

Abstract
Nicotinamide adenine dinucleotide (NAD+) is an essential cofactor for multiple cellular metabolic reactions and has a central role in energy production. Brain ischemia depletes NAD+ pools leading to bioenergetics failure and cell death. Nicotinamide mononucleotide (NMN) is utilized by the NAD+ salvage pathway enzyme, nicotinamide adenylyltransferase (Nmnat) to generate NAD+. Therefore, we examined whether NMN could protect against ischemic brain damage. Mice were subjected to transient forebrain ischemia and treated with NMN or vehicle at the start of reperfusion or 30 min after the ischemic insult. At 2, 4, and 24 h of recovery, the proteins poly-ADP-ribosylation (PAR), hippocampal NAD+ levels, and expression levels of NAD+ salvage pathway enzymes were determined. Furthermore, animal’s neurologic outcome and hippocampal CA1 neuronal death was assessed after six days of reperfusion. NMN (62.5 mg/kg) dramatically ameliorated the hippocampal CA1 injury and significantly improved the neurological outcome. Additionally, the post-ischemic NMN treatment prevented the increase in PAR formation and NAD+ catabolism. Since the NMN administration did not affect animal’s temperature, blood gases or regional cerebral blood flow during recovery, the protective effect was not a result of altered reperfusion conditions. These data suggest that administration of NMN at a proper dosage has a strong protective effect against ischemic brain injury.
Keywords: Nicotinamide mononucleotide, nicotinamide dinucleotide, poly-ADP-ribose, ischemia, brain, mouse
1. Introduction
One of the pathologic outcomes after ischemic insult is the free radical induced DNA damage that activates the nuclear enzyme poly(ADP-ribose) polymerase 1 (PARP1) (Endres et al., 1997; Strosznajder et al., 2003), for review see (Chiarugi, 2005). This enzyme utilizes NAD+ as a substrate to form the poly-ADP-ribose (PAR) polymer. It has been proposed that uncontrolled PARP1 activation can deplete intracellular NAD+ and consequently ATP, leading to cell death (Lo et al., 1998; Szabo and Dawson, 1998). NAD+ is an important cofactor involved in multiple metabolic reactions (Brennan et al., 2006). NAD+ and NADH have central roles in cellular energy production as electron-accepting and electron-donating cofactors. Therefore, maintenance of normal cellular NAD+ levels is essential for tissue bioenergetic metabolism and several cell functions. A prominent role for NAD+ catabolism in cell death mechanisms is supported by the observation that following excitotoxic insult or in vivo models of brain ischemia, epilepsy and Alzheimer’s disease, a significant decrease in total cellular NAD+ levels occurs prior to neuronal death (Endres et al., 1997; Liu et al., 2009).
NAD+ can be generated in cells either by de novo synthesis from tryptophan or it is re-synthesized from nicotinamide (Nam) via a salvage pathway (Belenky et al., 2007; Owens et al., 2013b). Majority of the NAD+ is replenished by the salvage pathway since Nam is the byproduct of the NAD+ catabolizing enzymes (Magni et al., 1999; Imai, 2009; Owens et al., 2013b). The salvage pathway represents two enzymatic reactions. In the first step, Nam is converted to nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (Nampt) (Revollo et al., 2004), also known as pre-B-cell colony-enhancing factor (PBEF). NMN is then adenylylated to form NAD+ by nicotinamide nucleotide adenylyltransferase (Nmnat) (Belenky et al., 2007).
To maintain cellular NAD+ levels following an ischemic insult, one can either inhibit the NAD+ catabolizing enzymes (PARP1, CD38) or facilitate NAD+ generation by the salvage pathway via administration of a NAD+ precursor. It was shown that administration of Nam increases tissue NAD+ levels and improves bioenergetics following ischemia (Yang et al., 2002a). Alternatively, one can directly stimulate the NAD+ synthesis by administration of NMN that will not require the activity of Nampt, the rate-limiting enzyme in NAD+ synthesis. Additionally, NMN does not cause the same unfavorable flushing side effect associated with Nam activating the GPR109A receptor (Canto et al., 2012; Benyo et al., 2005).
Application of NMN leads to an increase in cellular NAD+ levels by a one-step enzymatic reaction where NMN is converted to NAD+ by Nmnat (Belenky et al., 2007). Incubation of brain sections with NMN prevented a rapid NAD+ catabolism in the tissue (Balan et al., 2010). Furthermore, decreased activity of Nampt enzyme can significantly aggravate post-ischemic brain damage (Zhang et al., 2010). Thus, heterozygous Nampt knockdown animals manifested aggravated brain damage following photothrombosis-induced focal ischemia (Zhang et al., 2010). Transgenic mice with neuron-specific overexepression of Nampt show reduced infarct size when compared to wild type animals (Jing et al., 2014). Similarly, the adverse effect of Nampt inhibitor FK866 was reversed by intraventricular NMN injection (Wang et al., 2011). Taken together, these data suggest that post-ischemic NMN administration could improve bioenergetic metabolism of the post-ischemic tissue and ameliorate the brain damage. Therefore, the objective of this study was to examine the effect of NMN on brain tissue NAD+ catabolism, PAR formation, and histologic and neurologic outcomes following transient global cerebral ischemia.
2. Materials and methods
2.1. Animals and experimental groups
All the experimental procedures and treatment protocols were performed according to the laboratory practices described in (Lapchak et al., 2013), and (Landis et al., 2012). All mice procedures were conducted in AAALAC approved facilities following current ethical regulations from the Guide for the Care and use of Laboratory Animals of the National Institutes of Health international guidelines and were approved by the Animal Care and Use Committee of the University of Maryland Baltimore. Adult, 3 month old male C57Bl6 mice (Jackson Laboratories) were used. The animals were maintained in a 12-hours light/dark cycle, and were housed in groups of 2 to 5 mice per cage in a temperature (22 ± 1 °C) and humidity (55 ± 15%) controlled room. All mice were allowed free access to water and a maintenance diet (Teklad mouse diet, Envigo). All cages contained bedding and nesting material (Nestlets, Ancare) for environmental enrichment. The animal’s health status was monitored throughout the experiments according to the protocol approved by the Animal Care and Use Committee of the University of Maryland Baltimore. The mice were free of all viral, bacterial, and parasitic pathogens. The animals were tested quarterly for agents associated with pathogens.
Five experimental sets were designed for this study. Experiment I was performed to determine the effect of the NMN treatments on the histologic and neurologic outcome. We randomized 84 mice into seven groups: sham, forebrain ischemia + vehicle, forebrain ischemia +31.25 mg/kg NMN treatment, forebrain ischemia +62.5 mg/kg NMN treatment, forebrain ischemia +125 mg/kg NMN treatment, forebrain ischemia +250 mg/kg NMN treatment and forebrain ischemia +500 mg/kg NMN treatment. All groups were perfusion-fixed at 6 days of recovery. Experiment II was performed in a separate set of animals that received vehicle or 62.5 mg/kg NMN and their neurologic outcome was assessed at 6 days of recovery. In experiment III we repeated the animal’s treatment with vehicle or NMN at a dose of 62.5 mg/kg that were administered at 30 min of recovery. After 6 days of reperfusion these animals then underwent behavioral testing before they were perfusion fixed and their brains processed for histological evaluation of cell death.
Experiment IV included a separate set of animals with the same groups as described in Experiment II, but the tissue extracted at 2, 4, and 24 h of recovery were used for immunoblot analysis. Experiment V was performed in separate animal groups as mentioned in Experiment II, but the core temperature, blood gases and blood glucose levels or cerebral blood flow were measured during ischemia and the early reperfusion period.
