

Abstract
The enzyme 15-prostaglandin dehydrogenase (15-PGDH) catalyzes the first step in the degradation of prostaglandins including PGE2. It is a negative regulator of tissue repair and regeneration in multiple organs. Accordingly, inhibitors of 15-PGDH are anticipated to elevate in vivo levels of PGE2 and to promote healing and tissue regeneration. The small molecule SW033291 (1) inhibits 15-PGDH with Ki = 0.1 nM in vitro, doubles PGE2 levels in vivo, and shows efficacy in mouse models of recovery from bone marrow transplantation, ulcerative colitis, and partial hepatectomy. Here we describe optimized variants of 1 with improved solubility, drug-like properties and in vivo activity.
Graphical abstract
Introduction
Prostaglandin E2 (PGE2) is an endogenous signaling molecule involved in pain, inflammation, and cell proliferation.1 It is produced from arachidonic acid that is released from membranes in response to stress, cytokines or trauma (Scheme 1). The enzymes cyclooxygenase 1 or 2 (COX1/2) oxidize and cyclize arachidonic acid to prostaglandin H2, which is then converted to PGE2 by the action of prostaglandin E synthase (PGES). PGE2 is exported by dedicated transporters, and can then activate one of four G-protein coupled receptors, EP1–4. Binding of PGE2 to these receptors activates second messengers including cyclic-adenosine monophosphate and augments signaling through the Wnt pathway.1
Scheme 1.
Synthesis of pyridylthiophene inhibitors of 15-PGDH
Inhibitors of this pathway have been pursued as anti-inflammatory, analgesic and anticancer agents. However, we were interested in developing strategies to increase rather than decrease PGE2 levels in vivo. This objective emerged from the observation that PGE2 promotes growth, differentiation and healing in a variety of cellular settings.2 Accordingly, agents that elevated PGE2 levels might aid healing and tissue regeneration. In this context, PGE2 or the more metabolically stable analog 16,16-dimethyl-PGE2 (dmPGE2) augment hematopoiesis in zebrafish.3,4 Additionally, ex vivo exposure of murine bone marrow or primate cord blood to dmPGE2 enhances their effectiveness in bone marrow transplantation assays.5,6,7 A phase 1 study demonstrated that ex vivo treatment of human umbilical cord blood with dmPGE2 may accelerate neutrophil recovery in patients transplanted with the treated cells.8,9 Similarly, PGE2 has been shown to promote expansion of colonic stem cells in culture,10 and dmPGE2 has been shown to reduce disease severity in a murine colitis model.11 Collectively, these observations indicated that elevation of PGE2 levels in vivo may potentiate tissue regeneration and repair.2
PGE2 is degraded in vivo by the enzyme 15-prostaglandin dehydrogenase (15-PGDH). This enzyme catalyzes the transfer of the C15 hydride to NAD+, creating 15-keto-PGE2, which is unable to bind to prostaglandin receptors.12 We hypothesized that inhibitors of 15-PGDH would block the degradation of PGE2 and thereby elevate PGE2 levels in vivo. Encouragingly, we found that the 15-PGDH knockout mouse has approximately 2-fold higher levels of PGE2 within the colon, lung, liver and bone marrow. Moreover, 15-PGDH-KO mice are completely resistant to dextran sodium sulfate-induced colitis, display increased hematopoietic capacity, and regrow liver tissue more rapidly following partial resection compared to wild-type litter mates. 13,14
Several research groups have disclosed inhibitors of 15-PGDH (Figure 2A). For example, scientists at L’Oreal described a series of tetrazoles15 such as 2 that displayed partial enzyme inhibition at 50 μM and aminooxy amides16 including 3, which possessed an IC50 of 6 μM against the purified enzyme (Figure 2). Cho and colleagues have studied rhodanine alkylidenes such as compound 4.17 This inhibitor was active against the enzyme in vitro (IC50 = 20 nM) and in A549 cells at 5 μM. Additionally, compound 4 showed activity in a cell-based model of wound healing. Finally, a group from the NIH has disclosed several triazoles, exemplified by 5, and benzamidazoles, exemplified by 6, with IC50’s as low as 22 and 12 nM, respectively.18 In a cell culture experiment, these inhibitors displayed activities in the mid-nM range. While each of these lead compounds showed promising inhibition in vitro, none of them has been reported to show activity in any in vivo disease model.
Figure 2.

