreement with their acetyl-lysine mimicking properties. However, other aspects of their binding are different in line with their highly divergent scaffolds. Apart from both chemotypes directing a phenyl substituent to the WPF shelf, like the preceding discussion of the (+)-JQ1 binding mechanism, our compounds lack many of the hydrophobic and stacking interactions formed by I-BET151, reducing their respective affinities toward BRD4-1.
Finally, an inspection of other common BRD4 inhibitor chemotypes, including the quinazolones (e.g., RVX-208),36 diazobenzenes (e.g., MS436),37 and triazolopyridazines (e.g., bromosporine),38 shows that the prevailing commonality is the presence of an acetyl-lysine mimicking moiety which overlays with the pyrrolidinone of our N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamides. Interestingly, many members of these other chemotypes do not contain a pendant phenyl substituent directed toward the hydrophobic WPF shelf in a manner analogous to that of the 4-chlorophenyl and 2-methoxyphenyl moieties of (+)-JQ1 and PFI-1, respectively, despite retaining modest activity against BRD4-1, indicating that occupying the WPF shelf at this position with an aromatic substituent is not essential for potency. However, it is noteworthy that these compounds do have alternative aromatic and hydrophobic substituents, which can engage in other elaborate π–π stacking and van der Waals interactions with residues at the entrance of the acetyl-lysine binding pocket of BRD4-1, features which are absent from our compounds.
Taken together, the N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide chemotype reported in this work contains a pyrrolidinone moiety as an acetyl-lysine mimic, which establishes a direct hydrogen bond to the conserved N140 in BRD4-1. In this regard, our chemotype provides a novel chemical starting point to the development of structurally diverse BET inhibitors and illustrates the large chemical diversity accommodated by this protein pocket. However, unlike many of the most potent BET inhibitors such as (+)-JQ1 and I-BET151, our compounds lack many of the π–π stacking and hydrophobic interactions necessary for further stabilization inside of the acetyl-lysine binding site, likely explaining the reduced potency of this chemotype.
From the interaction diagrams (Figure 1 and Supplementary Figure 2) of our six cocrystal structures of N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide ligands, it is evident that all six compounds bind with the acetyl-lysine binding motif of bromodomain in a similar manner, presumably explaining the relatively narrow range of the observed IC50 values (11–44 μM) (Table 1). However, in the case of 8302, the presence of an additional H bond acceptor (−OMe group at the ortho position of the terminal phenyl ring, with respect to the sulfonamide functionality) appears to provide improved binding because of better stabilization of the water network. Adding relatively small halogen atoms, for example, F (in cases of 8110, 8200, and 8236) and Cl (8200), to the terminal phenyl ring does not change the binding significantly. However, adding bulky atoms such as Br, especially at the ortho position (8152) of the phenyl ring, seems to weaken the binding because of steric hindrance. Future SAR and optimization to include substituents enabling interactions observed with the most potent inhibitors (as described above) would be predicted to significantly improve potency.
Experimental Section
BRD4 Biochemical Activity Data
The activity data of known BRD4 inhibitors were extracted from the ChEMBL18 database (May 7, 2014) and also curated from the recent literature.27,30,31,39−43 We utilize only compounds with reported concentration response activity data (IC50/Kd/Ki). Chemical structures were standardized including protonation states (neutral form, protonate acids, and deprotonate bases), tautomer standardization, removal of salts/addends, and assigning the correct stereochemical and geometric configuration using Pipeline Pilot 8.0 (Accelrys) (ambiguous stereochemistry or geometric configuration is handled by LigPrep, see below); unique IDs were assigned corresponding to unique structure representations. Activity data were p-transformed (−log10) and aggregated by unique compound ID using the median activity p-value. We provide aggregate and standardized activity data of compounds tested in the Library of Integrated Network-based Cellular Signatures (LINCS) program (http://www.lincsproject.org/), which is available via the LINCS data portal (http://lincsportal.ccs.miami.edu/).
