


Abstract
A novel dNTP pyrophosphatase, Mj0226 from Methanococcus jannaschii, which catalyzes the hydrolysis of nucleoside triphosphates to the monophosphate and PPi, has been characterized. Mj0226 protein catalyzes hydrolysis of two major substrates, dITP and XTP, suggesting that the 6-keto group of hypoxanthine and xanthine is critical for interaction with the protein. Under optimal reaction conditions the kcat /Km value for these substrates was ∼10 000 times that with dATP. Neither endonuclease nor 3′-exonuclease activities were detected in this protein. Interestingly, dITP was efficiently inserted opposite a dC residue in a DNA template and four dNTPs were also incorporated opposite a hypoxanthine residue in template DNA by DNA polymerase I. Two protein homologs of Mj0226 from Escherichia coli and Archaeoglobus fulgidus were also cloned and purified. These have catalytic activities similar to Mj0226 protein under optimal conditions. The implications of these results have significance in understanding how homologous proteins, including Mj0226, act biologically in many organisms. It seems likely that Mj0226 and its homologs have a major role in preventing mutations caused by incorporation of dITP and XTP formed spontaneously in the nucleotide pool into DNA. This report is the first identification and functional characterization of an enzyme hydrolyzing non-canonical nucleotides, dITP and XTP.
INTRODUCTION
DNA is at risk of damage from UV light, ionizing radiation, active oxygen species and chemical mutagens (1). Oxidative deamination of DNA primarily converts DNA base amino groups to keto groups (2). Thus, adenine is converted to hypoxanthine (HX), guanine to xanthine (X) and cytosine to uracil (2–4). These bases can trigger an increased mutation rate. HX is biologically important because A:T→G:C transitions are induced at the site when DNA is damaged by oxidative deamination (5,6). Most organisms contain repair enzymes that are specific for these deaminated bases. Endonuclease V attacks DNA containing HX or X (7,8), whereas uracil-DNA N-glycosylase deals effectively with uracils in DNA (9). In addition, two other enzymes found in several organisms, including Escherichia coli, methylpurine DNA N-glycosylase and 3-methyladenine DNA glycosylase, are also active on HX-containing DNA substrates (10,11). However, oxidative deamination of DNA bases occurs not only in duplex DNA strands but also in the free nucleotide pool (12). Previously, some evidence for the presence of ITP (13,14) and an enzyme hydrolyzing ITP, dITP and XTP was presented for human cells (15) and rabbit tissues (16). A possible role for this enzyme in cells is to protect the human genome from incorporation of rogue nucleotides, such as ITP, dITP and XTP, into DNA and RNA (16). The model for the formation and degradation of ITP and IMP, the so-called inosinate cycle, was demonstrated in human cells (17). Moreover, as it is conceivable that oxidative damage to DNA bases occurs even more frequently in the nucleotide pool of organisms than in their chromosomal DNA, HX residues in DNA could be generated by misincorporation of dIMP from dITP in the nucleotide pool during replication (8,18,19).
The initial characterization of a hypothetical protein Mj0226 from a hyperthermophile, Methanococcus jannaschii, identified a novel dNTP pyrophosphatase by a protein structure-based approach (20). This enzyme hydrolyzes non-canonical nucleotides, including dITP, under physiological conditions, suggesting that Mj0226 is an important factor in preventing the misincorporation of aberrant nucleotides into DNA, as are MutT (21,22) and dUTPase (23–25), eliminating 8-oxo-dGTP and dUTP from the nucleotide precursor pools, respectively. Studies of these mutagenic bases have revealed a complex system that both repairs such lesions already in DNA and sanitizes the nucleotide precursor pool, from which 8-oxo-dGTP and dUTP can be incorporated into DNA (22,26). As dITP is also misincorporated into DNA by DNA polymerases (6,19,27), two factors blocking HX misincorporation were presumed to exist. The first is a sanitizing enzyme hydrolyzing the triphosphate nucleotide to the nucleoside monophosphate and pyrophosphate. Secondly, a base excision enzyme, HX N-glycosylase or endonuclease V, removing the HX base. The latter enzyme is already known in many organisms (8,10,11,28), whereas the protein hydrolyzing non-canonical nucleotides such as dITP and XTP to their monophosphates has yet to be identified. In this study we explore the substrate specificity of Mj0226 protein and its biological role in preventing mutagenesis by dITP and XTP.
MATERIALS AND METHODS
Optimal conditions for the nucleotide hydrolysis activity of Mj0226
The nucleotide hydrolysis activity of Mj0226 protein was analyzed in the pH range 6.0–11.5, with dITP as substrate. Mj0226 protein was incubated in various buffers at 80°C. MES buffer was used for pH 6.0–6.5, HEPES buffer for pH 7.0–7.5, Tris–Cl buffer for pH 8.0–8.5, CHES buffer for pH 9.0–9.5 and CAPS buffer for pH 10.0–11.5. The dependence of the nucleotide hydrolysis activity of Mj0226 on temperature was also investigated. Assays of a possible temperature effect were performed at various temperatures (30–90°C). The effects of various metal ions on enzyme activity were also investigated. Mj0226 was incubated with various metal ions at 4 mM concentration at 80°C. All reaction mixtures were analyzed using a Hypersil SAX 5 µ HPLC column (ThermoHypersil, USA) as described previously (20).
Determination of thermal denaturation and thermostability
Thermal denaturation of Mj0226 protein was measured by far-UV circular dichroism (CD) at 222 nm. A J-715 spectropolarimeter (Jasco, Japan) equipped with a temperature controlled liquid system was used. Measurements were performed in a pH 7.5 solution containing 20 mM potassium phosphate and 0.1 mg/ml Mj0226 protein. The thermal melting curve was measured at 1°C intervals with an average time of 60 s at each temperature and the change in ellipticity was monitored at 222 nm. The raw residue molar ellipticity data were transformed into fraction of protein unfolded.
