

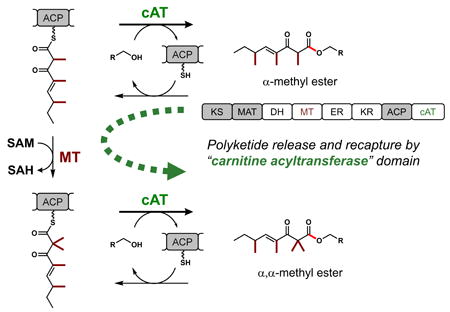
Abstract
Fungal polyketides have significant biological activities, yet the biosynthesis by highly reducing polyketide synthases (HRPKSs) remains enigmatic. An uncharacterized group of HRPKSs was found to contain a C-terminal domain with significant homology to carnitine O-acyltransferase (cAT). Characterization of one such HRPKS (Tv6-931) from Trichoderma virens showed that the cAT domain is capable of esterifying the polyketide product with poly-alcohol nucleophiles. This process is readily reversible as confirmed through the holo ACP-dependent transesterification of the released product. The methyltransferase (MT) domain of Tv6-931 can perform two consecutive α-methylation steps on the last β-keto intermediate to yield an α,α-gem-dimethyl product, a new programing feature among HRPKSs. Recapturing of the released product by cAT domain is suggested to facilitate complete gem-dimethylation by the MT.
Keywords: Polyketide, Biosynthesis, Methyltransferase, Acyltransferase, Heterologous Expression
Graphical abstract
Fungal polyketides constitutes an important family of natural products that have significant pharmaceutical value.[1] The biosynthesis of reduced fungal polyketides, such as lovastatin, by the iterative highly reducing polyketide synthase (HRPKS) is a highly programmed and enigmatic process.[2] This is mainly due to the iterative and permutative use of a single set of elongation and tailoring domains to synthesize a precisely modified polyketide chain. Recent studies showed that tailoring domains, which typically include methyltransferase (MT), ketoreductase (KR), dehydratase (DH) and enoylreductase (ER), have well-defined substrate preferences towards growing polyketide intermediates, and are kinetically synchronized during each iteration.[3] Furthermore, offloading of the completed polyketide chain, which represents the termination step in the iterative process, is also well coordinated with the rest of the domains to ensure a product with the correct length and chemical features is released.[4] Notwithstanding these new insights, prediction of product structures from the large number of HRPKS enzymes identified from fungal genome sequences remains unfeasible at the current time. Therefore, discovery and understanding of new HRPKS programming features remain important objectives towards genome mining and biosynthetic engineering.
We searched through 581 sequenced ascomycetes and basidiomycetes genomes and identified ∼4984 genes that are annotated as containing ketosynthase (KS) domains.[5] Based on phylogenetic classification (Figure S1A), we identified an uncharacterized clade of approximately ∼100 members that contains a C-terminal domain with strong sequence homology to carnitine O-acyltransferase (cAT, Figure 1A). cAT is important in the shuttling of short to long-chain fatty acid molecules to the mitochondrial matrix by catalyzing acylation with carnitine (Figure 1B).[6] The fusion of cAT with HRPKS therefore represents a potentially new mode of product release. Fungal HRPKS product release, either hydrolysis or transesterification, is canonically catalyzed by a separate, in trans thioesterase (TE) domains that uses an active site nucleophilic serine and proceeds through an TE-bound oxyester intermediate (Figure 1B).[7] In contrast, cAT domains use an active site histidine as a general base to deprotonate the carnitine hydroxyl group, followed by direct transesterification.[8] This reaction is readily reversible to yield the CoA thioester with an equilibrium constant of ∼1.[8b, 9]
Figure 1.