A. Inhibitors of 15-PGDH. B. Potential binding model for 15-PGDH with PGE2 and inhibitor 1.
We recently reported the discovery and characterization of the sulfoxide SW033291 (1) as a tight binding inhibitor of 15-PGDH with an apparent Ki of 0.1 nM.14 In mice, 1 doubled PGE2 levels in lungs, liver, colon and bone marrow at 3 h after a dose of 10 mg/kg. Furthermore, we found that it 1) accelerated recovery of neutrophils, platelets and red blood cells following bone marrow transplantation (BMT) in lethally irradiated mice; 2) ameliorated the severity of colitis induced by dextran sodium sulfate in mice; and 3) increased the rate and extent of liver regeneration following partial liver resection in mice. In the mouse BMT model, 15-PGDH inhibitor 1 accelerated neutrophil recovery by approximately one week, with similar effects on platelets and erythrocytes. In humans, this activity is anticipated to reduce morbidity and mortality associated with BMT by reducing the risk of infection, minimizing bleeding, and reducing the requirement for blood transfusion. Finally, compound 1 showed no adverse effects on weight, activity, blood counts or blood chemistry following 1 week of administration of 20 mg/kg, which is four-fold above the efficacious dose. Likewise, the 15-PGDH knockout mouse is healthy and lives a normal lifespan.13 These studies suggested that optimized inhibitors of 15-PGDH would safely elevate PGE2 levels in vivo and hasten recovery of multiple tissues.
Sulfoxide 1 is potent in vitro, active in cells, and efficacious in multiple mouse models of tissue regeneration. Nonetheless, we recognized opportunities to improve its physicochemical properties. For example, its high lipophilicity (cLogP = 5.8)19 was associated with low solubility and high plasma protein binding. We were particularly eager to identify a highly soluble inhibitor of 15-PGDH because intravenous (IV) administration is preferred for drugs used to treat patients receiving bone marrow or other hematopoietic stem cell transplants. We therefore targeted discovery of a potent, safe inhibitor of 15-PGDH with high aqueous solubility that would be suitable for IV administration.
No crystal structure of 15-PGDH bound to any inhibitor or PGE2 has been described, although the X-ray crystal structure of human 15-PGDH complexed with NAD+ was solved by Simeonov and coworkers.18a Consistent with mutagenesis studies, they proposed that the enzymatic processing of PGE2 involves deprotonation of the 15-OH by active site tyrosine 151 concurrent with hydride transfer to bound NAD+ (Figure 2B).12 This mechanism is anticipated to form a partial negative charge on the alcohol oxygen and a partial positive charge on the C15 carbon. In this context, we noted that the sulfoxide functionality of 1 features a charge distribution that might mimic the charge build-up in the transition state for oxidation of PGE2. Additionally, the sulfoxide side chain might fill a hydrophobic binding site occupied by the C16-C20 alkyl chain of PGE2. With this binding hypothesis in mind, we sought to decipher the structural requirements for inhibition of 15-PGDH and to discover optimized inhibitors of 15-PGDH suitable for use in humans.
Results and Discussion
Chemistry
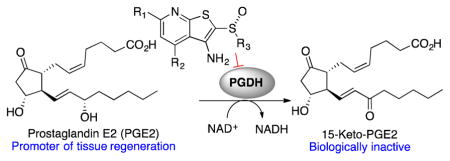
The pyridine scaffold was assembled through an annulation of 2-cyanothioacetamide with either β-diketones (7) or enones (8, Scheme 1A). The reactions with enones were usually carried out under either air or oxygen balloon. Under an inert atmosphere, the thiopyridone 9 was accompanied by a thiopyridone lacking the nitrile. Additionally, we occasionally observed conjugate addition of the thiopyridone to the enone. Alkylation on sulfur with α-halo carbonyl compounds under basic conditions led to cyclization onto the nitrile to yield the thienopyridines 10. Similarly, alkylation of thiopyridone 9 with chloromethyl thioethers yielded the dithianes 11. Mono-oxidation to the sulfoxide followed by cyclization with hydroxide or tert-butoxide base constructed the thienopyridine scaffold (12). When the final cyclization was performed on gram scale, it was possible to isolate the corresponding sulfones 14 as minor side-products. Alternatively, the sulfoxide of 12 could be reduced to a sulfide with TiCl4 and zinc dust (13). Single enantiomers of selected sulfoxides were accessed with preparative HPLC using a chiral solid support.
Once installed, the sulfoxide group tended to dominate all chemistry. For this reason, we frequently made changes to the periphery of the inhibitors prior to oxidation to the sulfoxide. For example, acetate 15 was hydrolyzed to the corresponding alcohol and mesylated. Nucleophilic substitution proceeded oxidation to the sulfoxide and final ring closure (Scheme 1B). In this way the terminal –CH3 of 1 was replaced with hydroxyl, halogens or a nitrile (16).
Methyl ester 17a also emerged as a versatile intermediate (Scheme 2). Reduction with LiBH4 provided the primary alcohol 18, which could be diverted down several paths. First, azidation with diphenylphosphoryl azide returned the benzylic azide 19. Subsequent reduction and acylation led to amides and carbamates 21. Alternatively, azide 19 could be oxidized and cyclized prior to Staudinger reduction to form amine 24. Sulfonylation provided the sulfonamide 25. A second set of analogs derived from benzylic alcohol 18 was formed through addition to isocyanates to form carbamates 22 after oxidation and cyclization. Alternatively, sulfoxidation of 18 and base-mediated cyclization yielded the primary alcohol 23a, which could be acylated with carboxylic acids in the presence of a carbodiimide to form the corresponding esters.
Scheme 2.