The surgeon performing the forebrain ischemia, sham procedures, and drug administration was blinded to the final identity of the groups. Furthermore, histologic and neurologic outcomes were assessed by an investigator who was naïve to the experimental groups and the order of the drug administration.
2.2. Transient forebrain ischemia
Forebrain ischemia was induced by bilateral common carotid artery (CCA) occlusion with concomitant reduction of mean arterial blood pressure (MABP) (Onken et al., 2012). Before the surgery, the animals were fasted with free access to tap water. Mice were intubated and mechanically ventilated with 1.5% isoflurane in N2O:O2 (70:30). A temperature probe was placed subcutaneously adjacent to the skull to monitor the pericranial temperature. Body temperature was monitored using a rectal probe. Both core and head temperatures were maintained at 37.0 ± 0.5 °C during the surgery. After the CCA were isolated, the forebrain ischemia was induced by increasing the isoflurane levels to 5% and clamping both CCA with micro-vessel clamps (Onken et al., 2012). The 10 min ischemic period was ended by reducing isoflurane levels to 0% and releasing the artery clamps. After surgery, the animals were placed into a temperature-controlled incubator for 3 h before they were moved to pre-heated cages for 24 h to avoid hypothermia. Sham operated animals went trough the same surgical procedure including the isoflurane protocol (exposed to 5% isoflurane for the same period of time as vehicle and NMN treated animals), however the common carotid arteries were not occluded.
In mouse models of global cerebral ischemia, the cell death matures at 3 days of reperfusion with no further increase in number of dead cells at 5 days after the ischemic insult (Onken et al., 2012; Sheng et al., 1999; Wellons et al., 2000). Therefore, to insure the maximum post-ischemic damage, the mice underwent 6 days of recovery before their neurologic status was evaluated and then they were perfusion fixed.
Of the total of 176 mice that were subjected to ischemic insult, 18 died before the individual experiments were performed. In the majority of cases, this was due to damage to the vagus nerve during the surgical procedure involving separation of common carotid artery from the nerve.
2.3. Monitoring of blood gasses
In a separate group of animals, an additional incision was made to expose and cannulate the femoral artery (PE 10 tubing) for heparin administration (300 IU·kg−1) and blood sampling. To properly setup the ventilator blood gases were measured before the onset of ischemia, and during the ischemic insult (Onken et al., 2012). Additionally, we also determined the blood gases in vehicle and NMN treated animals 10 min after the intraperitoneal (i.p.) injection.
2.4. Measurement of regional cerebral blood flow (rCBF)
Relative changes in regional blood flow were assessed by laser-Doppler flowmetry using a pencil 1 mm fiber-optic extension probe with blood flow meter unit connected to ADInstruments Power Lab 8/35 system. The tip of the probe was affixed to the surface of the skull over a brain region devoid of major vasculature 2 mm posterior to bregma and 1 mm lateral to midline, contralateral to the thermal probe. The rCBF recording started 5 min before onset of the 5% isoflurane concentration in the respiratory gases and continued during the 10 min of common carotid arteries occlusion and the first 10 min of reperfusion. Vehicle and NMN treated animals were used to determine the ischemia/reperfusion-induced changes in regional cerebral blood flow.
2.5. Histology
Mice were perfusion-fixed transcardially with PBS followed by 4% paraformaldehyde in PBS under deep anesthesia. After perfusion-fixation, the brains were removed from the scull and post-fixed in cold paraformaldehyde overnight and transferred into 30% sucrose solution for 2–3 days before sectioning. Coronal sections of 40 μm were cut on a freezing microtome and collected at 240 μm intervals for cresyl violet staining.
2.6. Cresyl violet staining
Sections were mounted on slides and dried for an hour. The slides were immersed in acetone for 10 min for defatting purpose, dipped in distilled water 3 times and dried at room temperature for 1 h. Then, the slides were immersed in filtered cresyl violet (FD Neurotechnologies, PS102-02) for 2 min and dipped again in distilled water. After, the slides were dehydrated in 95% ethanol with 0.1% glacial acetic acid for 1 min then in 100% ethanol for 2 min twice. Following 5 min immersion in xylene, the slides were coverslipped with DPX. Examination of cresyl violet staining was performed using light microscopy.
2.7. Stereologic quantifications
Counting of CA1 uninjured neurons was performed in 4 sections per hippocampus starting at 1.2 mm posterior to bregma encompassing the lesion volume of CA1 sub-region (−1.2, −1.44, −1.68, −1.92 mm from bregma). Quantitative analysis was performed on a computer-assisted image analysis system consisting of a Nikon Eclipse 800 photomicroscope equipped with a computer-controlled motorized stage, and a computer utilizing the Stereo Investigator program, a custom-designed morphology and stereology software (Vereczki et al., 2006). The total number of surviving neurons in the CA1 subregion of the hippocampus was counted using the optical fractionator method (Larsson et al., 2001). The sampled region for CA1 subfield was demarcated and surviving cresyl violet neuronal cell bodies were counted. The volume of the hippocampal subfield was measured using the Cavalieri estimator method. The estimated number of surviving neurons in each field was divided by the volume of the region of interest to obtain the cellular density expressed in counts/mm3.
2.8. Measurement of tissue NAD+ levels
NAD+ content in the tissue samples was determined according to (Balan et al., 2010). Mouse hippocampi at designated recovery times were dissected on ice and homogenized in 500 μl of 7% cold perchloric acid (PCA). The homogenate was centrifuged at 10,000 g for 10 min. The supernatant was then neutralized and the NAD+ levels were determined fluorimetrically using a cycling enzymatic assay that generates a fluorescent product (Kristian and Fiskum, 2004). The NAD+ data were normalized to the protein content of the tissue samples.
2.9. Western blotting
Hippocampi were dissected on ice and homogenized in lysis buffer (NaCl 150 mM, Tris 10 mM, 1% Triton X-100, 0.5% nonidet p-40) with protease inhibitor cocktail (EMD Milipore). Fifty μg of proteins were gel separated, transferred to immobilon PVDF-FL membrane. Membranes were incubated in Odyssey blocking buffer (Licor Biosciences) for an hour at room temperature. This was followed by incubation of membranes with primary antibody overnight at 4 °C (anti-PAR (10H), 1:1000 Enzo; anti-Nampt (PBEF), 1:1000 abcam; anti-Nmnat2, 1:1000 abcam, anti-β-actin at 1:10,000; Cell Signaling). After washing the membranes in PBS with 0.1% tween-20, they were incubated with the appropriate infrared fluorophore conjugated secondary antibody (Licor) for 30 min at room temperature. The membranes were scanned and the bands were quantified with Odyssey infrared image system (Licor). The β-actin signal was used for normalization of the data detected from bands of the proteins of interest.
2.10. NMN administration
Nicotinamide mononucleotide (NMN) was prepared in sterile PBS and was administered to mice in doses of 31.25, 62.5, 125, 250, and 500 mg/kg. The drug or vehicle solution (PBS) of the same volume (200 μl) was injected intraperitoneally using 29G needles at the onset of reperfusion or at 30 min of recovery (only 62.5 mg/kg dose) after 10 min of forebrain ischemia. NMN doses and vehicle solution were prepared the same day as administered. At 6 days of recovery, the animals were perfusion fixed and their brains were processed for histological outcome assessment. Another group of vehicle or NMN treated animals were decapitated after 2, 4, and 24 h of recovery. Then their brains were removed, hippocampal tissue was dissected on ice and processed for NAD+ measurement or western blots.