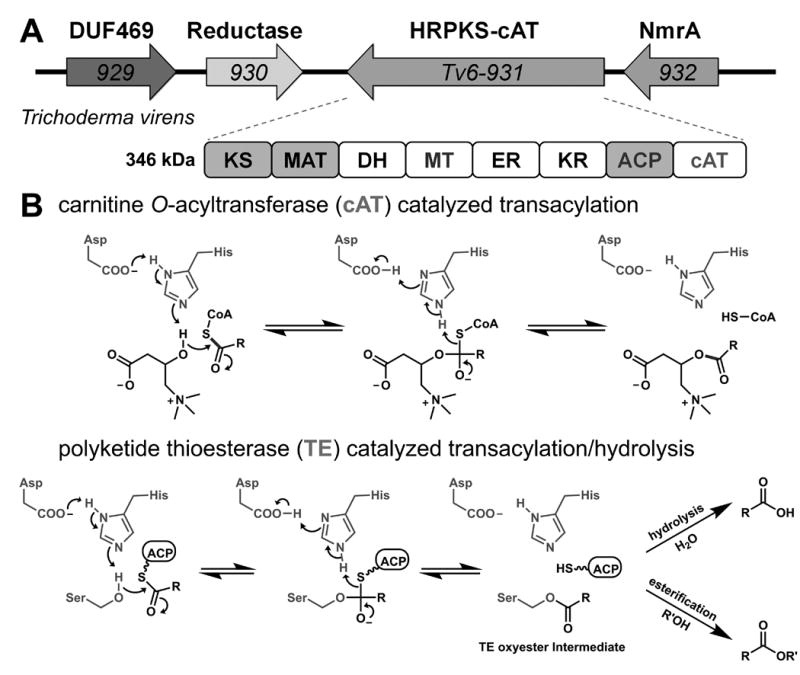
HRPKSs containing a C-terminal carnitine O-acyltransferase like domain. (A) Tv6-931 is a HRPKS-cAT; (B) Mechanistic comparison of reactions catalyzed by cAT and the canonical thioesterase (TE).
A phylogenetic tree of the ∼100 members of cAT containing HRPKSs is shown in Figure S1B, among which two members are found in gene clusters that have been associated with the biosynthesis of AF-toxin and sordarin.[10] The role of the C-terminal cAT-like domain in this large group of uncharacterized HRPKSs is therefore intriguing, especially its functional relationship to the programming rules of the upstream HRPKS.
One member of this family of HRPKS is Tv6-931 encoded in the genome of Trichoderma virens (Figure 1A), and is well-conserved across several Trichoderma species. The HRPKS contains all of the reductive domains, as well as a MT domain. Genes surrounding the HRPKSs do not provide additional insights into a plausible product structure. Due to difficulties associated with working with T. virens, we used heterologous expression to investigate the potential product. The continuous coding sequence was synthesized, assembled, and cloned into either a yeast 2μ vector (pLFH19) or an Aspergillus vector (pLFH25) (Table S2). Transformation of the vectors into the corresponding Saccharomyces cerevisiae (BJ5464-NpgA)[11] or Aspergillus nidulans A1145, however, did not yield any new products (Figure 2A, ii and v). Addition of genes surrounding Tv6-931 into yeast also did not lead to new metabolites.
Figure 2.

Functional characterization of Tv6-931 (Total Ion Count). (A) Heterologous expression in S. cerevisiae and A. nidulans lead to 1 and 2 in the presence of THME. (B) Biochemical assays using purified enzyme revealed the polyketide chain structure and list of offloading nucleophiles.