Synthesis of derivatives of 1 with substitution on the phenyl ring
A diverse set of amides arose from the para-methyl ester 17a and meta-methyl ester 17b (Scheme 2B). In particular, following formation of the thiophene ring (26), ester hydrolysis formed the carboxylic acid 27, which could be converted to amides 28 using HATU. Similarly, three alcohols (30–32) were accessible through late-stage manipulation of the corresponding esters 29 (Scheme 2c). Specifically, addition of MeLi generated the tertiary alcohol 30 while reduction with LiBH4 yielded primary alcohols 31 and 32.
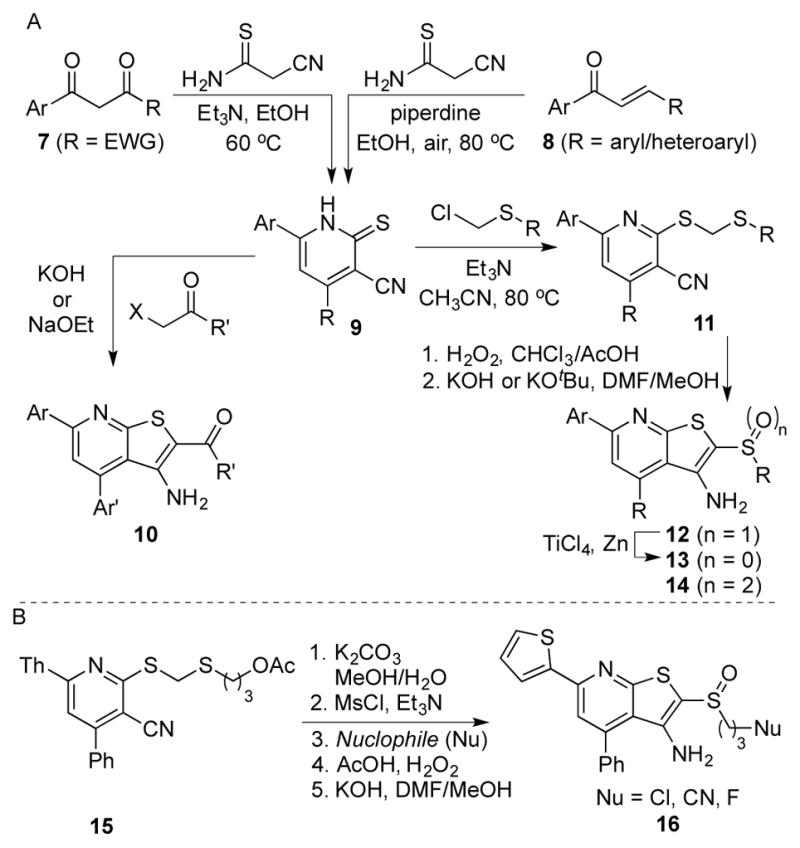
An alternative synthesis of the thienopyridine scaffold is shown in Scheme 3. This route proved particularly useful for analogs featuring a methyl or hydrogen at the 4-position of the pyridine (38, R′ = H or CH3). In detail, an SNAr/β-elimination sequence between chloropyridine 33 and β-mercaptopropionate methyl ester introduced the requisite sulfur atom in thiopyridone 36. Alternatively, the same ring system could be accessed from dichloropyridine 34. Thus, amination or Suzuki coupling proved regioselective at the site distal to the nitrile to provide chloropyridine 35. The sulfur was installed as before, and alkylation, oxidation and cyclization proceeded as described for other thienopyridines to yield analogs 38.
Scheme 3.
Alkylidene malononitrile 39 served as the precursor to several inhibitors featuring a pyrimidine ring (Scheme 4A). It underwent a condensation with thioamides 40 to form thiopyrimidone 41. Alkylation on sulfur, oxidation and cyclization proceeded analogously to what was observed in the pyridine series to provide pyrimidine inhibitors of 15-PGDH (44). Alternatively, commercially available dichloropyrimidine 45 provided the opportunity to generate inhibitors lacking the exocyclic –NH2 moiety. Thus, Suzuki coupling introduced two phenyl rings (46). Lithiation and trapping with dibutyldithiane introduced the side chain prior to final oxidation to sulfoxide 48. Alternatively, SNAr reaction with piperdine introduced an aliphatic heterocycle (49). Subsequent installation of the sulfoxide side chain and phenyl ring generated the pyrimidine 51.
Scheme 4.

