

Abstract
DNA double-strand breaks (DSBs) are deleterious DNA lesions that must be properly repaired to maintain genome stability. Agents, generated both exogenously (environmental radiation, dental X-rays, etc.) and endogenously (reactive oxygen species, DNA replication, V(D)J recombination, etc.), induce numerous DSBs every day. To counter these DSBs, there are two major repair pathways in mammalian cells, nonhomologous end joining (NHEJ) and homologous recombination (HR). NHEJ directly mediates the religation of the broken DNA molecule and is active in all phases of the cell cycle. HR directs repair via the use of a homologous DNA sequence as a template and is primarily active in only S/G2 phases owing to the availability of a DNA template via a sister chromatid. As NHEJ and HR are active in multiple cell cycle phases, there is significant interest in how a cell chooses between the two DSB repair pathways. Therefore, it is essential to utilize assays to study DSB repair that can distinguish between the two DSB repair pathways and the different phases of the cell cycle. In this chapter, we describe methods to measure the contribution of DNA repair pathways in different phases of the cell cycle. These methods are simple, can be applied to most mammalian cell lines, and can be used as a broad utility to monitor cell cycle-dependent DSB repair.
1. INTRODUCTION
The human genome is constantly under attack from a variety of agents that generate tens of thousands of DNA lesions per day. The most deleterious of these lesions is the DNA double-strand break (DSB). Two major pathways direct repair of DSBs in mammalian cells, homologous recombination (HR) and nonhomologous end joining (NHEJ) (Goodarzi & Jeggo, 2013; Hoeijmakers, 2001; Jackson & Bartek, 2009; Schipler & Iliakis, 2013). HR drives DSB repair by using a homologous DNA sequence as a template to guide error-free restoration of the DNA molecule. Since an accessible homologous template is found on a sister chromatid, error-free HR is believed to be primarily active in mid-S phase to early G2 phase of the cell cycle. NHEJ functions by directly religating the two broken DNA strands. As NHEJ does not require a homologous template, it is not restricted to a particular cell cycle phase. It should be noted that there is also an alternative end-joining (Alt-EJ) pathway, which is believed to primarily be a backup pathway for both HR and NHEJ. Alt-EJ typically utilizes microhomologies distant from the DSB site to drive repair (Schipler & Iliakis, 2013).
Since there are multiple DSB repair processes, a cell must properly choose the specific pathway to repair a broken DNA molecule. The cell cycle phase likely plays a role in this process as HR is primarily active in mid-S to early G2 phase of the cell cycle. However, NHEJ is also active in these cell cycle phases and thus there must be a process that assists the cell in choosing the appropriate DSB repair pathway. In particular, due to the high replication activity and the formation of single-ended replication fork-associated breaks in S phase and the critical G2 phase preceding the subsequent division in M phase, error-free repair of DSBs in S/G2 is paramount. Importantly, it has been shown that the majority of breaks are still repaired by NHEJ in early S phase with activities transitioning to the HR pathway from mid-S phase (Karanam, Kafri, Loewer, & Lahav, 2012). Thus, it is also important to distinguish and demarcate different subphases within the S phase to decipher DNA repair activity and pathway contributions accurately.
In this chapter, we will describe protocols that can be used to examine DSB repair processes in a cell cycle-specific manner. These methods were originally developed by other groups and later modified by us and utilized in various publications (Davis et al., 2015; Davis, So, & Chen, 2010; Lee et al., 2016; Shao et al., 2012). The protocols include: examining real-time dynamics of repair proteins localizing and dissociating from DSBs (Jackson & Bartek, 2009); immunofluorescence-based methods to monitor NHEJ, DNA end resection, and ongoing HR (Schipler & Iliakis, 2013); and determining overall repair capacity (Goodarzi & Jeggo, 2013).
2. DYNAMICS OF REPAIR PROTEINS TO LASER-GENERATED DSBS
The cellular response to DSBs initiates with the recognition of the ends of the broken DNA molecule. This DSB recognition results in the recruitment of a significant number of factors to the DSB site and the surrounding area. In this section, we will describe a technique that utilizes a microlaser system to generate DSBs coupled with live-cell microscopy to examine the recruitment and dynamics of a yellow fluorescent protein (YFP)-tagged protein to DSBs. To allow differentiation of cells in S phase and non-S phase, DsRed-tagged PCNA is monitored, as PCNA shows a faint and even distribution in non-S phase cells and forms a distinct punctate patterning in S phase (Fig. 1) (Shao et al., 2012). Here, we will outline the recruitment and kinetics of the NHEJ factor Ku80 to laser-generated DSBs, but this technique has also been successfully used to examine the localization and dynamics of other repair proteins to laser-generated DSBs, including DNA-PKcs, ATM, MDC1, and MRE11 (Davis & Chen, 2010; Kim et al., 2005; So, Davis, & Chen, 2009). It should be noted that the method outlined here is specific for our laser/microscope setup and appropriate adjustments will have to be made for other setups.
Fig. 1.
