

Abstract
[18F]THK-523 and [18F]807 are promising radioligands for imaging neurofibrillary tangles (NFTs) with positron emission tomography (PET) in neurodegenerative diseases, such as Alzheimer’s disease (AD) and traumatic brain injury. Although [18F]THK-523 and [18F]T807 are considered high-affinity selective radioligands for NFTs, uncertainty has existed as to whether PET radioligands for imaging NFTs bind to the same molecular site because in vitro assays for ligands binding to NFTs have been lacking. We labeled THK-523 and T807 with tritium to serve as reference radioligands for in vitro binding assays with AD brain homogenates for newly synthesized ligands. With these radioligands, we identified two distinct binding sites for small molecules, one site with high affinity for THK-523 and the other with high affinity for T807. Moreover, binding assays with [3H]PIB confirmed that the two newly identified binding sites are also distinct from the thioflavin-T binding site where all current clinically useful PET radioligands for imaging β-amyloid plaque bind with high affinity. The two newly identified binding sites are considered to reside on NFTs rather than on β-amyloid plaques. Furthermore, we applied all three binding assays to a set of newly prepared compounds, based on chain modifications to THK-523. Some compounds with high affinity and selectivity for the THK-523 binding site emerged from this set, including one with amenability to labeling with fluorine-18, namely, ligand 10b.
Keywords: Radioligand, in vitro assay, tangle, tau, Alzheimer’s disease, PET
Graphical abstract
Alzheimer’s disease (AD) and other tauopathies constitute an increasingly enormous societal burden in terms of mortality, morbidity and cost, amidst a continuing absence of effective treatment strategies. Brain neurofibrillary tangles (NFTs) and neuritic plaques are two pathological hallmarks of AD.1 NFTs are generated from abnormally hyperphosphorylated tau protein, whereas neuritic plaques are comprised primarily of β-amyloid. Human postmortem studies indicate that NFT deposition, unlike β-amyloid plaque deposition, correlates well with neurodegeneration and cognitive impairment.2–7 Furthermore, abundant NFTs are not observed in individuals who are cognitively unimpaired,5 in contrast to β-amyloid plaques which are nonetheless observed in some nondemented people.8–11 Moreover, tau and phospho-tau (ptau181) residing in cerebrospinal fluid are known to be useful biomarkers for the diagnosis of AD.12–15
NFTs are also characteristic of many other tauopathies besides AD, such as sporadic corticobasal degeneration, progressive supranuclear palsy, Pick’s disease, frontotemporal dementia Parkinsonism linked to chromosome 17, and Guamanian Parkinsonism dementia-amyotrophic lateral sclerosis complex.16 Tau aggregates are also the pathological hallmark of chronic traumatic encephalopathy, a neurodegenerative disorder that occurs as a consequence of traumatic brain injury.17 Aggregation of tau also arises from numerous forms of neuronal injury, including hypoxia, hyperglycemia, and heat shock.
Positron emission tomography (PET) may detect and quantify a low-density protein in living human brain when this imaging technique is used with an effective radioligand that penetrates brain to provide a robust and adequately high specific signal. Such radioligands are invariably labeled with either of two cyclotron-produced short-lived positron-emitters, namely, carbon-11 (t1/2 = 20.4 min) or fluorine-18 (t1/2 = 109.8 min). Over the past decade and a half, significant progress has been made on developing radioligands for PET imaging of β-amyloid plaques in diseased human brain (Chart 1). The radioligand from Eli Lilly, Amyvid (Florbetapir; [18F]AV-45),18,19 is the first approved by the United States Food and Drug Administration (FDA) for human use. GE’s Vizamyl (Flutemetamol; [18F]3′-F-PIB; GE-067)20 is also now approved by the FDA. NeuraCeq (Florbetaben; [18F]AV-1; [18F]BAY94-9172)21 and [18F]AZD469422 are also proving clinically useful. Each of these radioligands, and also the progenitor radioligand [11C]PIB,23 binds with high affinity to the so-called “thioflavin-T binding site” on β-amyloid plaques.
Chart 1.
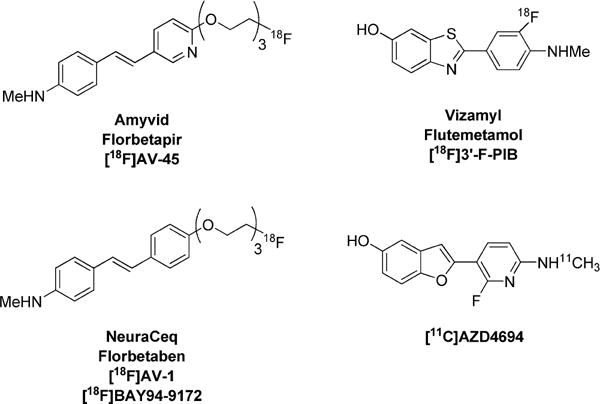
Clinically Useful PET Radioligands for Imaging β-Amyloid Plaques
By contrast with the PET imaging of β-amyloid plaques, the PET imaging of NFTs is much less advanced, mainly because candidate PET radioligands are still limited in efficacy.24,25 A high-performing PET radioligand for imaging brain NFTs would be useful not only to identify areas of neuronal damage but also to monitor the effectiveness of experimental therapies in AD, CTE, and other tauopathies. Indeed, several drugs that block phosphorylation and/or aggregation of tau are being developed for the possible treatment of these diseases.
Recent publications on [11C]PBB3,26 [18F]T807 (AV-1451),27 [18F]T808,28 and [18F]THK-52325 (Chart 2) suggest that these radioligands have at least moderate selectivity for binding to NFTs over β-amyloid plaques, primarily on the basis of evidence gleaned from in vitro autoradiography of postmortem human brain. [11C]PBB3,26 [18F]T807,27 and [18F]T80828 have also shown moderate NFT-specific signals in PET imaging of human subjects. However, [11C]PBB3 suffers from photoisomerization, which poses difficulties for regular practical application. Based on the initial report,25 [18F]THK-523 gives a low PET signal and may suffer some other undesirable properties, such as low brain entry and radiodefluorination. However, radioligands that are structurally related to [18F]THK-523, namely, [18F]THK-511729 and [18F]THK-535130 (Chart 2), have recently entered clinical trials. Therefore, we also selected THK-523 as a lead compound in our search for more promising PET radioligands for imaging NFTs.
Chart 2.
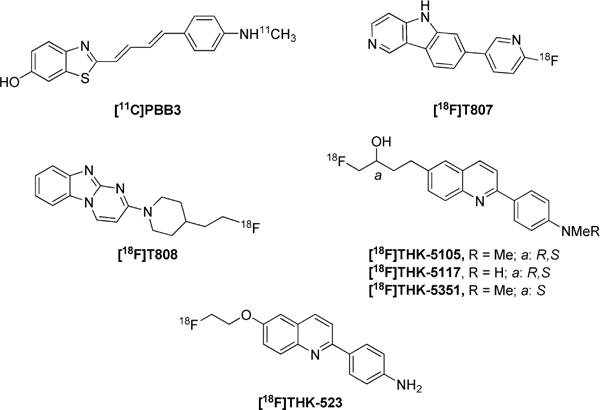
Candidate PET Radioligands for Imaging NFTs
Multiple binding sites for PET radioligands that target β-amyloid plaques have been identified.31,32 This suggested to us that more than one ligand binding site might also exist in NFTs. Therefore, we produced both [3H]T807 and [3H]THK-523 to serve as reference radioligands for our in vitro assays on new candidate ligands for NFTs. As a result, we discovered that the binding sites for these two radioligands are distinct from each other, and that they are also distinct from the β-amyloid plaque binding site for thioflavins, such as [3H/11C]PIB. In addition, a limited medicinal chemistry effort, based on chain extension and modification of THK-523, provided ligands with moderate lipophilicity and with high affinity and selectivity for the identified THK-523 binding site, including 10b which is amenable to labeling with fluorine-18.
RESULTS
Chemistry
Ligand Synthesis
Specifically, we wished to explore whether the affinity of THK-523 for binding to NFTs could be enhanced by extension of its pendant 6-(2-fluoroethoxy) group and by structural variation at its terminus. The use of oxyethylene units to extend chains has been implemented previously as a promising strategy in PET radioligand development for imaging β-amyloid plaques, as apparent in the structures of Florbetapir19 and Florbetaben21 (Chart 1). Especially, this strategy allows radioligand lipophilicity to be kept within a desirably moderate range and can also be used to retain a terminal site for labeling with fluorine-18. We further considered that variation in the terminal substituent of the pendant chain, especially with a bulkier group having hydrogen bond acceptors or donors, might enhance binding affinity with NFTs through extra binding interactions. Therefore, we devised a general strategy to prepare THK-523 derivatives featuring chains composed of a defined number of oxyethylene units (1–6) and with a fluoro, hydroxy, or larger substituent as a chain terminus. Our synthetic strategy was based on commercially available 2-chloro-6-methoxyquinoline (1) as starting material.
The first stage in the overall synthesis program was to replace the methoxy group in 1 with one or more oxyethylene units (Scheme 1). The phenol 2 was obtained in moderate yield (47%) by demethylation of 1 with boron tribromide. Various poly(oxyethylene)-bis-alcohols were monotosylated to give 3b–f in low to moderate yields (29–53%). Treatment of 2 with the bromo compound 3a or with one of the tosylates gave the hydroxyl terminated compounds with the desired number of oxyethylene units (4a–f) in good to excellent yields, except for 4a (27%). This approach was also explored for preparing analogous compounds with a fluoro terminus, but succeeded only for compound 5 having a pendant 6-(2-fluoroethoxy) chain. Nonetheless, 5 was obtained in virtually quantitative yield (Scheme 1).
Scheme 1.
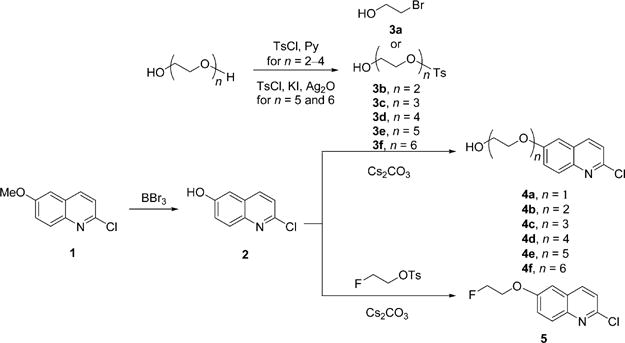
Synthesis of Hydroxy Compounds 4a–f and Fluoro Compound 5
The next stage aimed to cross couple the prepared chloroquinolines (4a–f and 5) with 4-(Boc-amino)-phenylboronic acid through Suzuki reactions. The preparation of the methoxy compound 6 from 1 (Scheme 2) was used as a model for establishing effective coupling conditions. We tried various combinations of phosphine ligand, palladium compound, and solvent, including 2′-phenyl-phenyl di-t-butyl phosphine plus Pd(OAc)2 in THF or DMF, 1,1′-bis-(diisopropylphosphino)ferrocene plus Pd2(dba)3 in toluene, and Pd(PPh3)4 plus K2CO3 in DME-H2O. The latter combination was most effective, giving virtually quantitative yield of 6. K2CO3 was necessary for the reaction to proceed, and the proportion of water had to be kept below 50%. These coupling conditions delivered the N-protected hydroxy-terminated compounds (7a–f) plus the N-protected fluoro compound 8 in good to excellent yields (60–99%). Compounds 7a–f were readily converted into their tosylates (9a–f) in excellent yields (83–90%) by treatment with tosyl chloride. Our prior attempts to insert the tosylate group into these compounds before Suzuki coupling did not succeed. Treatment of 8 with TFA gave THK-523 (10a) in quantitative yield (Scheme 2).
Scheme 2.
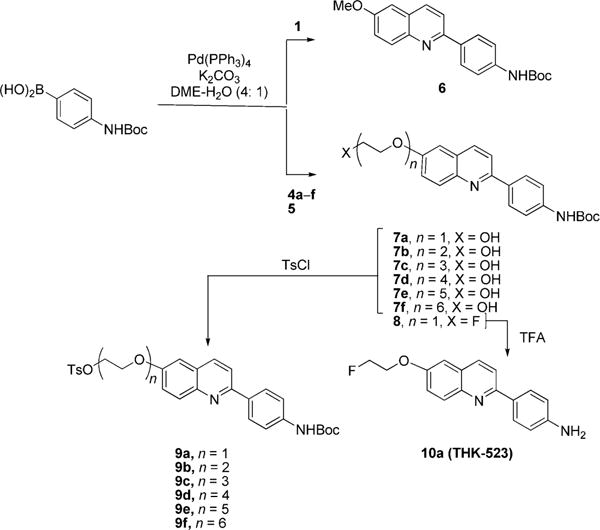
Syntheses of Tosylates 9a–f and THK-523 (10a)
Treatment of the N-protected tosylates 9b–f with TBAF in THF gave the target fluoro ligands 10b–f in moderate to high yield (40–92%), without the need for a separate deprotection reaction (Scheme 3). The small amount of water in the commercial reagent was found to be critical for the success of these syntheses. If water was rigorously excluded from the reactions, products were obtained with a lower number of oxyethylene units.
Scheme 3.
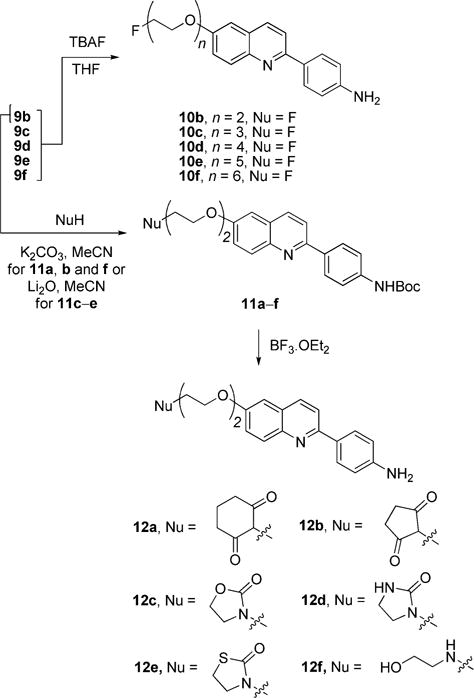
Syntheses of THK-523 Analogues with Poly(oxyethylene) Chains and Various Terminal Groups
Treatment of the Boc-protected tosylate 9b with cyclohexane-1,3-dione, cyclopentane-1,3-dione or 2-aminoethanol in acetonitrile with potassium carbonate as base gave the variously chain-terminated compounds 11a (79%), 11b (59%) and 11f (21%), respectively (Scheme 3). Treatment of 9b with base plus one of the ketones, oxazolidin-2-one, imidazolidin-2-one or thiazoline-2-one gave the compounds 11c–e with alternative bulky terminal groups in a similar range of yields (21–59%). The Boc group in each of the compounds 11a–f was removed readily with BF3·OEt2 to give the target ligands 12a–f in 35–72% yields (Scheme 3).
Synthesis of Halo Precursors for Tritiation
Treatment of THK-523 (10a) with NIS in chloroform–methanol at room temperature gave compound 13 with an iodo group at each position ortho to the free amino group (Scheme 4). Any protective group at the amino group, such as an acetyl or formyl group, prevented iodination. We found that the two iodo substituents in 13 could be removed with hydrogen in the presence of Pd/C as catalyst to regenerate THK-523. Thus, [3H]THK-523 was obtained by treating 13 with tritium gas as a custom synthesis (Moravek Biochemicals & Radiochemicals, Inc.) (Scheme 4).
Scheme 4.
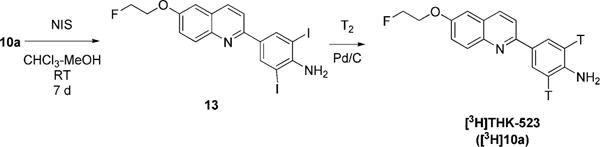
Synthesis of [3H]THK-523 ([3H]10a)
Attempt at an analogous iodination of T807 did not generate any tractable compound, and no further effort was made to synthesize an iodinated derivative of T807. Bromination of T807 with NBS generated the tribromo substituted derivative 14 (Scheme 5). We found that the three bromo substituents in 14 could be removed with hydrogen in the presence of Pd/C as catalyst to regenerate T807. Thus, [3H]T807 was obtained by treating 14 with tritium gas as a custom synthesis (Moravek Biochemicals & Radiochemicals, Inc.) (Scheme 5).
Scheme 5.
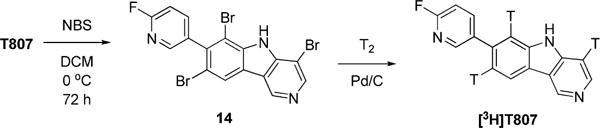
Synthesis of [3H]T807
18F-Labeling of THK-523 and Derivatives
Because [18F] 10b–f might be considered candidate PET radioligands, we were interested to test the feasibility of their labeling with no-carrier-added (NCA) cyclotron-produced [18F] fluoride ion. The N-Boc-protected tosylates, 9b–f, were used as precursors for trial 18F-labeling (Scheme 6). After successful substitution of the tosylate group with [18F]fluoride ion, the Boc group was removed under acidic conditions. The labeled THK-523 derivatives [18F]10b–f were each neutralized and isolated with HPLC in 20–30% decay-corrected radiochemical yields.