Synthesis of pyrimidines
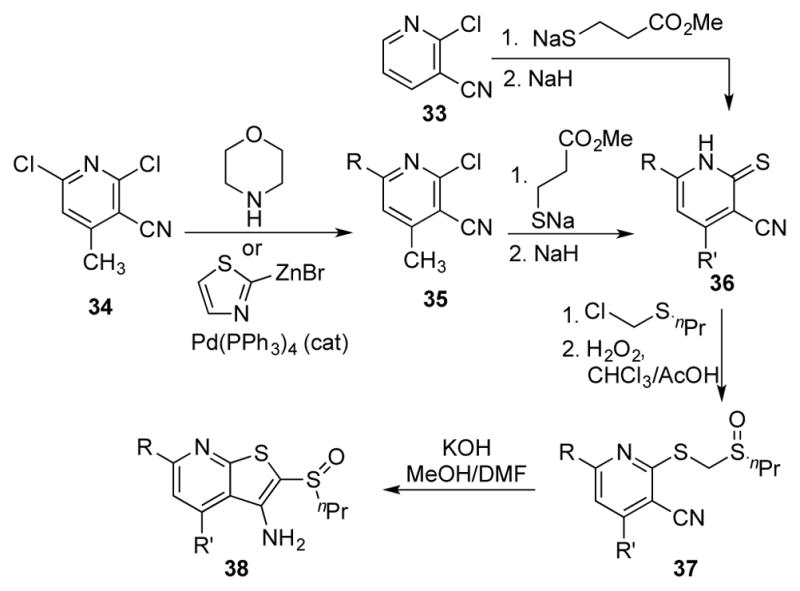
To evaluate replacements for the thiophene ring within 1, we synthesized two alternative scaffolds as shown in Scheme 5. Known mercaptothiazole 5220 was alkylated with butyl bromide, and then subjected to Suzuki coupling conditions to introduce the thiophene. Final oxidation with hydrogen peroxide provided thiazole sulfoxide 53. Using a similar strategy, chloropyridine 54 was coupled with 2-thiophene boronic acid prior to reduction of the nitro group to yield diaminopyridine 55. Condensation with thiourea in the melt formed the thioimidazolone ring, while alkylation and oxidation led to the final imidazole sulfoxide 57.
Scheme 5.
Synthesis of alternative pyridyl-fused heterocycles
Biological activity
Potential inhibitors were evaluated using an in vitro enzyme assay, a cell based assay, and an in vivo pharmacodynamic experiment. The in vitro experiment used recombinant human 15-PGDH, assayed at an enzyme concentration of approximately 3 nM. Sulfoxide 1 is a tight binding inhibitor, and its IC50 approximates half the enzyme concentration. Similar potencies were observed for many of the optimized compounds shown below. In general, IC50 values less than 3 nM are indistinguishable from each other, while inhibitors with IC50 values greater than 3 nM are considered weaker inhibitors than 1. Promising inhibitors of 15-PGDH were further profiled in a cell-based assay using adenocarcinoma A549 cells. In brief, addition of interleukin 1β (IL-1β) to these cells stimulates PGE2 production. Inhibition of PGE2 metabolism leads to a further increase in PGE2 levels in the culture media, which can be quantified with an ELISA assay. The data are reported as a fold-increase at a low dose (20 nM) and at a saturating dose (2.5 μM). Illustrative results for inhibitor 1 are shown in Figure 3: at 20 nM, sulfoxide 1 elevated PGE2 levels from 660 pg/mL to 1440 pg/mL, a 2.2-fold increase. In all experiments in A549 cells, we included 1 as a positive control. While the relative potency of inhibitors was consistent across many experiments, the magnitude of PGE2 induction varied. For this reason, in each experiment we adjusted performance of compound 1 to its average results from 12 independent experiments (1.9- and 2.7-fold PGE2 induction at 20 nM and 2.5 μM, respectively), and applied the same normalization factors to all other compounds tested at the same time. Un-adjusted data is presented in Table S1 in the supporting information. Compounds showing encouraging activity in vitro were further profiled for solubility and for in vitro and in vivo ADME characteristics.
Figure 3.

Inhibitors of 15-PGDH elevate PGE2 levels in the media of IL-1β-treated A549 cells. Cells were treated with IL-1β and compound and PGE2 levels in the cell culture media were determined after 16 h.
Our first experiments probed the requirement for a sulfoxide side chain (Table 1). The initial characterization of 1 utilized the racemic mixture, but we found that the enzyme activity resided predominantly in the positive, R enantiomer.21 While the single enantiomer inhibitor (+)-1 showed greater potency in the cell-based assay as expected, it showed markedly reduced solubility in crystalline form compared to amorphous racemate. The corresponding sulfide (13a) lost substantial activity, and the sulfone (14a) was around 10-fold less active than the sulfoxide. Attempts to replace the sulfoxide with carbonyls were not successful, as a similarly sized ketone (10a), amide (10b), ester (10c) and acid (10d) were all inactive against the recombinant enzyme.
Table 1.
Structure-Activity Relationship for Sulfoxide Side Chain.