Thermostability of Mj0226 protein was also determined. Mj0226 protein (0.1 mg/ml) was incubated in 20 mM potassium phosphate buffer, pH 7.5, containing 300 mM NaCl for various times at 95°C. Aliquots were withdrawn at periodic intervals and kept on ice. The residual nucleotide hydrolysis activity of aliquots was measured after all samples had been collected. The relative activity of the enzyme was defined as the residual activity of the enzyme compared to the activity of the unheated enzyme.
Preparation of nucleotide substrates
Nucleoside triphosphates (dATP, dTTP, dGTP and dCTP) and 8-oxo-dGTP were from Amersham Pharmacia Biotech (USA). Inosine triphosphate (ITP), dITP, dUTP and 7-deaza-dGTP were purchased from BMS (Germany). Xanthosine triphosphate (XTP), dideoxy-GTP (ddGTP), 7-methyl-GTP, 8-bromo-dATP (8-Br-dATP), 5-Br-dCTP, 5-Br-dUTP, 5′-guanylylimidodiphosphate (GMP-PNP) and guanosine 5′-O-(2-thiotriphosphate) (GTP-γ-S) were products of Sigma (USA). 8-Br-dGTP, N6-etheno-ATP and purine triphosphate (deoxynebularine triphosphate) were obtained from Biolog (France). Other nucleotide analogs were purchased from Trilink Biotech (USA). 6-N-hydroxylaminopurine deoxynucleoside triphosphate (dHAPTP) was synthesized from 6-chloropurine deoxyribose by the method described previously (29), with slight modifications, and purified by two successive column chromatography steps; a Hypersil SAX 5 µ HPLC column and a HiTrap desalting column (Amersham Pharmacia Biotech). About 10 µmol of dHAPTP was finally obtained and stored at –20°C.
Substrate specificity of Mj0226 protein
The specificity of nucleotide hydrolysis by Mj0226 protein was compared using various nucleoside triphosphates. These reactions were performed in 40 µl of reaction mixture containing 25 mM CAPS buffer, pH 10.5, 4 mM MgCl2 and 4.5 pmol Mj0226 protein at 80°C. Rates of hydrolysis of nucleoside triphosphates were determined by time course experiments with 10 different concentrations of the substrates. Amounts of products were measured using a Hypersil SAX 5 µ HPLC column (20). The kinetic parameters were calculated using the program ENZIFITTER (Biosoft).
Endonuclease and 3′→5′ exonuclease activity assays
The endonuclease activity of Mj0226 protein was investigated. A 32mer oligonucleotide containing a single HX residue at position 16 was purchased from Bioneer (Korea). The oligonucleotide duplex sequence (duplex A) used in this work is shown in Table 1. The complementary sequences with A, T, G and C opposite HX were also obtained from Bioneer. The HX-containing oligonucleotide was labeled on the 5′-end with [γ-32P]ATP (Amersham Pharmacia Biotech) using T4 polynucleotide kinase (Takara, Japan) at 37°C. Unincorporated [γ-32P]ATP was removed following purification of the oligonucleotide using a Microspin G-50 column (Amersham Pharmacia Biotech). The duplexes were prepared by annealing with an unlabeled complementary strand at 1.5-fold molar excess in 20 mM Tris–Cl, pH 7.4, 100 mM NaCl, 1 mM DTT, 1 mM EDTA and 3% glycerol. To generate duplex molecules the annealing reaction was heated to 75°C for ∼5 min and slowly cooled to room temperature. The annealed DNA was eluted by ethanol precipitation, dried and resuspended in double-distilled water. DNA cleavage reactions with Mj0226 protein were performed in 40 µl reaction mixtures with Mj0226 protein and 100 pmol radiolabeled 32mer oligonucleotide duplexes. After termination of the reaction with phenol/chloroform and ethanol precipitation, the oligonucleotides were resuspended in 20 µl of formamide loading buffer (containing 0.05% bromophenol blue and 0.05% xylene cyanol). The samples were heated for 5 min at 90°C and subjected to electrophoresis on a denaturing 15% polyacrylamide gel containing 7 M urea in 1× TBE buffer (89 mM Tris, 89 mM boric acid and 2 mM EDTA). The gel was dried and placed on an imaging plate. Intensities of bands in autoradiograms were measured using a BAS2000 image analyzer (Fuji, Japan). Escherichia coli endonuclease V (Trevigen) was used as a control for these reactions.
Table 1. Sequences of oligonucleotides used for the enzyme activity test.
 |
H, hypoxanthine (deoxyinosine); N, one of the four natural deoxynucleosides dA, dT, dG or dC, respectively.
The 3′→5′ exonuclease activity of Mj0226 protein was also investigated. A 5′-end-labeled 21mer oligonucleotide containing a single HX residue at the 3′-end was annealed with a 32mer complementary strand as above (Table 1, duplex B). Exonuclease reactions of Mj0226 protein were performed in the same way as endonuclease reactions. At the same time, Klenow fragment (Promega) was used as a positive control for this reaction.
Insertion fidelity of DNA polymerase I
dITP incorporation into DNA was examined with DNA polymerase I (exonuclease–) (Promega). 5′-End-labeling of 15mer primer DNA and annealing of the primer to template DNA (32mer) were as described above (Table 1, duplex C). DNA synthesis (dITP incorporation) on the template containing dN was carried out using 1 U DNA polymerase I with 100 µM dITP in 20 µl of reaction mixture at 37°C for 15 min. Reaction mixtures were separated on a denaturing 15% polyacrylamide gel containing 7 M urea and autoradiograms were obtained using a BAS2000 image analyzer (Fuji, Japan).
dNTP incorporation opposite HX residues in the DNA template was also tested. 5′-End-labeling of 16mer primer DNA and annealing with template DNA (32mer) containing HX were as described (Table 1, duplex D). dNTP incorporation on the template containing HX was carried out using 1 U Klenow fragment in 20 µl of reaction mixture at 37°C for 15 min. Reaction mixtures contained varying concentrations of dATP, dTTP, dGTP and dCTP.