Scheme 6.
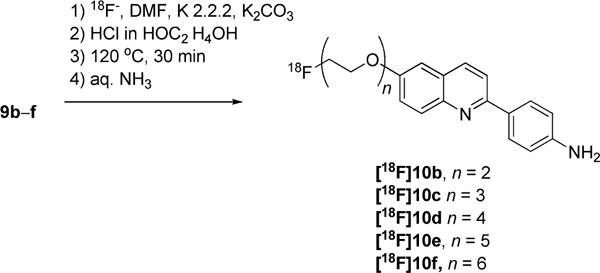
Radiosyntheses of [18F]10b–f
Binding Assays for Human AD β-Amyloid Plaques and NFTs
We used three tritium-labeled compounds as reference radioligands (Chart 3) to measure the in vitro inhibition constants of new compounds with human AD brain homogenate as protein source. Protocols similar to those that have been used previously for assays of [3H]PIB binding to AD brain tissue did not generate any discernible inhibition curves for [3H]THK523 or [3H]T807 binding. We tried a variety of ways to improve the binding assays, including use of a variety of assay mediums, such as PBS, aq. DMSO (20%), aq. DMSO (40%), neat DMSO, aq. ethanol (20%), aq. ethanol (40%), neat ethanol, aq. BSA (0.1%), or Tween-20 (0.1%) in PBS, and a variety of washing solutions, such as PBS, Tris, aq. ethanol (20%), aq. ethanol (40%), or neat ethanol. The combination of conditions that gave reproducible results was as follows: (a) use of AD brain tissue homogenate that was rehomogenized after thawing the stock homogenate; (b) treatment of the rehomogenized suspension with a high-power ultrasonic probe; and (c) use of Tween-20 (0.1%) in PBS instead of PBS as the assay medium. After the best conditions were implemented, the washing solution was found to have little effect on measured Ki values (Figure 1).
Chart 3.
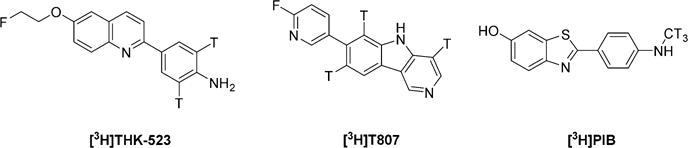
Three Reference Radioligands Used for in Vitro Binding Assays on AD Brain Tissue Homogenates
Figure 1.
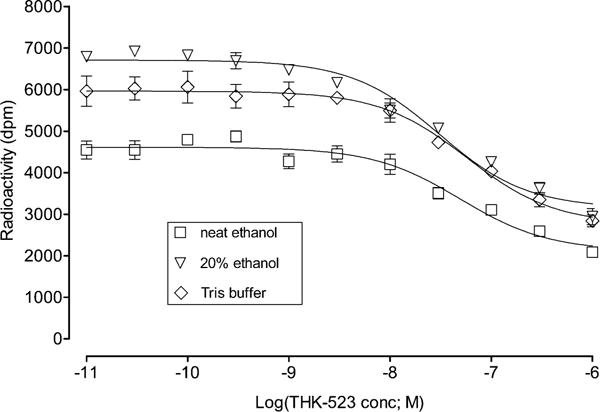
Lack of effect of different washing agents on the Ki measured for THK-523 in the in vitro binding assay with [3H]THK-523 as reference radioligand. The data points are means ± SD of three measurements (error bars not shown are within symbol size). The determined Ki values are 45 ± 12 nM with neat ethanol wash; 28 ± 6 nM for 20% ethanol wash; and 52 ± 14 nM for Tris-buffer (pH 7.2) wash.
Titration of AD Brain Homogenate with [3H]THK-523
The binding of the AD brain homogenate with [3H]THK-523 at different concentrations of THK-523 followed a typical saturation curve for a radioligand with a large proportion of nonspecific binding (Figure 2). The curve was fitted to the equation
where [BF] is the concentration of bound radioligand, Bmax is the concentration of maximal binding sites, [F] is the concentration of free radioligand plus THK-523, KD is the equilibrium dissociation constant, NS is the coefficient of nonspecific binding, and c is the concentration of bound radioligand in background. The best fit was obtained with KD = 3.5 ± 3.4 nM, Bmax = 1576 ± 499 dpm, NS = 321 ± 7, and c = 17.38 ± 173.
Figure 2.
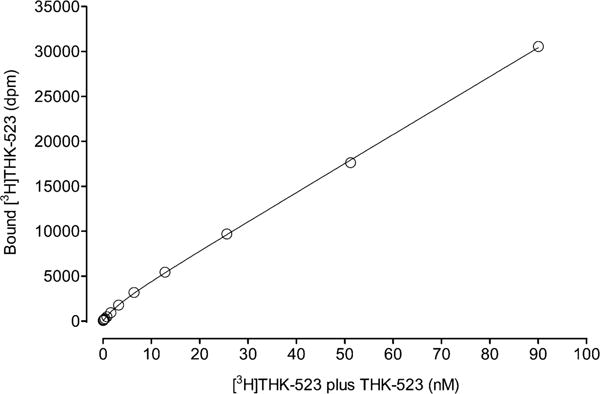
Binding of [3H]THK-523 to AD brain homogenate versus concentration of [3H]THK-523 plus THK-523. The data points are means of four measurements, and SDs are within symbol size. The curve was fitted to the equation [BF] = Bmax[F]/(KD + [F]) + (NS[F]) + c, giving KD = 3.5 ± 3.4 nM.
Titration of AD Brain Homogenate with [18F]THK-523
The binding of the AD brain homogenate with [18F]THK-523 at different concentrations of THK-523 also followed a typical saturation curve for a radioligand with a large proportion of nonspecific binding (Figure 3). The curve was fitted as in the analogous experiment with [3H]THK-523. The best fit was obtained with KD = 4.6 ± 1.8 nM, Bmax = 1639 ± 235 dpm, NS = 169 ± 3, and c = 57.7 ± 65.2.
Figure 3.
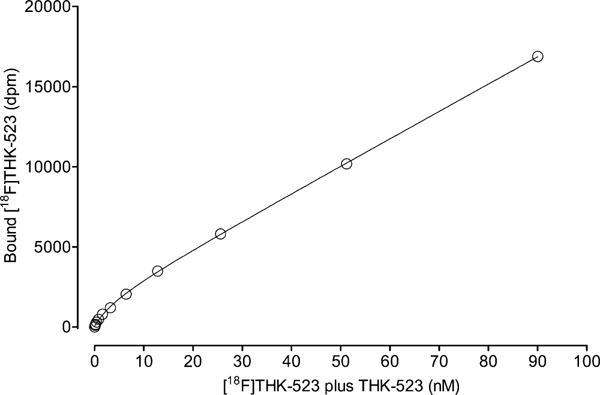
Binding of [18F]THK-523 to AD brain tissue homogenate versus concentration of THK-523 plus [18F]THK-523. The data are means from four measurements, and SDs are within symbol size. The curve was fitted to the equation [BF] = Bmax[F]/(KD + [F]) + (NS[F]) + c, giving KD = 4.6 ± 1.8 nM.
Inhibition of Binding to Human AD β-Amyloid Plaques and NFTs
When we used the three binding assays with AD brain homogenates to screen compounds for inhibition of radioligand binding at 1 μmol concentration, we found that the reference radioligands occupied distinctly different binding sites. Thus, T807 failed to inhibit [3H]THK-523 binding (Figure 4), and THK-523 failed to inhibit [3H]T807 binding (Figure 5). Similar mutually exclusive inhibition behavior was also observed for [3H]PIB (data not shown). These graphical demonstrations of multiple binding sites were also confirmed by the Ki values measured for THK-523, T807 and the newly synthesized compounds at the three different binding sites (Table 1). For the new fluoro compounds 10b–f, only the compound with two oxyethylene units (10b) showed high affinity (Ki = 28 nM) and selectivity for binding to the “THK binding site”. None of these compounds showed high affinity for the “T807 binding site”, whereas compound 12e, which has five oxyethylene units, showed high affinity (Ki = 43 nM) and selectivity for binding to the “PIB binding site”. Some variations in the terminal group were quite well tolerated among other compounds having two oxyethylene units. Thus, whereas compound 12a and 12b showed greatly decreased Ki values, compounds 12c–f retained both high affinity and selectivity for the THK binding site. Notably, 12e and 12f showed higher affinities (lower Ki values) than THK-523 itself in the same assay.
Figure 4.
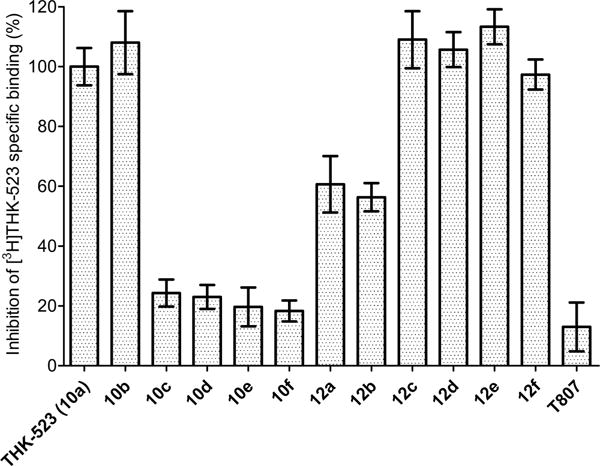
Percentages of specific binding of [3H]THK-523 to AD brain tissue homogenate inhibited by each of 13 compounds (10a–f, 12a–f, and T807) at 1 μM concentration. THK-523 (10a) gave full inhibition, whereas T807 gave very low inhibition. The data are mean ± SD from three measurements.
Figure 5.
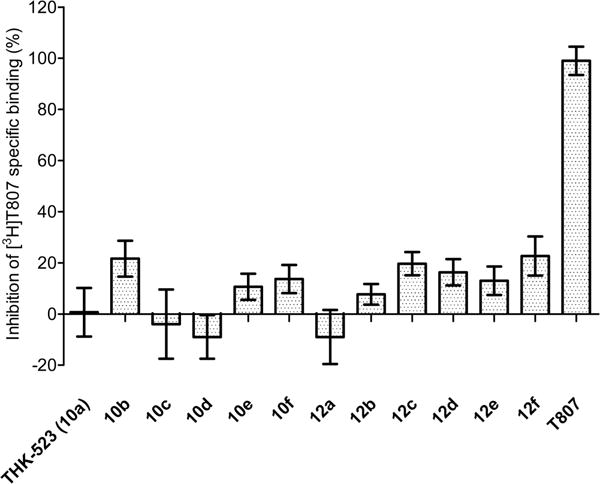
Percentages of specific binding of [3H]T807 to AD brain tissue homogenate inhibited by each of 13 compounds (10a–f, 12a–f, and T807) at 1 μM concentration. T807 gave full inhibition, whereas THK-523 (10a) gave very low inhibition. The data are mean ± SD from three measurements.
Table 1.
cLogD and Ki Values for Ligands at Three Binding Sites in AD Brain Tissue Homogenatesa
| Compound | Structure | cLogD |
Ki (nM) at
|
||
|---|---|---|---|---|---|
| THK-523 site | T807 site | PIB site | |||
| T807 |
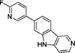
|
2.91 | >1000 | 2.6 ± 0.7 | >1000 |
| PIB |
|
2.30 | >1000 | >1000 | 34 ± 3 |
| THK-523 (10a) |
|
3.30 | 51 ± 22 | 425 ± 383 | >1000 |
| 10b |
|
3.74 | 28 ± 8 | >1000 | >1000 |
| 10c |
|
3.33 | >1000 | >1000 | >1000 |
| 10d |
|
2.62 | >1000 | >1000 | >1000 |
| 10e |
|
2.70 | >1000 | N.d. | 43 ± 39 |
| 10f |
|
2.29 | >1000 | N.d. | >500 |
| 12a |
|
−0.38 | 358 ± 381 | >1000 | 502 ± 143 |
| 12b |
|
1.79 | >1000 | >1000 | >1000 |
| 12c |
|
2.87 | 35 ± 19 | >1000 | >1000 |
| 12d |
|
2.16 | 78 ± 7 | >1000 | >1000 |
| 12e |
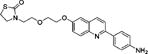
|
3.30 | 21 ± 6 | >1000 | >1000 |
| 12f |
|
0.38 | 22 ± 3 | >1000 | >1000 |
Ki values are average of five measurements for THK-523, T807, and PIB. Ki values for other compounds are mean ± SD from three measurements.
N.d. = Ki, not determined; see Figure 5 for percentage inhibition of [3H]T807 binding at 1 μmol concentration.
Computed Lipophilicity
Computation indicated that clogD for all the prepared target ligands was held within a moderate range (1.3–3.74), except for compounds 12a and 12f.
DISCUSSION
We overcame many obstacles to establish in vitro assays for measuring the relative binding affinities of small molecules for NFTs. The challenges that we met came from three main issues: (1) Ligand solubility is low in PBS; for example, THK-523 is insoluble in water or PBS; even its high fluorescence is not observed in water, reflecting its limited solubility in common media used for binding assays. (2) NFTs from AD brain homogenate are poorly accessible to small molecules; they exist as aggregates within neurons, unlike β-amyloid plaques, which exist outside neurons. (3) NFTs are highly heterogeneous, and on standing they precipitate within 5–10 min.
Effective conditions for binding assays on human AD brain tissue were achieved by taking three measures: (1) substituting PBS with 0.1% Tween-20 in PBS as the assay medium; (2) rehomogenizing the brain tissue homogenate before each experiment; and (3) ultrasound treatment of the tissue after the rehomogenization. With regard to the ligand solubility issue, we tried adding organic solvent, such as ethanol or DMSO, at various concentrations, to the binding assay medium. We also tried adding BSA. However, we did not obtain an appreciable signal for specific binding of radioligand, and the small dubious signal was also irreproducible. Although a low concentration of Tween-20 (0.001%) in PBS was ineffective, it was found that a much higher concentration 0.1% promoted complete dissolution of the ligands in aqueous media. The other two measures were designed to address the issue of poor NFT accessibility to small molecules. Rehomogenization before an assay experiment helped to break down the chunk of brain tissue into smaller pieces, thereby exposing individual neurons to the highest degree. Subsequent ultrasound treatment then helped to break the neuronal cell membranes, exposing the NFTs to small molecules.
During our initial efforts to work up assay conditions, we hypothesized that the specific radioligand binding signal was obscured by high nonspecific binding to the brain tissue homogenate. Therefore, various washing agents were tested to reduce radioligand nonspecific binding, such as PBS or Tris-buffer, or various concentrations of ethanol, DMSO, acetic acid, or formic acid in water. However, it was found that, after deploying the revised combination of conditions for the binding assay, the nature of the washing agent no longer affected the Ki measurement (Figure 2).
The KD value for THK-523 for binding to AD brain homogenate was also measured by titration. KD was found to be 3.5 ± 3.4 and 4.6 ± 1.8 nM with [3H]THK-523 (Figure 3) and [18F]THK-523 (Figure 4) as radioligands, respectively. These measurements are clearly in good agreement. However, the Ki value measured from the competitive inhibition experiment was much lower at 51 ± 22 nM. This discrepancy may derive from error that is intrinsic to the titration experiments, where the specific binding signals are only a small fraction of the total binding.
The principal discovery of this study is the plurality of distinct binding sites on NFTs. Both [18F]THK-523 and [18F]T807, and their close analogues, are considered to be radioligands that bind selectively to NFTs, based on ample evidence from autoradiography, immunohistochemistry, and the use of brain tissue from different disease origins.25,28,33,34 Therefore, in this study, we assumed that the binding of THK-523 analogues to AD tissue homogenate was entirely to binding sites on NFTs, rather than to other binding sites on β-amyloid plaques or to other proteins in AD brain homogenate. When we analyzed the binding data, we assumed a single-site model, to which the data fit quite well. No compelling reason to fit the data to two or more binding sites was found. The mutually exclusive binding behaviors of [3H]THK-523, [3H]T807 and of some of the prepared ligands (e.g., 10b, 12c, 12e; Table 1) showed that they occupy different binding sites within NFTs. Although multiple binding sites have been observed on β-amyloid plaques,31,32,35 only radioligands that bind to the thioflavin T site (Chart 1) have so far become useful for clinical PET imaging. Nonetheless, for biomedical research on the various NFTs in tauopathies, radioligands that bind to different sites on NFTs may well find distinct applications.
From only a small set of synthesized THK-523 derivatives, three compounds (10b, 12e, and 12f) emerged with high affinity (<50 nM) and selectivity for the THK-523 binding site (Table 1). These contained two oxyethylene units. Some five-membered rings were well tolerated as chain terminal groups, as in 12c and 12e. Compounds with more than two oxyethylene units showed no appreciable binding at the THK-523 site (Ki > 1000 nM), although 10e with five oxyethylene units showed high affinity and selectivity for binding to the PIB site (Ki> 4.3 nM). Compound 10b appears of special interest because of (i) its demonstrated amenability to labeling with fluorine-18, (ii) high affinity (Ki = 28 nM) on par with that of THK-523 itself in the same assay (Ki = 51 nM), and (iii) a moderate lipophilicity (logD, 3.74) that computes to be only 0.44 clogD units greater than that of THK-523 (Table 1).