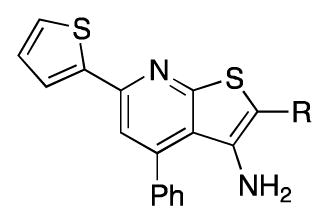
| |||||
|---|---|---|---|---|---|
| Compound | R | IC50 (nM) | Fold PGE2 increase at | Aq. Solubility (μg/mL)a,b | |
| 20 nM | 2500 nM | ||||
| 1 | -S(O)-nBu | 2.2 | 1.9 | 2.7 | 0.3 |
| (R)-1 | (R)-S(O)-nBu | 1.5 | 2 | 2.9 | 0.007c |
| (S)-1 | (S)-S(O)-nBu | 450 | 1.5 | 1.7 | |
| 13a | -S-nBu | >80d | 0.9 | 2.1 | |
| 14a | -SO2-nBu | 25 | |||
| 10a | -C(O)-nBu | >7500 | |||
| 10b | -C(O)NHnPr | >7500 | |||
| 10c | -CO2Et | >7500 | |||
| 10d | CO2H | >7500 | |||
| 12a | -S(O)CH3 | 125 | |||
| 12b | -S(O)nPr | 2.1 | 1.6 | 2.8 | |
| 12c | -S(O)iPr | 3.2 | 1.7 | 2.4 | |
| 12d | -S(O)-nPent | 2.6 | |||
| 12e | -S(O)-nHex | 18 | |||
| 12f | -S(O)(CH2)3OCH3 | 2.1 | 1.9 | 2.6 | 4.7 |
| 16a | -S(O)(CH2)3OH | 37 | 1.1 | 1.5 | |
| 16b | -S(O)(CH2)3Cl | 4 | 1.8 | 2.1 | |
| 16c | -S(O)(CH2)3F | 6.1 | 1.2 | 2.2 | |
| 16d | -S(O)(CH2)3CN | 8.7 | 1.7 | 2.7 | |
Fold-increase in PGE2 levels in A549 cell culture media relative to DMSO. Data color-coded to indicate more active (blue), less active (red) or equally active (white) as compound 1.
Amorphous solid unless otherwise indicated. Solubility in pH 7 citrate buffer, 0.1 M.
Crystalline solid.
Observed inhibition likely from trace 1.
The shape and composition of the sulfoxide side chain affected activity profoundly. For example, replacing the butyl chain with a methyl group decreased activity approximately 100-fold (12a), whereas an isopropyl, n-propyl and n-pentyl (12b-d) side chain showed similar activity, although not better, than the n-butyl side chain of 1. A larger n-hexyl replacement was deleterious (12e). In an attempt to add polarity to the side chain to enhance solubility, we explored addition of terminal methoxy (12f), hydroxyl (16a), halogen (16b, c) and nitrile (16d) groups. Encouragingly, we were able to incorporate an ether into the side chain while maintaining robust activity both in vitro and in A549 cells (12f). Additionally, this modest chemical change was accompanied by a roughly 10-fold increase in aqueous solubility. By contrast, other polar groups on the sulfoxide side chain decreased inhibitor potency in vitro and in cells.
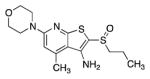
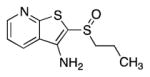
Table 2 outlines studies aimed at identifying a more polar replacement for the thiophene ring. We anticipated that N-heterocycles might improve solubility and minimize the chance of forming reactive metabolites such as epoxides. In this regard, we discovered that the corresponding thiazole (12g) and oxazole (12h) maintained full activity in vitro and were only slightly less active than 1 in the cell-based PGE2 assay. Encouragingly, thiazole 12g and oxazole 12h were roughly 4- and 9-fold more soluble than 1, respectively. By contrast, the imidazole 12i was a less potent inhibitor of 15-PGDH. As shown below, the phenyl ring of 1 could be replaced with either a hydrogen or a methyl group. In this context, the poor IC50’s of morpholine 38a and unsubstituted pyridine 38b reveal the benefit of a heteroaryl substituent at the 6 position of the pyridine ring. Nonetheless, the fact that the simplified analog 38b retains substantial inhibition of 15-PGDH indicates that the thienopyridyl sulfoxide scaffold contributes most of the binding energy. Taken together with the des-phenyl derivative of 1 (i.e. 12j, see Table 4), these results indicate that the heteroaryl ring at the 6-position increases inhibitory activity by two orders of magnitude.
Table 2.
Structure-Activity Relationship for 6-Pyridyl Substituent
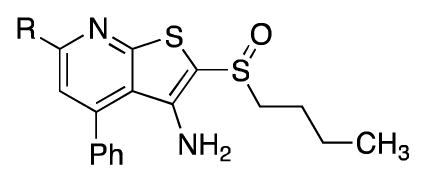
| |||||
|---|---|---|---|---|---|
| Compound | R/Inhibitor | IC50 (nM) | PGE2 increase at | Aq. Solubility (μg/mL)b | |
| 20 nMa | 2.5 μMa | ||||
| 1 | 2-Thiophene | 2.2 | 1.9 | 2.7 | 0.3 |
| 12g | 2-Thiazole | 1.4 | 1.7 | 2.5 | 1.1 |
| 12h | 2-oxazole | 1.4 | 1.9 | 2.4 | 2.8 |
| 12i | 2-(N-Me-imidazole) | 6.7 | 1.4 | 2.6 | 1.9 |
| 38a |
|
136 | |||
| 38b |
|
151 | |||
Fold-increase in PGE2 levels in A549 cell culture media relative to DMSO. Data color-coded to indicate more active (blue), less active (red) or equally active (white) as compound 1.
pH 7 citrate buffer, 0.1 M.