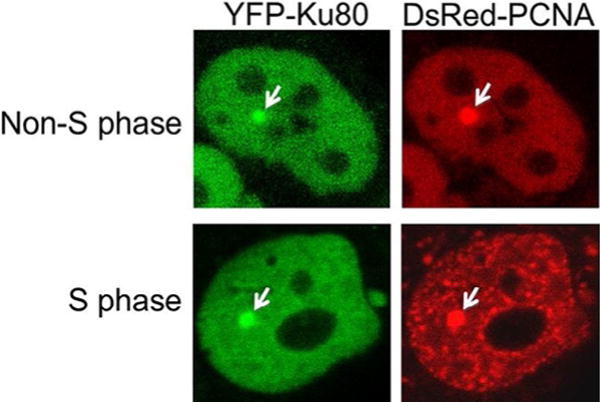
Differentiating S and non-S phase cells. YFP-tagged Ku80 and DsRed-tagged PCNA were transiently expressed in the Ku80-deficient CHO cell line Xrs5. S phase and non-S phase cells are differentiated by the localization pattern of DsRed-PCNA. Localization of Ku80 and PCNA to laser-generated DSBs is marked by a white arrow.
2.1 Transient Expression of YFP-Tagged Ku80 and DsRed-Tagged PCNA
2.1.1 Equipment
Amaxa Biosystems Nucleofector II (Lonza)
2.1.2 Buffers and Reagents
Full-length wild-type Ku80 cDNA was subcloned into a modified pcDNA3 vector that carries an YFP tag upstream of the multiple cloning site to generate YFP-tagged Ku80 (YFP-Ku80).
Full-length wild-type PCNA cDNA was subcloned into pDsRed-Monomer-C1 vector that carries DsRed tag upstream of the multiple cloning site to generate DsRed-tagged PCNA (DsRed-PCNA).
U2OS or HT1080 cells. It should be noted that these experiments are also performed in rodent cell lines deficient for the YFP-tagged protein of interest. For example, when testing YFP-Ku80, experiments can be performed in the K80-deficient Chinese Hamster Ovary (CHO) cell line Xrs5 or Xrs6 or Ku80-deficient mouse embryonic fibroblasts (MEFs).
Alpha-minimum Eagle’s medium (Fisher, #SH3026502) supplemented with 10% fetal bovine serum and fetal calf serum (1:1 ratio).
CO2-independent medium (Invitrogen, #19045088).
Transfection reagent such as Lipofectamine 2000 reagent (Invitrogen) or electroporation with Nucleofector (Lonza).
35-mm Glass-bottom culture dishes (MatTek, P35G-0-14-C).
2.1.3 Procedure
Cotransfect the YFP-Ku80 and DsRed-PCNA constructs via Nucleofector or Lipofectamine 2000 following the manufacturer’s protocol into U2OS or HT1080 cells. If using Nucleofector, use protocol X-001 with solution V to electroporate YFP-Ku80 and DsRed-PCNA into U2OS and HT1080 cells. For rodent cells, use protocol U-27 or U-23 with solution T.
After transfection, split the cells and plate 1 × 105 cells onto a 35-mm glass-bottom culture dish, allow to attach, and grow for 48h. Check expression via microscopy.
2.1.4 Notes
To amplify the Ku80 constructs, transform 50–100ng of plasmid into DH5α competent cells, and then plate on LB+AMP plate for overnight incubation at 37°C. Pick a single colony (choose small, not large colonies) and grow in 200mL LB+AMP in a shaker incubator at 37°C for 24h. Next day, purify the plasmid DNA by the Qiagen midi-prep kit according to manufacturer’s instructions. A typical yield for the Ku80 cDNA-containing vectors is approximately 300μg. It should be noted that expression constructs large in size, such as those that contain full-length cDNAs of DNA-PKcs, ATM, 53BP1, and BRCA1, tend to recombine during DNA amplification; therefore, Escherichia coli strains deficient in endonucleases and recombination (e.g., XL10) are recommended to improve the quality of the DNA preparation.
We typically use cells that are stably expressing the YFP-tagged protein of interest in laser microirradiation assays as this allows for more consistent experimental results. To make stable cell lines, linearize the vector with a restriction enzyme and verify the complete digestion via agarose gel electrophoresis. Purify the linearized vector by phenol extraction (twice) followed by ethanol precipitation and wash with 70% ethanol and allow the pellet to air dry. Resuspend the pellet in TE or water and measure the DNA concentration. Perform transfection as described. Following transfection, plate 1 × 104 cells on 100mm (4 total dishes) with normal growth medium. 24h posttransfection, add the selection drug G418 (500 μg/mL) and incubate the cells for 10 days. Pick single colonies and check expression of YFP-Ku80 via microscopy (YFP signal) and Western blot analysis (antibodies that recognize the YFP tag and/or Ku80).
2.2 Microscope and Laser-Irradiation Setup
2.2.1 Equipment
An inverted Axiovert 200M microscope equipped with a Plan-Apochromat 63×/NA 1.40 oil immersion objective and an AxioCam HRm digital camera (Carl Zeiss MicroImaging, Inc.) for time-lapse imaging. Computer with Carl Zeiss AxioVision 4.8.2 software.
Laser microirradiation unit. Spectra-physics nitrogen laser (Andor Technology, VSL-337ND-S): The Spectra-Physics VSL-337ND-S nitrogen laser emits 4-ns pulses in the UV at 337nm. The pulse repetition rate may be varied from 1 to 60Hz with a pulse energy of up to 300 μJ. The nitrogen laser is directly coupled to the epifluorescence path of the microscope through a Micropoint Laser Illumination and Ablation System (Photonic Instruments, Inc.) and focused through a Plan-Apochromat 63× oil immersion objective. The nitrogen wavelength can be changed from 337 to 365nm by BPBD 365 dye solution. Power output is controlled by the Micropoint System ranged from 1% to 89%.