To estimate enrichment and determine thresholds for docking score significance, the 246 unique BRD4 inhibitors from ChEMBL were submitted to generate decoy sets from the directory of useful decoys, enhanced (DUD-E).44 DUD-E generated 50–100 decoy molecules per submitted ligand SMILES. The protocol for preprocessing the 246 original ligands and the corresponding 15 250 decoy compounds (15 496 compounds total) for docking is below.
Ensemble Docking Protocol and Data Fusion
BRD4 Docking Models
We used the Schrödinger 2014.2 software suite for structure-based manipulations and simulations unless otherwise indicated.26 Seven BRD4 cocrystal structures were selected from the PDB from 65 available structures (January 2014). Three crystal structures for the extensively characterized BRD4 inhibitors, namely, JQ1 (3MXF), I-BET151 (3ZYU), and I-BET762 (3P5O), were chosen. Other PDB crystal structures included 4HXS, 4C67, 4LR6, and 4LYW. Overall, the selection criteria included the atomic resolution of the X-ray crystal structure, diversity in the cocrystal ligand scaffold, and ligand interactions in the binding site. From the seven crystal structures chosen, we identified conserved waters found in/around the ligand binding sites to incorporate likely ligand–protein water bridges into our models. To do this, we aligned all BRD4 crystal structures and determined which waters fell within 5 Å of the active site. We then kept these waters specifically in the preparation process of two out of the seven crystal structures, in addition to those that we had already prepared. This amounted to nine prepared crystal structures with which to produce docking models as described previously.
BRD4 protein structures were preprocessed using the protein preparation workflow in Maestro 9.5 to assign bond orders and refine the structure including hydrogen bond optimization and constrained minimization. Where needed, missing side chains were added using Schrödinger Prime. For each structure, one protein chain with the cocrystal ligand was kept, and water molecules were deleted beyond 5 Å from heteroatom groups. In addition, the internal hydrogen bond network was optimized, followed by constrained energy minimization.
For the resulting nine structure representations, docking grids were generated around the cocrystal binding sites using Schrödinger Glide in the default settings. Redocking each cocrystal ligand into its corresponding structure validated models; in each case, the cocrystal pose was reproduced with RMSD values <1 Å.
Ligand Preparation
Ionization states, tautomeric forms, and 3D conformations for 15 496 SMILES (246 known BRD4 ligands and 15 250 decoy compounds) were generated using LigPrep (Schrödinger 2014.2). The default conditions were maintained, except for the ionization states which were generated at the target pH of 7 ± 2 using Epik including the original state. The resulting 3D ligand representations were exported as structure data files, and unique IDs were assigned based on unique canonical structures to facilitate postdocking hierarchical data fusion as previously described.
Docking Protocol
Ligand representations obtained via LigPrep for all (BRD4 known active and decoy) compounds were docked against all nine prepared BRD4(1) models using Glide (Schrödinger 2014.2) in standard precision (SP) with the default settings, except writing out at most 5 poses per ligand representation and including 25 poses per ligand for postdocking minimization. All docking results were exported from Maestro 9.5 as delimited text files. The docking scores were aggregated hierarchically at three levels, at each level generating average and top docking scores as follows: (i) for each unique ligand representation structure, the scores of all corresponding docking poses were aggregated; (ii) pose-aggregate scores of unique ligand representations (generated in LigPrep) were aggregated by unique (original) compound structure (across ionization states and tautomers); and (iii) aggregate compound scores were combined across all protein structures. At the ligand representation level, we considered top and average top 3 and average top 5 pose scores; at the compound level, the top and average of all corresponding ligand representations; and at the protein level, the top and average top 3 and top 5 scores, giving a total of 18 aggregation methods. On the basis of the model evaluation results for all aggregation methods, for the final results we used the top scores obtained across all levels; for each ligand structure, this corresponds to the best pose for the best ligand representation in the best protein-docking model.