Table 4.
Structure-Activity Relationship for the 4-Phenyl Ring
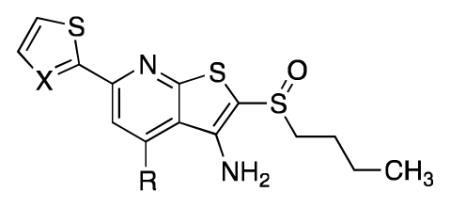
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | X | R | IC50 (nM) | PGE2 increase at | Aq. Solubility (μg/mL)b | ||
| 20 nMa | 2.5 μMa | ||||||
| 1 | CH | Ph | 2.2 | 1.9 | 2.7 | 0.3 | |
| 12j | CH | H | 1.0 | 1.6 | 2.3 | 3.6 | |
| 38c | N | -CH3c | 5.3 | 1.5 | 1.9 | 7.7 | |
| 12k | CH | -CO2Etc | 2.0 | 1.5 | 2.5 | ||
| 121 | CH | -CO2Hc | 71 | ||||
| 12m | CH | -CONMe2 | 14.7 | 0.8 | 2 | ||
| 12n | N | 2-thiazole | 1.8 | 2.6 | 3.5 | 0.3 | |
| 12o | N | 3-pyridyl | 2.6 | 1.7 | 2.6 | 2.8 (pH 7) 17.9 (pH 3) |
|
| 12p | N | 2-(N-methyl-imidazole) | 1.8 | 2.6 | 3.2 | 7.5 (pH 7) 280 (pH 3) |
|
| (R)-12q | N |
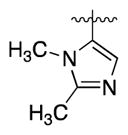
|
1.3 | 2.1 | 2.8 | ||
| (S)-12q | N | 165 | 1.5 | 1.8 | 34 (pH 7) >1400 (pH 4) 4300 (HCl salt)d |
||
| 12r | N |
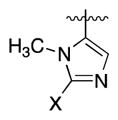
|
X = iPr | 20.5 | 1.5 | 2.4 | |
| (+)-12s | N | X = c Pr | 2 | 2.1 | 2.8 | 4 (pH 7)e 493 (pH 3)e |
|
| 12t | N | X = Cl | 3.2 | 1.4 | 2.4 | ||
| 12u | N |
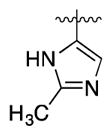
|
2.3 | 1.4 | 2.2 | 4.1 | |
Fold-increase in PGE2 levels in A549 cell culture media relative to DMSO. Data color-coded to indicate more active (blue), less active (red) or equally active (white) as compound 1.
Amorphous solid unless otherwise indicated. Solubility in pH 7 citrate buffer, 0.1 M. pH 7 citrate buffer, 0.1 M.
n-Pr sulfoxide in place of n-Bu sulfoxide.
Solubility of the HCl salt in pH 7 water; final pH = 5.
Solubility of racemate.
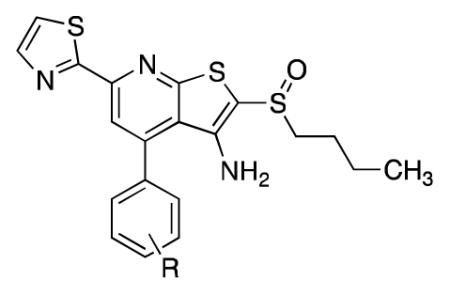
We investigated substitution on the phenyl ring of lead compound 1 with the intention of identifying groups that would improve solubility while maintaining high activity (Table 3). Installing an ester or amide in the 4-position of the phenyl decreased enzyme inhibition substantially (26a, 28a). The corresponding carboxylic acid (27a) was a potent inhibitor and displayed high aqueous solubility. However, it performed poorly in the cell-based assay. More broadly, inhibitors carrying either a positive or negative charge at physiological pH have not performed as well as 1 in cell-based assays to date. The benzylic alcohol (23a) and its corresponding ester (23b) were highly active both in vitro and in the A549 cell-based assay. Moreover, the alcohol showed approximately 50-fold improved solubility compared to 1 and offered the potential to form a pro-drug. Unfortunately, both inhibitors suffered from rapid degradation in the presence of mouse liver S9 fractions, presumably due to ester hydrolysis and oxidation to the corresponding aldehyde (23a: t1/2 = 13 min; 23b: t1/2 = 2 min). We attempted to identify a more stable inhibitor that maintained the activity profile of 23a and 23b by synthesizing the analogous amine (24), amide (21a), urea (21b), carbamate (22a) and sulfonamide (25). Among those, the amide (21a) and urea (21b) maintained high enzyme inhibitory properties, but both induced PGE2 only 80% as well as 1 at 2.5 μM, Similarly, a tertiary alcohol, 30, was a potent enzyme inhibitor, but was not as active as 1 in cells. Reasoning that the rapid metabolism of 23a resulted in part from oxidation of the benzylic alcohol, we prepared the ethanol derivative 31. This compound displayed promising activity against the enzyme, but it was less active than 1 in the cell-based assay. While the variation between in vitro activity and cellular activity remains confusing, we were gratified to find that an inhibitor containing an ethylene glycol moiety (32) was highly active both against the enzyme and in cells. As we had observed with 1, the 15-PGDH inhibitor activity resided predominantly in the (+)-enantiomer. Significantly, introduction of polar functionality on the phenyl ring improved solubility to >10-fold to 5 μg/ml and opened the possibility of generating pro-drugs.