Digital temperature control system with a heating insert P (PeCon, #0426.100) and a Tempcontrol 37-2 control unit (PeCon, #0503.000).
2.2.2 Procedure
Switch on temperature control at least 15min prior to use for live-cell imaging (37°C).
Turn on Stage Controller/MCU28.
Turn on laser via the key switch. Set repetition rate to 12 or 4 o’clock position depending on the experiment.
Switch on Micropoint and set number to 75 for DSB induction.
Turn on microscope and the EXFO: UV lamp.
Select Objective on 63× (oil immersion).
Use 70/30 beam splitter for laser damage along with GFP filter on the UV adaptor.
Filter set selection: select #5 filter for laser damage (FITC_TexasRed).
Replace normal growth medium with heated CO2-independent medium for microirradiation experiment.
Apply a drop of oil on the objective and lower objective with “focus down” bottom before placing the sample on it.
Attach the 35-mm dish onto the heated dish holder to stabilize the dish and then place this onto the heating insert.
Focus a cell and put “Cross mark” (only on the right side-eye) in the center of the nucleus.
- Set up imaging parameter utilizing AxioVision software. The parameters utilized for multidimensional acquisition are:
- “Dye”—YFP
- “Exposure”—“fixed”—“400ms”
- “Hardware setting”
- During acquisition—“workgroup: FITC_TexasRed”
- After acquisition—“workgroup: shutter closed”
- For initial accumulation (up to 10min) utilize “Time lapse setting”
- Interval—20s
- Cycles—32
- For 2h kinetics, utilize a hand-held timer and take images at specific time points. Typically, images are acquired before irradiation and then 3, 10, 20, 30, 60, 90, and 120min postirradiation
2.3 DSB Repair Kinetics With Laser Microirradiation
2.3.1 Procedure
DSBs are induced in the nucleus of cultured cells by microirradiation with a pulsed nitrogen laser (Spectra-Physics; 365nm, 10Hz pulse). Set the output of the laser power at 75% of the maximum.
Change the complete alpha-MEM media with CO2-independent medium heated to 37°C.
Identify multiple cells expressing YFP-Ku80 and DsRed-PCNA. DsRed-PCNA is used to differentiate S and non-S phase cells as PCNA forms a distinct punctate pattern in S phase, whereas it shows faint and even expression in non-S phase cells (Fig. 1). Select at least 10 cells in S phase and non-S phase.
- Initial accumulation (10min) of YFP-Ku80 to laser-generated DSBs.
- Start image acquisition before laser microirradiation to obtain an image of the unirradiated cell.
- After first image acquisition, induce DSBs in cell nuclei by microirradiation with pulsed nitrogen laser allowing 8 laser pulses to hit a defined region of a single nucleus.
- As described in Section 2.2.2, Note 13, the defined parameters in AxioVision software will acquire images before and after irradiation via a 400-ms exposure time.
- Capture images every 20s for the duration of the 10-min time course.
- Convert signal intensities of accumulated YFP at the microirradiated site into a numerical value by the use of the Carl Zeiss AxioVision software (see below).
- Kinetics of YFP-Ku80 at laser-induced DSBs (2h).
- Apply the same procedure as described above for 2-h microirradiation and time-lapse imaging except capture images at defined time points (3, 10, 20, 30, 60, 90, and 120min) post microirradiation.
- Set a fixed exposure time to capture all images before and after irradiation.
- Convert signal intensities of accumulated YFP at the microirradiated site into a numerical value by the use of the Carl Zeiss AxioVision software (see below).
2.4 Calculation of Relative Fluorescent Intensity for Protein Recruitment Kinetics
2.4.1 Procedure
Open AxioVision software and the appropriate image set for each individual cell.
Circle damage site (Dt, white circles in Fig. 2) and a background area (Bt, yellow circles in Fig. 2) at each time point. The circle of the DNA damage site (accumulation site of YFP-Ku80) is typically set at 1μm, but may need to be adjusted based on the size of the spot. The background site is typically 2μm.
Obtain densitometric mean (fluorescence of the spot) via the AxioVision software, which is based on the fluorescence intensity and area size. Perform all calculations by utilizing Microsoft Excel. The densitometric means are placed in Excel for calculations.
To eliminate the background fluorescence, laser-induced fluorescence intensity accumulated at the damaged spot Dt is subtracted with the background fluorescence intensity Bt at an undamaged spot in the same nuclei at each time point (Fig. 2). The fluorescence intensity It = Dt–Bt.
Compensate nonspecific photobleaching and UV lamp output fluctuation by normalizing the absolute fluorescence intensity I(t) accumulation at the damaged spot of each time point based on background intensity prior to laser damage using the formula: normalized fluorescence intensity NIt = (It/Bt)*D0, where Bt represents the undamaged site background intensity of each time point, and D0 represents the intensity of the damaged spot prior to irradiation.
Calculate relative fluorescence intensity (RFI) by using the formula: RFIt = (NIt NI0)/(NImax – NI0), where NI0 means normalized fluorescence intensity of the damaged spot prior to laser irradiation and NImax is the maximum normalized fluorescence intensity of the damaged spot.
Each data point is the average of 10 independent measurements.
Average the relative intensity to make a graph (Fig. 3).
Fig. 2.