We then turned to enzymatic assays using Tv6-931 purified by Ni-NTA chromatography to determine its function (Figure S2; See SI). Assay of the enzyme in the presence of only malonyl-CoA yielded triacetic acid lactone, indicating the KS, malonyl-CoA:ACP transacylase (MAT) and acyl carrier protein (ACP) domains are all functional. Addition of NADPH led to synthesis of the conjugated α-pyrone 3 (Figure 2B, ii),[4b, 11] indicating the reductive domains (KR, DH and ER) are active. Unexpectedly, addition of SAM led to no detectable polyketide products (Figure 2B, i). The absence of any α-pyrone product such as 3 suggested that the MT domain may be active, but the offloading mechanism was not fully reconstituted. Addition of carnitine and related compounds did not lead to formation of esterified products (Figure S3). Serendipitously, when repeating the same assay (+NADPH, +SAM) in Tris buffer, or PBS buffer containing glycerol, we detected formation of two new products, 4 (MW 315) and 5 (MW 286), respectively (Figure 2B, iii and iv, and Figure S5). Subtracting the mass of either Tris or glycerol from the molecular weights of 4 and 5, respectively, yielded the same residual molecular weight of 195 (C12O2H19), hinting that the same polyketide fragment is offloaded by Tris or glycerol. To gain insights into polyketide length, we performed the assay in the presence of [2-13C]-malonate and MatB to generate labeled [2-13C]-malonyl-CoA in situ.[12] A mass increase of 4 mu in 4 and 5 was observed (Figure S4), indicating the product is a tetraketide (C8). The remaining four carbon atoms in the molecule must therefore be derived from SAM, yet only three possible Cα-methylation positions are present in a tetraketide.
We assayed additional amino and hydroxyl containing substrates as offloading nucleophiles using the in vitro assay, including linear and branched alcohols (Figure S3). While several substrates supported the formation of the polyketide adduct to different degrees, two polyol compounds: 1,1,1-tris(hydroxymethyl)ethane (THME) and pentaerythritol (PE) led to significantly improved product turnover in 2 and 7, respectively (Figure 2B, iv and v). In each of these assays, we also observed a second product that is 14 mu less, 1 and 6. 13C-malonate assay revealed that both 1 and 6 contain one less SAM-derived methyl group. Based on product yield, the order of releasing nucleophile preference by Tv6-931 is THME∼PE>Tris>glycerol (Figure S3).
We repeated the heterologous expression experiment with the addition of 1% releasing substrate. Both yeast and A. nidulans produced predominantly 2 and minor 1 upon addition of THME (Figure 2A, iii and vi). Similarly, addition of glycerol or PE yielded 5 or 6 and 7, respectively (Figure S6). We purified 1, 2, and 5 from yeast and elucidated the structures with NMR (See SI). As expected, both 1 and 2 are tetraketides esterified with THME, while 5 is esterified through a primary alcohols of glycerol. The polyketide portion of 1 is 2,4,6-trimethyl-3-oxooct-4-enoate, with three SAM-derived methyl groups. The polyketide portion of 2 and 5, however, contains a gem-dimethyl at the last α-carbon. This is consistent with the suggestion of four SAM-derived methyl group from 13C labeling studies described above. The gem-dimethyl group in polyketides are relatively rare, most examples are from bacterial polyketides,[13] including the anticancer drugs epothilone and precursor of epoxyketone.[14] Keasling and coworkers showed that MT domains in these PKSs can gem-dimethylate either the β-keto polyketide intermediate, or malonyl-ACP prior to KS-catalyzed condensation.[13h] Keatinge-Clay and coworkers showed the GphH MT domain from the gephyronic acid biosynthetic pathway can gem-dimethylate acetoacetyl-S-N-acetylcysteamine (∼1 turnover in 72 hours).[13i] Fungal polyketides containing gem-dimethyl groups are rarer,[15] with no responsible MT identified so far.