CONCLUSIONS
We developed reliable in vitro binding assays to measure the binding affinities of small molecules proposed as possible radioligands for PET imaging of NFTs. Through tritium-labeling of THK-523 and T807, we identified two different kinds of binding sites on NFTs. These two binding sites are also different from that of PIB. Use of these three assays expands the scope for developing new PET radioligands for NFTs by bringing a variety of binding sites into consideration. Moreover, chain modifications in THK-523 were successful in producing compounds with high affinity and selectivity for the THK binding site, accompanied by moderate lipophilicity compatible with further development as PET radioligands.
METHODS
Materials
Water was purified with a system comprising two filters, one Rio, one reservoir, and one Milli-Q synthesis system (Millipore; Bedford, MA). Common solvents were obtained from Fisher Scientific (Pittsburgh, PA). Literature methods were used to prepare PIB,23 T807,36 2-fluoroethyl 4-methylbenzenesulfonate,37 and 2-hydroxyethyl 4-methylbenzenesulfonate.38 [3H]PIB was purchased from Moravek Biochemicals Inc. (Brea, CA) as a custom radiosynthesis. 2-Chloro-6-methoxyquinoline (1) was obtained from Ark Pharma Inc. (Liberty-ville, IL). Other chemicals, including 2-bromoethan-1-ol (3a) were purchased from Aldrich Chemical Co. (Milwaukee, WI), Fluka Chemical Co. (Milwaukee, WI), Acros (Hampton, NH), or Strem Chemicals (Newburyport, MA), and were used without further purification.
Instruments and General Methods
Analytical TLC was performed on precoated aluminum plates (silica gel 60, F254) and compounds were visualized under UV light at 254 or 365 nm. Reaction products were purified with flash column chromatography on silica gel 60 (230–400 mesh), and EtOAc-hexane-MeOH was used as mobile phase. Analytical HPLC was performed with a reversed phase column (X-bridge C18; 5 μm; 10.0 × 250 mm; Waters Corp.; Milford, MA) eluted with c. NH3 (0.025%) in MeCN-H2O at 6.2 mL/min. The chromatography system was fitted with a continuous wavelength UV-vis detector (System Gold 168 Detector; Beckman; Fullerton, CA) and an autosampler (System Gold 508; Beckman). For semi-preparative HPLC, a reversed phase column (Atlantis C18; 5 μm; 30 × 150 mm; Waters Corp.) or silica gel column (10 μm; 30 × 250 mm; Phenomenex; Torrance, CA) was eluted at 30 mL/min. The HPLC system was fitted with a manual injector (5 mL loop) and a third delivery pump using MeCN or EtOAc as mobile phase at 1 mL/min. The purities of compounds were determined with HPLC monitored for absorbance at 254 nm and expressed as area percentage of all peaks. All compound purities exceeded 95%.
The 1H and 13C NMR spectra of all compounds were acquired on a DRX 400 instrument (400 MHz 1H and 100 MHz 13C; Bruker; Billerica, MA), using the chemical shifts of residual deuterated solvent as the internal standard; chemical shift (5) data for the proton and carbon resonances are reported in parts per million (ppm) downfield relative to the signal for TMS (δ = 0). Mass spectra were acquired using a Velos Pro LC-MS instrument (Thermo Fisher Scientific; Waltham, MA) fitted with a Luna C18 column (5 μm; 2.0 × 150 mm; Phenomenex) eluted at 150 μL/min with a MeOH-H2O mixture. High-resolution mass spectra (HRMS) were acquired from the Mass Spectrometry Laboratory, University of Illinois at Urbana-Champaign (Urbana, IL). Either electron spray ionization (ESI) or electron ionization (EI) was used. Melting points were measured with a Mel-Temp (Electrothermal; Thermo Fisher Scientific) or SMP-30 (Stuart; Staffordshire, UK) manual melting point apparatus and were uncorrected. cLogD was computed with Pallas for Windows software version 3.8 in default option (CompuDrug; Bal Harbor, FL). A Discover apparatus (CEM; Matthews, NC) was used for microwave synthesis.
Brain Tissue
Postmortem brain tissues from an autopsy-confirmed AD case and a healthy control were obtained from the Human Brain Collection Core (HBCC), NIH.
Ligand Syntheses
2-Chloroquinolin-6-ol (2)
2-Chloro-6-methoxyquinoline (1; 10 g; 52 mmol) was dissolved in DCM (200 mL), and BBr3 (12 mL; 126 mmol) was added through a large needle syringe. Some off-white precipitate formed quickly and accumulated during the reaction. The mixture was stirred at RT overnight, and then quenched slowly by adding MeOH until heat and gas generation stopped. The solution was basified by adding NaHCO3 until gas generation stopped. The mixture was then extracted with DCM (200 mL × 3). The collected organic phase was dried (MgSO4), and the solvent was removed to give 2 as a brown solid (4.8 g; 47%) that was pure enough for subsequent reactions: lH NMR (CD3OD) δ 8.11 (d, 1H, 3JHH = 8.6 Hz, ArH), 7.78 (d, 1H, 3JHH = 9.2 Hz, ArH), 7.38 (m, 2H, ArH), 7.15 (d, 4JHH = 2.6 Hz, 1H, ArH); m/z (ESI-MS): 180.3 (100%, [M + H]+), 182.2 (30%, [M + H]+), 190.1 (22%); HRMS (TOF+): calcd for C9H7ClNO [M + H]+, 180.0216; found, 180.0215. Error (ppm): −2.5.
2-(2-Hydroxyethoxy)ethyl 4-Methylbenzenesulfonate (3b)
2,2′-Oxybis(ethan-1-ol) (1.7 g, 16 mmol) was dissolved in DCM (10 mL). p-TsCl (1.0 g, 5.3 mmol) and pyridine (1.2 mL, 16 mmol) were added sequentially. The reaction mixture was stirred at RT for 2 days, and then partitioned between DCM (100 mL) and brine (100 mL). The aq. phase was extracted twice with DCM (100 mL). The combined organic phase was dried (MgSO4), and the solvent was removed to leave a colorless oil, which was dissolved in DCM. Silica gel (4 mL) was added, and the mixture was dried by rotary evaporation. The dry powder was loaded onto a silica gel column with silica gel–hexane (100 mL). Oxybis(ethane-2,1-diyl) bis(4-methylbenzenesulfonate) eluted in EtOAc-hexane (1:3). Elution with EtOAc-hexane (1:1) followed by removal of solvent gave 3b as a colorless oil (0.57 g; 42%): 1H NMR (CDCl3) δ 7.77 (dm, 3JHH = 8.3 Hz, 2H, ArH), 7.32 (dm, 3JHH = 8.6 Hz, 2H, ArH), 4.17–4.15 (m, 2H, OCH2), 3.67–3.63 (m, 4H, OCH2), 2.42 (s, 3H, CH3); 13C NMR (CDCl3) δ 145.1, 133.0, 130.0, 128.0, 72.7, 69.4, 68.8, 61.8, 21.8 (s, 1C, CH3); m/z (ESI-MS): 371.1 (30%, [TsO(C2H4O)4H + Na]+), 349.1 (30%, TsO(C2H4O)4H + H]+), 283.1 (100%, [M + Na]+), 278.1 (34%, [M + H2O]+), 261.1 (100%, [M + H]+), 235.1 (30%), 89.1 (40%, [CH2CH2OCH2CH2OH]+); HRMS (TOF+) calcd for C11H16O5S, 261.0797 [M + H]+; found, 261.0792. Error (ppm): −1.9.
2-(2-(2-Hydroxyethoxy)ethoxy)ethyl 4-Methylbenzenesulfonate (3c)
2,2′-(Ethane-1,2-diylbis(oxy))bis(ethan-1-ol) (15.5 g, 103 mmol) was dissolved in DCM (50 mL). p-TsCl (6.2 g, 32.5 mmol) and pyridine (8.0 mL, 99 mmol) were added sequentially. The reaction mixture was stirred at RT for 2 days, and then silica gel (60 mL) was added. The mixture was dried by rotary evaporation. The dry powder was loaded onto a silica gel column with silica gel–hexane (600 mL). The bis-tosylate eluted with EtOAc-hexane (1:3). Elution with EtOAc-hexane (1:1) followed by removal of solvent gave 3c as a colorless oil (5.2 g; 53%). 1H NMR (CDCl3) δ 7.79 (d, 3JHH = 8.4 Hz, 2H), 7.35 (d, 3JHH = 8 Hz, 2H), 4.16 (m, 2H), 3.69 (m, 4H), 3.59 (s, 4H), 3.56 (m, 2H), 2.85 (s, 1H), 2.45 (s, 3H); 13C{1H} NMR (CDCl3) δ 144.9, 132.8, 129.9, 127.9, 72.5, 70.7, 70.2, 69.3, 68.6, 61.6, 21.6; m/z (ESI-MS): 459.2 (50%, [TsO(C2H4O)6H + Na]+), 454.1 (26%, [TsO(C2H4O)6H + H2O]+), 437.2 (28%, [TsO(C2H4O)6H + H]+), 415.2 (11%), 343.1 (16%), 327.1 (97%, [M + Na]+), 454.1 (34%, [M + H2O]+), 305.1 (100%, [M + H]+), 301.1 (20%), 199.0 (17%), 181.0 (14%), 133.1 (23%); HRMS (TOF+) calcd for C13H21O6S, 305.1059 [M + H]+; found, 305.1056. Error (ppm): −1.0.
2-(2-(2-(2-Hydroxyethoxy)ethoxy)ethoxy)ethyl 4-Methylbenzene-sulfonate (3d)
The method for 3c was applied to 2,2′-((oxybis-(ethane-2,1-diyl))bis(oxy))bis(ethan-1-ol) (10.2 g, 52.5 mmol) and gave 3d as a colorless oil (3.2 g; 43%): 1H NMR (CDCl3) δ 7.79 (d, 3JHH = 8.4 Hz, 2H), 7.35 (d, 3JHH = 8 Hz, 2H), 4.16 (m, 2H), 3.65 (m, 14H), 2.61 (s, 1H), 2.45 (s, 3H); 13C NMR (CDCl3) δ 144.9, 132.9, 129.8, 128.0, 72.5, 70.7, 70.6, 70.4, 70.3, 69.3, 68.7, 61.7, 21.6; m/z (ESI-MS): 547.2 (27%, [TsO(C2H4O)8H + Na]+), 542.3 (34%, [TsO(C2H4O)8H + H2O]+), 525.2 (28%, [TsO(C2H4O)8H + H]+), 371.1 (70%, [M + Na]+), 349.1 (100%, [M + H]+); HRMS (TOF+) calcd for C15H25O7S, 349.1321 [M + H]+; found, 349.1323. Error (ppm): +0.6.
14-Hydroxy-3,6,9,12-tetraoxatetradecyl 4-Methylbenzenesulfonate (3e)
p-TsCl (19.15 g, 108 mmol), KI (557 mg, 3.36 mmol), and silver(I) oxide (9.34 g, 40.3 mmol) were added to a solution of pentaethylene glycol (8 g, 33.6 mmol) in DCM (150 mL), and the mixture was stirred overnight and then filtered over Celite. The filtrate was washed with H2O and extracted three times with DCM, dried (MgSO4), and filtered. The solvent was evaporated off from the filtrate and the residue was purified with flash chromatography (acetone-hexane-triethylamine; 39.9:60:0.1) to give 3e as a colorless oil (4.08 g; 31%): 1H NMR (CDO3) δ 7.79 (d, 3JHH = 8.4 Hz, 2H), 7.35 (d, 3JHH = 8 Hz, 2H), 4.16 (m, 2H), 3.65 (m, 18H), 2.88 (s, 1H), 2.45 (s, 3H); 13C NMR (CDCl3) δ 144.8, 132.9, 129.8, 127.9, 72.5, 72.5, 70.7, 70.5, 70.5, 70.5, 70.2, 69.3, 68.6, 61.6, 21.6. HRMS (TOF+) calcd for C17H29O8S, 393.1583 [M + H]+; found, 393.1591. Error (ppm): +2.0.
17-Hydroxy-3,6,9,12,15-pentaoxaheptadecyl 4-Methylbenzene-sulfonate (3f)
The method for 3e was applied to hexaethylene glycol (8 g, 28.3 gave 3f as a colorless oil (3.57 g; 29%): 1H NMR (CDCl3) δ 7.79 (m, 2H), 7.35 (d, J =8 Hz, 2H), 4.16 (m, 2H), 3.66 (m, 22H), 2.81 (s, 1H), 2.45 (s, 3H); 13C NMR (CDCl3) δ 144.81, 132.94, 129.83, 127.96, 72.50, 70.69, 70.58, 70.53, 70.51, 70.49, 70.30, 69.28, 68.64, 61.68, 21.64; HRMS (TOF+) calcd for C19H33O9S, 437.1845 [M + H]+; found, 437.1845. Error (ppm): +0.0.
2-((2-Chloroquinolin-6-yl)oxy)ethan-1-ol (4a)
Compound 2 (0.10 g, 0.56 mmol), 2-bromoethanol (3a; 0.20 mL, 1.7 mmol), and Cs2CO3 (0.9 g, 2.8 mmol) were suspended in DMF (1.0 mL) and stirred at RT for 2 d. Conversion of the starting material was 90%. The mixture was microwaved (100c5m50w250 psi). After the reaction was found to be 95% complete by HPLC, the mixture was partitioned between EtOAc and brine. The combined organic phase was dried (MgSO4). After removing solvent, the crude product was dissolved in DCM and mixed with silica gel (4.0 mL). Solvent was then removed. Chromatography of the residual powder on silica gel (EtOAc-hexane,1:1) gave 4a as a white solid (34 mg; 27%): 1H NMR (CDCl3) δ 7.96 (d, 1H, 8.8 Hz, ArH), 7.91 (d, 1H, 3JHH = 9.2 Hz, ArH), 7.37 (dd, 1H, 3JHH = 9.2 Hz, 4JHH = 2.8 Hz, ArH), 7.33 (d, 3JHH = 8.6 Hz, 1H, ArH), 7.07 (d, 4JHH = 2.8 Hz, 1H, ArH), 4.21–4.19 (m, 2H, OCH2), 4.07–4.05 (m, 2H, OCH2), 2.35 (brs, 1H, OH); 13C NMR (CD3OD) δ 157.1, 147.0, 143.0, 138.7, 128.0, 127.4, 123.3, 122.4, 106.8, 70.0, 59.4; m/z (ESI-MS): 226.0, 224.0 (100%, [m + H]+), 182.0, 180.0 (10%, [M + H–C2H4O]+); HRMS (TOF+): calcd for C11H10ClNO2 [M + H]+, 224.0478; found, 224.0481. Error (ppm): +1.3.
2-(2-((2-Chloroquinolin-6-yl)oxy)ethoxy)ethan-1-ol (4b)
Compound 2 (0.10 g, 0.56 mmol), 3b (0.26 g, 1.0 mmol), and Cs2CO3 (0.55 g, 1.7 mmol) were suspended in DMF (1.0 mL) within a nitrogen-filled glovebox. The mixture was microwaved (100c30m50w250 psi). HPLC showed greater than 90% conversion of starting material. The mixture was partitioned between EtOAc and brine. The combined organic phase was dried (MgSO4). The solvent was removed to give 4b as an oil (0.15 g; 100%): 1H NMR (CDCl3) δ 7.91 (d, 3JHH = 8.5 Hz, 1H, ArH), 7.85 (d, 3JHH = 9.2 Hz, 1H, ArH), 7.34 (dd, 3JHH = 9.2 Hz, 4JHH = 2.8 Hz, 1H, ArH), 7.27 (d, 3JHH = 8.6 Hz, 1H, ArH), 7.01 (d, 4JHH = 2.8 Hz, 1H, ArH), 4.20–4.17 (m, 2H, OCH2), 3.89–3.87 (m, 2H, OCH2), 3.76–3.73 (m, 2H, OCH2), 3.67–3.64 (m, 2H, OCH2); 13C NMR (CDCl3) δ 157.3, 148.3, 143.9, 137.9, 130.1, 127.9, 123.3, 122.7, 106.4, 72.9, 69.6, 67.9, 61.9; m/z (ES-MS): 310.1 (10%), 279.1 (12%), 268.1 (100%, [M + H]+), 270.1 (34%, [M + H]+); HRMS (TOF+) calcd for C13H15ClNO3, 268.0740 [M + H]+; found, 268.0747. Error (ppm): +2.6.