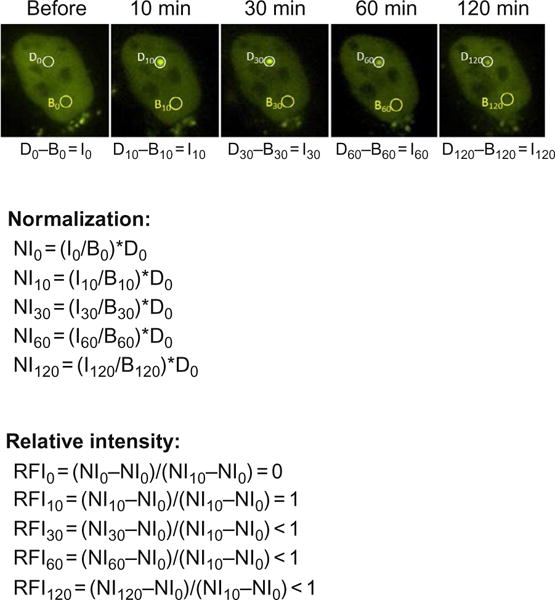
Calculation of relative fluorescence intensity for recruitment kinetics. A panel of images showing the localization of YFP-tagged Ku80 to laser-generated DSBs in a 2-h time course. Relative intensities of YFP-tagged Ku80 at sites of laser damage at each time point is calculated by using the following equations. Dt, fluorescence intensity accumulated at the damaged spot at a given time; Bt, background fluorescence intensity at an undamaged spot a given time; It, fluorescence intensity of the damaged spot at a given time; NIt, normalized fluorescence intensity of the damaged spot at a given time; RFIt, relative fluorescence intensity of the damaged spot at a given time compared to that of maximum normalized fluorescence intensity.
Fig. 3.
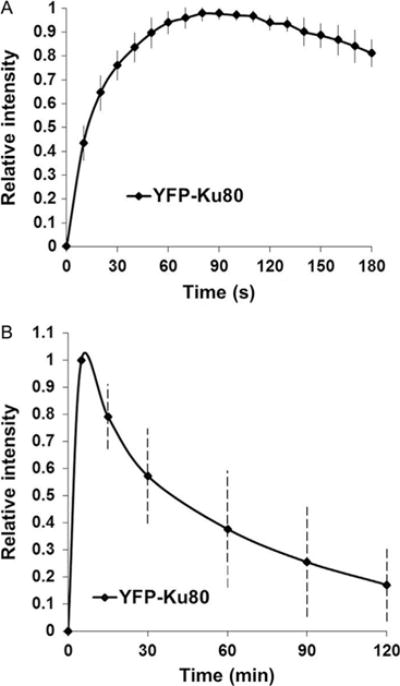
Localization and kinetics of YFP-tagged Ku80 to laser-generated DSBs. Initial localization (A) and 2 h kinetics (B) of relative fluorescence intensity of YFP-tagged Ku80 to laser-induced DSB in Xrs5 cells.
2.5 Enhancement of Laser Microirradiation With DNA Photosensitizer
DNA photosensitizers, such as Hoechst 33258 and bromodeoxyuridine (BrdU), can also be used to assist in increasing the fluorescent signal due to increased DNA damage.
For sensitization with Hoechst 33258, change to CO2-independent medium, then add 1μL of 10mg/mL Hoechst 33258 in 2.5mL medium and incubate the cells for 10min before initiating the experiment.
For BrdU sensitization, treat cells in regular medium with 10μM BrdU for 16–24h, then change to CO2-independent medium, followed by laser microirradiation.
3. CELL CYCLE-SPECIFIC IMMUNOFLUORESCENCE ASSAYS TO EXAMINE NHEJ, DNA END RESECTION, AND ONGOING HR
A powerful tool utilized in the DSB repair field is an immunofluorescence-based technique to monitor DSB-induced foci formation and resolution of repair proteins. This technique uses immunodetection of specific proteins in response to DNA damage and can be coupled with prelabeling cells with EdU (5-ethynyl-2′-deoxyuridine) to allow differentiation of cell cycle stages. Here, we will describe methods to examine NHEJ, DNA end resection, and ongoing HR in a cell cycle-specific manner using immunofluorescence coupled with microscopy.
3.1 Pulse-Labeling Cells With EdU to Allow Differentiation of Cell Cycle Stages
3.1.1 Buffers and Reagents
Falcon™ Culture Slides (4 chambers) (Fisher Scientific, Catalog no: 08-774-209).
Fisherfinest™ Premium Cover Glasses (50 × 20mm) (Fisher Scientific, Catalog no: 12-548-5E).
Click-iT® EdU Alexa Fluor® 555 Imaging Kit (ThermoFisher Scientific, Catalog no: C10338).
U2OS, HT1080, or rodent cell lines.
3.1.2 Procedure
Seed an appropriate number of cells on the Falcon culture slides (typically 10–50 × 103 cells) and allow to grow for 24–48h. Make sure the cells are healthy and well dispersed.
Prepare a 2× working solution of EdU from the Click-iT kit (Component A) in complete medium.
Prewarm the 2× EdU solution, then add an equal volume of the 2× EdU solution to the volume of the media currently on the cells. Replacing all of the media is not recommended as this could affect the rate of cell proliferation. Protect from light.
The final concentration of EdU typically used is 50 μM (35 μM for rodent cell lines).
Incubate for 30min to allow the incorporation of EdU into replicating DNA.
Following the incubation with EdU, wash the cells 2× with complete medium and then replace with fresh medium. The cells can now be exposed to a DNA damaging agent and the experiment performed. EdU detection will be performed in the middle of the procedures outlined in Sections 3.2 and 3.3. Fig. 4 shows a representation of EdU labeling.