To investigate the activity of the Tv6-931 MT domain, we used simplified triketide (3-oxohexanoyl-SNAC) and tetraketide (3-oxooctanoyl-SNAC, 8) to perform in vitro methylation assays. Both the full length Tv6-931 and a truncated version lacking the cAT domain (Tv6-931 ΔcAT) were used to probe any potential role of the cAT domain in the successive Cα-methylation reactions. In the presence of the triketide substrate, Tv6-931 catalyzed only one methylation step to yield 2-methyl-3-oxohexanoyl-SNAC (Figure S7). In contrast, when incubated with 8, we detected the conversion to both the 2-methyl-3-oxooctanoyl-SNAC 9 (matched to standard), and a new compound with mass corresponding to 2,2-dimethyl-3-oxooctanoyl-SNAC 10 (Figure 3, i). Direct assay of Tv6-931 with 9 also led to formation of 10 (Figure 3, ii). The identity of 10 was confirmed by NMR characterization (Table S7). The truncated Tv6-931 ΔcAT also catalyzed the methylation reactions (Figure 3, v and vi). These results therefore confirmed that the MT domain is specifically programmed to differentiate between triketide and tetraketide substrates. Independent of the cAT domain, MT can indeed catalyze two successive Cα-methylation reactions on the tetraketide substrate 8 to 10. The catalytic efficiency of ∼1 turnover per hour, albeit still low as expected with SNAC substrates, is significantly higher than that reported for GphH.13
Figure 3.
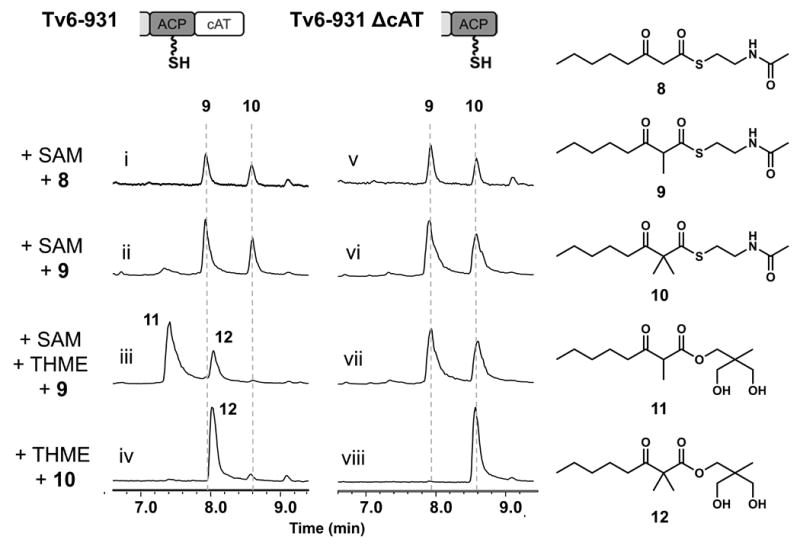
Characterization of MT and cAT domains of Tv6-931 using SNAC substrates (Total Ion Count). Reaction conditions: 15 μM enzyme (Tv6-931 or Tv6-931 ΔcAT), 5 mM SAM, 2 mM SNAC substrate, 5 mM offloading nucleophile when added, PBS buffer, pH 7.4, 16 hours of reaction.
We also recapitulated the activities of the cAT domain using the SNAC substrates. When THME, SAM and 9 were added to Tv6-931, we observed a majority of 9 readily converted to the ester 11 with a small amount to the 2,2-dimethyl-3-oxooctanoyl-THME ester 12 (Figure 3, iii). When 10 was directly added to Tv6-931 with THME, nearly complete conversion to 12 was seen (Figure 3, iv). In contrast, the truncated Tv6-931 ΔcAT enzyme was unable to transesterify the products as THME esters (Figure 3, vii and viii). Addition of the standalone recombinant cAT domain in trans at increasing concentrations progressively restored the release of ester products (Figure S9). Collectively, these experiments confirmed the role of the cAT domain in Tv6-931 is responsible for product offloading. Comparison of the active site residues of Tv6-931 cAT domain with authentic carnitine-O-acyltransferases revealed conservation of the catalytic histidine and residues that coordinate to a water molecule in the active site (Figure S9-10). A serine residue proposed to stabilize the oxyanion is also conserved. Residues that interact with carnitine are not conserved, expected since carnitine was not accepted as an offloading nucleophile.