2-(2-(2-((2-Chloroquinolin-6-yl)oxy)ethoxy)ethoxy)ethan-1-ol (4c)
The method for 4b was applied to 3c (0.34 g, 1.1 mmol) and gave 4c as an oil (0.18 g; 100%): 1H NMR (CDCl3) δ 7.96 (d, 3JHH = 8.4 Hz, 1H), 7.90 (d, 3JHH = 9.2 Hz, 1H), 7.41 (dd, 3JHH = 6.8 Hz, 6.8 Hz, 1H), 7.32 (d, 3JHH = 8.4 Hz, 1H), 7.07 (d, 4JHH = 2.4 Hz, 1H), 4.24 (m, 2H), 3.93 (m, 2H), 3.73 (m, 6H), 3.62 (t, 3JHH = 4 Hz, 2H), 2.68 (s, 1H); 13C NMR (CDCl3) δ 157.2, 148.1, 143.8, 137.7, 129.9, 127.8, 123.3, 122.5, 106.2, 72.5, 70.9, 70.4, 69.6, 67.7, 61.7; HRMS (TOF+) calcd for C13H19ClNO4, 312.1003 [M+ + H]+; found, 312.1010. Error (ppm): +2.2.
2-(2-(2-((2-Chloroquinolin-6-yl)oxy)ethoxy)ethoxy)ethan-1-ol (4d)
The method for 4b was applied to 3d (0.39 g, 1.1 mmol) and gave 4d as an oil (0.16 g; 78%): 1H NMR (CDCl3) δ 7.97 (d, 3JHH = 8.4 Hz, 1H), 7.90 (d, 3JHH = 9.2 Hz, 1H), 7.41 (dd, 3JHH = 9.6 Hz, 9.6 Hz, 1H), 7.32 (d, 3JHH = 8.8 Hz, 1H), 7.08 (d, 4JHH = 2.8 Hz, 1H), 4.24 (m, 2H), 3.93 (m, 2H), 3.68 (m, 12H), 2.91 (s, 1H); 13C NMR (CDCl3) δ 157.2, 148.1, 143.8, 137.7, 129.8, 127.8, 123.3, 122.5, 106.2, 72.5, 70.8, 70.6, 70.6, 70.3, 69.6, 67.8, 61.7; m/z (ESI-MS): 536.2 (7%, [M − Cl + (OCH2CH2)4OH + Na]+), 532.2 (16%, [M − Cl + (OCH2CH2)4OH + H2O + H]+), 514.3 (28%, [M − Cl + (OCH2CH2)4OH + H]+), 378.1 (32%, [M + Na]+), 380.1 (11%), 356.1 (100%, [M + H]+), 358.1 (30%), 320.2 (9%, [M − Cl]+); HRMS (TOF+) calcd for C17H23ClNO5, 356.1265 [M + H]+; found, 356.1267. Error (ppm): +0.6.
14-((2-Chloroquinolin-6-yl)oxy)-3,6,9,12-tetraoxatetradecan-1-ol (4e)
Compound 3e (4.0 g, 10.2 mmol) and Cs2CO3 (16.6 g, 51 mmol) were added to a solution of 2 (1.53 g, 8.05 mmol) in DMF (15 mL) in a microwave tube loaded with a stirrer bar, and the mixture was microwaved (95c60w30m) with stirring. The reaction mixture was washed with brine and extracted three times with EtOAc, dried (MgSO4), and filtered. The solvent was evaporated off, and the residue was purified with flash chromatography (EtOAc-hexane-MeOH; 90:5:5) to give 4e as a yellowish-white solid (2.07 g, 52%): 1H NMR (CDCl3) δ 7.98 (d, 3JHH = 8.4 Hz, 1H), 7.91 (d, 3JHH = 9.2 Hz, 1H), 7.41 (dd, 3JHH = 9.2 Hz, 9.2 Hz, 1H), 7.33 (d, 3JHH = 8.4 Hz, 1H), 7.09 (d, 4JHH = 2.4 Hz, 1H), 4.25 (m, 2H), 3.93 (m, 2H), 3.76 (m, 2H), 3.68 (m, 12H), 3.60 (m, 2H), 2.68 (s, 1H); 13C NMR (CDCl3) δ 157.3, 148.1, 143.8, 137.7, 129.9, 127.9, 123.4, 122.5, 106.2, 72.5, 70.9, 70.6, 70.6, 70.6, 70.5, 70.3, 69.6, 67.8, 61.7; HRMS (TOF+) calcd for C19H27ClNO6, 400.1527 [M + H]+; found, 400.1527. Error (ppm): +0.0.
17-((2-Chloroquinolin-6-yl)oxy)-3,6,9,12,15-pentaoxaheptadecan-1-ol (4f)
The method for 4e was applied to 3f (819 mg, 1.87 mmol). Flash chromatography (EtOAc-MeOH-hexane; 85:10:5) of the crude product gave 4f as a yellow-white solid (373 mg, 54%): 1H NMR (CDCl3) δ 7.98 (d, 3JHH = 8 Hz, 1H), 7.89 (d, 3JHH = 12 Hz, 1H), 7.41 (dd, 3JHH = 6.8 Hz, 3JHH = 6.8 Hz, 1H), 7.32 (d, 3JHH = 8 Hz, 1H), 7.32 (d, 4JHH = 4 Hz, 1H), 7.07 (d, 4JHH = 2.4 Hz, 1H), 4.24 (m, 2H), 3.92 (m, 2H), 3.93 (m, 20H), 2.68 (s, 1H); 13C NMR (CDCl3) δ 157.3, 148.1, 143.8, 137.7, 130.0, 127.9, 123.4, 122.5, 106.2, 72.6, 70.9, 70.6, 70.6, 70.5, 70.5, 70.2, 69.6, 67.8, 61.7; HRMS (TOF+) calcd for C21H31ClNO7, 444.1789 [M + H]+; found, 444.1784. Error (ppm): −1.1.
2-Chloro-6-(2-fluoroethoxy)quinoline (5)
Compound 2 (0.50 g, 2.8 mmol), 2-fluoroethyl tosylate (0.90 g, 4.1 mmol), and Cs2CO3 (4.6 g, 14.1 mmol) were suspended in THF (5.0 mL). The mixture was stirred overnight. After the reaction was found to be complete with HPLC, the mixture was partitioned between EtOAc and brine. The combined organic phase was dried (MgSO4). After removing solvent from the filtrate, the crude product was dissolved in DCM and mixed with silica gel (4.0 mL). Solvent was then removed. Chromatography of the residual powder on silica gel (EtOAc-hexane) gave 5 as a white solid (0.63 g; 100%): 1H NMR (CD3OD) δ 7.97 (d, 1H, 3JHH = 8.5 Hz, ArH), 7.92 (d, 1H, 3JHH = 9.2 Hz, ArH), 7.42 (dd, 1H, 3JHH = 9.2 Hz, 4JHH = 2.8 Hz, ArH), 7.33 (d, 3JHH = 8.6 Hz, 1H, ArH), 7.07 (d, 4JHH = 2.8 Hz, 1H, ArH), 4.88–4.86 (m, 1H, FCH2), 4.76–4.74 (m, 1H, FCH2), 4.36–4.34 (m, 1H, OCH2), 4.29–4.27 (m, 1H, OCH2); m/z (ESI-MS): 272.0, 270.0 (40%, [M − Cl + Br]), 226.2 (100%, [M + H]+), 228.2 (30%, [M + H]+); HRMS (TOF+): calcd for C11H9ClFNO [M + H]+, 226.0435; found, 226.0437. Error (ppm): +0.9.
t-Butyl (4-(6-Methoxyquinolin-2-yl)phenyl)carbamate (6)
Compound 1 (0.10 g, 0.52 mmol), (4-((t-Boc)amino)phenyl)boronic acid (0.137 g, 0.67 mmol), Pd(PPh3)4 (0.026 g, 0.023 mmol), and K2CO3 (0.122 g, 1.04 mmol) were suspended in DME-H2O (4:1; 10 mL). The mixture was microwaved (80c60m50w250 psi). After the reaction was found to be complete by HPLC, the mixture was partitioned between EtOAc and brine (100 mL × 3). The combined organic phase was dried (MgSO4). After the solvent was removed, the crude product was dissolved in DCM, and silica gel (4 mL) was added, and dried. Chromatography of the residual powder on silica gel (100 mL; EtOAc-hexane, 1:3) gave 6 as a white solid (0.16 g; 100%): 1H NMR (acetone-d6) δ 8.58 (brs, 1H, NH), 8.25 (d, 1H, 3JHH = 8.9 Hz, ArH), 8.22 (dm, 2H, 3JHH = 9.2 Hz, ArH), 8.01 (d, 1H, 3JHH = 8.7 Hz, ArH), 7.97 (d, 3JHH = 9.2 Hz, 1H, ArH), 7.72 (d, 3JHH = 8.9 Hz, 1H, ArH), 7.38 (dd, 3JHH = 9.5 Hz, 4JHH = 2.6 Hz, 1H, ArH), 7.32 (d, 4JHH = 2.9 Hz, 1H, ArH), 3.95 (s, 3H, OCH3), 1.51 (s, 9H, CH3); 1H NMR (CDCl3) δ 8.07 (dm, 2H, 3JHH = 8.7 Hz, ArH), 8.05 (d, 1H, Hz, ArH), 8.01 (d, 1H, 3JHH = 8.9.2 Hz, ArH), 7.78 (d, 1H, 3JHH = 8.6 Hz, ArH), 7.49 (d, 2H, 3JHH = 8.6 Hz, ArH), 7.34 (dd, 3JHH = 9.2 Hz,, 4JHH = 2.8 Hz, 1H, ArH), 7.05 (d, 4JHH = 2.8 Hz, 1H, ArH), 6.62 (s, 1H, NH), 3.92 (s, 3H, OCH3), 1.52 (s, 9H, CH3); 13C NMR (CDCl3) δ 157.7, 154.6, 152.8, 144.5, 141.3, 139.4, 135.7, 134.5, 131.2, 128.2, 128.1, 122.4, 119.1, 118.6, 105.2, 80.9, 77.9, 55.7, 29.9, 28.6; m/z (ESI-MS): 350.9 (100%, [M + H]+), 295.2 (38%, [M − C(CH3)3 + H]+); HRMS (TOF+): calcd for C21H23N2O3 [M + H]+, 351.1709; found, 351.1704. Error (ppm): 1.4.
t-Butyl (4-(6-(2-Hydroxyethoxy)quinolin-2-yl)phenyl)carbamate (7a)
The method for 6 was applied to 4a (0.700 g, 3.13 mmol) and gave 7a as a white solid (0.714 g; 60%): 1H NMR (CDCl3) δ 8.09 (d, 2H, 3JHH = 8.8 Hz, ArH), 8.05 (d, 2H, 3JHH = 9.2 Hz, ArH), 7.81 (d, 1H, 3JHH = 8.8 Hz, ArH), 7.51 (d, 3JHH = 8.8 Hz, 1H, ArH), 7.39 (dd, 3JHH = 9.2 Hz, 4JHH = 2.8 Hz, 1H, ArH), 7.10 (d, 4JHH = 2.8 Hz, 1H, ArH), 6.60 (brs, 1H, NH), 4.24–4.21 (m, 2H, OCH2), 4.06–4.04 (m, 2H, OCH2), 1.55 (s, 9H, CH3); m/z (ESI-MS): 381.2 (100%, [M + H]+), 325.1 (81%, [M − C4H8 + H]+); HRMS (TOF+): calcd for C22H24N2O4 [M + H]+, 381.1814; found, 381.1812. Error (ppm): −0.5.
t-Butyl (4-(6-(2-(2-Hydroxyethoxy)ethoxy)quinolin-2-yl)phenyl)-carbamate (7b)
The method for 6 was applied to 4b (0.15 g, 0.56 mmol) and gave 7b as a white solid (0.24 g; 99%): 1H NMR (CDCl3) δ 8.06 (d, 2H, 3JHH = 8.6 Hz, ArH), 8.04 (d, 2H, 3JHH = 9.0 Hz, ArH), 8.03 (d, 1H, 3JHH = 8.7 Hz, ArH), 8.02 (d, 3JHH = 9.1 Hz, 1H, ArH), 7.77 (d, 3JHH = 8.7 Hz, 1H, ArH), 7.48 (d, 3JHH = 8.5 Hz, 1H, ArH), 7.37 (dd, 4JHH = 9.2 Hz, 4JHH = 2.7 Hz, 1H, ArH), 7.06 (d, 4JHH = 2.7 Hz, 1H, ArH), 6.62 (brs, 1H, NH), 4.26–4.24 (m, 2H, OCH2), 3.94–3.92 (m, 2H, OCH2), 3.79–3.76 (m, 2H, OCH2), 3.71–3.69 (m, 2H, OCH2), 1.52 (s, 9H, CH3); 13C NMR (CDCl3) δ 156.8, 154.8, 152.8, 144.6, 139.5, 135.7, 134.5, 131.3, 128.2, 128.1, 122.6, 119.1, 118.7, 106.3, 81.0 (CO), 72.9 (CO), 69.8 (CO), 67.9 (CO), 62.0 (CO), 28.6 (CH3); m/z (ESI-MS): 425.2 (93%, [M + H]+), 310.1 (10%), 279.1 (100%); HRMS (TOF+): calcd for C24H29N2O5 [M + H]+, 425.2076; found, 425.2073. Error (ppm): −0.7.
t-Butyl (4-(6-(2-(2-(2-Hydroxyethoxy)ethoxy)ethoxy)quinolin-2-yl)phenyl)carbamate (7c)
The method for 6 was applied to 4c (0.18 g, 0.56 mmol) and gave 7c as a white solid (0.26 g; 99%): 1H NMR (CDCl3) δ 8.02 (m, 4H), 7.71 (d, 3JHH = 8.8 Hz, 1H), 7.50 (d, 3JHH = 8.4 Hz, 2 H), 7.37 (dd, 3JHH = 6.8 Hz, 6.8 Hz, 1H), 7.02 (d, 4JHH = 2.4 Hz, 1H), 6.89 (s, 1H), 4.24 (m, 2H), 3.91 (m, 2H), 3.73 (m, 6H), 3.62 (m, 2H), 2.71 (s, 1H), 1.53 (s, 9H); 13C NMR (CDCl3) δ 156.5, 154.5, 152.7, 144.3, 139.4, 135.5, 134.2, 130.9, 127.9, 127.8, 122.4, 118.8, 118.5, 106.0, 80.6, 72.5, 70.9, 70.4, 69.7, 67.6, 61.7, 28.4; m/z (ESI-MS): 469.2 (100%, [M + H]+), 470.2 (30%); HRMS (TOF+): calcd for C26H33N2O6 [M + H]+, 469.2339; found, 469.2331. Error (ppm): −1.7.
t-Butyl (4-(6-(2-(2-(2-Hydroxyethoxy)ethoxy)ethoxy)quinolin-2-yl)phenyl)carbamate (7d)
The method for 6 was applied to 4d (0.18 g, 0.45 mmol) and gave 7d as a white solid (0.23 g; 99%): 1H NMR (CDCl3) δ 8.05 (m, 4H), 7.79 (d, J = 8.8 Hz, 1H), 7.50 (d, J = 8.4 Hz, 2 H), 7.39 (dd, J = 6.8 Hz, 6.8 Hz, 1H), 7.09 (d, J = 2.4 Hz, 1H), 6.63 (s, 1H), 4.27 (m, 2H), 3.93 (m, 2H), 3.68 (m, 12H), 1.53 (s, 9H); 13C NMR (CDCl3) δ 154.7, 152.7, 150.7, 142.5, 137.4, 133.6, 132.4, 129.1, 126.1, 126.0, 120.6, 117.0, 116.6, 104.2, 78.8, 70.7, 69.0, 68.8, 68.7, 68.4, 67.8, 65.8, 59.9, 26.5; m/z (ESI-MS): 535.2 (9%, [M + Na]+), 513.3 (100%, [M + H]+); HRMS (TOF+): calcd for C28H37N2O7, [M + H]+, 513.2601; found, 513.2595. Error (ppm): −1.2.
t-Butyl (4-(6-((14-Hydroxy-3,6,9,12-tetraoxatetradecyl)oxy)-quinolin-2-yl)phenyl)carbamate (7e)
4-(N-Boc-amino)-phenylboronic acid (1.42 g, 6.02 mmol), K2CO3 (1.38 g, 10 mmol), and Pd(PPh3)4 (288 mg, 0.25 mmol) were added to a solution of 4e (2.0 g, 5 mmol) in DME-H2O (4:1; 15 mL) and then microwaved (80c40m80w) with stirring. The reaction mixture was washed with brine and extracted with three times with EtOAc, dried (MgSO4), and filtered. The solvent was evaporated off from the filtrate, and the residue was purified by flash chromatography (EtOAc–hexane–MeOH; 90:7:3) to give 7e as a yellowish-white solid (1.86 g, 67%): 1H NMR (CDCl3) δ 8.02 (m, 4H), 7.71 (d, 3JHH = 8.8 Hz, 1H), 7.50 (d, 3JHH = 8.4 Hz, 2 H), 7.37 (dd, 3JHH = 6.8 Hz, 6.8 Hz, 1H), 7.02 (d, 4JHH = 2.4 Hz, 1H), 6.89 (s, 1H), 4.24 (m, 2H), 3.91 (m, 2H), 3.73 (m, 6H), 3.62 (m, 2H), 2.71 (s, 1H), 1.53 (s, 9H); 13C NMR (CDCl3) δ 156.5, 154.5, 152.7, 144.3, 139.4, 135.5, 134.2, 130.9, 127.9, 127.8, 122.4, 118.8, 118.5, 106.0, 80.6, 72.5, 70.9, 70.4, 69.7, 67.6, 61.7, 28.4; HRMS (TOF+): calcd for C30H41N2O8, [M + H]+, 557.2863; found, 557.2869. Error (ppm): +0.6.