Expression and purification of protein homologs of Mj0226
Two homologous proteins, Af2237 and an E.coli homolog (accession no. P52061), among several hypothetical proteins homologous to Mj0226, were cloned and purified. All purification procedures for Af2237 from Archaeoglobus fulgidus were the same as those for Mj0226 (20). In the case of the homologous E.coli protein (termed Ec197) the maltose-binding protein (MBP) fusion vector pMal-c2× (New England Biolabs) was used for expression and purification. The amplified DNA fragment of Ec197, obtained using genomic DNA, was digested with EcoRI and SalI and ligated to the pMal-c2× expression vector. The complete coding sequence of Ec197 was amplified by PCR, with the DNA primer pair 5′-GCCTCACGTGAATTCCAAAAAGTTGTCCTCGCA-3′ (underlining indicates the EcoRI site) and 5′-GCTAGGCATGTCGACTTAACCATTACGTAAAGCGTC-3′ (underlining indicates the SalI site). The recombinant plasmid (pMal-Ec197) was introduced into E.coli TB1 for protein expression. Escherichia coli TB1 cells harboring the pMal-Ec197 plasmid were induced to express the MBP–Ec197 fusion protein by addition of IPTG to the growth medium. MBP–Ec197 fusion protein was purified by amylose resin affinity chromatography (New England Biolabs). The fusion protein was cleaved with Factor Xa and Ec197 protein was further purified by MonoQ anion-exchange and Sephacryl S200 gel filtration (0.5 × 150 cm) chromatography. The protein concentration of a homogeneous preparation was determined by the method of Bradford (30), with bovine serum albumin as the standard.
RESULTS
Optimal conditions for the nucleotide hydrolysis activities of Mj0226 protein
We have shown previously that Mj0226 protein has a dNTP pyrophosphatase activity that efficiently hydrolyzes non-canonical nucleotides to dNMP and PPi under physiological conditions (20). In order to investigate the optimal conditions of reaction, the effects of pH, temperature and various metal ions were examined. The effects of pH on the nucleotide hydrolysis activities of Mj0226 protein were analyzed under various pH conditions with the substrate dITP (Fig. 1A). The nucleotide hydrolysis activity of Mj0226 protein has a sharp optimum at alkaline pH. In particular, the reaction rates under neutral conditions were <30% of maximum (Fig. 1A). Dependence of the nucleotide hydrolysis activities of Mj0226 protein on temperature was also investigated. The optimal reaction temperature for Mj0226 protein was 80°C, which is similar to the optimum temperature for growth of M.jannaschii. At low temperature (30°C) Mj0226 protein showed an ∼5-fold decrease in activity compared to the maximum activity at 80°C (Fig. 1B). Nucleotide hydrolysis by Mj0226 protein required divalent cations (Fig. 1C). Magnesium ions were required for optimal activity, with Mn2+, Zn2+ and Ni2+ supporting <50% of the maximum rate.
Figure 1.



Optimal conditions for the nucleotide hydrolysis activities of Mj0226 protein. (A) The activity assay in various pH buffers was performed at 80°C. The following buffers were used: MES buffer, pH 6.0–6.5; HEPES buffer, pH 7.0–7.5; Tris–Cl buffer, pH 8.0–8.5; CHES buffer, pH 9.0–9.5; and CAPS buffer, pH 10.0–11.5. (B) The activity assay was performed at various temperatures in CAPS buffer, pH 10.5. (C) The effect of metal ions was assayed at 80°C with each metal ion (4 mM). Control, reaction without Mj0226 protein; +Mj0226, reaction with Mj0226 protein.
Thermal denaturation and thermostability of Mj0226 protein
Thermal denaturation of Mj0226 protein was examined using a far-UV CD spectropolarimeter. The thermal melting curve was measured at 1°C intervals with an average time of 60 s at each temperature and the change in ellipticity was monitored at 222 nm. Mj0226 protein was stable at high temperatures, but was irreversibly denatured above 83°C (Fig. 2A) when no salt was added to the Mj0226 protein.
Figure 2.


Thermal denaturation and thermostability of Mj0226 protein. (A) Thermal denaturation using CD. Measurements were performed in pH 7.5 buffer containing 20 mM potassium phosphate and 0.1 mg/ml Mj0226 protein at 222 nm. Thermal melting curves were measured at 1°C intervals with an averaging time of 60 s at each temperature and the change in ellipticity was monitored. The raw residue molar ellipticity values were transformed into the fraction of protein unfolded. (B) Thermostability of Mj0226 protein. Mj0226 protein (0.1 mg/ml) was incubated in a pH 7.5 buffer solution containing 20 mM potassium phosphate and 300 mM NaCl at 95°C. Aliquots were withdrawn at periodic intervals and kept on ice and then nucleotide hydrolysis assay was carried out by HPLC. The relative activity is presented as the residual activity of the enzyme compared to the activity of the unheated enzyme control.
The thermostability of Mj0226 protein was investigated by incubation at 95°C, which is a higher temperature than the optimal growth condition of M.jannaschii, in 20 mM potassium phosphate buffer containing 300 mM NaCl for variable times. Aliquots were taken at periodic intervals and the residual activity of each aliquot was measured. As shown in Figure 2B, Mj0226 protein is relatively stable against heat inactivation. The half-life of Mj0226 at 95°C is ∼200 min. Purified Mj0226 protein was found to be remarkably resistant to heat, as are several other proteins from hyperthermophiles (31,32). The optimum salt concentration for thermostability of Mj0226 was examined between 0.02 and 1 M NaCl. The thermostability of Mj0226 protein was highest near 300 mM NaCl (data not shown). An extrinsic factor such as presence of salt could play a role in stabilization of the protein.