Fig. 4.
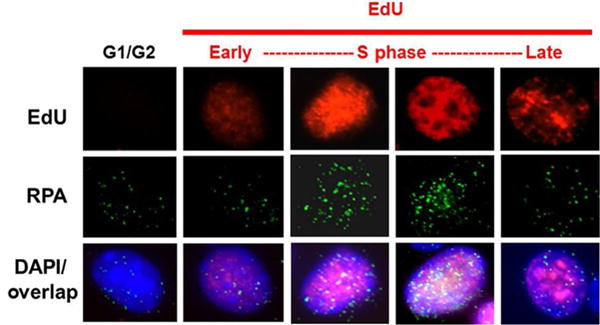
EdU labeling allows identification of cells in S phase. Panel of images depicting cells in different cell cycle stages, in particular, cells in early-, mid-, and late-S phase via differences in nuclear EdU staining. The images also include an overlay of nuclear EdU staining with RPA foci and DAPI stain.
3.2 Monitoring NHEJ in G1 Phase of the Cell Cycle
3.2.1 Equipment
Gamma irradiator (Cs137)
3.2.2 Buffers and Reagents
Phosphate-buffered saline (PBS), pH 7.4.
4% Paraformaldehyde solution (PFA): Dissolve 4g of paraformaldehyde in 50mL of water and 1mL of 1 M NaOH (heat at 65°C in a water bath until powder is completely dissolved). Cool to room temperature. Add 10mL of 10× PBS. Adjust the pH to 7.4 using HCl. Make up the volume to 100mL with water. Filter through 0.2μm filter. Store at −20°C in aliquots.
Triton X-100 (0.5%) in 1× PBS, store at 4°C and use within 2–3 weeks.
5% Normal goat serum in 1× PBS, store at 4°C and use within 2–3 weeks.
Wash buffer (1% BSA in 1× PBS), store at 4°C and use within 2–3 weeks.
Click-iT® EdU Alexa Fluor® 555 Imaging Kit (ThermoFisher Scientific, Catalog no: C10337).
Vectashield with DAPI (Vector Labs, Catalog no: H-1200).
Anti-53BP1 rabbit polyclonal antibody (Santa Cruz, Catalog no: sc-22760).
Anti-DNA-PKcs (phospho S2056) rabbit polyclonal antibody (Abcam, Catalog no: ab18192).
FITC-488-antirabbit secondary antibody.
3.2.3 Procedure
Follow the pulse labeling of cells with EdU protocol from Section 3.1.
Expose cells to 1Gy of γ-rays and place in incubator.
At different time points after irradiation (30, 60, 120, 240, and 480min post-IR), wash cells twice with ice-cold 1× PBS. (Remove the buffer with pipette to avoid cell loss.)
Add 4% paraformaldehyde (in 1× PBS) to fix cells and allow to incubate for 20min at room temperature.
Wash cells five times with ice-cold 1× PBS (final two washes are 5-min washes with rocking). See Section 3.2.4, Note 1.
Incubate the cells in ice-cold 0.5% Triton X-100 (in PBS) on ice for 10min.
Wash the cells five times with ice-cold 1× PBS (final two washes are 5-min washes with rocking).
Incubate the cells in blocking solution (5% goat serum in 1× PBS) for 2h or overnight. See Section 3.2.4, Note 1.
Wash the cells once with ice-cold 1× PBS.
Add the DNA-PKcs phospho-2056 (1:200 dilution) or 53BP1 (1:500 dilution) rabbit polyclonal antibody diluted in 5% normal goat serum in 1× PBS to the cells. See Section 3.2.4, Note 2.
Incubate the cells with the antibody at room temperature for 2–4h.
Wash the cells three times with ice-cold wash buffer (each wash for 5min with rocking).
Perform the Click-iT reaction for EdU detection following the manufacturer’s established protocol. In total, the Click-iT reaction typically takes 60–90min.
Once the Click-iT reaction is completed, wash the cells five times with ice-cold wash buffer.
Incubate the cells with FITC-488-antirabbit secondary antibody (diluted 1:1000) in 1% BSA and 2.5% normal goat serum in 1× PBS for 1h at room temperature (in the dark).
Wash the cells five times with ice-cold wash buffer (each wash for 5min with rocking).
After the last wash, remove the entire wash buffer completely and allow the cells to air dry.
Remove the chamber partition and mount the cells in VectaShield mounting medium containing DAPI.
Acquire images and count foci (see Section 3.4).
3.2.4 Notes
At this step, the cells can be stored at 4°C for a few days.
G1 phase nuclei can be distinguished from S and G2 phase nuclei by their lack of EdU staining, low intensity of DAPI staining, and smaller size of the nuclei.
Both DNA-PKcs phospho-2056 and 53BP1 focus formation can be used to monitor NHEJ and either can be used in an individual experiment.
3.3 Monitoring DNA End Resection or Ongoing HR in Mid-S Phase of the Cell Cycle
3.3.1 Equipment
Gamma irradiator (Cs137)
3.3.2 Reagents and Buffers
Phosphate-buffered saline (PBS), pH 7.4.
4% Paraformaldehyde (PFA).
Triton X-100 (0.5%) in PBS.
5% Normal goat serum in PBS.
Wash buffer (1% BSA in 1× PBS).