Optimized Docking Models
We generated optimized docking models from three MD simulation frames of the three most active sulfonamide compounds (8302, 8110, and 8200) at 40, 45, and 50 ns in the simulation, respectively. Docking was performed using Glide (Schrödinger 2014.2) in SP with the default settings (keeping cocrystal waters within 5 Å of the ligand), except writing out at most 5 poses per ligand representation and including 25 poses per ligand for postdocking minimization. Maestro 9.5 allows export of all docking results as delimited text files. The docking scores were aggregated first by pose for each ligand representation, then across all representations for each ligand (e.g., tautomers and protonation states), and then across the protein structure (docking) models for 8302, 8110, and 8200, at each level generating average and top docking scores as described previously.26
MM-GBSA Energy Calculations
Prime MM-GBSA calculations were carried out using the initial and MD-optimized receptor–ligand complexes. All default parameters were used, including the VSGB 2.0 solvation model. Calculations were performed using the Schrödinger 2016.3 software suite.
Evaluation and Characterization of Computational Predictions
The docking method optimization was evaluated by the ROC score using the known BRD4 inhibitors and the large decoy dataset (described above). The ROC score show sensitivity over 1—specificity, that is, true positive rate over the false positive rate. On the basis of the model evaluation results for all permutations of top and average scores across poses, ligand representations, and protein structure models, we ended up using the top scores obtained across all levels; for each ligand structure, this corresponds to the best pose for the best ligand representation in the best protein-docking model termed. ROC curves for the initial and MD-optimized (best) docking scores are shown in Supplementary Figure 4. Additionally, EF at 1% top scoring compounds and correlation of aggregate docking scores and activity data were calculated for the 908 commercially available compounds for which activity information had been determined (Supplementary Figure 5, Supplementary Table 3). EF at 1% is [true positives (top 1%)/number of samples (top 1%)]/[positives (total)/number of samples (total)], that is, the ratio of true positives detected in the subset divided by the fraction of overall (total) positives. Also, a log–enrichment curve was calculated for the initial and MD-optimized docking models for predicted commercially available compounds (Supplementary Figure 6).
MD Simulations
All-atom (OPLS2005 force field) explicit water MD simulations were performed using the Desmond 2014.2 software suite (D. E. Shaw Research) via Maestro 9.8.12,14,45 MD systems were built using the highest scoring docking pose for each sulfonamide compound. Specifically, PDB structures 4HXS for 8236 and 8228, 3MXF for 8110, and 4LR6 for 8302, 8152, and 8200. The system boundary distances were specified as 10.0 Å in each dimension in an orthorhombic box. For all simulations, the Desmond default OPLS_2005 force field with simple point charge water model and NPT (temperature (T), pressure (P), and the number of particles(N)) ensemble was used. Desmond automatically places Na+ ions to neutralize the system and a salt concentration of 0.15 M and performs multiple rounds of energy minimization and model relaxation before the production simulation. Each 50 ns simulation was run on the Pegasus cluster from the Center for Computational Science at the University of Miami (http://ccs.miami.edu/hpc/) using 48 processors and completed in approximately 50 h. Simulation analysis was performed using the Desmond trajectory analysis software.
The 500 ns MD simulation of ligand 8302 in BRD4 (BD1) (starting from the best pose docked into 4LR6 as above) was performed using the same parameters as described above, but it was run on an Exxact Quantum TXR410-0128R equipped with Nvidia Tesla M40 GPUs using the GPU-accelerated Desmond software (v2016.3) and one GPU.
Analysis of MD Simulations
MD result analysis consisted of computing both the simulation event analysis and simulation interaction diagrams and exporting the results to a spreadsheet and diagram for analysis. Calculation of the trajectory ligand RMSD differences relative to the target experimental cocrystal pose (in contrast to the default starting docking pose) RMSD was as follows
| E1 |
where Pi refers to the ligand pose as the time index i, Pn is the final ligand pose at the end of the simulation, Po is the starting ligand pose, that is, the docking pose, and Pr is the experimentally determined reference ligand pose.