Despite confirming the activity of the cAT domain in Tv6-931, it remains unclear what is the chemical strategy behind Nature's use of the in cis cAT domain as an offloading catalyst, instead of the more canonical TE domain. A particular important feature of the acyl transfer reaction between acyl CoA-thioesters and carnitine catalyzed by carnitine-O-acyltransferase is that the reaction is readily reversible, which allows shuttling of acyl-CoA across the mitochondria membrane.[8b, 9] We therefore hypothesized that the reversibility feature could be advantageous to the programming rules of Tv6-931, especially with respect to the gem-dimethylation activities of the MT domain.
To test the reversibility of the Tv6-931 cAT domain, we performed a post-release transesterification assay in which the THME esters 1 or 2 is mixed with an equal molar amount of the free alcohol PE, a kinetically comparable substrate to cAT as THME. Interestingly, we observed the transesterification readily occurs with the final distributions between THME (1 or 2) and PE esters (6 or 7) to be close to 1:1 (Figure 4, ii and iii). In contrast, the reaction does not occur when Tv6-931 ΔcAT was used (Figure 4, v and vi). However, the standalone cAT does not catalyze the interconversion, suggesting the reversible reaction is dependent on the HRPKS, namely the thiol-terminating phosphosphopantheinyl arm of the ACP domain. This was readily confirmed by expressing the apo form of Tv6-931 using a yeast strain without an integrated phosphopantetheinyl transferase NpgA. No transesterification can be detected using the apo enzyme (Figure 4, viii and ix).
Figure 4.
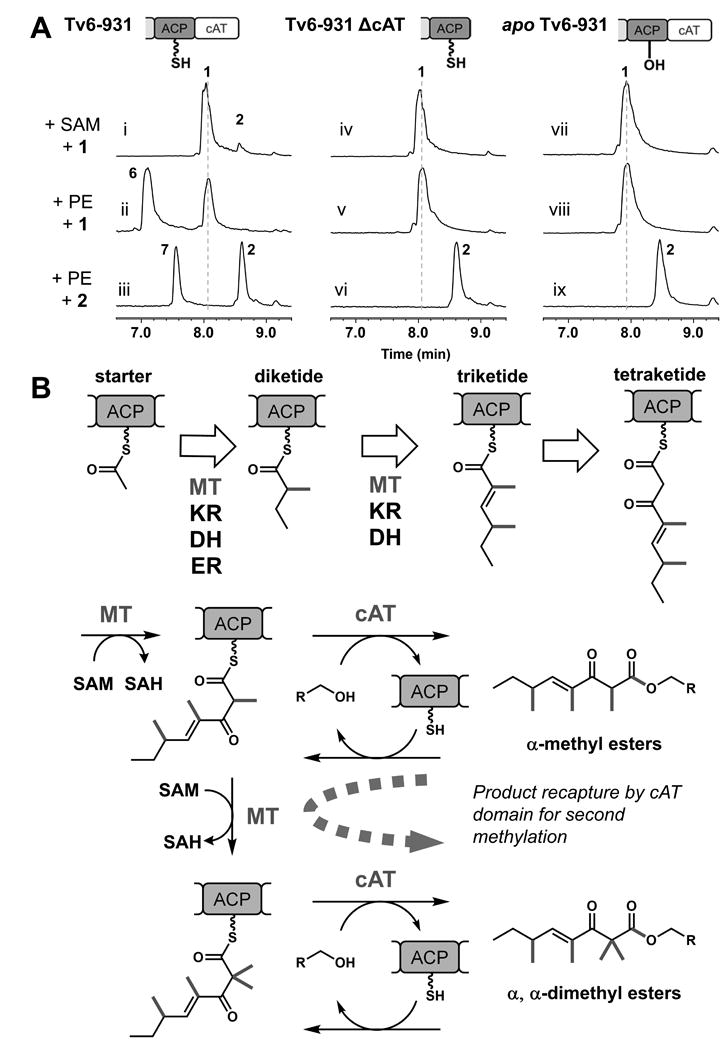
Reversible product offloading by the cAT domain in Tv6-931. (A) Assay demonstrating post-release methylation; (B) Proposed mechanism and interplay between the cAT and MT domains.