Substrate specificity of Mj0226 determined with various nucleotide analogs
Previously, hydrolysis reactions by Mj0226 protein were examined with substrates containing the four canonical nucleotides (20). As Mj0226 protein efficiently hydrolyzed non-canonical nucleotides, we suggested a DNA repair function for Mj0226 protein (20). To support this hypothesis, the reactions of various nucleotide substrates with Mj0226 were tested. The kinetic parameters for hydrolysis of nucleoside triphosphates by Mj0226 protein were measured (Table 2). Of the various nucleoside triphosphates and their analogs, purine nucleotides are the favored substrates for Mj0226 protein, followed by pyrimidine nucleotides. In particular, the hydrolytic efficiency (kcat/Km) for dITP (or ITP) and XTP was >10 000 times higher than that for dATP. Mj0226 protein hardly hydrolyzed the pyrimidine dNTPs, dCTP and dTTP, whereas the purine dGTP (or GTP) was hydrolyzed with modest efficiency. We also examined whether Mj0226 protein showed activity for 8-oxo-dGTP and dUTP, specifically hydrolyzed in vitro by the DNA repair proteins MutT (21,22) and dUTPase (23,24), respectively, but these activities were extremely low (Table 2). Thus, dITP (or ITP) and XTP are the preferred substrates of Mj0226.
Table 2. Comparison of substrate specificity of Mj0226 protein.
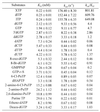 |
Reactions were performed in 40 µl of reaction mixture containing 25 mM CAPS buffer, pH 10.5, 4 mM MgCl2 and 4.5 pmol Mj0226 protein at 80°C. Rates of hydrolysis of nucleoside triphosphates were determined by time course experiments with 10 different concentrations of the substrates. Amounts of products were also measured using a Hypersil SAX 5 µ HPLC column. The kinetic parameters were calculated using the program ENZIFITTER (Biosoft).
Besides the substrates in Table 2, pyrophosphate (for inorganic pyrophosphatase activity), triphosphate (for triphosphatase activity), trimetaphosphate (for trimetaphosphatase activity), FAD (for FAD pyrophosphatase activity) and NAD (for NAD pyrophosphatase activity), which are substrates for other hydrolases acting on phosphorous-containing anhydrides (EC 3.6.1), were examined as substrates for Mj0226 protein, and were not hydrolyzed (data not shown). As Mj0226 protein has an optimum activity under alkaline conditions, the activity of alkaline phosphatase (EC 3.1.3.1), which is active at high pH and has wide specificity, was also tested, with no activity being detected (data not shown).
Does Mj0226 have a DNA endonuclease or error correction function?
Base analogs such as HX (the base of dITP) and X (the base of XTP) can arise in DNA from adenine and guanine by oxidative deamination (2,33). HX residues are mutagenic, since they give rise to A:T→G:C transitions (5,6,34). Mj0226 may act on DNA containing HX residues in two ways; either as an exonuclease (editing activity) removing dIMP from a 3′-terminal mismatch in newly replicated DNA or as an endonuclease, including DNA glycosylase/lyase activity, hydrolyzing the phosphodiester linkage in the DNA duplex. Whether Mj0226 protein has 3′-terminal exonuclease activity was investigated using duplex substrates (Table 1, duplex B) containing dIMP at the 3′-terminus of one strand (Fig. 3A). Since the oligonucleotide containing dIMP was labeled on the 5′-end with [γ-32P]ATP, the distinct electrophoretic mobilities of the oligonucleotide products could be observed. It could be seen that dIMP was not removed from the DNA strand by Mj0226 (Fig. 3A, lanes 3–6), whereas Klenow fragment efficiently hydrolyzed it (Fig. 3A, lane 1).
Figure 3.
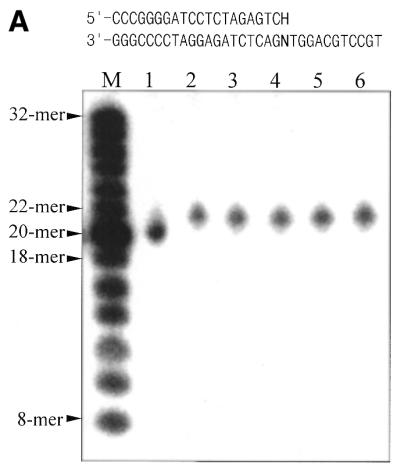
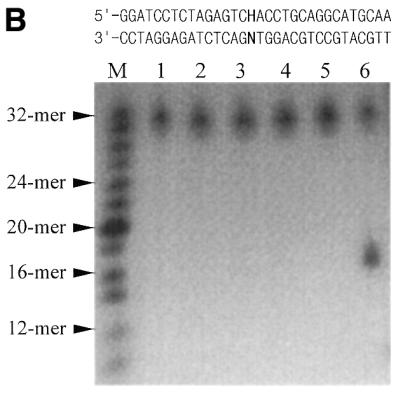
Exonuclease and endonuclease activity assay of Mj0226 protein. (A) 3′-Exonuclease activity assay of Mj0226. Mj0226 protein was incubated with 100 pmol 5′-labeled HX-containing duplex (top sequences) for 30 min. The products were analyzed by denaturing PAGE using a BAS2000 image analyzer. Lane M, size marker; lane 1, HX:C incubated with Klenow fragment; lane 2, HX:C only; lanes 3–6, HX:A, T, G and C, respectively, incubated with Mj0226. (B) Endonuclease activity assay of Mj0226. Mj0226 protein was incubated with 100 pmol 5′-labeled HX-containing duplex (top sequences) for 30 min. The products were analyzed by denaturing PAGE using a BAS2000 image analyzer. Lane M, size marker; lane 1, HX:T only; lanes 2–5, HX:A, T, G and C, respectively, incubated with Mj0226; lane 6, HX:T incubated with E.coli endonuclease V.