2.11. Y-maze test
To test the spatial working memory of mice, we used the Y-maze test as described in (Park et al., 2008). We chose the y-maze test because it is less stressful and more consistent with the natural behavior of mice than other spatial memory tests like the Morris water maze (Sarnyai et al., 2000). There are several advantages to use this test for mouse studies (Dellu et al., 2000). First, it is based on the natural behavior of mice with no requirement of learning new rules and thus enables specific testing of working memory. Second, locomotor activity, recorded as number of arm visits, can be evaluated. Importantly, the influence of locomotor activity on memory performance is limited in this procedure since the dependent measure is principally based on the mouse curiosity (alternation rate). Third, the Y-maze takes advantage of the propensity of rodents to explore new environments (Dellu et al., 2000). Finally, measurement of behavior is quick, precise, and entirely automated, permitting a detailed analysis of performance. This test has particular utility for pharmacological studies enabling to examine the effect of drugs on behavioral deficits associated with hippocampal functions (Conrad et al., 1996, 1997; Dellu et al., 1997). Before and 6 days following forebrain ischemia, each mouse was put at the end of one arm and allowed to freely explore the Y-maze for 5 min. The sequence and number of all arm entries were recorded for each animal by an investigator blinded to the experimental treatments for the mice. Alternation rate was calculated based on the total number of arm entries (E), and number of alternation (A), which was marked when the mouse entered all three arms consecutively (see Park et al., 2008).
2.12. Statistical analysis
Histologic outcome was considered as the primary outcomes to calculate the sample size in each group to provide 80% power at alfa = 0.05. From our previous experience with mice model of transient forebrain ischemia, we anticipated 50% ± 15% cell loss of the CA1 pyramidal neurons. Treatment with NMN was hypothesized to reduce the cell death by 25 to 80% ± 15%. A sample size of 11 in each group would provide 80% power to detect the main effect due to NMN treatment. Furthermore, based on our experience, mortality of about 10% was anticipated for some groups over the survival period of 48 h. Therefore, 12 mice were randomized to each group for evaluation of the histologic outcome. Test of normality was performed by the Shapiro-Wilk test since the sample number in experimental groups is <30 (Ghasemi and Zahediasl, 2012). For each test, the experimental unit was an individual animal. Data among individual experimental groups were compared by one-way ANOVA followed by appropriate post hoc test. Values are presented as mean ± SEM. The p values of <0.05 were considered to be statistically significant. The normality calculation and statistical analysis was performed using SigmaPlot software from Systat (SPSS).
3. Results
3.1. Assessment of physiological parameters
To assure that the blood gases are at physiological levels before and during induction of forebrain ischemia six mice were subjected to physiological study. The ventilator was adjusted to give pO2 and pCO2 values within normal levels. As Table 1 shows, the blood gas data, including pCO2, pO2, and pH showed normal physiological levels with a ventilator setting of 120 strokes per min and about 0.3 cm3 of stroke volume. There were no significant changes in blood gas levels during the ischemic insult when compared to pre-ischemic data.
Table 1.
Blood gases before and during the ischemic insult.
| Blood gases | pO2 (mm Hg) | pCO2 (mm Hg) | pH |
|---|---|---|---|
| Pre-ischemia | 120 ± 7.6 | 35 ± 2.0 | 7.34 ± 0.03 |
| Intra-ischemia | 111 ± 6.5 | 37 ± 0.7 | 7.28 ± 0.02 |
Values are mean ± SEM, n = 6.
3.2. Dose-response study
It was shown that administration of Nam increases tissue NAD+ levels (Klaidman et al., 1996; Yang et al., 2002a, Sadanaga-Akiyoshi et al., 2003) and ameliorates brain damage induced by focal ischemia (Yang et al., 2002a). The most effective protection was observed at relatively high, 500 mg/kg dose of Nam administration (Yang et al., 2002a, b). Therefore, to determine whether NMN has any effect on ischemia-induced brain damage, we started with the same dose of NMN (500 mg/kg). At this high dosage, there was a tendency towards reduced cell death of CA1 neurons, however the number of surviving neurons was not significantly increased when compared to vehicle treated animals (p = 0.0642) (Fig. 1B). Since recently it was shown that accumulated NMN in neurons can have adverse effect on axonal regeneration (Di Stefano et al., 2014), we decided to lower the NMN dose and examine the effect of this compound at 250, 125, 62.5 and 31.25 mg/kg. In contrast to mice treated with vehicle, the brain damage in mice treated with lower doses of NMN was reduced. Protection increased by lowering the MNM doses from 500 to 62.5 mg/kg, except that the number of surviving CA1 neurons at the lowest dose (31.25 mg/kg) decreased when compared to 62.5 mg/kg treatment group. Thus, the most effective treatment was with 62.5 mg/kg of NMN. There were no signs of NMN toxicity, in terms of mortality. However, with higher dosage than 62.5 mg/kg, the NMN manifested an adverse effect on survival of CA1 pyramidal neurons. These experimental results provide proof of principle for using NMN as treatment for global cerebral ischemia. To increase a translational relevance of this approach we repeated the administration of the most efficient NMN dose (62.5 mg/kg) at 30 min after start of reperfusion. Fig. 2 shows that NMN treatment at 30 min of recovery also had dramatic protective effect against the ischemia-induced hippocampal CA1 damage.
Fig. 1.
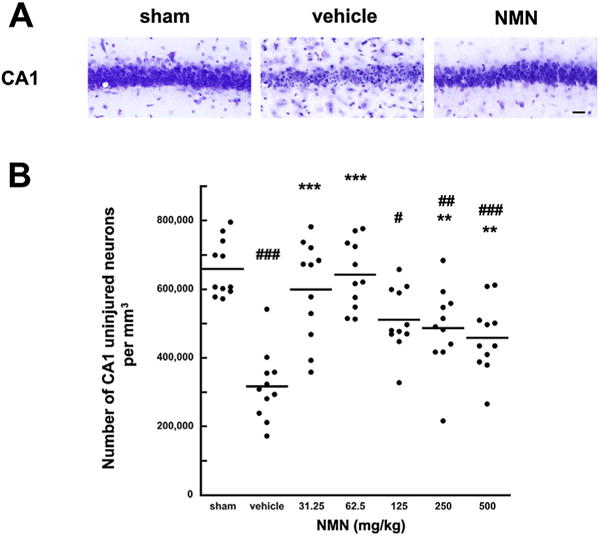
Effect of NMN on CA1 neuronal cell death induced by global cerebral ischemia. Cresyl violet staining of the post-ischemic hippocampal CA1 sector following 6 days of recovery (A). There is a major loss of CA1 pyramidal neurons in vehicle treated animals. In sham operated animals all the CA1 neurons appeared normal. NMN (62.5 mg/kg) treated animals also show normal neuronal morphology. Scale bar represents 50 μm. Quantification of uninjured CA1 neurons in hippocampus (B) shows that there is about 50% cell death at 6 days of recovery in vehicle treated animals. NMN administration at the start of recovery significantly improves the cells survival. The horizontal lines represent the mean value. The most efficient dose was 62.5 mg/kg (p < 0.0001). *p < 0.05; **p < 0.01, ***p < 0.001 when compared to vehicle treated group (n = 11), #p < 0.05, ##p < 0.01, ###p < 0.001 when compared to sham (n = 11), ANOVA followed by Bonferroni’s test.