The definition of RMSD in the course of an MD simulation is
| E2 |
where pj′(ti) corresponds to the position atom j of the ligand with N atoms at time ti, in our case Pi, and pj(tref) corresponds to the position of the ligand atom at a reference time point (usually time 0). E1 transforms RMSD of Pi relative time point 0 (Po) into RMSD relative to the absolute (simulation independent) reference pose Pr, the experimental ligand conformation. These calculations were implemented in Python after exporting RMSD data from Maestro for all six MD trajectories.
Ligand Shape Similarity
The ROCS 3.2.0 shape similarity screening software (Open Eye Scientific)29,46 was utilized to evaluate ligand 3D shape and electrostatic similarity between the experimentally determined active N-[3-(2-oxo-pyrrolidinyl)phenyl]-benzenesulfonamide compounds to all known BRD4 active compounds. The ROCS query was created from six aligned sulfonamide compounds exported from their best BRD4 docking pose (see above). The option to merge color atoms was selected, best hits defined as 100, shape only set to false, rank by set to Tanimoto Combo, and random starts set to 50. The Tanimoto Combo score was the main metric evaluated by our study.
Experimental Determination of Binding Activity
BRD4(1) AlphaScreen and DSF Assays
To perform the BRD4(1) AlphaScreen and DSF assays, we followed the same protocol described previously.26 Compounds were obtained from Enamine, LLC, and the supplier provided purity confirmed ≥95% for each compound based on high-performance liquid chromatography; the traces can be found in the Supporting Information. To confirm assay results, we perform a PerkinElmer TruHits counter screen with the same assay system to determine if any compounds were interacting with the assay components, allowing for the determination of possible false positives.
X-Ray Crystallography and Structure Determination
Crystals of BRD4-1 were grown in the presence of 1 mM ligand and 10% (v/v) DMSO from vapor-diffusion hanging drops using reservoir as described previously,47 harvested in cryoprotectant (reservoir containing 25% (v/v) ethylene glycol and 0.5 mM ligand) and flash frozen in a stream of nitrogen gas. X-ray diffraction data were recorded at −180 °C in the Moffitt Cancer Center Structural Biology Core using Cu Kα X-rays generated by a Rigaku MicroMax 007-HF X-ray generator, focused by mirror optics, and equipped with a Rigaku CCD Saturn 944 system and a Rigaku R-AXIS HTC imager. Data were reduced and scaled with XDS;48 PHENIX49 was employed for phasing and refinement, and model building was performed using Coot.50 The structure was solved by molecular replacement using Phaser51 and the monomer of PDB entry 4O7A(47) as the search model. Initial models for the small-molecule ligands were generated using MarvinSketch (ChemAxon, Cambridge, MA) with ligand restraints from eLBOW of the PHOENIX suite. All structures were validated by MolProbity52 and phenix.model_vs_data.53 Figures were prepared using PyMOL (Schrödinger, LLC).
Accession Codes
Atomic coordinates and structure factors for complexes of BRD4(BD1) with compounds 8110, 8152, 8200, 8228, 8236, and 8302 have been deposited in the PDB under accession codes 5TI2, 5TI3, 5TI4, 5TI5, 5TI6, and 5TI7, respectively.
Acknowledgments
The authors would like to thank ChemAxon for providing the academic research license for their Cheminformatics software tools including JChem for Excel and the Marvin tools. The authors thank D. E. Shaw Research and Schrödinger for the Molecular Dynamics (MD) simulation package and Open Eye Scientific Software for their academic research licenses. S.M. gratefully acknowledges the receipt of CREST fellowship award (BT/IN/DBT-CREST Awards/29/SM/2012-13) from the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India. S.C.S. acknowledges computational resources of the Drug Discovery program of the Center for Computational Science at the University of Miami. The authors thank the Moffitt Structural Biology Core for use of the X-ray crystallography facility.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00553.