Our results demonstrate the reversible release and recapture of the polyketide product back on to the ACP domain, catalyzed by the appended cAT domain (Figure 4B). The involvement of ACP-SH to form the reloaded acyl thioester is consistent with the reaction mechanism of canonical carnitine-O-acyltransferase. While this may appear to be a futile cycle in terms of polyketide biosynthesis, we propose that assembly line reentry may be a strategy to recycle prematurely offloaded 2-monomethyl products back on to the ACP and to allow the second methylation to take place. One observation from in vitro assays suggests such a kinetic competition between offloading by cAT and the second methylation step by MT (Figure 2B, iii-vi): 2-monomethyl products such as 1 and 6 are more abundant with better offloading nucleophiles such as PE and THME; while 2-dimethyl products are major products for slower substrates such as Tris and glycerol. While we do not yet know the natural offloading substrate of Tv6-931, it is likely to be a kinetically competent one that will compete with the second methylation step. To test the product recapture hypothesis shown in Figure 4B, we directly added 1 to different versions of Tv6-931 in the presence of SAM. We were able to observe the conversion of 1 to 2 (Figure 4A, i) only in the presence of holo Tv6-931. The identity of the product was verified using crude 1H-NMR and compared to purified 2 (Figure S11). In contrast, the ΔcAT and apo versions did not support any conversion of 1 to 2. We verified the MT and cAT domains of the apo form were active using 9 and 10 (Figure S12), thereby confirming the lack of activity in the apo protein is solely due to the absence of the phosphopantetheinyl prosthetic group (Figure S13).
The post-release methylation of 1 to 2 via cAT and ACP provides a complementary route to the direct iterative methylation to produce the gem-dimethyl polyketide 2 (Figure 4B). Given that the methylation steps are essentially irreversible, the recycling mechanism facilitated should lead to the gem-dimethyl products as the final products. This mechanism may explain that under in vivo conditions in A. nidulans (Figure 2A, vi), the predominant product was 2, while under in vitro conditions where equilibrium favors the offloaded product, a significant fraction of products remained the monomethylated 1.
In conclusion, the cAT domain studied here can catalyze reversible release and recapturing of the polyketide product, consistent with the mechanism of carnitine-O-acyltransferase in primary metabolism. A remaining unanswered question is the natural nucleophile of the cAT domain, as the substrates we discovered here are not expected to be present in T. virens. It is evident that the natural substrate should be unique to T. virens, as it is absent in both S. cerevisiae and A. nidulans. Active metabolomics work is ongoing to discover the authentic natural product of Tv6-931. Hence, our work illustrates a potential bottleneck of genome mining efforts when a required substrate present in the natural host is unavailable in model heterologous organisms.
Supplementary Material
Acknowledgments
This work was supported by the by NIH 1R35GM118056 and 1DP1GM106413 to Y.T; and 1U01GM110706 to M.E.H. L. H. and N. L are supported by NIH Training Grants T32GM007185 and T32GM067555, respectively.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Leibniz Hang, Department of Chemistry and Biochemistry, Department of Chemical and Biomolecular Engineering, University of California Los Angeles, CA 90095 (USA).
Dr. Man-Cheng Tang, Department of Chemistry and Biochemistry, Department of Chemical and Biomolecular Engineering, University of California Los Angeles, CA 90095 (USA).
Dr. Colin J. B. Harvey, Stanford Genome Technology Center, Stanford University, Palo, California 93404 (USA)
Claire G. Page, Department of Chemistry and Biochemistry, Department of Chemical and Biomolecular Engineering, University of California Los Angeles, CA 90095 (USA)
Jian Li, Stanford Genome Technology Center, Stanford University, Palo, California 93404 (USA).