Table 3.
Structure-Activity Relationship for the 4-Phenyl Ring
| |||||
|---|---|---|---|---|---|
| Compound | R | IC50 (nM) | PGE2 increase at | Aq. Solubility (μg/mL)b | |
| 20 nMa | 2.5 μMa | ||||
| 21a | 4-CH2NHAc | 2.5 | 1.7 | 2.2 | 2.16 |
| 21b | 4-CH2NHC(O)NHEt | 2.1 | 1.6 | 2.2 | 8.17 |
| 22a | 4-CH2OC(O)NHEt | 3.4 | 1.8 | 2.1 | |
| 23a | 4-CH2OH | 2.5 | 2.2 | 2.8 | 12.5 |
| 23b | 4-CH2OC(O)CH2NMe2 | 3.5 | 2.3 | 2.9 | 6 |
| 23c | 3-CH2OH | 4.3 | 1.4 | 2 | |
| 24 | 4-CH2NH2 | 11.5 | |||
| 25 | 4-CH2NHSO2CH3 | 4.6 | 1.6 | 1.8 | |
| 26a | 4-CO2Me | 15.1 | 1.5 | 2.5 | |
| 26b | 3-CO2Me | 9 | 1.3 | 2.1 | |
| 27a | 4-CO2H | 3.4 | 1.2 | 1.8 | 208 |
| 28a | 4-CONMe2 | 5.9 | 0.9 | 1.8 | |
| 28b | 3-CONMe2 | 18.5 | 1.2 | 2 | |
| 28c | 3-CONH(CH2)2OH | 3.4 | 1.7 | 2.8 | 45 |
| 30 | 4-C(CH3)2OH | 3.6 | 1.8 | 2.4 | |
| 31 | 4-(CH2)2OH | 2.9 | 1.6 | 2.3 | |
| (+)-32 | 4-OCH2CH2OH | 1.9 | 2.1 | 2.8 | 5c |
| (−)-32 | 4-OCH2CH2OH | 158 | |||
Fold-increase in PGE2 levels in A549 cell culture media relative to DMSO. Data color-coded to indicate more active (blue), less active (red) or equally active (white) as compound 1.
Amorphous solid unless otherwise indicated. Solubility in pH 7 citrate buffer, 0.1 M. pH 7 citrate buffer, 0.1 M.
Solubility of racemate.
Substitution on the phenyl ring meta to the pyridine was also explored. The primary alcohol (23c) methyl ester (26b), tertiary amide (28b) and were less active than 1, but the secondary amide 28c showed good enzyme inhibition, robust activity in cells and excellent solubility.
In parallel with efforts to add substituents to the phenyl ring of 1, we looked to replace this group with more polar substituents to increase solubility. We were surprised to discover that removing the phenyl ring altogether did not impact enzyme inhibition (12j, Table 4). However, this inhibitor was nearly inactive in cells and was rapidly metabolized in the presences of mouse liver S9 fractions (t1/2 = 13 min). Likewise replacing the phenyl ring with a methyl group (38c) or an ethyl ester (12k) maintained good potency against 15-PGDH, but displayed lower activity than 1 in cells. The corresponding carboxylic acid (12l) and tertiary amide (12m) showed decreased inhibitory activity relative to the ester (12k). We next replaced the phenyl ring of 1 with a variety of heterocyclic rings. The bis-thiazole 12n showed excellent potency against 15-PGDH and the highest activity we have observed in cells. Surprisingly, however, its solubility was not notably better than 1, despite a substantial increase in polarity (cLogP = 3.3 vs. 5.0).
Our first clue towards developing highly soluble inhibitors of 15-PGDH came with compound 12o, which features a 3-pyridyl appendage. It showed good activity both in vitro and in cells, and a ~10-fold improvement in solubility relative to 1. More significantly, it showed pH-dependent solubility such that at pH 3 it was >50-fold more soluble than 1. Accordingly, we incorporated a more basic heterocycle and obtained the 2-substituted imidazole 12p. This inhibitor was highly active and showed better solubility than 12o at both pH 7 and pH 3. An isomeric imidazole, 12q, was even more soluble, exceeding 1 mg/mL at pH 4. Additionally, the corresponding HCl salt was soluble up to 4.3 mg/mL in neutral water. We attribute the improved solubility of 12q vs. 12p to an increase in the basicity of the imidazole with a 2-methyl group (12q) compared to a 2-pyridyl group (12p). With compound 1, the single enantiomer crystalline form was >10-fold less soluble than the amorphous racemate. Interestingly, however, optically active 12p showed a similar solubility profile as the racemic compound. Further modifying 12q, we replaced the 2-methyl group with isopropyl (2r), cyclopropyl (2s) and chloro groups (2t). The isopropyl group appeared too big for the binding site, but both the cyclopropyl and chloro groups were tolerated. However, only the cyclopropyl-containing imidazole showed high activity in cells. Finally, removing the N-methyl group from 12q decreased both solubility and cellular activity compared to 1 (12u).