Endonuclease activities of Mj0226 protein were also tested, similarly to the exonuclease experiments. The DNA strand cleavage activity of Mj0226 was examined using a 32mer oligonucleotide duplex (Table 1, duplex A) containing a single HX:N pair (Fig. 3B). Strand cleavage activity of Mj0226 protein was not observed for any of the HX-containing strands (HX:A, HX:T, HX:G and HX:C) (Fig. 3B, lanes 2–5), whereas E.coli endonuclease V showed HX-containing strand cleavage with the HX:T pair-containing duplex oligonucleotide (Fig. 3B, lane 6).
DNA replication fidelity of DNA polymerase I
As HX is mutagenic in some organisms (2,5,34), we tested the behavior of dITP in a polymerase system. Using the polymerase subunit of E.coli DNA polymerase I (exonuclease–), incorporation of dITP into synthetic DNA templates was examined. 5′-End-labeled 15mer primer DNA was annealed to template DNA (Table 1, duplex C) and polymerized with dITP by DNA polymerase I. dITP was efficiently inserted opposite dC residues in the template (Fig. 4A), with no evidence of incorporation of dITP opposite dA, dT or dG. This result agrees with that reported by Thomas et al. (27).
Figure 4.


DNA replication fidelity of DNA polymerase I. (A) dITP incorporation opposite dN residues of template DNA by DNA polymerase I. 5′-End labeling of 15mer primer DNA annealed to template DNA (top sequences) was used. dITP incorporation into the dN template was with 1 U DNA polymerase (exonuclease–) and 100 µM dITP in 20 µl of reaction mixture at 37°C for 15 min. Reaction mixtures were separated by denaturing PAGE and autoradiograms were obtained using a BAS2000 image analyzer. (B) dNTP incorporation opposite HX residues of template DNA by DNA polymerase I. 5′-End-labeling of 16mer primer DNA annealed to template DNAs (top sequences) was used in this experiment. dNTP incorporation into the HX template was with 1 U Klenow fragment and 1–100 µM dNTP in 20 µl of reaction mixture at 37°C for 15 min. Lane C, control; lane 1, 1 µM dNTP; lane 2, 10 µM dNTP; lane 3, 100 µM dNTP.
dNTP incorporation opposite HX residues in template DNA was also tested. dNTP incorporation by DNA polymerase I was assayed with 5′-end-labeled 16mer primer DNA annealed to template DNA containing HX (Table 1, duplex D). In experiments using the Klenow fragment all four dNTPs were incorporated opposite HX (Fig. 4B). The 3′→5′ exonuclease proof-reading function of DNA polymerase I does not recognize dN:HX during incorporation of dNTPs (Fig. 4B).
Nucleotide hydrolysis activity of homologs of Mj0226
By analysis of multiple sequence alignments, protein homologs of Mj0226 were found in three superkingdoms (eukaryotes, prokaryotes and Archaea) (20). The soluble proteins A.fulgidus Af2237 and E.coli Ec197, which are homologs of Mj0226, were purified (Fig. 5). All expression and purification steps for Af2237 protein were the same as for Mj0226, as previously reported (20). To obtain the soluble fraction of Ec197 protein, a pMal-c2× MBP fusion vector system was used as described in Materials and Methods, because it was insoluble in the pET system. The final preparations of all proteins produced single bands on SDS–PAGE with >95% purity (Fig. 5).
Figure 5.

Purification of homologs of Mj0226. Two homologs of Mj0226 from E.coli and A.fulgidus were cloned and purified as described in Materials and Methods. Lane M, molecular weight marker; lane 1, Mj0226 from M.jannaschii; lane 2, Af2237 from A.fulgidus; lane 3, Ec197 from E.coli.
The nucleotide hydrolysis activities of these homologous proteins were also tested against several nucleotide substrates. As expected, these proteins possess nucleotide hydrolysis activities and their specificities are similar to that of Mj0226 protein (Table 3). Like Mj0226 protein, these homologous proteins showed maximum activity under alkaline conditions (data not shown). As a result, the biochemical function of Mj0226 protein was confirmed with certainty and these hypothetical proteins, including Mj0226, may be indispensable for a part of cellular metabolism in all organisms.
Table 3. Nucleotide hydrolysis activity of protein homologs of Mj0226.
 |
Af2237 and Ec197 are hypothetical proteins from A.fulgidus and E.coli, respectively. The activity assays of Mj0226 and Af2237 were performed at 80°C. The activity of Ec197 was measured at 37°C.
DISCUSSION
It has been observed that Mj0226 protein has pyrophosphate-releasing activity by hydrolysis of nucleoside triphosphates (20). An analogous reaction has been shown with the enzymes 8-oxo-dGTPase (MutT protein) (21,22) and dUTPase (23–25), removing inappropriate members from the nucleoside triphosphate pool. It is notable that these enzymes release PPi from their respective substrates in the same way as Mj0226. The importance of the dUTPase function in E.coli and eukaryotic systems has been firmly established (23–25), whereas MutT is important in preventing misincorporation of 8-oxo-dGTP (21). 8-oxo-dGTP is hydrolyzed faster than dGTP, making it the likely preferred biological substrate for MutT (22). Mj0226 protein acts like these repair enzymes, so the preferred substrates of this protein are likely to be in the nucleotide pool, before they are misincorporated into DNA. Therefore, identification of the specific substrate of Mj0226 protein is of great significance.