Click-iT® EdU Alexa Fluor® 555 Imaging Kit (ThermoFisher Scientific, Catalog no: C10338).
Vectashield with DAPI (Vector Labs, Catalog no: H-1200).
Anti-RPA2 mouse monoclonal antibody (Millipore, Catalog no: NA19L).
Anti-Rad51 rabbit polyclonal antibody (Santa Cruz, Catalog no: sc-8349).
Extraction Buffer (CSK Buffer): 10mM HEPES, pH 7.4, 300mM sucrose, 100mM NaCl, 3mM MgCl2, 0.1% Triton X-100. Prepare fresh.
3.3.3 Procedure
Follow the pulse labeling of cells with EdU protocol from Section 3.1.
Expose cells to 8Gy of γ-rays and place in incubator.
At different time points (Mock, 2, 4, 8, and 12h) post-IR, wash the cells twice with ice-cold 1× PBS.
Add ice-cold Extraction Buffer (CSK Buffer) to the cells and incubate for 7–8min on ice.
Wash the cells five times with ice-cold 1× PBS (remove the buffer with pipette to avoid cell loss).
Fix the cells with 4% paraformaldehyde (in 1× PBS) for 20min at RT.
Wash the cells five times with ice-cold 1× PBS (final two washes are 5-min washes with rocking).
Incubate the cells in ice-cold 0.5% Triton X-100 (in PBS) on ice for 10min.
Wash the cells five times with ice-cold 1× PBS (final two washes are 5-min washes with rocking).
Incubate the cells in blocking solution (5% goat serum in 1× PBS) for 2h or overnight. See Section 3.2.3, Note 1.
Wash the cells once with ice-cold 1× PBS.
Add the RPA2 (1:500 dilution) mouse monoclonal antibody or Rad51 (1:800 dilution) rabbit polyclonal antibody diluted in 5% normal goat serum in 1× PBS to the cells. Incubate for 2–3h.
Incubate the cells with the antibody at room temperature for 2–4h.
Wash the cells three times with ice-cold wash buffer (each wash for 5min with rocking).
Perform the Click-iT reaction for EdU detection following the manufacturer’s established protocol. In total, the Click-iT reaction typically takes 60–90min.
Once the Click-iT reaction is completed, wash the cells five times with ice-cold wash buffer.
Incubate the cells with FITC-488-antimouse secondary antibody for RPA (diluted 1:1000) or FITC-488-antirabbit secondary antibody for Rad51 (diluted 1:1000) in 1% BSA and 2.5% normal goat serum in 1× PBS for 1h at room temperature (in the dark).
Wash the cells five times with ice-cold wash buffer (each wash for 5min with rocking).
After the last wash, remove the entire wash buffer completely and allow the cells to air dry.
Remove the chamber partition and mount the cells in VectaShield mounting medium containing DAPI.
Acquire images and count foci (see Section 3.4). Fig. 4 shows a representation of EdU labeling and RPA foci staining.
3.4 Quantification of Foci
3.4.1 Equipment
Fluorescence or confocal microscope
Imaris or ImageJ image analysis software
3.4.2 Procedure
Acquire images using fluorescence or confocal microscope with a magnification of 60× or higher.
For mid-S phase cells, select cells with even pan-nuclear EdU stained nuclei. See Fig. 4.
Preferentially, acquire Z stacks and count foci on maximum intensity projection (all Z sections merged) images so that all foci are visualized at the same plane.
Foci numbers per nuclei can be enumerated using Imaris image analysis software or ImageJ software.
4. DETERMINATION OF DNA REPAIR CAPACITY IN DIFFERENT PHASES OF THE CELL CYCLE
Colony formation or cell survival assays test cell proliferation after being challenged with a DNA damaging agent. It is a well-established and accurate method to determine the sensitivity of cells to DNA damaging agents like ionizing radiation and chemotherapeutic agents. Typically, cell survival assays are performed with asynchronous cell populations (Munshi, Hobbs, & Meyn, 2005), which may skew the results as cells have differential radiosensitivities throughout the cell cycle (more radioresistant in S phase and more radiosensitivity in mitosis), and it does not allow for the ability to compare radioresponses in different phases of the cell cycle. Thus, in order to accurately determine radiosensitivity of all subpopulations within a specific cell type, survival assays with synchronized population of cells are imperative. Here, we describe a method to amalgamate a simple synchronization method utilizing double-thymidine block (Bootsma, Budke, & Vos, 1964), where cell cycle progression is hindered by excess thymidine leading to feedback inhibition of nucleotide synthesis, with a cell survival assay to examine radiosensitivity of cells in G1 and S phase of the cell cycle (Lee et al., 2016).
4.1 Cell Synchronization Utilizing Double-Thymidine Block
4.1.1 Buffers and Reagents
Thymidine (T9250, Sigma Aldrich).
Complete DMEM (D6429, Sigma Aldrich) (with 10% fetal bovine serum plus penicillin–streptomycin).
1× PBS, pH 7.4.
Cells of interest. This protocol has been optimized specifically for mouse ear fibroblasts and MEFs and should be modified for other cell lines.
4.1.2 Procedure
Plate cells in DMEM at 30% confluency 1 day prior to thymidine treatment.
First thymidine block. Add thymidine to the cells to a final concentration of 2mM and incubate for 8h.
Wash the cells three times with 1× PBS.
Add complete DMEM for 4h to release the cells from the thymidine block.