Input coordinates of MD simulations and various other structures and result files are available in the Github repository: https://github.com/schurerlab/molecular-modeling (PDF)
Author Contributions
B.K.A. and S.M. performed computational modeling, molecular dynamics, data curation, and data integration; B.K.A. developed code and performed biological experiments. S.W.J.E., J.-Y.Z., and E.S. conducted X-ray diffraction and DSF experiments. B.K.A. and S.C.S. performed data analysis. S.C.S. designed the study, and S.C.S. and N.G.A. advised the project. B.K.A., S.W.J.E., S.M., and S.C.S. wrote the manuscript. All authors contributed to and reviewed the manuscript.
This work was in part supported by grants U54CA189205 (Illuminating the Druggable Genome Knowledge Management Center, IDG-KMC) and U54HL127624 (BD2K LINCS Data Coordination and Integration Center, DCIC). The IDG-KMC is a component of the Illuminating the Druggable Genome (IDG) project (https://commonfund.nih.gov/idg) awarded by the NCI. The BD2K LINC DCIC is awarded by the National Heart, Lung, and Blood Institute through funds provided by the trans-NIH Library of Integrated Network-based Cellular Signatures (LINCS) Program (http://www.lincsproject.org/) and the trans-NIH Big Data to Knowledge (BD2K) initiative (http://www.bd2k.nih.gov). Both IDG and LINCS are NIH Common Fund projects. This work was in part supported by NS067289 to NGA.
The authors declare no competing financial interest.
Supplementary Material
References
- Allen B. K.; Stathias V.; Maloof M. E.; Vidovic D.; Winterbottom E. F.; Capobianco A. J.; Clarke J.; Schurer S.; Robbins D. J.; Ayad N. G. Epigenetic pathways and glioblastoma treatment: insights from signaling cascades. J. Cell. Biochem. 2015, 116, 351–363. 10.1002/jcb.24990. [DOI] [PubMed] [Google Scholar]
- Zoghbi H. Y.; Beaudet A. L. Epigenetics and Human Disease. Cold Spring Harbor Perspect. Biol. 2016, 8, a019497. 10.1101/cshperspect.a019497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J.; Ramírez-Torres A.; Encarnación-Guevara S. Lysine acetylation and cancer: A proteomics perspective. J. Proteomics 2017, 150, 297–309. 10.1016/j.jprot.2016.10.003. [DOI] [PubMed] [Google Scholar]
- Gallenkamp D.; Gelato K. A.; Haendler B.; Weinmann H. Bromodomains and their pharmacological inhibitors. ChemMedChem 2014, 9, 438–464. 10.1002/cmdc.201300434. [DOI] [PubMed] [Google Scholar]
- Muvva C.; Singam E. R. A.; Raman S. S.; Subramanian V. Structure-based virtual screening of novel, high-affinity BRD4 inhibitors. Mol. BioSyst. 2014, 10, 2384–2397. 10.1039/c4mb00243a. [DOI] [PubMed] [Google Scholar]
- Bandopadhayay P.; Bergthold G.; Nguyen B.; Schubert S.; Gholamin S.; Tang Y.; Bolin S.; Schumacher S. E.; Zeid R.; Masoud S.; Yu F.; Vue N.; Gibson W. J.; Paolella B. R.; Mitra S. S.; Cheshier S. H.; Qi J.; Liu K.-W.; Wechsler-Reya R.; Weiss W. A.; Swartling F. J.; Kieran M. W.; Bradner J. E.; Beroukhim R.; Cho Y.-J. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. 10.1158/1078-0432.ccr-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastori C.; Daniel M.; Penas C.; Volmar C.-H.; Johnstone A. L.; Brothers S. P.; Graham R. M.; Allen B.; Sarkaria J. N.; Komotar R. J.; Wahlestedt C.; Ayad N. G. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 2014, 9, 611–620. 10.4161/epi.27906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Wang P.; Chen H.; Wold E. A.; Tian B.; Brasier A. R.; Zhou J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem. 2017, 60, 4533–4558. 10.1021/acs.jmedchem.6b01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwoski G.; Kothiwale S.; Meiler J.; Lowe E. W. Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teague S. J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discovery 2003, 2, 527–541. 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- Ma B.; Kumar S.; Tsai C.-J.; Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999, 12, 713–720. 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- Bowers K. J.; Dror R. O.; Shaw D. E. The midpoint method for parallelization of particle simulations. J. Chem. Phys. 2006, 124, 184109. 10.1063/1.2191489. [DOI] [PubMed] [Google Scholar]
- Ge H.; Wang Y.; Li C.; Chen N.; Xie Y.; Xu M.; He Y.; Gu X.; Wu R.; Gu Q.; Zeng L.; Xu J. Molecular dynamics-based virtual screening: accelerating the drug discovery process by high-performance computing. J. Chem. Inf. Model. 2013, 53, 2757–2764. 10.1021/ci400391s. [DOI] [PubMed] [Google Scholar]
- Bowers K. J.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), 2006.