The data in Table 2 show the results for several nucleoside triphosphates in order to assess the relative importance of the various functional groups on hydrolysis rate. Of the non-standard nucleotides, Mj0226 showed a remarkably higher activity for dITP (or ITP) and XTP, suggesting that the O6 atom (6-keto group) of these nucleotides is critical for the reactivity of Mj0226 (Fig. 6). The O6 atoms of X, HX and guanine can form a hydrogen bond to the Nδ2 of Asn19 of Mj0226 with a slight conformational change (20). Although dGTP also has a 6-keto group, its hydrolysis rate is ∼150- to 180-fold less relative to those of dITP and XTP (Fig. 6), indicating that the exocyclic 2-amino group of guanine likely reduces reactivity of dGTP with the protein.
Figure 6.

Comparison of base structure of nucleoside triphosphates tested as substrates for Mj0226. The values in parentheses (%) are activity of Mj0226 with the substrates relative to that of dITP. PuTP, purine triphosphate.
The sugar ring structure of the substrate is less important, as indicated by the similar reactivities of dITP and ITP (Table 2). With respect to the phosphate linkage, Mj0226 protein is highly specific for a phosphoanhydride bond of triphosphate form. The modified form of the phosphate linkage, as in GMP-PNP and GTP-γ-S, was not hydrolyzed by Mj0226 (Table 2). The di- and monophosphates are entirely resistant and Mj0226 cannot hydrolyze the phosphodiester linkages in an oligonucleotide strand containing a HX residue (data not shown). Thus, dITP (or ITP) and XTP are the specific substrates for Mj0226 protein.
Mj0226 protein also showed no activity with some chemical mutagens, such as 2-aminopurine triphosphate, 2,6-diaminopurine triphosphate and dHAPTP (Table 2 and Fig. 6). These nucleotide analogs, it is believed, induce mistakes during replication (35–38). Although the structure of these nucleotide analogs is similar to dITP (or XTP), they were not hydrolyzed by Mj0226 protein due to lack of a 6-keto group.
While the mutagenic mechanism of hypoxanthine already incorporated into DNA is clear (2,33), the precise mutagenic mechanisms of incorporation of dITP into DNA templates opposite dN and vice versa remain interesting questions. dITP was incorporated into DNA by DNA polymerase I in vitro instead of dGTP and, less efficiently, instead of dATP, dCTP and dTTP (Fig. 4A). These results confirm those of Thomas et al. (27), while in earlier reports an in vitro interaction between HX and dN in synthetic DNA has also been observed (39–41). The basis for these results is that the HX residue can form base pairs with multiple bases as well as cytosine. We also observed that HX, when present in template DNA, directs incorporation of all four dNTPs by Klenow fragment with similar rates (Fig. 4B). It is proposed that initial incorporation of dITP opposite dC leads to mutation generation in the next replication cycle with high probability, as random dNTPs would be incorporated opposite HX. Thus it is supposed that dITP incorporation would generate transversion as well as transition mutations. However, HX in DNA can occur not only by spontaneous deamination of dA residues but also by misincorporation of dIMP from dITP during replication by DNA polymerases (6,19). Also, the mutagenic properties of dITP have been ascertained by site-specific mutagenensis in vivo (5,34). Therefore, HX mutagenesis might also occur by incorporation of dITP, although it is likely to be quantitatively less important. These results suggest that a major biological function of Mj0226 protein is to remove dITP from the nucleotide pool. Since the fidelity of the polymerization reaction may in part depend on the fidelity of various DNA polymerases, studies on the fidelity of dITP incorporation with a variety of DNA polymerases are on-going. Although DNA polymerase insertion data using an oligonucleotide containing xanthosine were not obtained, we presume that DNA polymerase fidelity with XTP is similar to that with dITP, as some evidence of mutagenesis by xanthine has been reported (42–45).
How does Mj0226 protein act on DNA? The first possibility is that it could act as an exonuclease, removing dIMP from a terminal dN:dI mismatch. This was examined as described in Figure 3A. It can be seen that dIMP was not removed from the oligonucleotide duplex by Mj0226 protein, whereas Klenow fragment did hydrolyze it. Thus, Mj0226 protein does not have an error correction function. The second possibility is that Mj0226 protein has endonuclease activity, namely HX-specific endonuclease activity. The endonuclease activity of Mj0226 protein was also tested, similarly to the exonuclease experiments, but Mj0226 does not have endonuclease activity (Fig. 3B). This HX residue can be removed by some DNA repair enzymes, DNA glycosylases (10,11) and endonuclease V (8,28,46). A third possibility is that Mj0226 protein prevents formation of dN:dI mispairs during DNA synthesis by sanitizing dITP in the nucleotide pool. This sanitizing reaction has been seen for MutT (21,22,47) and dUTPase (23–25,48), removing unwelcome nucleotides from the nucleotide pool. From the distinct nucleoside triphosphate hydrolysis by Mj0226 protein it seems that the biological function of Mj0226 protein is to prevent misincorporation of dITP and XTP into DNA.
We have shown, using multiple alignment, that hypothetical homologs of Mj0226 exist in other organisms (20). Their functions have been unknown until now and, interestingly, the amino acid sizes of these proteins are similar and several regions are conserved (20). These results suggest that the biological function of the homologous proteins is likely similar to that of Mj0226 protein. Two homologs of Mj0226 were purified (Fig. 5) and their biochemical functions were verified (Table 3). Many organisms have a significant proportion of their gene products involved in a variety of repair mechanisms. It seems that Mj0226 protein and its homologs from other organisms have a major role in preventing mutation caused by misincorporation into DNA of dITP and XTP, which are formed spontaneously in the nucleotide pool.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Clifford D. Mol of the Scripps Research Institute for his participation in preparation of the manuscript. This work was supported by an International Cooperation Research Program of the Ministry of Science and Technology, Korea.