Second thymidine block. Add thymidine to the cells to a final concentration of 2mM and incubate for 12h. This should result in the cells to be fully synchronized at the G1/S phase border. For G1 phase survival assay, cells can be used directly at this stage (see Section 4.1.3, Note 2). Take a small aliquot of cells for cell cycle analysis (see Section 4.2). For collecting S phase cells for survival assay, proceed to step 6.
Wash the cells 3× with 1× PBS.
Add complete DMEM to release the cells from the thymidine block and incubate at 37°C for 2.5h to achieve a maximum population of cells in the mid- to late-S phase of the cell cycle. See Section 4.1.3, Note 3.
Place the cells on ice to stop cell cycle progression.
Take a small aliquot of cells for cell cycle analysis (see Section 4.2).
4.1.3 Notes
The synchronization protocol described here is specific for rodent fibroblasts with a doubling time of 12–14h.
Before attempting synchronization, it is essential to know the doubling time of the experimental cell line. Most common cell lines have established protocols for synchronization that can be readily used. If not available, BrdU incorporation assay should be performed to estimate the duration of different phases of the cell line and then synchronization attempted.
For G1 phase synchronization, approximately 75%–80% cells are in G1/S border using thymidine double block method.
For S phase synchronization, typically, approximately 73%–76% of the cells will be in S phase, 14%–16% in G1 phase, and 10%–11% in G2 phase of the cell cycle.
4.2 Cell Cycle Analysis by Propidium Iodide Staining Followed by Flow Cytometry
4.2.1 Equipment
Flow cytometer (AMNIS FlowSight® Imaging Flowcytometer)
Gamma irradiator (Cs137)
Biohazard hood
4.2.2 Buffers and Reagents
70% Ethanol (in DI water)
1× PBS, pH 7.4
Propidium Iodide (PI) (50μg/mL) in 1× PBS
Ribonuclease A (RNase A) (100μg/mL) in 1× PBS
4.2.3 Procedure
Harvest cells by trypsinization or scraping and wash with 1× PBS.
Fix cells by adding ice-cold 70% ethanol (approx. 1mL 70% ethanol to 1 million cells). Add dropwise to the cell pellet while vortexing to ensure fixation of all cells and minimize clumping.
Incubate for at least 30min at 4°C to ensure full fixation. See Section 4.2.4, Note 1.
Wash the fixed cells twice in 1× PBS.
Spin at 2000rpm and discard the supernatant. See Section 4.2.4, Note 2.
Add 50μL of 100μg/mL RNase to the cells. This will ensure that only the DNA will be stained by the PI.
Add 400μL of PI per million cells and mix well.
Incubate the cells for 5–10min in a 37°C water bath or 30min at room temperature.
Analyze samples by flow cytometry and measure at least 10,000 single cells. See Section 4.2.4, Note 3.
4.2.4 Notes
Once fixed, the samples can be stored for several weeks at 4°C or −20°C.
Care should be taken to avoid cell loss when discarding the supernatant, especially the 70% ethanol fixation step.
The cells can be directly analyzed in the PI/RNaseA solution and thus there is no need to wash the cells.
4.3 Survival Assay With Various Fractions of Synchronous Cells Obtained From Double-Thymidine Block Method
4.3.1 Buffers and Reagents
Synchronized cells (see Section 4.1).
Complete DMEM (with 10% fetal bovine serum plus penicillin–streptomycin).
Trypsin–EDTA, to make single-cell suspensions from monolayer cultures. Store at 4°C.
60-mm Tissue culture dishes.
15-mL Conical tubes.
1× PBS, pH 7.4.
0.5% Crystal Violet (made in 100% methanol). Store at room temperature in a dark plastic bottle.
4.3.2 Procedure
Label the 60-mm dishes and 15-mL conical tubes for each sample. One will need 60-mm dishes in triplicate and two cell numbers will be plated for each dose of radiation, i.e., 6 total dishes per dose (see Section 4.3.3, Note 1). Label the bottom of each dish and not on the lid as the lids will be discarded during staining.
Add 5mL of growth medium to the flasks and keep them aside in a hood.
Make sure that the cells are in single-cell suspension and obtain an accurate cell count using an automated cell counter or a hemocytometer.
Count the cells and divide them into 5 dishes corresponding to 0, 1, 2, 4, 6Gy of IR, while still maintaining on ice.
Irradiate the dishes marked 1–6Gy with the corresponding doses of IR.
After the irradiation treatment, make serial dilutions to obtain the number of cells to be plated for each radiation dose. Plate the cells in triplicate and place in the incubator for 10–12 days to allow colony formation. For example, plate 50 and 100 cells in triplicate for 0Gy control.
Remove the medium from the plates and add 0.5% Crystal Violet stain to the dishes. Allow to sit for 3–4min.
Remove the Crystal Violet stain and rinse the stained plates upside down in a pan with lukewarm water. Rinsing the plates upside down in the pan prevents the colonies from loosening and washing off.
Let the plates air dry overnight and then count the colonies using a dissecting microscope under a magnified field. A cluster of blue-staining cells is considered a colony if it comprises at least 50 cells. Note the numbers for both the A and B dilutions and make a chart.
- Use the nonirradiated control cells to obtain a plating efficiency. Average the three colony counts for each dilution A and B and divide the mean by the number of cells plated. This will give the PE:
- Following determination of PE, calculate the fraction of cells surviving a given treatment. First, normalize all the plating efficiencies of the treated samples to that of the control unirradiated plates, considering that to be 100%. The surviving fraction (SF) is determined by dividing the PE of the treated cells by the PE of the controls, and then multiplying by 100:
Plot the data on an Excel spreadsheet with the dose of radiation on the x-axis and survival on the y-axis.