Fig. 2.
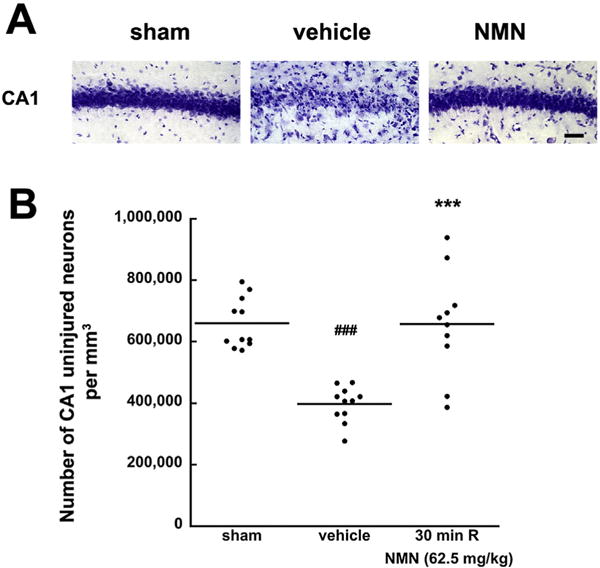
Effect of delayed NMN treatment on CA1 neuronal cell death induced by global cerebral ischemia. Cresyl violet staining of the post-ischemic hippocampal CA1 sector following 6 days of recovery (A). In sham operated animals all the CA1 neurons appeared normal. Animals treated with vehicle and NMN (62.5 mg/kg) at 30 min after start of reperfusion. There is a major loss of CA1 pyramidal neurons in vehicle treated animals, while NMN treated animals show normal neuronal morphology. Scale bar represents 50 μm. (B) Shows that there is about 50% cell death at 6 days of recovery in delayed vehicle treated animals. NMN administration at 30 min of recovery significantly improves the cells survival. The horizontal lines represent the mean value. ***p < 0.001 when compared to the vehicle treated group (n = 10), ###p < 0.001 cell loss when compared to sham (n = 10), ANOVA followed by Tukey HSD test.
3.3. NMN administration improves neurological deficit of post-ischemic animals
Once we determined the most efficient dose, we decided to examine whether the neuroprotection exerted by NMN treatment will improve the animal’s post-ischemic neurologic outcome. To provide evidence of cognitive improvement by NMN treatment, we examined the performance of mice at the Y-maze test that assesses hippocampus-dependent spatial memory (Ellen and Deloache, 1968). Mice treated with vehicle or the most efficient dose (62.5 mg/kg) of NMN were tested before and 6 days after transient forebrain ischemia. Vehicle treated animals showed reduced spontaneous alterations (Fig. 3A) suggesting hippocampal dysfunction (Kiyota et al., 1991). This trend was reversed in the animals treated with NMN. Thus, there was no significant difference in the animal’s performance before and after the ischemic insult in the NMN treated group. No motor deficit was observed in animals after the ischemic insult (Fig. 3B). As the behavioral data show, there was rather inclination for hyperactivity after ischemia as reflected in increased total number of arm entries (Fig. 3B).
Fig. 3.
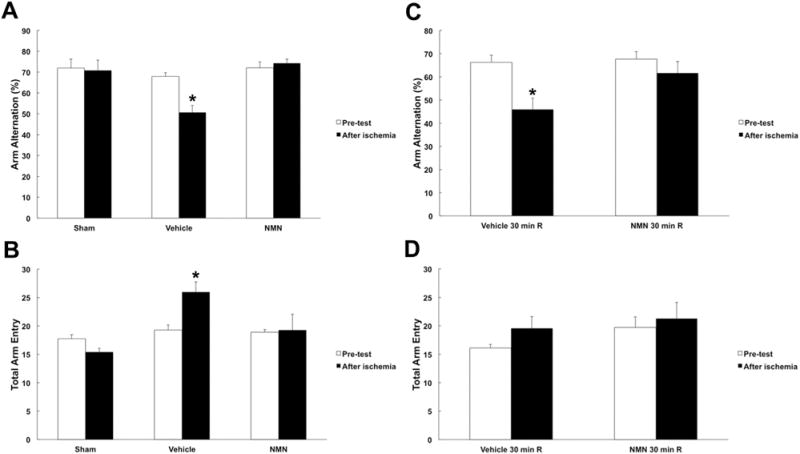
Effect of NMN on post-ischemic neurologic outcome. Treatment of animals with NMN restored spatial working memory loss induced by forebrain ischemia. (A) Y-maze arm entry alteration for vehicle and NMN (62.5 mg/kg) treated animals before and 6 days after forebrain ischemia. The NMN administration prevented the decrease in arm entry alternations, as well as the total number of arm entries (B). There was an increase in motor activity in vehicle treated post-ischemic animals (n = 10). (C) Y-maze arm entry alternation for animals treated with vehicle or NMN (62.5 mg/kg) 30 min after forebrain ischemia. NMN administration significantly prevented the decrease in arm entry alternations compared to vehicle. (D) There was a trend showing increased motor activity in vehicle treated post-ischemic animals. However, this increase was not significance. *p < 0.05 (n = 10), ANOVA followed by Tukey HSD test.
Mice receiving NMN at 30 min recovery showed similar neurologic outcome (Fig. 3C, D). Vehicle treated animals at 30 min reperfusion displayed significantly lower spontaneous alterations (Fig. 3C). The NMN administration resulted in no significant difference in animal’s behavior between pre-, and post-insult tests.
3.4. NMN does not affect animal’s temperature or rCBF during reperfusion
To ensure that the protective effect of NMN is not due to induction of hypothermia, we monitored the animal’s core temperature during the first hour after the NMN administration. As Fig. 4B shows, there was no significant difference in body temperature between vehicle and NMN treated mice. Mouse body temperature was tightly controlled and kept warm for 24 h after the ischemic insult.
Fig. 4.
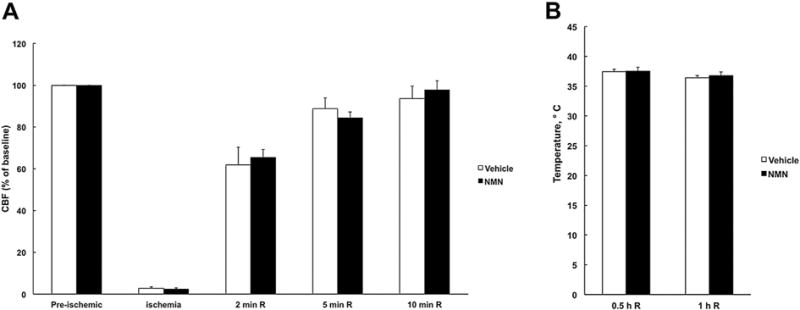
NMN does not affect the animal’s regional cerebral blood flow or temperature during recovery. (A) Regional cerebral blood flow (rCBF) changes in vehicle and NMN treated animals during ischemia and reperfusion. NMN administration at the start of reperfusion did not affect the rCBF recovery (n = 8). (B) Mice body temperature after vehicle or NMN administration (n = 4).
Similarly, to examine the effect of NMN treatment on cortical tissue blood flow following ischemia, we monitored the rCBF during the first 10 min of reperfusion. As Fig. 4A shows, there was no significant difference in blood flow recovery between the vehicle and NMN treated groups. Finally, we measured the blood gases and glucose levels in the vehicle and NMN treated animals. Table 2 shows the NMN treatment did not alter animal’s blood pCO2, pO2 or glucose levels.