References
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 2.Shapiro R. and Pohl,S.H. (1968) The reaction of ribonucleosides with nitrous acid. Side products and kinetics. Biochemistry, 7, 448–455. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T. (1979) DNA glycosylases, endonucleases for apurinic/apyrimidinic sites and base excision-repair. Prog. Nucleic Acid Res. Mol. Biol., 22, 135–192. [DOI] [PubMed] [Google Scholar]
- 4.Schuster H. (1960) The reaction of nitrous acid with deoxyribonucleic acid. Biochem. Biophys. Res. Commun., 2, 320–323. [Google Scholar]
- 5.Hill-Perkins M., Jones,M.D. and Karran,P. (1986) Site-specific mutagenesis in vivo by single methylated or deaminated purine bases. Mutat. Res., 162, 153–163. [DOI] [PubMed] [Google Scholar]
- 6.Karran P. and Lindahl,T. (1980) Hypoxanthine in deoxyribonucleic acid: generation by heat-induced hydrolysis of adenine residues and release in free form by a deoxyribonucleic acid glycosylase from calf thymus. Biochemistry, 19, 6005–6011. [DOI] [PubMed] [Google Scholar]
- 7.Yao M. and Kow,Y.W. (1997) Further characterization of Escherichia coli endonuclease V. J. Biol. Chem., 272, 30774–30779. [DOI] [PubMed] [Google Scholar]
- 8.He B., Qing,H. and Kow,Y.W. (2000) Deoxyxanthosine in DNA is repaired by Escherichia coli endonuclease V. Mutat. Res., 459, 109–114. [DOI] [PubMed] [Google Scholar]
- 9.Duncan B.K. (1981) DNA glycosylases. In Boyer,P.D. (ed.), The Enzymes, Vol. 14. Academic Press, New York, NY, pp. 565–586.
- 10.Asaeda A., Ide,H., Asagoshi,K., Matsuyama,S., Tano,K., Marakami,A., Takamori,Y. and Kubo,K. (2000) Substrate specificity of human methylpurine DNA N-glycosylase. Biochemistry, 39, 1959–1965. [DOI] [PubMed] [Google Scholar]
- 11.Saparbaev M., Mani,J.C. and Laval,J. (2000) Interactions of the human, rat, S.cerevisiae and E.coli 3-methyladenine DNA glycosylases with DNA containing dIMP residues. Nucleic Acids Res., 28, 1332–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bianchi V., Pontis,E. and Reichard,P. (1986) Changes of deoxyribonucleoside triphosphate pools induced by hydroxyurea and their relation to DNA synthesis. J. Biol. Chem., 261, 16037–16042. [PubMed] [Google Scholar]
- 13.Vanderheiden B.S. (1969) Genetic studies of human erythrocyte inosine triphosphatase. Biochem. Genet., 3, 289–297. [DOI] [PubMed] [Google Scholar]
- 14.van Waeg G., Niklasson,F., Ericson,A. and de Verdier,C.H. (1988) Purine metabolism in normal and ITP-pyrophosphohydrolase-deficient human erythrocytes. Clin. Chim. Acta, 171, 279–292. [DOI] [PubMed] [Google Scholar]
- 15.Holmes S.L., Turner,B.M. and Hirschhorn,K. (1979) Human inosine triphosphatase: catalytic properties and population studies. Clin. Chim. Acta, 97, 143–153. [DOI] [PubMed] [Google Scholar]
- 16.Wang J.K. and Morris,A.J. (1974) The distribution of nucleoside triphosphate pyrophosphohydrolase in the tissues of the rabbit. Arch. Biochem. Biophys., 161, 118–124. [Google Scholar]
- 17.Duley J.A., Simmonds,H.A., Hopkinson,D.A. and Levinsky,R.J. (1990) Inosine triphosphate pyrophosphohydrolase deficiency in a kindred with adenosine deaminase deficiency. Clin. Chim. Acta, 188, 243–252. [DOI] [PubMed] [Google Scholar]
- 18.Seeberg E. and Kleppe,K. (1981) Chromosome Damage and Repair. Plenum Press, New York, NY.