t-Butyl (4-(6-((17-Hydroxy-3,6,9,12,15-pentaoxaheptadecyl)oxy)-quinolin-2-yl)phenyl)carbamate (7f)
The method for 6 was applied to 4f (1.225 g, 2.04 mmol) and gave 7f as a yellowish white solid (1.01 g, 83%): 1H NMR (CDCl3) δ 8.03 (m, 4H), 7.73 (d, 3JHH = 8.8 Hz, 1H), 7.52 (d, 3JHH = 8.8 Hz, 2H), 7.38 (dd, 3JHH = 9.2 Hz, 3JHH = 9.2 Hz, 1H), 7.04 (m, 2H), 4.22 (m, 2H), 3.9 (m, 2H), 3.69 (m, 18H), 3.57 (m, 2H), 1.53 (s, 9H); 13C NMR (CDCl3) δ 156.6, 154.5, 152.7, 144.2, 139.5, 135.5, 134.1, 130.8, 127.9, 127.8, 122.5, 118.8, 118.5, 106.0, 80.6, 72.7, 70.8, 70.6, 70.6, 70.5, 70.5, 70.4, 70.2, 69.6, 67.7, 61.6, 28.4. HRMS (TOF+): calcd for C32H45N2O9, [M + H]+, 601.3125; found, 601.3143. Error (ppm): +3.0.
t-Butyl (4-(6-(2-Fluoroethoxy)quinolin-2-yl)phenyl)carbamate (8)
The method for 6 was applied to compound 5 (0.727 g, 3.22 mmol) and gave 0.831 g; 67%): 1H NMR (acetone-d6) δ 8.59 (brs, 1H, NH), 8.25 (d, 1H, 3JHH = 9.7 Hz, ArH), 8.23 (dm, 2H, 3JHH = 9.0 Hz, ArH), 8.02 (d, 1H, 3JHH = 8.8 Hz, ArH), 8.00 (d, 3JHH = 10.4 Hz, 1H, ArH), 7.72 (d, 3JHH = 8.7 Hz, 1H, ArH), 7.44 (dd, 3JHH = 9.2 Hz, 4JHH = 2.8 Hz, 1H, ArH), 7.36 (d, 4JHH = 2.8 Hz, 1H, ArH), 4.92–4.91 (m, 2JHF = 43.9 Hz, 1H, FCH2), 4.81–4.79 (m, 2JHF = 43.9 Hz, 1H, FCH2), 4.49–4.47 (m, 2JHF = 29.1 Hz, 1H, OCH2), 4.42–4.4 (m, 2JHF = 29.1 Hz, 1H, OCH2), 1.52 (s, 9H, CH3); 13C NMR (CDCl3) δ 156.5, 155.0, 152.8, 144.7, 139.5, 135.7, 134.4, 131.4, 128.0, 122.5, 119.2, 118.7, 106.4, 82.1 (2JCF = 169.8 Hz, CF), 81.0 (CO), 67.6 (3JCF = 20.7 Hz, CCF), 28.6 (CH3); m/z (ESI-MS): 383.2 (100%, [M + H]+), 327.2 (20%, [M − C4H8 + H]+); HRMS (TOF+): calcd for C22H24FN2O3 [M + H]+, 383.1771; found, 383.1770. Error (ppm): −0.3.
2-((2-(4-((t-Boc)amino)phenyl)quinolin-6-yl)oxy)ethyl 4-methyl-benzenesulfonate (9a)
Compound 7a (0.21 g, 0.55 mmol) and TsCl (0.24 g, 1.3 mmol) were dissolved in DCM (10 mL). Triethylamine (0.5 mL, 3.6 mmol) was added slowly. The mixture was stirred overnight. After the reaction was found to be complete by HPLC, the mixture was partitioned between EtOAc and brine (50 mL × 3). The combined organic phase was dried (MgSO4). After the solvent was removed, the crude product was dissolved in DCM, and silica gel (2 mL) was added, and dried. Chromatography of the residual powder on silica gel (50 mL; EtOAc–hexane,1:3) gave 9a as a white solid (0.26 g; 89%): 1H NMR (CDCl3) δ 8.09 (d, 2H, 3JHH = 8.8 Hz, ArH), 8.02 (vt, 2H, 3JHH = 9.4 Hz, ArH), 7.83 (d, 2H, 3JHH = 8.0 Hz, ArH), 7.80 (d, 1H, 3JHH = 8.8 Hz, ArH), 7.51 (d, 3JHH = 8.8 Hz, 2H, ArH), 7.33 (d, 3JHH = 8.0 Hz, 2H, ArH), 7.21 (dd, 3JHH = 8.8 Hz, 4JHH = 3.2 Hz, 1H, ArH), 6.97 (d, 4JHH = 2.8 Hz, 1H, ArH), 6.62 (s, 1H, NH), 4.47–4.44 (m, 2H, OCH2), 4.29–4.27 (m, 2H, OCH2), 2.42 (s, 3H, CH3), 1.54 (s, 9H, CH3); 13C NMR (CDCl3) δ 155.8 (CO), 154.8, 152.5, 145.0, 144.5, 139.4, 135.5, 134.2, 132.9, 131.2, 130.0, 128.03, 127.99, 127.7, 122.1, 119.0, 118.4, 106.1, 80.8 (COtBu), 68.0, 65.6, 28.4 (C(CH3)3), 21.7 (CH3); m/z (ESI-MS): 589.1 (10%), 535.2 (100%, [M + H]+), 479.1 (12%), 263.1 (14%); HRMS (TOF+): calcd for C31H35N2O7S, [M + H]+, 579.2165; found, 579.2155. Error (ppm): −1.7. m/z (ESI-MS): 535.2 (100%, [M + H]+), 536.2 (34%); HRMS (TOF+): calcd for C29H31N2O6S [M + H]+, 535.1903; found, 535.1899. Error (ppm): −0.7.
2-(2-((2-(4-((t-Boc)amino)phenyl)quinolin-6-yl)oxy)ethoxy)ethyl 4-methylbenzenesulfonate (9b)
The method for 9a was applied to 7b (0.24 g, 0.55 mmol) and gave 9b as a white solid (0.29 g; 90%): 1H NMR (CDCl3) δ 8.12 (d, 2H, 3JHH = 8.9 Hz, ArH), 8.09 (d, 1H, 3JHH = 9.2 Hz, ArH), 8.05 (d, 1H, 3JHH = 9.5 Hz, ArH), 7.83 (d, 1H, 3JHH = 8.5 Hz, ArH), 7.81 (d, 3JHH = 8.2 Hz, 2H, ArH), 7.53 (d, 3JHH = 9.2 Hz, 2H, ArH), 7.37 (dd, 3JHH = 9.5 Hz, 4JHH = 3.0 Hz, 1H, ArH), 7.30 (d, 3JHH = 9.2 Hz, 2H, ArH), 7.08 (d, 4JHH = 3.0 Hz, 1H, ArH), 6.60 (s, 1H, NH), 4.24 (m, 2H, OCH2), 4.20 (m, 2H, OCH2), 3.89 (m, 2H, OCH2), 3.82 (m, 2H, OCH2), 2.40 (s, 3H, CH3), 1.56 (s, 9H, CH3); 13C NMR (CDCl3) δ 156.5 (CO), 154.6, 152.6, 144.8, 144.4, 139.3, 137.7, 135.5, 134.3, 133.0, 131.1, 129.8, 128.0, 127.9, 122.4, 118.9, 118.4, 106.1, 80.7 (COtBu), 69.8, 69.2, 69.0, 67.7, 28.4 (C(CH3)3), 21.6 (CH3); m/z (ESI-MS): 579.2 (100%, [M + H]+), 580.2 (36%), 422.1 (9%); HRMS (TOF+): calcd for C31H35N2O7S, [M + H]+, 579.2165; found, 579.2155. Error (ppm): −1.7.
2-(2-(2-((2-(4-((t-Boc)amino)phenyl)quinolin-6-yl)oxy)ethoxy)-ethoxy)ethyl 4-methylbenzenesulfonate (9c)
p-TsCl (3.05g, 16 mmol) and excess triethylamine were added to a solution of 7c (300 mg, 0.64 mmol) in DCM (100 mL) and stirred at RT overnight. The reaction mixture was washed with brine and extracted three times with EtOAc, dried (MgSO4), and filtered. Solvent was evaporated off. Flash chromatography (EtOAc–hexane 1:1) of the residue gave 9c as a yellowish-white solid (354 mg, 89%): 1H NMR (CDCl3) δ 8.04 (m, 4H), 7.77 (m, 3H), 7.51(d, 3JHH = 8.8 Hz, 2H), 7.37 (dd, 3JHH = 6.4 Hz. 6.4 Hz, 1H), 7.29 (d, 3JHH = 8 Hz, 2H), 7.06 (d, 4JHH = 2.8 Hz, 1H), 6.74 (s, 1H), 4.23 (m, 2H), 4.15 (m, 2H), 3.89 (m, 2H), 3.70 (m, 4H), 3.64 (m, 2H), 2.40 (s, 3H), 1.54 (s, 9H); 13C NMR (CDCl3) δ 156.6, 154.5, 152.6, 144.8, 144.4, 139.3, 135.5, 134.3, 132.9, 131.0, 129.8, 128.0, 127.9, 127.9, 80.7, 70.8, 70.8, 69.7, 69.3, 68.8, 67.7, 28.4, 21.6; m/z (ESI-MS): 623.2 (100%, [M + H]+), 624.2 (38%); HRMS (TOF+): calcd for C33H39N2O8S, [M + H]+, 623.2427; found, 623.2418. Error (ppm): −1.4.
2-(2-(2-(2-((2-(4-((t-Boc)amino)phenyl)quinolin-6-yl)oxy)ethoxy)-ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (9d)
The method for 9c was applied to 7d (1.18 g, 2.3 mmol). Flash chromatography (acetone–hexane–triethylamine; 30:69:1) of the crude product gave 9d as a yellowish-white solid (1.26 g, 83%): 1H NMR (CDCl3) δ 8.06 (m, 4H), 7.77 (d, 3JHH = 8.4 Hz 3H), 7.51(d, 3JHH = 8.4 Hz, 2H), 7.38 (dd, 3JHH = 9.2 Hz. 9.2 Hz, 1H), 7.3 (d, 3JHH = 8 Hz, 2H), 7.07 (d, 4JHH = 2.8 Hz, 1H), 6.73 (s, 1H), 4.24 (m, 2H), 4.14 (m, 2H), 3.92 (m, 2H), 3.75 (m, 2H), 3.67 (m, 4H), 3.59 (m, 4H), 2.40 (s, 3H), 1.54 (s, 9H); 13C NMR (CDCl3) δ 156.7, 154.4, 152.6, 144.8, 144.0, 139.4, 135.8, 134.0, 132.9, 130.7, 128.0, 122.7, 118.9, 118.4, 106.0, 80.7, 70.9, 70.8, 70.7, 70.6, 69.7, 69.3, 68.7, 67.7, 28.4, 21.6. HRMS (TOF+): calcd for C35H43N2O9S, [M + H]+, 667.2689; found, 667.2675. Error (ppm): −2.1.
14-((2-(4-((t-Boc)amino)phenyl)quinolin-6-yl)oxy)-3,6,9,12-tetraoxatetradecyl 4-methylbenzenesulfonate (9e)
The method for 9c was applied to 7e (1.8 g, 3.2 mmol) and after flash chromatography (EtOAc–hexane–triethylamine; 80:19:1) of the crude product gave 9e as a yellowish-white solid (1.88 g, 83%): 1H NMR (CDCl3) δ 8.04 (m, 4H), 7.77 (m, 3H), 7.51(d, 3JHH = 8.4 Hz, 2H), 7.38(dd, 3JHH = 9.2 Hz, 3JHH = 9.2 Hz, 1H), 7.30 (d, 3JHH = 8 Hz, 2H), 7.06 (d, 4JHH = 2.8 Hz, 1H), 6.71 (s, 1H), 4.24 (m, 2H), 4.14 (m, 2H), 3.92 (m, 2H), 3.76 (m, 2H), 3.65 (m, 8H), 3.56 (m, 4H), 2.41 (s, 3H), 1.54 (s, 9H); 13C NMR (CDCl3) δ 156.8, 154.3, 152.5, 144.8, 139.6, 136.1, 133.0, 130.4, 129.8, 128.1, 128.0, 127.9, 122.9, 119.0, 118.4, 106.0, 80.8, 70.9, 70.7, 70.7, 70.6, 70.5, 69.7, 69.3, 68.7, 67.8, 28.4, 21.6; HRMS (TOF+): calcd for C37H47N2O10S, [M + H]+, 711.2951; found, 711.2932. Error (ppm): −2.7 ppm.
17-((2-(4-((t-Boc)amino)phenyl)quinolin-6-yl)oxy)-3,6,9,12,15-pentaoxaheptadecyl 4-methylbenzenesulfonate (9f)
The method for 9c was applied to 7f (300 mg, 0.50 mmol) and after flash chromatography (EtOAc–hexane–MeOH; 8:1:1) of the crude product gave 9f (316 mg, 84%) as a yellowish-white solid: 1H NMR (CDCl3) δ 8.04 (m, 4H), 7.77 (m, 3H), 7.51(d, 3JHH = 8.8 Hz, 2H), 7.38 (dd, 3JHH = 6.4 Hz, 3JHH = 6.4 Hz, 1H), 7.31 (d, 3JHH = 8 Hz, 2H), 7.06 (d, 4JHH = 2.8 Hz, 1H), 6.82 (s, 1H), 4.24 (m, 2H), 4.13 (m, 2H), 3.92 (m, 2H), 3.76 (m, 2H), 3.63 (m, 16H), 2.42 (s, 3H), 1.54 (s, 9H); 13C NMR (CDCl3) δ 156.6, 154.5, 152.6, 144.8, 144.3, 142.8, 141.5, 139.4, 135.5, 134.2, 132.9, 130.9, 129.8, 129.7, 128.0, 127.9, 127.9, 125.4, 125.3, 122.5, 118.8, 118.4, 106.0, 80.6, 70.9, 70.7, 70.6, 70.6, 70.6, 70.6, 70.5, 69.9, 69.7, 69.3, 68.7, 67.7, 28.4, 21.7; HRMS (TOF+): calcd for C39H51N2O11S, [M + H]+, 755.3214; found, 755.3209. Error (ppm): −0.7 ppm.
4-(6-(2-Fluoroethoxy)quinolin-2-yl)aniline (10a)
Compound 8 (0.21 g; 0.549 mmol) was dissolved in DCM (12 mL) and TFA (1 mL) was added. The mixture became a pale brown solution. After the reaction was confirmed to be complete, the mixture was partitioned between EtOAc and brine (100 mL × 3). The combined organic phase was dried (MgSO4). Removal of solvent gave 10a as a white solid (0.155 g; 100%): 1H NMR (DMSO-d6) δ 8.17 (d, 1H, 3JHH = 8.7 Hz, ArH), 7.96 (d, 2H, 3JHH = 8.6 Hz, ArH), 7.92 (d, 1H, 3JHH = 8.8 Hz, ArH), 7.87 (d, 1H, 3JHH = 9.1 Hz, ArH), 7.38 (dd, 1H, 3JHH = 9.1 Hz, 4JHH = 2.8 Hz, ArH), 7.34 (d, 1H, 4JHH = 2.7 Hz, ArH), 6.68 (d, 1H, 3JHH = 8.6 Hz, ArH), 5.49 (brs, 2H, NH2), 4.82 (dm, 2JHH = 47.8 Hz, 2H, CH2F), 4.36 (dm, 3JHH = 30.0 Hz, 2H, CH2O); 13C NMR (DMSO-d6) δ 155.3, 154.5, 150.2, 143.6, 135.4, 130.2, 127.9, 127.1, 126.0, 121.7, 118.0, 113.7, 106.7, 82.1 (2JHH = 166 Hz, CF), 67.4 (3JCF = 18.9 Hz, CCF); m/z (ESI-MS): 283.1 (100%, [M + H]+), 284.1 (27%); HRMS (TOF+): calcd for C17H16FN2O, [M + H]+, 283.1247; found, 283.1240. Error (ppm): −2.5.