Table 2.
Effect of NMN on physiological parameters.
| Group | Vehicle | NMN (62.5 mg/kg) |
|---|---|---|
| pH | 7.40 ± 0.02 | 7.39 ± 0.03 |
| PaCO2 (mm Hg) | 35.4 ± 1.1 | 35.7 ± 1.5 |
| PaO2 (mm Hg) | 117 ± 5.2 | 123 ± 1.7 |
| Blood glucose (mg/dl) | 123.2 ± 7.1 | 119.4 ± 5.5 |
| Weight (g) | 24.2 ± 1.41 | 26.1 ± 1.22 |
Values are mean ± SEM, n = 6.
3.5. NMN administration prevents post-ischemic depletion of hippocampal NAD+ pools by inhibiting proteins poly-ADP-ribosylation
We assessed how the post-ischemic hippocampal NAD+ catabolism is affected by NMN administration. Fig. 5A shows that following forebrain ischemia, the hippocampal tissue NAD+ levels are significantly reduced to about 50% of control at 24 h of recovery. In the NMN treated group, the NAD+ catabolism was inhibited, resulting in preservation of the hippocampal tissue NAD+ at control, pre-ischemic levels.
Fig. 5.
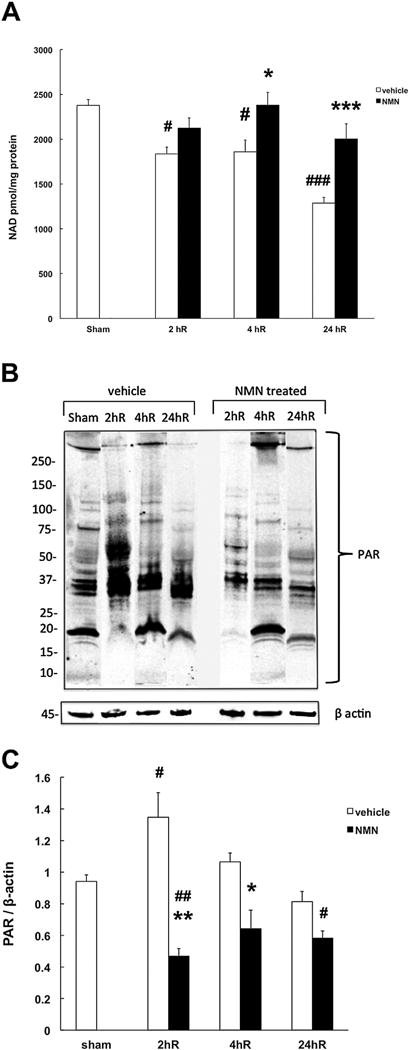
NMN prevents post-ischemic NAD+ catabolism and increased poly-ADP-ribosilation. (A) NMN inhibits ischemia-induced NAD+ degradation in mouse hippocampus. (B) Effect of NMN on post-ischemic PAR formation. β actin was used as the loading control. Mice underwent forebrain ischemia or sham procedure and hippocampal samples were collected at 2, 4, or 24 h post insult. Mice subjected to ischemic insult were either treated with NMN (62.5 mg/kg) or vehicle (PBS) at the start of reperfusion. (C) Quantification of poly-ADP-ribosylation (PAR). NMN treated mice showed significantly lower PAR levels when compared to samples from vehicle treated animals at 2 hR. The NMN treated samples were also significantly lower than sham, 2 hR samples from untreated animals were significantly higher than sham. (*p < 0.05, ***p < 0.001, #p < 0.05, ##p < 0.01, ###p < 0.001, (n = 6), ANOVA followed by Bonferroni’s test.
The NAD+ levels can be maintained by NMN through preventing the post-ischemic NAD+ catabolism due to inhibition of the NAD+ consuming enzyme poly-ADP-ribose polymerase 1 (PARP1) or by facilitating the NAD+ synthesis via the NAD+ salvage pathway. Therefore, we assessed the effect of NMN on post-ischemic PAR formation. As Fig. 5B shows, the highest levels of poly-ADP-ribosylated proteins in the hippocampal tissue were detected at the 2 h of recovery. At later reperfusion times, the PAR levels were gradually reduced towards the pre-ischemic values. Administration of NMN at the start of reperfusion reduced the PARP1 activity as reflected in significantly lower proteins poly-ADP-ribosylation when compared to control, vehicle treated animals (Fig. 5C).
The cellular NAD+ levels are determined by the balance between the activity of NAD+ consuming enzymes and the activity of the NAD+ synthesizing enzymes. Therefore, we examined the post-ischemic levels of the rate-limiting enzyme in the NAD+ salvage pathway, Nampt. As Fig. 6A and C shows, Nampt expression levels were significantly increased at 2 h after ischemia (about 35% when compared to control). At later reperfusion times, the Nampt gradually returned to pre-ischemic levels. Interestingly, animals treated with NMN show no significant changes in Nampt expression levels after the ischemic insult. Similarly, the expression of the second enzyme in the NAD+ salvage pathway, Nmnat2 was slightly elevated at 2 h recovery, however the increase was not significant (p = 0.184). The NMN administration reduced the ischemia-induced changes in expression levels of these enzymes (Fig. 6B, D).
Fig. 6.
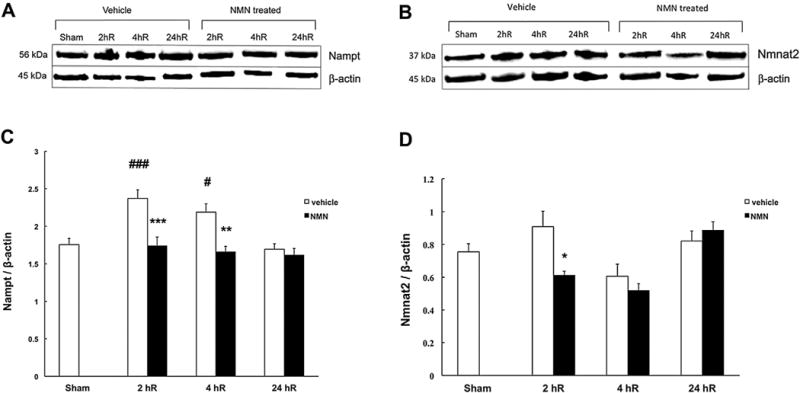
Post-ischemic NMN treatment prevents overexpression of NAD+ salvage pathway enzymes. Immunoreactivity of nicotinamide phosphoribosyl transferase (Nampt) and cytosolic Nicotinamide mononucleotide adenylyl transferase (Nmnat2) in mouse hippocampus. Animals were subjected to forebrain ischemia and treated with NMN (62.5 mg/kg) or vehicle (PBS) at the start of reperfusion. The rate-limiting enzyme of the NAD+ salvage pathway Nampt showed increased expression at early 2 h recovery period. The expression of the Nmnat2 enzyme showed tendency for increase, however the change was not significant when compared to sham animals. The ischemia-induced increase in expression was suppressed in NMN treated animals (A, B). Quantification of Nampt and Nmnat2 expression levels normalized to β actin (C, D). *p < 0.05, **p < 0.01 when compared to vehicle treated group, ##p < 0.01, ###p < 0.001 when compared to sham group (n = 8), ANOVA followed by Bonferroni’s test.