- 19.Myrnes B., Guddal,P.H. and Krokan,H. (1982) Metabolism of dITP in HeLa cell extracts, incorporation into DNA by isolated nuclei and release of hypoxanthine from DNA by a hypoxanthine-DNA glycosylase activity. Nucleic Acids Res., 10, 3693–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang K.Y., Chung,J.H., Kim,S.-H., Han,Y.S. and Cho,Y. (1999) Structure-based identification of a novel NTPase from Methanococcus jannaschii. Nature Struct. Biol., 6, 691–696. [DOI] [PubMed] [Google Scholar]
- 21.Bridges B.A. (1997) MutT prevents leakiness. Science, 278, 78–79. [DOI] [PubMed] [Google Scholar]
- 22.Maki H. and Sekiguchi,M. (1992) MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature, 355, 273–275. [DOI] [PubMed] [Google Scholar]
- 23.McIntosh E.M., Ager,D.D., Gadsden,M.H. and Haynes,R.H. (1992) Human dUTP pyrophosphatase: cDNA sequence and potential biological importance of the enzyme. Proc. Natl Acad. Sci. USA, 89, 8020–8024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gadsden M.H., McIntosh,E.M., Game,J.C., Wilson,P.J. and Haynes,R.H. (1993) dUTP pyrophosphatase is an essential enzyme in Saccharomyces cerevisiae. EMBO J., 12, 4425–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larsson G., Nyman,P.O. and Kvassman,J.-O. (1996) Kinetic characterization of dUTPase from Escherichia coli. J. Biol. Chem., 271, 24010–24016. [DOI] [PubMed] [Google Scholar]
- 26.Warner H.R., Duncan,B.K., Garrett,C. and Neuhard,J. (1981) Synthesis and metabolism of uracil-containing deoxyribonucleic acid in Escherichia coli. J. Bacteriol., 145, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas K.R., Manlapaz-Ramos,P., Lundquist,R. and Olivera,B.M. (1978) Formation of okazaki pieces at the Escherichia coli replication fork invitro. Cold Spring Harbor Symp. Quant. Biol., 43, 231–237. [DOI] [PubMed] [Google Scholar]
- 28.Yao M., Hatahet,Z., Melamede,R.J. and Kow,Y.W. (1994) Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3′-endonuclease, from Escherichia coli. J. Biol. Chem., 269, 16260–16268. [PubMed] [Google Scholar]
- 29.Abdul-Masih M.T. and Bessman,M.J. (1986) Biochemical studies on the mutagen, 6-N-hydroxylaminopurine. J. Biol. Chem., 261, 2020–2026. [PubMed] [Google Scholar]
- 30.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of dye-binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 31.DeDecker B.S., O’Brien,R., Fleming,P.J., Geiger,J.H., Jackson,S.P. and Sigler,P.B. (1996) The crystal structure of a hyperthermophilic archaeal TATA-box binding protein. J. Mol. Biol., 264, 1072–1084. [DOI] [PubMed] [Google Scholar]
- 32.Klump H., DiRuggiero,J., Kessel,M., Park,J.B., Adams,M.W. and Robb,E.T. (1992) Glutamate dehydrogenase from the hyperthermophilic Pyrococcus furiosus: thermal denaturation and activation. J. Biol. Chem., 267, 22681–22685. [PubMed] [Google Scholar]
- 33.Karran P. and Lindahl,T. (1978) Enzymatic excision of free hypoxanthine from polydeoxynucleotides and DNA containing deoxyinosine monophosphate residues. J. Biol. Chem., 253, 5877–5879. [PubMed] [Google Scholar]
- 34.Kamiya H., Miura,H., Kato,H., Nishimura,S. and Ohtsuka,E. (1992) Induction of mutation of a synthetic c-Ha-ras gene containing hypoxanthine. Cancer Res., 52, 1836–1839. [PubMed] [Google Scholar]
- 35.Ronen A. (1979) 2-Aminopurine. Mutat. Res., 75, 1–47. [DOI] [PubMed] [Google Scholar]
- 36.Barrett J.C. (1981) Induction of gene mutation in and cell transformation of mammalian cells by modified purines: 2-aminopurine and 6-N-hydroxylaminopurine. Proc. Natl Acad. Sci. USA, 78, 5685–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray V. (1987) Transversion-specific purine analogue mutagens and the mechanism of hydroxylaminopurine mutagenesis. Mutat. Res., 177, 189–199. [DOI] [PubMed] [Google Scholar]
- 38.Shcherbakova P.V., Noskov,V.N., Pshenichnov,M.R. and Pavlov,Y.I. (1996) Base analog 6-N-hydroxylaminopurine mutagenesis in the yeast Saccharomyces cerevisiae is controlled by replicative DNA polymerases. Mutat. Res., 369, 33–44. [DOI] [PubMed] [Google Scholar]
- 39.Leonard G.A., Booth,E.D., Hunter,W.M. and Brown,T. (1992) The conformational variability of an adenosine-inosine base pair in a synthetic DNA dodecamer. Nucleic Acids Res., 20, 4753–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oda Y., Uesugi,S., Ikehara,M., Kawase,Y. and Ohtsuka,E. (1987) NMR studies for identification of dI:dG mismatch base-pairing structure in DNA. Nucleic Acids Res., 19, 5263–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uesugi S., Oda,Y., Ikehara,M., Kawase,Y. and Ohtsuka,E. (1987) Identification of I:A mismatch base-pairing structure in DNA. J. Biol. Chem., 262, 6965–6968. [PubMed] [Google Scholar]
- 42.Michelson A.M. and Monny,C. (1966) Polynucleotide analogues. IX. Poly-xanthylic acid. Biochim. Biophys. Acta, 129, 460–474. [PubMed] [Google Scholar]
- 43.Vanderbilt A.S. and Tessman,I. (1970) Identification of the altered bases in mutated single-stranded DNA. IV. Nitrous acid induction of the transitions guanine to adenine and thymine to cytosine. Genetics, 66, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eritja R., Horowitz,D.M., Walker,P.A., Ziehler-Martin,J.P., Boosalis,M.S., Goodman,M.F., Itakura,K. and Kaplan,B.E. (1986) Synthesis and properties of oligonucleotides containing 2′-deoxynebularine and 2′-deoxyxanthosine. Nucleic Acids Res., 14, 8135–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamiya H., Sakaguchi,T., Murata,N., Fujimuro,M., Miura,H., Ishikawa,H., Shimizu,M., Inoue,H., Nishimura,S., Matsukage,A. et al. (1992) In vitro replication study of modified bases in ras sequence. Chem. Pharm. Bull., 40, 2792–2795. [DOI] [PubMed] [Google Scholar]
- 46.Schouten K.A. and Weiss,B. (1999) Endonuclease V protects Escherichia coli against specific mutations caused by nitrous acid. Mutat. Res., 435, 245–254. [DOI] [PubMed] [Google Scholar]
- 47.Fowler R.G. and Schaaper,R.M. (1997) The role of the mutT gene of Escherichia coli in maintaining replication fidelity. FEMS Microbiol. Rev., 21, 43–54. [DOI] [PubMed] [Google Scholar]
- 48.Harris J.M., McIntosh,E.M. and Muscat,E.O. (1999) Structure/function analysis of a dUTPase: catalytic mechanism of a potential chemotherapeutic target. J. Mol. Biol., 288, 275–287. [DOI] [PubMed] [Google Scholar]