4-(6-(2-(2-Fluoroethoxy)ethoxy)quinolin-2-yl)aniline (10b)
TBAF solution (1 M in THF, 0.20 mL, 0.20 mmol) was added to a solution of 9b (100 mg, 0.16 mmol) in DMF (10 mL) and microwaved (120c20m80w). The reaction mixture was partitioned between ethyl acetate and brine, and the aqueous layer was extracted with EtOAc. The combined organic phase was dried (MgSO4) and filtered. The solvent was evaporated off. HPLC purification of the residue gave 10b as a white solid (45 mg, 75%): 1H NMR (CDCl3) δ 8.04 (d, 3JHH = 8.4 Hz, 2H), 7.99 (d, 3JHH = 8.8 Hz, 2H), 7.76 (d, 3JHH = 8.8 Hz, 1H), 7.38 (dd, 3JHH = 9.2 Hz, 3JHH = 2.8 Hz, 1H), 7.08 (d, 4JHH = 2.8 Hz, 1H), 6.80 (d, 3JHH = 8.8 Hz, 2H), 4.68 (m, 1H), 4.56 (m, 1H), 4.28 (m, 2H), 3.97 (m, 2H), 3.90 (m, 1H), 3.83 (m, 1H); 13C NMR (CDCl3) δ 156.4, 155.1, 147.6, 144.3, 135.6, 130.7, 130.0, 128.6, 127.6, 122.3, 118.7, 115.2, 106.2, 84.1, 82.4, 70.8, 70.6, 69.9, 67.8. m/z (ESI-MS): 415.2 (36%), 371.2 (21%), 355.1 (21%), 327.2 (100%, [M + H]+); HRMS (TOF+): calcd for C19H20N2O2F, [M + H]+, 327.1509; found, 327.1513. Error (ppm): +1.2.
4-(6-(2-(2-(2-Fluoroethoxy)ethoxy)ethoxy)quinolin-2-yl)aniline (10c)
TBAF solution (1 M in THF, 10 mg, 0.038 mmol) was added to a solution of 9c (20 mg, 0.032 mmol) in DMF (2 mL) and microwaved (120c20m80w). The reaction mixture was washed with H2O and extracted three times with EtOAc, dried (MgSO4), and filtered. Solvent was evaporated off. HPLC of the residue gave 10c as a white solid (9.1 mg, 77%): 1H NMR (CDCl3) δ 7.99 (m, 4H), 7.75 (d, 3JHH = 8.8 Hz, 1H), 7.37 (dd, 3JHH = 6.4 Hz. 6.4 Hz, 1H), 7.06 (d, 4JHH = 2.8 Hz, 1H), 6.79 (d, 3JHH = 8.4 Hz, 2H), 4.63 (m, 1H), 4.51 (m, 1H), 4.26 (m, 2H), 3.94 (m, 2H), 3.76 (m, 6H); 13C NMR (CDCl3) δ 156.4, 155.1, 147.5, 144.3, 135.4, 130.8, 130.0, 128.5, 127.5, 122.3, 118.6, 115.2, 106.1, 84.0, 82.3, 70.9, 70.9, 70.6, 70.4, 69.8, 67.7. HRMS (TOF+): calcd for C21H24FN2O3, [M + H]+, 370.1771; found, 371.1766. Error (ppm): −1.3.
4-(6-(2-(2-(2-(2-Fluoroethoxy)ethoxy)ethoxy)ethoxy)quinolin-2-yl)aniline (10d)
The method for 10c was applied to 9d (20 mg, 0.030 mmol) and gave 10d as a white solid (11.4 mg, 92%). 1H NMR (CDCl3) δ 8.39 (s, 1H), 8.12 (d, J = 8.4 Hz, 2H), 7.99 (t, J = 9.6 Hz, 2H), 7.75 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 1H), 7.39 (dd, J = 9.2 Hz. 9.2 Hz, 1H), 6.97 (d, J = 2.8 Hz, 1H), 4.58 (m, 1H), 4.46 (m, 1H), 4.18 (m, 2H), 3.92 (m, 2H), 3.73 (m, 10H); 13C NMR (CDCl3) δ 156.90, 153.50, 144.17, 137.35, 135.99, 135.70, 130.93, 128.09, 122.82, 120.66, 118.65, 105.82, 83.95, 82.27, 70.90, 70.79, 70.77, 70.66, 70.48, 70.29, 69.67, 67.70; HRMS (TOF+): calcd for C23H28FN2O4, [M + H]+, 415.2033; found, 415.2034. Error (ppm): 0.2.
4-(6-((14-Fluoro-3,6,9,12-tetraoxatetradecyl)oxy)quinolin-2-yl)-aniline (10e)
The method for 10c was applied to 9e (20 mg, 0.028 mmol) and gave 10e as a white solid (8.7 mg, 68%): 1H NMR (CDCl3) δ 8.00 (m, 4H), 7.75 (d, 3JHH = 8.8 Hz, 1H), 7.37 (dd, 3JHH = 9.2 Hz, 3JHH = 9.2 Hz, 1H), 7.06 (d, 4JHH = 2.8 Hz, 1H), 6.80 (m, 2H), 4.63 (m, 1H), 4.49 (m, 1H), 3.93 (m, 2H), 3.75(m, 3H), 3.68 (m, 11H); 13C NMR (CDCl3) δ 156.4, 155.1, 147.5, 144.3, 135.4, 130.8, 130.0, 128.5, 127.5, 122.3, 118.6, 115.2, 106.1, 84.0, 82.3, 70.9, 70.8, 70.7, 70.7, 70.6, 70.5, 70.3, 69.7, 67.7. HRMS (TOF+): calcd for C25H32FN2O5, [M + H]+, 459.2295; found, 459.2294. Error (ppm): −0.1.
4-(6-((17-Fluoro-3,6,9,12,15-pentaoxaheptadecyl)oxy)quinolin-2-yl)aniline (10f)
The method for 10c was applied to 9f (20 mg, 0.039 mmol) and gave 10f as a white solid (7.8 mg, 40%): 1H NMR (DMSO-d6) δ 8.00 (m, 4H), 7.75 (d, 3JHH = 8.8 Hz, 1H), 7.37 (dd, 3JHH = 9.2 Hz, 3JHH = 9.2 Hz, 1H), 7.07 (d, 4JHH = 2.8 Hz, 1H), 6.80 (d, 3JHH = 8.4 Hz, 2H), 4.61 (m, 1H), 4.49 (m, 1H), 4.26 (m, 2H), 3.93 (m, 2H), 3.76 (m, 3H), 3.68 (m, 15H); 13C NMR (DMSO-d6) δ 155.6, 154.3, 150.1, 135.6, 130.0, 127.8, 127.1, 121.8, 117.9, 113.7, 106.6, 83.8, 82.2, 69.9, 69.8, 69.8, 69.5, 68.8, 67.4; HRMS (TOF+): calcd for C27H36FN2O6, [M + H]+, 503.2557; found, 503.2556. Error (ppm): −0.2.
t-Butyl (4-(6-(2-(2-(2,6-Dioxocyclohexyl)ethoxy)ethoxy)quinolin-2-yl)phenyl)carbamate (11a)
Compound 9a (21.2 mg, 0.0367 mmol), cyclohexane-1,3-dione (9.4 mg, 0.0838 mmol), and K2CO3 (67 mg, 0.485 mmol) were suspended in MeCN (1.0 mL). The mixture was microwaved (120c40m30w250 psi). After the reaction was found to be complete by HPLC, semipreparative HPLC gave 11a as a white solid (15 mg; 79%): 1H NMR (CDCl3) δ 8.07 (d, 2H, 3JHH = 9.1 Hz, ArH), 8.04 (d, 1H, 3JHH = 9.4 Hz, ArH), 8.01 (d, 1H, 3JHH = 9.4 Hz, ArH), 7.79 (d, 1H, 3JHH = 9.1 Hz, ArH), 7.49 (d, 3JHH = 8.1 Hz, 1H, ArH), 7.38 (dd, 3JHH = 9.4 Hz, 4JHH = 2.6 Hz, 1H, ArH), 7.07 (d, 4JHH = 2.6 Hz, 1H, ArH), 6.56 (s, 1H, NH), 5.35 (s, 1H, CH), 4.27 (m, 2H, OCH2), 4.01 (m, 2H, OCH2), 3.95 (m, 2H, OCH2), 3.90 (m, 2H, OCH2), 2.40 (m, 2H, CH2), 2.31 (m, 2H, CH2), 1.95 (m, 2H, CH2), 1.52 (s, 9H, CH3); 13C NMR (CDCl3) δ 199.9 (C=O), 177.9 (C=O), 156.7, 154.8, 152.8, 144.6, 139.5, 135.7, 134.5, 131.3, 128.2, 128.0, 122.6, 119.1, 118.6, 106.3, 103.2, 80.9 (CO), 70.1 (CO), 69.4 (CO), 68.0 (CO), 67.9 (CO), 36.9 (CH), 29.1 (CH2), 28.6 (CH3), 21.4 (CH2); m/z (ESI-MS): 519.2 (100%, [M + H]+), 520.3 (25%); HRMS (TOF+): calcd for C30H35N2O6, [M + H]+, 519.2495; found, 519.2495. Error (ppm): 0.0.
t-Butyl (4-(6-(2-(2-(2,5-Dioxocyclopentyl)ethoxy)ethoxy)quinolin-2-yl)phenyl)carbamate (11b)
The method for 11a was applied to 9b (27.0 mg, 0.0497 mmol) and cyclopentane-1,3-dione (12.8 mg, 0.130 mmol) under 120c60m100w250 psi conditions and gave 11b as a white solid (14 mg; 59%): 1H NMR (CDCl3) δ 8.07 (d, 2H, 3JHH = 8.7 Hz, ArH), 8.04 (d, 1H, 3JHH = 9.5 Hz, ArH), 8.02 (d, 1H, 10.2 Hz, ArH), 7.79 (d, 1H, 3JHH = 8.6 Hz, ArH), 7.49 (d, 3JHH = 8.6 Hz, 1H, ArH), 7.37 (dd, 3JHH = 9.2 Hz, 4JHH = 2.7 Hz, 1H, ArH), 7.07 (d, 4JHH = 2.8 Hz, 1H, ArH), 6.57 (s, 1H, NH), 5.31 (s, 1H, CH), 4.27 (m, 2H, OCH2), 4.15 (m, 2H, OCH2), 3.96 (m, 2H, OCH2), 3.92 (m, 2H, OCH2), 2.62 (m, 2H, CH2), 2.42 (m, 2H, CH2), 1.52 (s, 9H, CH3); 13C NMR (CDCl3) δ 205.9 (C=O), 190.0 (C=O), 156.5, 154.6, 152.6, 144.4, 139.3, 135.5, 134.2, 131.1, 128.0, 127.8, 122.3, 118.9, 118.4, 106.1, 105.1, 80.7 (CO), 71.1 (CO), 69.9 (CO), 69.1 (CO), 67.7 (CO), 34.1 (CH), 28.5 (CH2), 28.4 (CH3); m/z (ESI-MS): 505.2 (100%, [M + H]+), 506.2 (30%), 449.3 (35%), 450.3 (12%); HRMS (TOF+): calcd for C29H33N2O6, [M + H]+, 505.2339; found, 505.2327. Error (ppm): −2.4.
t-Butyl (4-(6-(2-(2-(2-Oxooxazolidin-3-yl)ethoxy)ethoxy)quinolin-2-yl)phenyl)carbamate (11c)
Compound 9c (12.1 mg, 0.0209 mmol), oxazolidin-2-one (5.3 mg, 0.0609 mmol), and Li2O (5 mg, 0.167 mmol) were suspended in DMF (1.0 mL). The mixture was microwaved (80c10m60w250 psi). After the reaction was found to be complete by HPLC, semipreparative HPLC gave 11c as a white solid (6.0 mg; 58%): 1H NMR (CDCl3) δ 8.07 (d, 2H, 3JHH = 8.9 Hz, ArH), 8.05 (d, 1H, 3JHH = 9.2 Hz, ArH), 8.01 (d, 1H, 3JHH = 9.2 Hz, ArH), 7.79 (d, 1H, 3JHH = 9.2 Hz, ArH), 7.49 (d, 3JHH = 8.9 Hz, 1H, ArH), 7.34 (dd, 3JHH = 9.2 Hz, 4JHH = 3.2 Hz, 1H, ArH), 7.06 (d, 4JHH = 2.9 Hz, 1H, ArH), 6.61 (s, 1H, NH), 4.26–4.21 (m, 2H, OCH2), 3.89–3.86 (m, 2H, OCH2), 3.75 (t, 3JHH = 5.2 Hz, 1H, CH2), 3.70–3.66 (m, 2H, OCH2), 3.48 (t, 3JHH = 5.2 Hz, 1H, CH2), 1.52 (s, 9H, CH3); 13C NMR (CDCl3) δ 158.6, 156.6, 154.6, 152.6, 144.4, 1393.3, 135.5, 134.2, 131.1, 128.0, 127.9, 122.3, 118.9, 118.4, 106.1, 80.7 (CO), 69.9 (CO), 69.4 (CO), 67.6 (CO), 62.0 (CO), 46.0 (CN), 44.1 (CN), 28.4 (CH3); m/z (ESI-MS): 494.2 (100%, [M + H]+), 495.2 (30%); HRMS (TOF+): calcd for C27H32N3O6, [M + H]+, 494.2291; found, 494.2298. Error (ppm): +1.4.
t-Butyl (4-(6-(2-(2-(2-Oxoimidazolidin-1-yl)ethoxy)ethoxy)-quinolin-2-yl)phenyl)carbamate (11d)
The method for 11c was applied to 9d (20.0 mg, 0.0346 mmol) and imidazolidin-2-one (15.0 mg, 0.174 mmol) and gave 11d as a white solid (3.5 mg; 21%): 1H NMR (CDCl3) δ 8.08 (d, 2H, 3JHH = 8.7 Hz, ArH), 8.06 (d, 1H, 3JHH = 9.0 Hz, ArH), 8.01 (d, 1H, 3JHH = 9.4 Hz, ArH), 7.79 (d, 1H, 3JHH = 9.0 Hz, ArH), 7.49 (d, 3JHH = 9.0 Hz, 1H, ArH), 7.36 (dd, 3JHH = 9.4 Hz, 4JHH = 2.9 Hz, 1H, ArH), 7.08 (d, 4JHH = 2.6 Hz, 1H, ArH), 6.58 (s, 1H, NH), 4.23 (tm, 3JHH = 4.5 Hz, 2H, OCH2), 3.88 (tm, 3JHH = 4.5 Hz, 2H, OCH2), 3.72 (tm, 3JHH = 5.3 Hz, 2H, OCH2), 3.56 (tm, 3JHH = 7.9 Hz, 2H, OCH2), 3.41 (tm, 3JHH = 5.3 Hz, 2H, OCH2), 3.33 (tm, 3JHH = 7.9 Hz, 2H, OCH2), 1.52 (s, 9H, CH3); 13C NMR (CDCl3) δ 162.9, 156.9, 154.8, 152.8, 144.6, 139.5, 135.7, 134.5, 131.3, 128.2, 128.1, 122.6, 119.1, 118.7, 106.3, 81.0 (CO), 70.6 (CO), 69.5 (CO), 67.9 (CO), 46.7 (CN), 43.6 (CN), 38.5 (CN), 28.6 (CH3); m/z (ESI-MS): 493.2 (100%, [M + H]+), 494.2 (28%), 437.3 (26%); HRMS (TOF+): calcd for C27H33N4O5, [M + H]+, 493.2451; found, 493.2441. Error (ppm): −2.0.
t-Butyl (4-(6-(2-(2-(2-Oxothiazolidin-3-yl)ethoxy)ethoxy)quinolin-2-yl)phenyl)carbamate (11e)
The method for 11c was applied to 9e (17.9 mg, 0.0310 mmol) and thiazolidin-2-one (24.6 mg, 0.239 mmol) and gave 11e as a white solid (11 mg; 70%): 1H NMR (CD3OD) δ 8.28 (d, 2H, 3JHH = 8.8 Hz, ArH), 8.02 (d, 1H, 3JHH = 8.9 Hz, ArH), 8.00 (d, 1H, 3JHH = 9.4 Hz, ArH), 7.92 (d, 1H, 3JHH = 8.9 Hz, ArH), 7.60 (d, 3JHH = 9.4 Hz, 1H, ArH), 7.44 (dd, 3JHH = 9.4 Hz, 4JHH = 2.8 Hz, 1H, ArH), 7.33 (d, 4JHH = 2.8 Hz, 1H, ArH), 4.30 (m, 2H, OCH2), 3.92 (m, 2H, OCH2), 3.82 (t, 3JHH = 7.9 Hz, 2H, NCH2), 3.71 (m, 2H, OCH2), 3.54 (m, 2H, SCH2), 3.26 (t, 3JHH = 7.9 Hz, 2H, SCH2), 1.54 (s, 9H, CH3); 13C NMR (CDCl3) δ 172.5, 156.8, 154.8, 152.8, 144.6, 139.5, 135.7, 134.4, 131.3, 128.2, 128.1, 122.5, 119.1, 118.6, 106.3, 81.0 (CO), 70.1 (CO), 69.7 (CO), 67.8 (CO), 50.2 (CN), 44.9 (CN), 28.6 (CH3), 26.2 (C–S); m/z (ESI-MS): 510.1 (100%, [M + H]+), 511.1 (30%), 454.3 (40%); HRMS (TOF+): calcd for C27H32N3O5S, [M + H]+, 510.2063; found, 510.2073. Error (ppm): +2.0.
t-Butyl (4-(6-(2-(2-((2-Hydroxyethyl)amino)ethoxy)ethoxy)-quinolin-2-yl)phenyl)carbamate (11f)
The method for 11a was applied to 9f (20 mg, 0.0346 mmol) and 2-aminoethanol (10 μL, 0.165 mmol) under microwave conditions (120c30m300w250 psi) and gave 11f as a white solid (12 mg; 74%): 1H NMR (CDCl3) δ 8.07 (d, 2H, 3JHH = 8.8 Hz, ArH), 8.04 (d, 1H, 3JHH = 9.5 Hz, ArH), 8.01 (d, 1H, 3JHH = 9.3 Hz, ArH), 7.78 (d, 1H, 3JHH = 8.6 Hz, ArH), 7.48 (d, 3JHH = 8.7 Hz, 1H, ArH), 7.37 (dd, 3JHH = 9.2 Hz, 4JHH = 2.8 Hz, 1H, ArH), 7.07 (d, 4JHH = 2.8 Hz, 1H, ArH), 6.58 (s, 1H, NH), 4.26–4.21 (m, 2H, OCH2), 3.91–3.49 (m, 2H, OCH2), 3.74–3.72 (m, 2H, OCH2), 3.68–3.64 (m, 2H, OCH2), 2.92–2.89 (m, 2H, NCH2), 2.86–2.82 (m, 2H, NCH2), 1.52 (s, 9H, CH3); 13C NMR (CDCl3) δ 156.5, 154.6, 152.6, 144.4, 139.3, 135.5, 134.3, 131.1, 128.0, 127.8, 122.3, 118.9, 118.5, 106.2, 105.1, 80.8 (CO), 69.6 (CO), 67.6 (CO), 59.9 (CO), 50.9 (CN), 48.4 (CN), 28.4 (CH3); m/z (ESI-MS): 468.3 (60%, [M + H]+), 437.7 (20%), 425.2 (27%), 311.1 (15%), 234.6 (18%), 206.6 (100%), 197.6 (14%); HRMS (TOF+): calcd for C26H34N3O5, [M + H]+, 468.2498; found, 468.2511. Error (ppm): +2.8.