4. Discussion
We show here that administration of NMN following transient forebrain ischemia has a dramatic protective effect against the cell death of CA1 pyramidal neurons. This was reflected in complete recovery of post-ischemic neurological scores and in prevention of the ischemia-induced NAD+ catabolism. Furthermore, NMN treated animals did not show any increase in poly-ADP-ribosylation of hippocampal proteins.
Our model of forebrain ischemia utilizes increased levels of isoflurane as tool to reduce the mean arterial blood pressure during the common carotids occlusion (Onken et al., 2012). This is a disadvantage of this model since it has been shown that isoflurane can reduce brain damage induced by global ischemic insult (Miura et al., 1998; Baughman et al., 1988; Blanck et al., 2000). However, the protective effect of anesthetics was observed only at short, 3 days recovery period (Elsersy et al., 2004), for discussion see (Onken et al., 2012). Furthermore, our model gives rise to similar extends of CA1 damage as observed in commonly used models of mouse forebrain ischemia (Hua et al., 2006; Zhen and Dore, 2007, Yonekura et al., 2004, Panahian et al., 1996). Additionally, it is unlikely that the NMN protective effect was due to increased intra-ischemic isoflurane levels since both vehicle and NMN treated animals were subjected to the same isoflurane protocol.
NAD+ catabolism and associated increase in poly-ADP-ribosylation was reported after both global and focal ischemia (Endres et al., 1997; Strosznajder et al., 2003, Cozzi et al., 2006). Similar to our mouse forebrain ischemia model, the PAR levels were increased during the first 2 h of reperfusion in hippocampal tissue of gerbils subjected to global cerebral ischemia (Strosznajder et al., 2003) and also following 2 h of middle cerebral artery occlusion (MCAO) in mouse cortex (Endres et al., 1997). The PAR formation was reduced by a PARP1 inhibitor, 3-aminobenzamide (3-AB), and was not detected in PARP1 null mice (Endres et al., 1997). PARP1 functions as a genomic stability enzyme and is activated by DNA oxidative damage caused by increased free radical production following ischemic insult (Eliasson et al., 1997). The increased PAR formation coincides with increased production of reactive oxygen species (ROS) that was detected during the first hour of reperfusion following ischemic insult (Piantadosi and Zhang, 1996, Chan et al., 1998; Kurinami et al., 2014).
We observed that the increase in PAR concurred with progressive decrease in hippocampal tissue NAD+ level that was reduced to about 50% of control at 24 h recovery. All these changes were negated by NMN administration, suggesting NMN is inhibiting the PARP1 dependent NAD+ catabolism. Since administration of PARP1 inhibitors significantly improved brain damage due to acute brain injury (Endres et al., 1997; Eliasson et al., 1997; Strosznajder et al., 2003, Kauppinen and Swanson, 2007), it is not surprising that NMN ameliorated ischemic hippocampal cell death and improved post-ischemic neurologic outcome. The inhibition of PAR formation in NMN treated animals suggests that this compound’s protective mechanism is not due to an increase in NAD+ synthesis via the salvage pathway but rather by inhibition of PARP1 activity following ischemic insult. However, since PAR is consumed by poly-ADP-ribose glycohydrolase (PARG) (Davidovic et al., 2001), the above statement is valid under the assumption that there is no effect of NMN on PAR degradation by PARG.
Administration of Nam also protects against excitotoxic and ischemic cell death (Ayoub et al., 1999; Yang et al., 2002a, Liu et al., 2009). It was reported that Nam exerts a number of pharmacological effects including prevention of ATP depletion (Yang et al., 2002b), inhibition of PARP1 (Klaidman et al., 1996; Yang et al., 2002b, Liu et al., 2009), lipid peroxidation (Liu et al., 2009; Klaidman et al., 2001), anti-inflammatory activity (Ungerstedt et al., 2003), and prevention of apoptosis (Klaidman et al., 1996, 2001).
Although Nam showed improved outcomes at 125 mg/kg dose, the best results were detected at much higher dose of 500 mg/kg (Yang et al., 2002a). The most efficient dose for NMN treatment was around 60 mg/kg. Interestingly, animals treated with NMN at 125 mg/kg and higher doses showed progressively less protection, suggesting that the NMN can exert an adverse effect on the post-ischemic neurons. Recently, it was shown that increased levels of NMN after axonal injury promote a degeneration process (Di Stefano et al., 2014). The activation of degenerative events seems to be triggered after NMN reaches a threshold level (see also (Di Stefano et al., 2014).
The important role of maintaining the tissue NAD+ pools for cell survival is implicated also by data showing that heterozygous knockdown of the Nampt enzyme leads to aggravated ischemic brain damage (Zhang et al., 2010). Nampt is highly expressed in neurons (see also Zhang et al., 2010) suggesting that under physiological conditions, there is a higher level and turnover rate of NAD+ in these cells when compared to astrocytes or microglia (see also (Balan et al., 2010). This probably reflects the high-energy metabolism of neurons when compared to other cell types in brain. Specific silencing of Nampt gene in hippocampal and cortical neurons caused atrophy of these brain structures, astrogliosis, microgliosis, and abnormal CA1 dendritic morphology by mature age (Stein et al., 2014). These histologic changes were accompanied by abnormal behavior: defects in memory skills, and reduced anxiety (Stein et al., 2014).
We detected a transient increase in Nampt expression levels during the early reperfusion period. This likely is a compensatory response to the increased NAD+ consumption in post-ischemic brain. Animals that received NMN at the start of recovery maintained both enzymes at the control pre-ischemic levels.
NMN is a product of the Nampt activity and is used to directly synthetize NAD+ by Nmnat (for review see Di Stefano and Conforti, 2013; Owens et al., 2013a). Therefore, by administering NMN to animals, one can directly support the NAD+ synthesis without requiring Nampt activity. NAD+ generation from NMN is more efficient and also less energy dependent since only one ATP molecule is consumed in contrast to two ATPs when the NAD+ is synthetized from Nam (Di Stefano and Conforti, 2013; Owens et al., 2013a). Thus, NMN is preserving the tissue NAD+ levels by inhibiting the NAD+ catabolism via inhibition of PARP1 activity and also by stimulating the NAD+ synthesis by feeding directly into Nmnat driven NAD+ generation.
Although a NMN transporter has not been identified in mammalian cells so far, the mechanism of NMN uptake involves an extracellular conversion to nicotinamide riboside (NR) that can be transported into the cells and then converted by intracellular NR kinases to NMN (Belenky et al., 2007). This notion is supported by the finding that NR levels increase following NMN administration, implying that some NMN might be converted to NR (Yoshino et al., 2011). After administration, NMN is immediately utilized and converted to NAD+ within 15 min, resulting in significant increase in NAD+ levels over a 60 min period (Yoshino et al., 2011). NMN did not affect the animal’s core temperature, blood gases or regional cerebral blood flow during reperfusion suggesting that the protective effect was not a result of altered reperfusion conditions.
The damage to CA1 neurons induced by global cerebral ischemia is associated with deficits in hippocampal function and behavioral performance (Yoshino et al., 2011). The NMN treatment improved the cognitive performance of post-ischemic animals as assessed by the Y-maze test, indicating preservation of hippocampal structures involved in spatial working memory (Ellen et al., 1973). This observation suggests that the CA1 neurons surviving after NMN treatments are not only morphologically intact but also are able to perform physiological functions.