Yiu-Sun Hung, Department of Chemistry and Biochemistry, Department of Chemical and Biomolecular Engineering, University of California Los Angeles, CA 90095 (USA).
Nicholas Liu, Department of Chemistry and Biochemistry, Department of Chemical and Biomolecular Engineering, University of California Los Angeles, CA 90095 (USA).
Dr. Maureen E. Hillenmeyer, Stanford Genome Technology Center, Stanford University, Palo, California 93404 (USA)
Dr. Yi Tang, Department of Chemistry and Biochemistry, Department of Chemical and Biomolecular Engineering, University of California Los Angeles, CA 90095 (USA)
References
- 1.Lim FY, Keller NP. Nat Prod Rep. 2014;31:1277–1286. doi: 10.1039/c4np00083h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Cox RJ. Org Biomol Chem. 2007;5:2010–2026. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]; b) Chooi YH, Tang Y. J Org Chem. 2012;77:9933–9953. doi: 10.1021/jo301592k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Cacho RA, Thuss J, Xu W, Sanichar R, Gao Z, Nguyen A, Vederas JC, Tang Y. J Am Chem Soc. 2015;137:15688–15691. doi: 10.1021/jacs.5b11814. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Roberts DM, Bartel C, Scott A, Ivison D, Simpson TJ, Cox RJ. Chem Sci. 2017;8:1116–1126. doi: 10.1039/c6sc03496a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liddle E, Scott A, Han C, Ivison D, Simpson TJ, Willis CL, Cox RJ. Chem Commun. 2017;53:1727–1730. doi: 10.1039/c6cc10172k. [DOI] [PubMed] [Google Scholar]
- 4.a) Zabala AO, Chooi YH, Choi MS, Lin HC, Tang Y. Acs Chem Biol. 2014;9:1576–1586. doi: 10.1021/cb500284t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu W, Chooi YH, Choi JW, Li S, Vederas JC, Da Silva NA, Tang Y. Angew Chem Int Ed. 2013;52:6472–6475. doi: 10.1002/anie.201302406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li YF, Tsai KJS, Harvey CJB, Li JJ, Ary BE, Berlew EE, Boehman BL, Findley DM, Friant AG, Gardner CA, Gould MP, Ha JH, Lilley BK, McKinstry EL, Nawal S, Parry RC, Rothchild KW, Silbert SD, Tentilucci MD, Thurston AM, Wai RB, Yoon YJ, Aiyar RS, Medema MH, Hillenmeyer ME, Charkoudian LK. Fungal Genet Biol. 2016;89:18–28. doi: 10.1016/j.fgb.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colucci WJ, Gandour RD. Bioorg Chem. 1988;16:307–334. [Google Scholar]
- 7.a) Tsai SC, Lu HX, Cane DE, Khosla C, Stroud RM. Biochemistry. 2002;41:12598–12606. doi: 10.1021/bi0260177. [DOI] [PubMed] [Google Scholar]; b) Korman TP, Crawford JM, Labonte JW, Newman AG, Wong J, Townsend CA, Tsai SC. P Natl Acad Sci USA. 2010;107:6246–6251. doi: 10.1073/pnas.0913531107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Jogl G, Tong L. Cell. 2003;112:113–122. doi: 10.1016/s0092-8674(02)01228-x. [DOI] [PubMed] [Google Scholar]; b) Hsiao YS, Jogl G, Tong L. J Biol Chem. 2006;281:28480–28487. doi: 10.1074/jbc.M602622200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Ramsay RR, Gandour RD, van der Leij FR. Bba-Protein Struct M. 2001;1546:21–43. doi: 10.1016/s0167-4838(01)00147-9. [DOI] [PubMed] [Google Scholar]; b) Ramsay RR, Naismith JH. Trends Biochem Sci. 2003;28:343–346. doi: 10.1016/S0968-0004(03)00137-3. [DOI] [PubMed] [Google Scholar]