A representative survival curve is shown in Fig. 5.
Fig. 5.
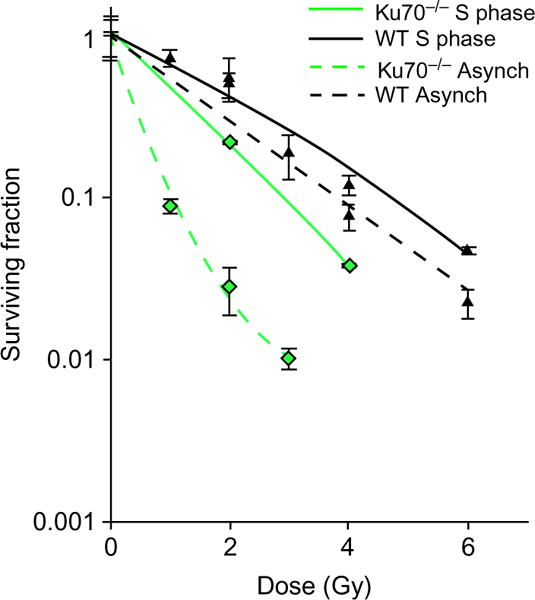
Cell cycle-specific IR survival assay. Colony formation assays were performed to compare radiation sensitivity of Ku70−/− mouse fibroblasts or Ku70−/− fibroblasts complemented with Ku70 wild type in synchronized S phase or as an asynchronous cell population. The cell lines were left cycling or synchronized by double-thymidine block method and then released. Subsequently, the cells were irradiated at the indicated doses and plated for analysis of survival and colony-forming ability. An increase in radio-resistance was observed in S phase of Ku70−/− cells as compared to its asynchronous counterpart. Error bars denote S.D. Note that the Ku70−/− mouse fibroblasts have higher rates of homologous recombination due to the absence of classical NHEJ machinery.
4.3.3 Notes
To determine the number of cells to be plated for each radiation dose, a preliminary experiment must be performed where increasing number of cells are plated for each dose of radiation, which yields distinct cell colonies for counting, preferably less than 100 colonies per dish. Typically, we plate X and 2X with the cell number being previously determined by colony-forming assays.
Staining plates with Crystal Violet stain is easy, but care must be taken not to get it on one’s clothes, because it is difficult to remove. It is suggested that a laboratory coat and double gloves be used for the staining procedure.
If possible, it is a good idea to dedicate an incubator to clonogenic cell survival. This avoids unnecessary bumping of colonies by other users when they open and close the door of the incubator. As the colonies grow, bumping the incubator or shelf can cause the cells to detach and settle as new colonies, thereby leading to an increase in the colony count and erroneous results. This is especially true for CHO cells.
Acknowledgments
The authors are supported by the National Institutes of Health grants (CA162804 and CA092584).
References
- Bootsma D, Budke L, Vos O. Studies on synchronous division of tissue culture cells initiated by excess thymidine. Experimental Cell Research. 1964;33:301–309. doi: 10.1016/s0014-4827(64)81035-1. [DOI] [PubMed] [Google Scholar]
- Davis AJ, Chen DJ. A role for ATM kinase activity and Mre11 in microhomology-mediated end-joining. Cell Cycle. 2010;9:3147–3148. doi: 10.4161/cc.9.16.12814. [DOI] [PubMed] [Google Scholar]
- Davis AJ, Chi L, So S, Lee KJ, Mori E, Fattah K, et al. BRCA1 modulates the autophosphorylation status of DNA-PKcs in S phase of the cell cycle. Nucleic Acids Research. 2015;42:11487–11501. doi: 10.1093/nar/gku824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AJ, So S, Chen DJ. Dynamics of the PI3K-like protein kinase members ATM and DNA-PKcs at DNA double strand breaks. Cell Cycle. 2010;9:2529–2536. doi: 10.4161/cc.9.13.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Jeggo PA. The repair and signaling responses to DNA double-strand breaks. Advances in Genetics. 2013;82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Molecular Cell. 2012;47:320–329. doi: 10.1016/j.molcel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Krasieva TB, Kurumizaka H, Chen DJ, Taylor AM, Yokomori K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. The Journal of Cell Biology. 2005;170:341–347. doi: 10.1083/jcb.200411083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Saha J, Sun J, Fattah KR, Wang SC, Jakob B, et al. Phosphorylation of Ku dictates DNA double-strand break (DSB) repair pathway choice in S phase. Nucleic Acids Research. 2016;44:1732–1745. doi: 10.1093/nar/gkv1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi A, Hobbs M, Meyn RE. Clonogenic cell survival assay. Methods inMolecular Medicine. 2005;110:21–28. doi: 10.1385/1-59259-869-2:021. [DOI] [PubMed] [Google Scholar]
- Schipler A, Iliakis G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Research. 2013;41:7589–7605. doi: 10.1093/nar/gkt556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Davis AJ, Fattah KR, So S, Sun J, Lee KJ, et al. Persistently bound Ku at DNA ends attenuates DNA end resection and homologous recombination. DNA Repair (Amst) 2012;11:310–316. doi: 10.1016/j.dnarep.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. The Journal of Cell Biology. 2009;187:977–990. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]