Stroke represents one of the most complex neurologic conditions. It causes bio-energetic failure, that triggers multiple mechanisms leading to the development of tissue death as a consequence of numerous ionic, biochemical, and cellular events (for review see Lipton, 1999, Dirnagl et al., 1999; Lo et al., 2003; Hossmann, 2006). During the last three decades multiple mechanisms and mediators of ischemic brain injury were defined. As a result, a plethora of neuroprotective compounds and therapeutic approaches were identified in pre-clinical research. Calcium channel blockers, glutamate antagonists, NMDA and AMPA receptor antagonists, GABA agonists, magnesium, phospholipid precursors, mitochondria protecting agents, antioxidants, nitric oxide signal transduction down-regulators, immunosuppressants and leukocyte inhibitors demonstrated neuroprotection in animal models of brain ischemia (for review see Ginsberg, 2008). The majority of these compounds target a particular mechanism and to be effective they were administered prior to ischemic conditions. Recently it was recognized that successful stroke treatment will require a strategy that can affect several targets/mechanisms in multiple brain cell types (Moskowitz et al., 2010). Reports in the literature and our data suggest that NMN protection against ischemic brain damage is due to its effect on several targets that are mainly related to NAD+ catabolism and possibly suppressing downstream mechanisms via PARP1 and CD38 inhibition (Balan et al., 2010), thus also modulating the inflammatory response and microglia overactivation (Choe et al., 2011; Malavasi et al., 2008).
Despite the large number of promising anti-ischemic compounds that were identified in pre-clinical research, the overwhelming majority of clinical trials focused on neuroprotection for ischemic stroke failed (for review see Ginsberg, 2008). Although many shortcomings of these clinical studies were identified (Ginsberg, 2008) it is clear that more rigorous pre-clinical research is needed before potential therapy candidate is considered for clinical trials (Lapchak, 2013).
In the current study, we examined the effect of NMN on post-ischemic brain damage when the compound was administered at the start of reperfusion and found that it exhibits clear protective effects at the dose of about 60 mg/kg. To increase the translational applicability of the NMN treatment we also administered the compound 30 min after the start of reperfusion. This protocol also resulted in dramatic protection against ischemic brain damage. In future studies, we will perform further time dependent studies with administration of NMN after ischemic insult to determine the window of opportunity for this compound and to better establish its potential clinical applicability. Moreover, we examined the protective effect of NMN in male mice. Since there are significant difference in NAD+ metabolism between male and females (see Siegel and McCullough, 2013, Owens et al., 2013b), in the next project we will determine the efficacy of this compound in female animals.
In summary, we have shown the administration of NMN following ischemic insult protects hippocampal CA1 neurons from ischemic cell death and preserves neuronal functions after global cerebral ischemia.
Acknowledgments
This work was supported by U.S. Veterans Affairs Merit grant BX000917 to TK.
Footnotes
Conflict of interest
TK has a pending patent application for use of NMN to modulate NAD+ activity in neuropathological conditions. JHP, KO and AL declare no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
References
- Ayoub IA, et al. Nicotinamide reduces infarction up to two hours after the onset of permanent focal cerebral ischemia in Wistar rats. Neurosci Lett. 1999;259:21–24. doi: 10.1016/s0304-3940(98)00881-7. [DOI] [PubMed] [Google Scholar]
- Balan IS, et al. Visualization and quantification of NAD(H) in brain sections by a novel histo-enzymatic nitrotetrazolium blue staining technique. Brain Res. 2010;1316:112–119. doi: 10.1016/j.brainres.2009.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman VL, et al. Neurologic outcome in rats following incomplete cerebral ischemia during halothane, isoflurane, or N2O. Anesthesiology. 1988;69:192–198. doi: 10.1097/00000542-198808000-00007. [DOI] [PubMed] [Google Scholar]
- Belenky P, et al. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Benyo Z, et al. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J Clin Invest. 2005;115:3634–3640. doi: 10.1172/JCI23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanck TJ, et al. Isoflurane pretreatment ameliorates postischemic neurologic dysfunction and preserves hippocampal Ca2+/calmodulin-dependent protein kinase in a canine cardiac arrest model. Anesthesiology. 2000;93:1285–1293. doi: 10.1097/00000542-200011000-00023. [DOI] [PubMed] [Google Scholar]
- Brennan AM, et al. NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. J Cereb Blood Flow Metab. 2006;26:1389–1406. doi: 10.1038/sj.jcbfm.9600292. [DOI] [PubMed] [Google Scholar]
- Canto C, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH, et al. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A. Poly(ADP-ribosyl)ation and stroke. Pharmacol Res. 2005;52:15–24. doi: 10.1016/j.phrs.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Choe CU, et al. CD38 exacerbates focal cytokine production, postischemic inflammation and brain injury after focal cerebral ischemia. PLoS One. 2011;6:e19046. doi: 10.1371/journal.pone.0019046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CD, et al. Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav Neurosci. 1996;110:1321–1334. doi: 10.1037//0735-7044.110.6.1321. [DOI] [PubMed] [Google Scholar]
- Conrad CD, et al. The effects of type I and type II corticosteroid receptor agonists on exploratory behavior and spatial memory in the Y-maze. Brain Res. 1997;759:76–83. doi: 10.1016/s0006-8993(97)00236-9. [DOI] [PubMed] [Google Scholar]
- Cozzi A, et al. Poly(ADP-ribose) accumulation and enhancement of postischemic brain damage in 110-kDa poly(ADP-ribose) glycohydrolase null mice. J Cereb Blood Flow Metab. 2006;26:684–695. doi: 10.1038/sj.jcbfm.9600222. [DOI] [PubMed] [Google Scholar]
- Davidovic L, et al. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- Dellu F, et al. Extension of a new two-trial memory task in the rat: influence of environmental context on recognition processes. Neurobiol Learn Mem. 1997;67:112–120. doi: 10.1006/nlme.1997.3746. [DOI] [PubMed] [Google Scholar]
- Dellu F, et al. Genetic differences in response to novelty and spatial memory using a two-trial recognition task in mice. Neurobiol Learn Mem. 2000;73:31–48. doi: 10.1006/nlme.1999.3919. [DOI] [PubMed] [Google Scholar]
- Di Stefano M, Conforti L. Diversification of NAD biological role: the importance of location. FEBS J. 2013;280:4711–4728. doi: 10.1111/febs.12433. [DOI] [PubMed] [Google Scholar]
- Di Stefano M, et al. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, et al. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Ellen P, Deloache J. Hippocampal lesions and spontaneous alternation behavior in the rat. Physiol Behav. 1968;3:857–860. [Google Scholar]
- Ellen P, et al. Pretraining effects on performance of rats with hippocampal lesions. J Comp Physiol Psychol. 1973;84:622–628. doi: 10.1037/h0034860. [DOI] [PubMed] [Google Scholar]
- Elsersy H, et al. Effects of isoflurane versus fentanyl-nitrous oxide anesthesia on long-term outcome from severe forebrain ischemia in the rat. Anesthesiology. 2004;100:1160–1166. doi: 10.1097/00000542-200405000-00018. [DOI] [PubMed] [Google Scholar]