- 10.a) Ruswandi S, Kitani K, Akimitsu K, Tsuge T, Shiraishi T, Yamamoto M. J Gen Plant Pathol. 2005;71:107–116. [Google Scholar]; b) Kudo F, Matsuura Y, Hayashi T, Fukushima M, Eguchi T. J Antibiot. 2016;69:541–548. doi: 10.1038/ja.2016.40. [DOI] [PubMed] [Google Scholar]
- 11.Ma SM, Li JW, Choi JW, Zhou H, Lee KK, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Science. 2009;326:589–592. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.An JH, Kim YS. Eur J Biochem. 1998;257:395–402. doi: 10.1046/j.1432-1327.1998.2570395.x. [DOI] [PubMed] [Google Scholar]
- 13.a) Molnar I, Schupp T, Ono M, Zirkle R, Milnamow M, Nowak-Thompson B, Engel N, Toupet C, Stratmann A, Cyr DD, Gorlach J, Mayo JM, Hu A, Goff S, Schmid J, Ligon JM. Chem Biol. 2000;7:97–109. doi: 10.1016/s1074-5521(00)00075-2. [DOI] [PubMed] [Google Scholar]; b) Young J, Stevens DC, Carmichael R, Tan J, Rachid S, Boddy CN, Muller R, Taylor RE. J Nat Prod. 2013;76:2269–2276. doi: 10.1021/np400629v. [DOI] [PubMed] [Google Scholar]; c) Miller DA, Luo L, Hillson N, Keating TA, Walsh CT. Chem Biol. 2002;9:333–344. doi: 10.1016/s1074-5521(02)00115-1. [DOI] [PubMed] [Google Scholar]; d) Sudek S, Lopanik NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. J Nat Prod. 2007;70:67–74. doi: 10.1021/np060361d. [DOI] [PubMed] [Google Scholar]; e) Pfeifer BA, Wang CCC, Walsh CT, Khosla C. Appl Environ Microb. 2003;69:6698–6702. doi: 10.1128/AEM.69.11.6698-6702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Piel J. P Natl Acad Sci USA. 2002;99:14002–14007. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Zhao CH, Coughlin JM, Ju JH, Zhu DQ, Wendt-Pienkowski E, Zhou XF, Wang ZJ, Shen B, Deng ZX. J Biol Chem. 2010;285:20097–20108. doi: 10.1074/jbc.M109.090092. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Poust S, Phelan RM, Deng K, Katz L, Petzold CJ, Keasling JD. Angew Chem Int Ed. 2015;54:2370–2373. doi: 10.1002/anie.201410124. [DOI] [PubMed] [Google Scholar]; i) Wagner DT, Stevens DC, Mehaffey MR, Manion HR, Taylor RE, Brodbelt JS, Keatinge-Clay AT. Chem Commun. 2016;52:8822–8825. doi: 10.1039/c6cc04418b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Liu J, Zhu XJ, Zhang WJ. Chembiochem. 2015;16:2585–2589. doi: 10.1002/cbic.201500496. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zettler J, Zubeil F, Kulik A, Grond S, Kaysser L. Chembiochem. 2016;17:792–798. doi: 10.1002/cbic.201500567. [DOI] [PubMed] [Google Scholar]
- 15.a) Poch GK, Gloer JB. J Nat Prod. 1989;52:257–260. doi: 10.1021/np50062a006. [DOI] [PubMed] [Google Scholar]; b) Martin J, Crespo G, Gonzalez-Menendez V, Perez-Moreno G, Sanchez-Carrasco P, Perez-Victoria I, Ruiz-Perez LM, Gonzalez-Pacanowska D, Vicente F, Genilloud O, Bills GF, Reyes F. J Nat Prod. 2014;77:2118–2123. doi: 10.1021/np500577v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.