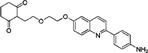
2-(2-(2-((2-(4-Aminophenyl)quinolin-6-yl)oxy)ethoxy)ethyl)-cyclohexane-1,3-dione (12a)
BF3·OEt2 (20 μL) was added to a solution of 11a (20.2 mg; 0.0390 mmol) in CHCl3 (1.0 mL). The mixture became an orange-yellowish solution. After the reaction was found to be complete by HPLC, the solution was partitioned between EtOAc and brine (10 mL × 3). The combined organic phase was dried (MgSO4). Removal of solvent gave 12a as a white solid (11 mg; 67%): 1H NMR (CDCl3) δ 8.84 (d, 2H, 3JHH = 8.4 Hz, ArH), 8.27 (d, 1H, 3JHH = 9.9 Hz, ArH), 8.12 (d, 1H, 3JHH = 8.4 Hz, ArH), 7.97 (d, 2H, 3JHH = 8.4 Hz, ArH), 7.78 (dd, 3JHH = 9.2 Hz, 4JHH = 2.7 Hz, 1H, ArH), 7.54 (d, 3JHH = 8.4 Hz, 2H, ArH), 7.43 (d, 4JHH = 2.7 Hz, 1H, ArH), 6.56 (s, 1H, NH), 4.39 (m, 2H, OCH2), 4.09 (m, 2H, OCH2), 3.98 (m, 2H, OCH2), 3.88 (m, 2H, CCH2), 2.89 (m, 2H, CH2), 2.70 (m, 2H, CH2), 2.18 (m, 2H, CH2); 13C NMR (CDCl3) δ 199.9, 177.9, 156.4, 155.4, 147.7, 144.6, 135.5, 131.1, 130.2, 128.7, 127.7, 122.3, 118.9, 115.4, 106.4, 103.2, 70.1, 69.4, 68.0, 67.9, 36.9, 29.2, 21.4; m/z (ESI-MS): 419.4 (100%, [M + H]+), 420.4 (20%); HRMS (TOF+): calcd for C25H27N2O4, [M + H]+, 418.1971; found, 418.1974. Error (ppm): 0.7.
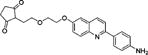
2-(2-(2-((2-(4-Aminophenyl)quinolin-6-yl)oxy)ethoxy)ethyl)-cyclopentane-1,3-dione (12b)
Compound 11b (21.2 mg; 0.0420 mmol) was suspended in dioxane (1.0 mL). After ultrasound treatment, a cloudy milky suspension formed. BF3·OEt2 (20 μL, 3.9 equiv) was added. The mixture became an orange-yellowish suspension. After 2 to 5 min a bright yellow solution formed and then a yellow precipitate. After the reaction was found to be complete by HPLC, c. NH3 solution (0.5 mL) was added to the mixture, which then changed from yellow to white, with some residue of a white solid. The mixture was partitioned between EtOAc and brine (10 mL × 3). The combined organic phase was dried (MgSO4). Removal of solvent gave 12b as a white solid (5.9 mg, 35%): 1H NMR (CDCl3) δ 8.01 (d, 1H, 3JHH = 8.6 Hz, ArH), 7.99 (d, 1H, 3JHH = 9.1 Hz, ArH), 7.96 (d, 2H, 3JHH = 8.6 Hz, ArH), 7.74 (d, 1H, 3JHH = 8.6 Hz, ArH), 7.35 (dd, 3JHH = 9.7 Hz, 4JHH = 2.8 Hz, 1H, ArH), 7.05 (d, 4JHH = 2.8 Hz, 2H, ArH), 6.78 (d, 3JHH = 8.6 Hz, 2H, ArH), 4.26 (m, 2H, OCH2), 4.16 (m, 2H, OCH2), 3.96–3.91 (m, 4H, OCH2 + CCH2), 2.60 (m, 2H, CCH2), 2.40 (m, 2H, CH2); 13C NMR (CDCl3) δ 205.8, 190.0, 156.2, 155.3, 147.5, 144.4, 135.3, 130.9, 130.0, 128.5, 127.5, 122.1, 118.7, 115.2, 106.2, 105.1, 71.1, 70.0, 69.1, 67.7, 34.1, 28.5; m/z (ESI-MS): 405.4 (100%, [M + H]+), 406.4 (30%); HRMS (TOF+): calcd for C24H25N2O4, [M + H]+, 404.1814; found, 404.1814. Error (ppm): 0.
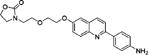
3-(2-(2-((2-(4-Aminophenyl)quinolin-6-yl)oxy)ethoxy)ethyl)-oxazolidin-2-one (12c)
Compound 11c (18.9 mg; 0.0383 mmol) was suspended in MeCN (1.0 mL) with the help of ultrasound to form a milky solution and BF3·OEt2 (24 μL, 5 equiv) was added in one portion. The mixture became an orange-yellowish solution and was stirred at RT for 1 h. The reaction was found to be complete by HPLC. c. NH3 in ethylene glycol (1:4; 0.5 mL) was added and stirred for 10 min. Preparative HPLC gave 12c as a white solid (10.9 mg; 72%): 1H NMR (DMSO-d6) δ 8.17 (d, 1H, 3JHH = 8.8 Hz, ArH), 7.95 (d, 2H, 3JHH = 8.6 Hz, ArH), 7.91 (d, 1H, 3JHH = 8.8 Hz, ArH), 7.86 (d, 1H, 3JHH = 9.0 Hz, ArH), 7.35 (dd, 3JHH = 9.0 Hz, 4JHH = 2.8 Hz, 1H, ArH), 7.33 (d, 4JHH = 2.7 Hz, 1H, ArH), 6.67 (d, 2H, 3JHH = 8.6 Hz, ArH), 5.48 (brs, 2H, NH2), 4.24–4.20 (m, 4H, NCH2 + OCH2), 3.83–3.81 (m, 2H, OCH2), 3.64 (t, 2H, 3JHH = 5.4 Hz, OCH2), 3.62–3.58 (m, 2H, OCH2), 3.36 (m, 2H, NCH2); 13C NMR (DMSO-d6) δ 206.8 (C=O), 158.0, 155.6, 154.4, 150.1, 143.6, 135.4, 130.1, 127.9, 127.1, 126.1, 121.8, 117.9, 113.7, 106.6, 68.5, 68.1, 67.4, 62.8, 61.7, 44.7, 43.3; m/z (ESI-MS): 394.2 (100%, [M + H]+), 395.2 (39%); HRMS (TOF+): calcd for C22H24N3O4, [M + H]+, 394.1767; found, 394.1760. Error (ppm): −1.8.
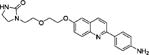
1-(2-(2-((2-(4-Aminophenyl)quinolin-6-yl)oxy)ethoxy)ethyl)-imidazolidin-2-one (12d)
Compound 11d (8.2 mg; 0.0166 mmol) was suspended in MeCN (1.0 mL) with the help of ultrasound to form a milky suspension, and BF3·OEt2 (11 μL, 5 equiv) was added in one portion. The mixture became an orange-yellowish solution. After stirring the mixture for 1 h at RT, the reaction was found to be complete by HPLC. c. NH3 in ethylene glycol (1:4; 0.5 mL) was added and stirred for 10 min. Preparative HPLC of the mixture gave 12d as a white solid (3.6 mg; 55%): 1H NMR (DMSO-d6) δ 8.29 (brd, 3JHH = 8.2 Hz, 1H, ArH), 8.00–7.92 (m, 4H, ArH), 7.44–7.41 (m, 2H, ArH), 6.71 (d, 3JHH = 8.5 Hz, 2H, ArH), 6.52 (brs, 1H, NH), 6.28 (brs, 1H, NH), 4.25–4.23 (m, 2H, OCH2), 3.83–3.80 (m, 2H, OCH2), 3.58 (t, 2H, 3JHH = 5.5 Hz, OCH2), 3.39 (t, 2H, 3JHH = 7.8 Hz, NCH2), 3.32 (t, 2H, 3JHH = 5.6 Hz, OCH2), 3.18 (t, 2H, 3JHH = 7.8 Hz, NCH2); m/z (ESI-MS): 393.2 (100%, [M + H]+), 394.2 (26%), 325.2 (34%), 223.9 (58%), 195.9 (33%), 178.9 (43%), 116.0 (45%); HRMS (TOF+): calcd for C22H25N3O3, [M + H]+, 393.1927; found, 393.1928. Error (ppm): 0.3.
3-(2-(2-((2-(4-Aminophenyl)quinolin-6-yl)oxy)ethoxy)ethyl)-thiazolidin-2-one (12e)
Compound 11e (20.1 mg; 0.0394 mmol) was added to MeCN (1.0 mL) with the help of ultrasound to form a milky suspension, and BF3·OEt2 in MeCN solution (1:9, 280 μL, 5 equiv) was added. The mixture became a yellowish-orange solution. The mixture was stirred for 1 h at RT for 1 h. The reaction was found to be complete by HPLC. c. NH3 in ethylene glycol (1:4; 0.5 mL) was added and stirred for 10 min. Preparative HPLC of the mixture gave 12e as a white solid (12.3 mg; 76%): 1H NMR (DMSO-d6) δ 8.50 (d, 1H, 3JHH = 8.7 Hz, ArH), 8.22 (d, 1H, 3JHH = 8.0 Hz, ArH), 8.13 (d, 1H, 3JHH = 8.8 Hz, ArH), 8.05 (d, 2H, 3JHH = 8.7 Hz, ArH), 7.55 (dd, 3JHH = 8.7 Hz, 4JHH = 2.0 Hz, 1H, ArH), 7.53 (s, 1H, ArH), 6.80 (d, 3JHH = 8.6 Hz, 2H, ArH), 4.26 (m, 2H, NCH2), 3.83 (m, 2H, SCH2), 3.70 (t, 2H, 3JHH = 7.3 Hz, OCH2), 3.63 (t, 2H, 3JHH = 5.4 Hz, OCH2), 3.42 (t, 2H, 3JHH = 5.4 Hz, NCH2), 3.26 (t, 2H, 3JHH = 7.3 Hz, OCH2); 13C NMR (DMSO-d6) δ 170.9, 162.3, 156.7, 152.7, 151.1, 139.5, 138.2, 129.7, 127.4, 125.7, 123.9, 119.4, 114.5, 107.4, 68.4, 68.0, 67.6, 48.8, 43.8, 25.4; m/z (ESI-MS): 410.2 (100%, [M + H]+), 411.2 (26%); HRMS (TOF+): calcd for C24H24N2O4, [M + H]+, 410.1538; found, 410.1535. Error (ppm): −0.7.
2-((2-(2-((2-(4-Aminophenyl)quinolin-6-yl)oxy)ethoxy)ethyl)-amino)ethan-1-ol (12f)
Compound 11f (12.4 mg; 0.0265 mmol) was added to MeCN (1.0 mL) to form a milky suspension and BF3·OEt2 (13 μL, 5 equiv) was added. The mixture became a yellowish-orange solution. After 2 h at RT, the concentration of the starting material was less than that of an impurity, as monitored by HPLC. Semipreparative HPLC of the reaction mixture gave 12f as a white solid (6.7 mg; 69%): 1H NMR (CDCl3) δ 8.01 (d, 1H, 3JHH = 8.8 Hz, ArH), 7.99 (d, 1H, 3JHH = 9.4 Hz, ArH), 7.96 (d, 2H, 3JHH = 8.8 Hz, ArH), 7.75 (d, 1H, 3JHH = 8.8 Hz, ArH), 7.36 (dd, 3JHH = 9.4 Hz, 4JHH = 3.0 Hz, 1H, ArH), 7.06 (d, 4JHH = 2.7 Hz, 1H, ArH), 6.79 (d, 3JHH = 9.1 Hz, 2H, ArH), 4.24 (m, 2H, OCH2), 3.89 (m, 2H, OCH2), 3.83 (s, 2H, OH +NH), 3.69 (m, 2H, OCH2), 3.61 (m, 2H, OCH2), 2.84 (m, 2H, NCH2), 2.79 (m, 2H, NCH2); 13C NMR (DMSO-d6) δ 155.5, 154.4, 150.1, 143.6, 135.4, 130.1, 127.9, 127.1, 126.0, 121.8, 118.0, 113.7, 106.6, 68.9, 67.3, 66.9, 57.3, 49.6, 46.8; m/z (ESI-MS): 368.3 (100%, [M + H]+), 369.3 (30%), 325.2 (5%), 281.1 (62%, [M − C2H4NHC2H4O + H]+), 237.1 (62%, [M − C2H4OC2H4NHC2H4O + H]+), 184.6 (82%), 175.6 (52%); HRMS (TOF+): calcd for C21H26N3O3, [M + H]+, 368.1974; found, 368.1974. Error (ppm): 0.
4-(6-(2-Fluoroethoxy)quinolin-2-yl)-2,6-diiodoaniline (13)
THK-523 (12a; 109 mg, 0.386 mmol) and NIS (175 mg, 0.778 mmol) were dissolved in CHCl3–MeOH (1:1; 5 mL) and stirred overnight. HPLC showed a 1:1 ratio of mono and di-iodo substituted products. More NIS (55 mg, 0.245 mmol) was added and the reaction mixture was stirred for another 5 days, or until the reaction was found to be complete by HPLC. The mixture was partitioned between EtOAc and brine (50 mL × 3). The combined organic phase was dried (MgSO4). After removal of solvent, the crude product was dissolved in DCM, and silica gel (2 mL) was added, and dried. Chromatography of the resultant powder on a silica gel column (50 mL; EtOAc–hexane: 1:1) gave 13 as a dark solid (0.11 g; 53%): 1H NMR (CDCl3) δ 8.46 (s, 2H, ArH), 8.10 (brs, 1H, ArH), 8.07 (d, 1H, 3JHH 8.6 Hz, ArH), 7.70 (d, 2H, 3JHH 8.5 Hz, ArH), 7.42 (dd, 1H, 3JHH 9.8 Hz, 4JHH 2.9 Hz, ArH), 7.08 (d, 4JHH 2.6 Hz, 1H, ArH), 4.89–4.87 (m, 2JHF 47.4 Hz, 1H, FCH2), 4.83 (brs, 2H, NH2), 4.77–4.75 (m, 2JHF 47.4 Hz, 1H, FCH2), 4.38–4.36 (m, 2JHF 27.6 Hz, 1H, OCH2), 4.31–4.29 (m, 2JHF 27.6 Hz, 1H, OCH2), 1.52 (s, 9H, CH3); 13C NMR (DMSO-d6) δ 156.0, 151.2, 148.1, 143.6, 137.7, 136.3, 131.0, 130.6, 127.9, 122.5, 118.4, 106.8, 82.3 (1JCF 166.2 Hz, CF), 81.9 (CI), 67.6 (2JCF 19.2 Hz, CCF); m/z (ESI-MS): 535.0 (100%, [M + H]+), 536.0 (18%); HRMS (TOF+): calcd for C17H14FI2N2O [M + H]+, 534.9180; found, 534.9185. Error (ppm): +0.9.