- Endres M, et al. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Ghasemi A, Zahediasl S. Normality tests for statistical analysis: a guide for non-statisticians. Int J Endocrinol Metab. 2012;10:486–489. doi: 10.5812/ijem.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossmann KA. Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol. 2006;26:1057–1083. doi: 10.1007/s10571-006-9008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua F, et al. The development of a novel mouse model of transient global cerebral ischemia. Neurosci Lett. 2006;400:69–74. doi: 10.1016/j.neulet.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Imai S. The NAD World: a new systemic regulatory network for metabolism and aging—Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem Biophys. 2009;53:65–74. doi: 10.1007/s12013-008-9041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Z, et al. Neuronal NAMPT is released after cerebral ischemia and protects against white matter injury. J Cereb Blood Flow Metab. 2014;34:1613–1621. doi: 10.1038/jcbfm.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. The role of poly(ADP-ribose) polymerase-1 in CNS disease. Neuroscience. 2007;145:1267–1272. doi: 10.1016/j.neuroscience.2006.09.034. [DOI] [PubMed] [Google Scholar]
- Kiyota Y, et al. Relationship between brain damage and memory impairment in rats exposed to transient forebrain ischemia. Brain Res. 1991;538:295–302. doi: 10.1016/0006-8993(91)90443-y. [DOI] [PubMed] [Google Scholar]
- Klaidman LK, et al. Nicotinamide as a precursor for NAD+ prevents apoptosis in the mouse brain induced by tertiary-butylhydroperoxide. Neurosci Lett. 1996;206:5–8. doi: 10.1016/0304-3940(96)12446-0. [DOI] [PubMed] [Google Scholar]
- Klaidman LK, et al. Oxidative changes in brain pyridine nucleotides and neuroprotection using nicotinamide. Biochim Biophys Acta. 2001;1525:136–148. doi: 10.1016/s0304-4165(00)00181-1. [DOI] [PubMed] [Google Scholar]
- Kristian T, Fiskum G. A fluorescence-based technique for screening compounds that protect against damage to brain mitochondria. Brain Res Brain Res Protoc. 2004;13:176–182. doi: 10.1016/j.brainresprot.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Kurinami H, et al. Prohibitin viral gene transfer protects hippocampal CA1 neurons from ischemia and ameliorates postischemic hippocampal dysfunction. Stroke. 2014;45:1131–1138. doi: 10.1161/STROKEAHA.113.003577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis SC, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–191. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapchak PA. Recommendations and practices to optimize stroke therapy: developing effective translational research programs. Stroke. 2013;44:841–843. doi: 10.1161/STROKEAHA.112.680439. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, et al. RIGOR guidelines: escalating STAIR and STEPS for effective translational research. Transl Stroke Res. 2013;4:279–285. doi: 10.1007/s12975-012-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson E, et al. Stereological assessment of vulnerability of immunocytochemically identified striatal and hippocampal neurons after global cerebral ischemia in rats. Brain Res. 2001;913:117–132. doi: 10.1016/s0006-8993(01)02762-7. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu D, et al. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neruomol Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, et al. Inhibition of poly(ADP-ribose) polymerase: reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke. 1998;29:830–836. doi: 10.1161/01.str.29.4.830. [DOI] [PubMed] [Google Scholar]
- Lo EH, et al. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Magni G, et al. Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol Biol. 1999;73:135–182 (xi). doi: 10.1002/9780470123195.ch5. [DOI] [PubMed] [Google Scholar]
- Malavasi F, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- Miura Y, et al. Differential effects of anesthetic agents on outcome from near-complete but not incomplete global ischemia in the rat. Anesthesiology. 1998;89:391–400. doi: 10.1097/00000542-199808000-00016. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, et al. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onken M, et al. Simple model of forebrain ischemia in mouse. J Neurosci Methods. 2012;204:254–261. doi: 10.1016/j.jneumeth.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens K, et al. Utilizing commercial microwave for rapid and effective immunostaining. J Neurosci Methods. 2013a;219:20–26. doi: 10.1016/j.jneumeth.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Owens K, et al. Mitochondrial dysfunction and NAD+ metabolism alterations in the pathophysiology of acute brain injury. Transl Stroke Res. 2013b;4:618–634. doi: 10.1007/s12975-013-0278-x. [DOI] [PubMed] [Google Scholar]
- Panahian N, et al. Attenuated hippocampal damage after global cerebral ischemia in mice mutant in neuronal nitric oxide synthase. Neuroscience. 1996;72:343–354. doi: 10.1016/0306-4522(95)00563-3. [DOI] [PubMed] [Google Scholar]
- Park L, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. (discussion 332) [DOI] [PubMed] [Google Scholar]
- Revollo JR, et al. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279(50):754–763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Sadanaga-Akiyoshi F, et al. Nicotinamide attenuates focal ischemic brain injury in rats: with special reference to changes in nicotinamide and NAD+ levels in ischemic core and penumbra. Neurochem Res. 2003;28:1227–1234. doi: 10.1023/a:1024236614015. [DOI] [PubMed] [Google Scholar]
- Sarnyai Z, et al. Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proc Natl Acad Sci U S A. 2000;97(14):731–736. doi: 10.1073/pnas.97.26.14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng H, et al. Characterization of a recovery global cerebral ischemia model in the mouse. J Neurosci Methods. 1999;88:103–109. doi: 10.1016/s0165-0270(99)00018-7. [DOI] [PubMed] [Google Scholar]
- Siegel CS, McCullough LD. NAD+ and nicotinamide: sex differences in cerebral ischemia. Neuroscience. 2013;237:223–231. doi: 10.1016/j.neuroscience.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, et al. Expression of Nampt in hippocampal and cortical excitatory neurons is critical for cognitive function. J Neurosci. 2014;34:5800–5815. doi: 10.1523/JNEUROSCI.4730-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strosznajder RP, et al. Poly(ADP-ribose) polymerase during reperfusion after transient forebrain ischemia: its role in brain edema and cell death. J Mol Neurosci. 2003;20:61–72. doi: 10.1385/JMN:20:1:61. [DOI] [PubMed] [Google Scholar]
- Szabo C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- Ungerstedt JS, et al. Nicotinamide is a potent inhibitor of proinflammatory cytokines. Clin Exp Immunol. 2003;131:48–52. doi: 10.1046/j.1365-2249.2003.02031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereczki V, et al. Normoxic resuscitation after cardiac arrest protects against hippocampal oxidative stress, metabolic dysfunction, and neuronal death. J Cereb Blood Flow Metab. 2006;26:821–835. doi: 10.1038/sj.jcbfm.9600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, et al. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann Neurol. 2011;69:360–374. doi: 10.1002/ana.22236. [DOI] [PubMed] [Google Scholar]
- Wellons JC, III, et al. A comparison of strain-related susceptibility in two murine recovery models of global cerebral ischemia. Brain Res. 2000;868:14–21. doi: 10.1016/s0006-8993(00)02216-2. [DOI] [PubMed] [Google Scholar]
- Yang J, et al. Nicotinamide therapy protects against both necrosis and apoptosis in a stroke model. Pharmacol Biochem Behav. 2002a;73:901–910. doi: 10.1016/s0091-3057(02)00939-5. [DOI] [PubMed] [Google Scholar]
- Yang J, et al. The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in Wistar rats. Neurosci Lett. 2002b;333:91–94. doi: 10.1016/s0304-3940(02)01005-4. [DOI] [PubMed] [Google Scholar]
- Yonekura I, et al. A model of global cerebral ischemia in C57 BL/6 mice. J Cereb Blood Flow Metab. 2004;24:151–158. doi: 10.1097/01.WCB.0000096063.84070.C1. [DOI] [PubMed] [Google Scholar]
- Yoshino J, et al. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, et al. Neuronal protective role of PBEF in a mouse model of cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:1962–1971. doi: 10.1038/jcbfm.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen G, Dore S. Optimized protocol to reduce variable outcomes for the bilateral common carotid artery occlusion model in mice. J Neurosci Methods. 2007;166:73–80. doi: 10.1016/j.jneumeth.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]