- Chen H.; Li S.; Hu Y.; Chen G.; Jiang Q.; Tong R.; Zang Z.; Cai L. An Integrated In Silico Method to Discover Novel Rock1 Inhibitors: Multi- Complex-Based Pharmacophore, Molecular Dynamics Simulation and Hybrid Protocol Virtual Screening. Comb. Chem. High Throughput Screening 2016, 19, 36–50. 10.2174/1386207319666151203001946. [DOI] [PubMed] [Google Scholar]
- Abbasi M.; Sadeghi-Aliabadi H.; Hassanzadeh F.; Amanlou M. Prediction of dual agents as an activator of mutant p53 and inhibitor of Hsp90 by docking, molecular dynamic simulation and virtual screening. J. Mol. Graphics Modell. 2015, 61, 186–195. 10.1016/j.jmgm.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Afzal O.; Kumar S.; Kumar R.; Firoz A.; Jaggi M.; Bawa S. Docking based virtual screening and molecular dynamics study to identify potential monoacylglycerol lipase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3986–3996. 10.1016/j.bmcl.2014.06.029. [DOI] [PubMed] [Google Scholar]
- Ali M. R.; Latif R.; Davies T. F.; Mezei M. Monte Carlo loop refinement and virtual screening of the thyroid-stimulating hormone receptor transmembrane domain. J. Biomol. Struct. Dyn. 2015, 33, 1140–1152. 10.1080/07391102.2014.932310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni A.; Bertini L.; De Gioia L.; Papaleo E. Inhibitors of the Cdc34 acidic loop: A computational investigation integrating molecular dynamics, virtual screening and docking approaches. FEBS Open Bio 2014, 4, 473–484. 10.1016/j.fob.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavasotto C. N.; Orry A. J. W.; Murgolo N. J.; Czarniecki M. F.; Kocsi S. A.; Hawes B. E.; O’Neill K. A.; Hine H.; Burton M. S.; Voigt J. H.; Abagyan R. A.; Bayne M. L.; Monsma F. J. Jr. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J. Med. Chem. 2008, 51, 581–588. 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- Aldeghi M.; Heifetz A.; Bodkin M. J.; Knapp S.; Biggin P. C. Accurate calculation of the absolute free energy of binding for drug molecules. Chem. Sci. 2016, 7, 207–218. 10.1039/c5sc02678d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappel D.; Hall M. L.; Lenselink E. B.; Beuming T.; Qi J.; Bradner J.; Sherman W. Relative Binding Free Energy Calculations Applied to Protein Homology Models. J. Chem. Inf. Model. 2016, 56, 2388–2400. 10.1021/acs.jcim.6b00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson B. M.; de Waal P. W.; Ramjan Z. H.; Xu H. E.; Rothbart S. B. A fast, open source implementation of adaptive biasing potentials uncovers a ligand design strategy for the chromatin regulator BRD4. J. Chem. Phys. 2016, 145, 154113. 10.1063/1.4964776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran T.; Zhang Z.; Liu K.; Lu Y.; Li H.; Xu J.; Xiong X.; Zhang Y.; Xu A.; Lu S.; Liu H.; Lu T.; Chen Y. Insight into the key interactions of bromodomain inhibitors based on molecular docking, interaction fingerprinting, molecular dynamics and binding free energy calculation. Mol. BioSyst. 2015, 11, 1295–1304. 10.1039/c4mb00723a. [DOI] [PubMed] [Google Scholar]