4,6,9-Tribromo-7-(6-fluoropyridin-3-yl)-5H-pyrido[4,3-b]indole (14)
A solution of T807 (66 mg; 0.25 mmol) in DCM (3 mL) was treated with NBS (1.0 mmol) at 0 °C. The reaction mixture was stirred at RT (48 h) until starting materials had disappeared, as evidenced by TLC. A small amount of silica gel was added and the solvent was evaporated off under reduced pressure. Flash chromatography of the residue on silica gel (EtOAc) gave 14 as a colorless solid (19 mg; 15%): mp 176–178 °C; 1H NMR (CDCl3): δ 7.10 (dd, 4JHH 4 Hz, 3JHH 8 Hz, 1H), 7.76 (dt, 4JHH 4, 3JHH 8 Hz, 1H), 8.19 (s, 1H), 8.42 (s, 1H), 8.61 (brs, 1H), 8.72 (s, 1H), 9.23 (s, 1H); HRMS (EI): m/z calcd for C16H8Br3FN3 (M+): 497.8257; found: 497.8252. Error (ppm): +1.0.
Tritiated Ligands
[3H]T807 (1850 GBq per mmol, radiochemical purity > 97%) and [3H]THK-523 (3[H]10a; 1665 GBq per mmol, radiochemical purity > 99%) were obtained through custom syntheses at Moravek Biochemicals & Radiochemicals, Inc. (Brea, CA) based on hydrogenation of the halo precursors 13 and 14, respectively.
In Vitro Binding Assays
Human AD brain tissue was homogenized in assay medium (0.1% Tween-20 in PBS) at 1:500 dilution in volume, and aliquots of this suspension (100 μL) were added to each of four tubes (total 48 in a rack). A solution of tritiated radioligand ([3H]THK-523, [3H]T807 or [3H]PIB); 10 Bq/μL) in assay medium (100 μL) was added to each tube. Nonradioactive ligand (THK-523, T807 or PIB) or test compound was dissolved in DMSO to give a 1 mM stock solution, which was further diluted with DMSO to give solutions ranging in concentration from 1 × 10−5 to 10−10 M. A volume of 10 μL of each solution was added to each tube. After assembly of the components listed above in each tube (made up to a total volume of 1.0 mL with assay medium), the tubes were vortexed and then incubated for 2 h at 37 °C. After separation of tube contents with a cell harvester, the filter paper (GF/B; Whatman; pretreated with 0.5% polyethylenimine solution) was washed with PBS (3 mL × 3). The filters were placed in 7 mL plastic vials and then scintillation fluid (4 mL) added to each. Each vial was vortexed until the filter was floating. After overnight incubation, the scintillation vials were counted for radioactivity. The collected data were analyzed with GraphPad Prism 4 version 4.03 (GraphPad Software; San Diego, CA) with “One site competition” curve-fitting. Ki values were calculated according to the Cheng–Prusoff equation:39
where [L] is the concentration (0.4 nM) and KD is the equilibrium dissociation constant of the reference radioligand. KD was determined with “Scatchard analysis of homologous displacement” from multiple runs with self-displacement from AD brain tissue.
Preparation of [18F]THK-523 ([18F]10a) and [18F]10b–f
The general method is exemplified for the radiosynthesis of [18F]THK-523 ([18F]10a), as follows. No-carrier-added (NCA) [18F]fluoride ion was obtained from the 18O(p,n)18F reaction by the proton irradiation of 18O-enriched water (>95 atom %)40 at the NIH Clinical Center. NCA [18F]fluoride ion (3.7–9.25 GBq in 0.25 mL of H218O) was added to kryptofix 2.2.2 (5 mg, 13.3 μmol) and K2CO3 (0.5 mg; 3.6 μmol) in MeCN–water (95:5; 0.1 mL), and dried azeotropically by four addition–evaporation cycles of MeCN (1 mL each cycle). Precursor 9a (2.0 mg) was loaded into a 1.5 mL closed vial. [18F]Fluoride ion-K+-kryptofix 2.2.2 in dry DMF (0.7 mL) was then added. The reaction mixture was heated at 120 °C for 15 min, and the solvent then removed by blowing nitrogen through the mixture at 120 °C for 5 min. A solution of c. HCl–ethylene glycol solution (0.5 mL; 1:9 v/v) was added, heated at 120 °C for 5 min, and then cooled to below 40 °C. The reaction was quenched with a solution (0.5 mL) of c. NH3 (33%)-water (1:4 v/v). The reaction mixture was loaded onto a HPLC column (X-Bridge C-18; 19 × 150 mm, 10 μm particle; Waters) eluted at 10.0 mL/min with a mixture of A (0.1 M ammonium formate) and B (MeCN), with B increased from 20 to 40% over 60 min. Eluate was monitored for absorbance at 350 nm and radioactivity (NaI(Tl) Flow Count detector; Bioscan). The radioactive fraction with the same retention time as the respective reference ligand was collected and diluted with water (∼100 mL). This solution was passed through a C-18 Sep-Pak cartridge (Waters; Milford, MA), which was washed at least twice with water. The radioactive product was washed out of the cartridge with absolute ethanol (1.0 mL) into saline (0.9% w/v; 10 mL) plus Ethanol USP (0.9 mL) containing Polysorbate 80 (20 mg), and then sterile-filtered into a sterile and pyrogen-free dose vial.
The formulated radioligand was analyzed with HPLC on an X-bridge C18 column (10 μm, 4.6 × 250 mm; Waters) eluted with 40% acetonitrile in water at 1.75 mL/min over 15 min. Eluate was monitored for absorbance at 350 nm and for radioactivity.
Acknowledgments
We gratefully thank Dr. Mary Herman [Human Brain Collection Core (HBCC), NIH] for postmortem human brain tissues and useful discussions. We also thank the NIH Clinical PET Center (Chief: Dr. P. Herscovitch) for fluorine-18 production.
Funding
This research was supported by the Intramural Research Program of the NIH, specifically the NIMH (ZIA-MH0023793), and by the Henry Jackson Foundation (Grant Award Number 306135-10.01-60855).
ABBREVIATIONS
- NCA
no-carrier-added
- NFTs
neurofibrillary tangles
- PIB
Pittsburgh Compound B
- RCY
decay-corrected radiochemical yield
- SD
standard deviation
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 3.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 4.Dickson DW. Neuropathological diagnosis of Alzheimer’s disease: a perspective from longitudinal clinicopathological studies. Neurobiol Aging. 1997;18:S21–S26. doi: 10.1016/s0197-4580(97)00065-1. [DOI] [PubMed] [Google Scholar]
- 5.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 6.Dèlaere P, Duyckaerts C, Brion JP, Poulain V, Hauw JJ. Tau, paired helical filaments and amyloid in the neocortex: a morphometric study of 15 cases with graded intellectual status in aging and senile dementia of Alzheimer type. Acta Neuropathol. 1989;77:645–653. doi: 10.1007/BF00687893. [DOI] [PubMed] [Google Scholar]
- 7.Duyckaerts C, Brion JP, Hauw JJ, Flament-Durand J. Quantitative assessment of the density of neurofibrillary tangles and senile plaques in senile dementia of the Alzheimer type. Comparison of immunocytochemistry with a specific antibody and Bodian’s protargol method. Acta Neuropathol. 1987;73:167–170. doi: 10.1007/BF00693783. [DOI] [PubMed] [Google Scholar]
- 8.Rowe CC, Ackerman U, Browne W, Mulligan R, Pike KL, O’Keefe G, Tochon-Danguy H, Chan G, Berlangieri SU, Jones G, Dickinson-Rowe KL, Kung HP, Zhang W, Kung MP, Skovronsky D, Dyrks T, Holl G, Krause S, Friebe M, Lehman L, Lindemann S, Dinkelborg LM, Masters CL, Villemagne VL. Imaging of amyloid β in Alzheimer’s disease with 18F-BAY94–9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 2008;7:129–135. doi: 10.1016/S1474-4422(08)70001-2. [DOI] [PubMed] [Google Scholar]
- 9.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O’Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 10.Dèlaere P, Duyckaerts C, Masters C, Beyreuther K, Piette F, Hauw JJ. Large amounts of neocortical βA4 deposits without neuritic plaques nor tangles in a psychometrically assessed, non-demented person. Neurosci Lett. 1990;116:87–93. doi: 10.1016/0304-3940(90)90391-l. [DOI] [PubMed] [Google Scholar]
- 11.Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, Renbing X, Peck A. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- 12.Hampel H, Blennow K, Shaw LM, Hoessler YC, Zetterberg H, Trojanowski JQ. Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp Gerontol. 2010;45:30–40. doi: 10.1016/j.exger.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampel H, Ewers M, Bürger K, Annas P, Mörtberg A, Bogstedt A, Frölich L, Schröder J, Schönknecht P, Riepe MW, Kraft I, Gasser T, Leyhe T, Möller HJ, Kurz A, Basun H. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70:922–931. [PubMed] [Google Scholar]
- 14.Ganzer S, Arlt S, Schoder V, Buhmann C, Mandelkow EM, Finckh U, Beisiegel U, Naber D, Müller-Thomsen T. CSF-tau, CSF-Aβ1–42, ApoE-genotype and clinical parameters in the diagnosis of Alzheimer’s disease: combination of CSF-tau and MMSE yields highest sensitivity and specificity. J Neural Transm. 2003;110:1149–1160. doi: 10.1007/s00702-003-0017-7. [DOI] [PubMed] [Google Scholar]
- 15.Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003;2:605–613. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- 16.Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 17.Barrio JR, Small GW, Wong KP, Huang SC, Liu J, Merrill DA, Giza CC, Fitzsimmons RP, Omalu B, Bailes J, Kepe V. In vivo characterization of chronic traumatic encephalopathy using [18F]FDDNP PET brain imaging. Proc Natl Acad Sci U S A. 2015;112:E2039–E2047. doi: 10.1073/pnas.1409952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carome M, Wolfe S. Florbetapir-PET imaging and postmortem β-amyloid pathology. JAMA. 2011;305:1857–1858. doi: 10.1001/jama.2011.579. [DOI] [PubMed] [Google Scholar]
- 19.Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, Pontecorvo MJ, Hefti F, Carpenter AP, Flitter ML, Krautkramer MJ, Kung HF, Coleman RE, Doraiswamy PM, Fleisher AS, Sabbagh MN, Sadowsky CH, Reiman EP, Zehntner SP, Skovronsky DM. Use of florbetapir-PET for imaging β-amyloid pathology. JAMA. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolk DA, Grachev ID, Buckley C, Kazi H, Grady MS, Trojanowski JQ, Hamilton RH, Sherwin P, McLain R, Arnold SE. Association between in vivo fluorine 18-labeled flutemetamol amyloid positron emission tomography imaging and in vivo cerebral cortical histopathology. JAMA Neurol. 2011;68:1398–1403. doi: 10.1001/archneurol.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rowe CC, Ackerman U, Browne W, Mulligan R, Pike KL, O’Keefe G, Tochon-Danguy H, Chan G, Berlangieri SU, Jones G, Dickinson-Rowe KL, Kung HP, Zhang W, Kung MP, Skovronsky D, Dyrks T, Holl G, Krause S, Friebe M, Lehman L, Lindemann S, Dinkelborg LM, Masters CL, Villemagne VL. Imaging of amyloid β in Alzheimer’s disease with 18F-BAY94–9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 2008;7:129–135. doi: 10.1016/S1474-4422(08)70001-2. [DOI] [PubMed] [Google Scholar]
- 22.Rowe CC, Pejoska S, Mulligan RS, Jones G, Chan JG, Svensson S, Cselényi Z, Masters CL, Villemagne VL. Head-to-head comparison of 11C-PIB and 18F-AZD4694 (NAV4694) for beta-amyloid imaging in aging and dementia. J Nucl Med. 2013;54:880–886. doi: 10.2967/jnumed.112.114785. [DOI] [PubMed] [Google Scholar]
- 23.Mathis CA, Wang YM, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 24.Okamura N, Suemoto T, Furumoto S, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, Fujiwara H, Nemoto M, Maruyama M, Arai H, Yanai K, Sawada T, Kudo Y. Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. J Neurosci. 2005;25:10857–10862. doi: 10.1523/JNEUROSCI.1738-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, Cao D, Rigopoulos A, Cartwright GA, O’Keefe G, Gong S, Adlard PA, Barnham KJ, Rowe CC, Masters CL, Kudo Y, Cappai R, Yanai K, Villemagne VL. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain. 2011;134:1089–1100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- 26.Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, Zhang MR, Trojanowski JQ, Lee VMY, Ono M, Masamoto K, Takano H, Sahara N, Iwata N, Okamura N, Furumoto S, Kudo Y, Chang Q, Saido TC, Takashima A, Lewis J, Jang MK, Aoki I, Ito H, Higuchi M. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–1108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W, Arteaga J, Cashion DK, Chen G, Gangadharmath U, Gomez LF, Kasi D, Lam C, Liang Q, Liu C, Mocharla VP, Mu F, Sinha A, Szardenings AK, Wang E, Walsh JC, Xia C, Yu C, Zhao T, Kolb HC. A highly selective and specific PET tracer for imaging of tau pathologies. J Alzheimer’s Dis. 2012;31:601–612. doi: 10.3233/JAD-2012-120712. [DOI] [PubMed] [Google Scholar]
- 28.Chien DT, Szardenings AK, Bahri S, Walsh JC, Mu F, Xia C, Shankle WR, Lerner AJ, Su MY, Elizarova A, Kolb HC. Early clinical PET imaging results with the novel PHF-Tau radioligand [F18]-T808. J Alzheimer’s Dis. 2014;38:171–184. doi: 10.3233/JAD-130098. [DOI] [PubMed] [Google Scholar]
- 29.Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N, Hiraoka K, Watanuki S, Shidahara M, Miyake M, Ishikawa Y, Matsuda R, Inami A, Yoshikawa T, Tago T, Funaki Y, Iwata R, Tashiro M, Yanai K, Arai H, Kudo Y. [18F]THK-5117 PET for assessing neurofibrillary pathology in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2015;42:1052–1061. doi: 10.1007/s00259-015-3035-4. [DOI] [PubMed] [Google Scholar]
- 30.Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N, Tago T, Hiraoka K, Watanuki S, Shidahara M, Miyake M, Ishikawa Y, Matsuda R, Inanmi A, Yoshikawa T, Funaki Y, Iwata R, Tashiro M, Yanai K, Arai H, Kudo Y. 18F-THK5351: A novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer disease. J Nucl Med. 2016;57:208–214. doi: 10.2967/jnumed.115.164848. [DOI] [PubMed] [Google Scholar]
- 31.Zhuang ZP, Kung MP, Hou C, Skovronsky DM, Gur TL, Plössl K, Trojanowski JQ, Lee VMY, Kung HF. Radioiodinated styrylbenzenes and thioflavins as probes for amyloid aggregates. J Med Chem. 2001;44:1905–1914. doi: 10.1021/jm010045q. [DOI] [PubMed] [Google Scholar]
- 32.Ni R, Gillberg PG, Bergfors A, Marutle A, Nordberg A. Amyloid tracers detect multiple binding sites in Alzheimer’s disease brain tissue. Brain. 2013;136:2217–2227. doi: 10.1093/brain/awt142. [DOI] [PubMed] [Google Scholar]
- 33.Lemoine L, Saint-Aubert L, Marutle A, Antoni G, Eriksson JP, Ghetti B, Okamura N, Nennesmo I, Gillberg PG, Nordberg A. Visualization of regional tau deposits using 3H-THK5117 in Alzheimer brain tissue. Acta Neuropathol Commun. 2015;3:40. doi: 10.1186/s40478-015-0220-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marquié M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, Klunk WE, Mathis CA, Ikonomovic MD, Debnath ML, Vasdev N, Dickerson BC, Gomperts SN, Growdon JH, Johnson KA, Frosch MP, Hyman BT, Gómez-Isla T. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015;78:787–800. doi: 10.1002/ana.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai LS, Innis RB, Pike VW. Radioligand development for PET imaging of β-amyloid (Aβ)-current status. Curr Med Chem. 2007;14:19–52. doi: 10.2174/092986707779313471. [DOI] [PubMed] [Google Scholar]
- 36.Szardenings AK, Kolb HC, Walsh JC, Chen G, Gangadharmath UB, Kasi D, Liu C, Sinha A, Wang E, Yu C, Zhang W, Chen K, Mocharla VP, Scott PJH. Carbazole derivatives as imaging agents for detecting neurological dysfunction and their preparation. 0302755. US Patent. 2012:A1.
- 37.van Wieringen JP, Shalgunov V, Janssen HM, Fransen PM, Janssen AGM, Michel MC, Booij J, Elsinga PH. Synthesis and characterization of a novel series of agonist compounds as potential radiopharmaceuticals for imaging dopamine D2/3 receptors in their high-affinity state. J Med Chem. 2014;57:391–410. doi: 10.1021/jm401384w. [DOI] [PubMed] [Google Scholar]
- 38.Miyata O, Takahashi S, Tamura A, Ueda M, Naito T. Novel synthesis of substituted pyrrolidines and piperidines via radical addition-ionic cyclization reaction of oxime ethers. Tetrahedron. 2008;64:1270–1284. [Google Scholar]
- 39.Cheng Y, Prusoff WH. Relationship between inhibition constant (K1) and concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic-reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 40.Stöcklin G, Pike VW. Radiopharmaceuticals for Positron Emission Tomography: Methodological Aspects. Kluwer Academic Publishers; Dordrecht, Boston: 1993. [Google Scholar]