- Bernstein F. C.; Koetzle T. F.; Williams G. J. B.; Meyer E. F. Jr.; Brice M. D.; Rodgers J. R.; Kennard O.; Shimanouchi T.; Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- Allen B. K.; Mehta S.; Ember S. W. J.; Schonbrunn E.; Ayad N.; Schürer S. C. Large-Scale Computational Screening Identifies First in Class Multitarget Inhibitor of EGFR Kinase and BRD4. Sci. Rep. 2015, 5, 16924. 10.1038/srep16924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish P. V.; Filippakopoulos P.; Bish G.; Brennan P. E.; Bunnage M. E.; Cook A. S.; Federov O.; Gerstenberger B. S.; Jones H.; Knapp S.; Marsden B.; Nocka K.; Owen D. R.; Philpott M.; Picaud S.; Primiano M. J.; Ralph M. J.; Sciammetta N.; Trzupek J. D. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J. Med. Chem. 2012, 55, 9831–9837. 10.1021/jm3010515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P.; Diallo H.; Goodacre J. D.; Gordon L.; Lewis A.; Seal J. T.; Wilson D. M.; Woodrow M. D.; Chung C.-w. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J. Med. Chem. 2012, 55, 587–596. 10.1021/jm201283q. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C. D.; Skillman A. G.; Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Leblanc Y.; Côté A.; Nasveschuk C. G.; Taylor A. M.; Harmange J.-C.; Audia J. E.; Pardo E.; Joshi S.; Sandy P.; Mertz J. A.; Sims R. J. 3rd; Bergeron L.; Bryant B. M.; Bellon S.; Poy F.; Jayaram H.; Sankaranarayanan R.; Yellapantula S.; Srinivasamurthy N. B.; Birudukota S.; Albrecht B. K. Discovery, Design, and Optimization of Isoxazole Azepine BET Inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. 10.1021/ml4001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O.; Gosmini R.; Toum J.; Clément C. A.; Barnathan M.; Brusq J.-M.; Mordaunt J. E.; Grimes R. M.; Crowe M.; Pineau O.; Ajakane M.; Daugan A.; Jeffrey P.; Cutler L.; Haynes A. C.; Smithers N. N.; Chung C.-w.; Bamborough P.; Uings I. J.; Lewis A.; Witherington J.; Parr N.; Prinjha R. K.; Nicodème E. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 2013, 56, 7501–7515. 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C.-w.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Da Costa D.; Thanasopoulou A.; Filippakopoulos P.; Fish P. V.; Philpott M.; Fedorov O.; Brennan P.; Bunnage M. E.; Owen D. R.; Bradner J. E.; Taniere P.; O’Sullivan B.; Muller S.; Schwaller J.; Stankovic T.; Knapp S. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res. 2013, 73, 3336–3346. 10.1158/0008-5472.can-12-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Prinjha R. K.; Dittmann A.; Giotopoulos G.; Bantscheff M.; Chan W.-I.; Robson S. C.; Chung C.-w.; Hopf C.; Savitski M. M.; Huthmacher C.; Gudgin E.; Lugo D.; Beinke S.; Chapman T. D.; Roberts E. J.; Soden P. E.; Auger K. R.; Mirguet O.; Doehner K.; Delwel R.; Burnett A. K.; Jeffrey P.; Drewes G.; Lee K.; Huntly B. J. P.; Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Wells C.; Felletar I.; Brotherton D.; Martin S.; Savitsky P.; Diez-Dacal B.; Philpott M.; Bountra C.; Lingard H.; Fedorov O.; Muller S.; Brennan P. E.; Knapp S.; Filippakopoulos P. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 19754–19759. 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Plotnikov A. N.; Rusinova E.; Shen T.; Morohashi K.; Joshua J.; Zeng L.; Mujtaba S.; Ohlmeyer M.; Zhou M.-M. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J. Med. Chem. 2013, 56, 9251–9264. 10.1021/jm401334s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Leonards K.; Lambert J.-P.; Dovey O.; Wells C.; Fedorov O.; Monteiro O.; Fujisawa T.; Wang C.-Y.; Lingard H.; Tallant C.; Nikbin N.; Guetzoyan L.; Ingham R.; Ley S. V.; Brennan P.; Muller S.; Samsonova A.; Gingras A.-C.; Schwaller J.; Vassiliou G.; Knapp S.; Filippakopoulos P. Promiscuous targeting of bromodomains by bromosporine identifies BET proteins as master regulators of primary transcription response in leukemia. Sci. Adv. 2016, 2, e1600760 10.1126/sciadv.1600760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Fedorov O.; Keller M.; Wrobel M.; Morgenstern O.; Bracher F.; Knapp S. Benzodiazepines and benzotriazepines as protein interaction inhibitors targeting bromodomains of the BET family. Bioorg. Med. Chem. 2012, 20, 1878–1886. 10.1016/j.bmc.2011.10.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidler L. R.; Filippakopoulos P.; Fedorov O.; Picaud S.; Martin S.; Tomsett M.; Woodward H.; Brown N.; Knapp S.; Hoelder S. Discovery of novel small-molecule inhibitors of BRD4 using structure-based virtual screening. J. Med. Chem. 2013, 56, 8073–8088. 10.1021/jm4011302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.-w.; Coste H.; White J. H.; Mirguet O.; Wilde J.; Gosmini R. L.; Delves C.; Magny S. M.; Woodward R.; Hughes S. A.; Boursier E. V.; Flynn H.; Bouillot A. M.; Bamborough P.; Brusq J.-M.; Gellibert F. J.; Jones E. J.; Riou A. M.; Homes P.; Martin S. L.; Uings I. J.; Toum J.; Clément C. A.; Boullay A.-B.; Grimley R. L.; Blandel F. M.; Prinjha R. K.; Lee K.; Kirilovsky J.; Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 2011, 54, 3827–3838. 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Cao D.; Chen T.; Wang Y.; Miao Z.; Xu Y.; Chen W.; Wang X.; Li Y.; Du Z.; Xiong B.; Li J.; Xu C.; Zhang N.; He J.; Shen J. Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain. J. Med. Chem. 2013, 56, 3833–3851. 10.1021/jm301793a. [DOI] [PubMed] [Google Scholar]
- Fedorov O.; Lingard H.; Wells C.; Monteiro O. P.; Picaud S.; Keates T.; Yapp C.; Philpott M.; Martin S. J.; Felletar I.; Marsden B. D.; Filippakopoulos P.; Müller S.; Knapp S.; Brennan P. E. [1,2,4]triazolo[4,3-a]phthalazines: inhibitors of diverse bromodomains. J. Med. Chem. 2014, 57, 462–476. 10.1021/jm401568s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Carchia M.; Irwin J. J.; Shoichet B. K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar D.; Williams J.; Wu Y.; Damm W.; Shelley J.; Sherman W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- Grant J. A.; Gallardo M. A.; Pickup B. T. A fast method of molecular shape comparison: A simple application of a Gaussian description of molecular shape. J. Comput. Chem. 1996, 17, 1653–1666. . [DOI] [Google Scholar]
- Ember S. W. J.; Zhu J.-Y.; Olesen S. H.; Martin M. P.; Becker A.; Berndt N.; Georg G. I.; Schönbrunn E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. 10.1021/cb500072z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/s0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Chen V. B.; Headd J. J.; Moriarty N. W.; Richardson J. S.; Richardson D. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. phenix.model_vs_data: a high-level tool for the calculation of crystallographic model and data statistics. J. Appl. Crystallogr. 2010, 43, 669–676. 10.1107/s0021889810015608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.