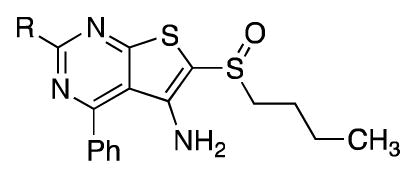
We also explored inhibitors with a pyrimidine core (Table 5). The direct analog to 1 (44a) retained good activity against the enzyme and in cells, although solubility was not improved. While replacing the thiophene appendage with a phenyl ring resulted in decreased potency (44b), replacing the thiophene with a thiazole led to 44c, an inhibitor with excellent activity in vitro and in cells. This inhibitor also showed improved solubility relative to 1. Incorporating either an imidazole or oxazole ring (44d, 44e) greatly improved the solubility and provided active inhibitors. Unfortunately, imidazole 44d had poor pharmacokinetic properties in mice, with only 1/3 the total exposure of 1 following an IP dose of 10 mg/kg body weight. Finally, the pyrimidine scaffold allowed us to more fully probe the requirements for activity. Removing the – NH2 from 44b to provide 48 was accompanied by an approximately 20-fold loss in potency against the enzyme. Replacement of the 4-phenyl ring with a saturated heterocycle was further accompanied by a >10-fold loss in potency (51).
Table 5.
Pyrimidine Inhibitors of 15-PGDH
| |||||
|---|---|---|---|---|---|
| Compound | R/inhibitor | IC50 (nM) | PGE2 increase at | Aq. Solubility (μg/mL)b | |
| 20 nMa | 2.5 μMa | ||||
| 44a |
|
2.4 | 1.8 | 2.5 | 0.3 |
| 44b | Ph | 4.9 | |||
| (+)-44c |
|
1.0 | 62.1 | 3.1 | 2.2c |
| 44d |
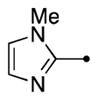
|
3.7 | 2.2 | 2.9 | 71 |
| 44e |
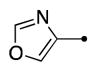
|
2.6 | 1.6 | 2.1 | 37 |
| 48 |
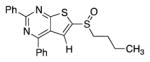
|
93 | |||
| 51 |
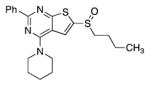
|
1320 | |||
Fold-increase in PGE2 levels in A549 cell culture media relative to DMSO. Data color-coded to indicate more active (blue), less active (red) or equally active (white) as compound 1.
Amorphous solid unless otherwise indicated. Solubility in pH 7 citrate buffer, 0.1 M. pH 7 citrate buffer, 0.1 M.
Solubility of racemate.
Based on the observations described above, we synthesized several derivatives that incorporated multiple structural changes from our previously reported 15-PGDH inhibitor 1 (Scheme 6). Addition of a methyl group to the thiazole ring of 12q provided 12v and only slightly decreased in vitro and cell-based activity. Similarly, introducing an ether into the sulfoxide side chain in (+)-12w or 12x maintained good activity against 15-PGDH and in cells. Moreover, this change dramatically improved solubility to >1 mg/mL at pH 7 for (+)-12w and increased the in vitro metabolic stability relative to 12q: Both 12w and 12x had half-lives greater than 4 h in the presence of mouse liver S9 fractions whereas the half-life of (+)-12q, featuring an n-butyl side chain, was 35 min under identical conditions. The increased metabolic stability of the ether side chain relative to the alkyl side chain could indicate that oxidation of the n-butyl group is a dominant mode of metabolism. Alternatively, the ether moiety could slow oxidation of the sulfoxide to a sulfone through inductive effects. As we observed with 1, the sulfone version of 12q was about 30-fold less active against 15-PGDH (14b). Finally, replacing the fused thiophene ring of 12j with either a thiazole (53) or imidazole (57) returned an inhibitor that was >1000X-fold less active against the enzyme.
Scheme 6. Additional 15-PGDH inhibitors.

aFold-increase in PGE2 levels in A549 cell culture media relative to DMSO. bSolubility of racemate
The pharmacokinetic properties of selected inhibitors of 15-PGDH were evaluated to identify compounds suitable for in vivo experiments (Table 1). The active enantiomer of our initial lead compound, (+)-1, was rapidly metabolized in the presence of mouse liver S9 fractions and highly protein bound. Indeed, we observed more rapid metabolism of the (+)-enantiomers with several inhibitors. For example, the half-lives for the (R)-(+)-12q and its enantiomer were 35 and 289 minutes, respectively. However, in human liver S9 fractions, the more active R-enantiomer proved to be at least as stable as the less active S-enantiomer. All of the compounds shown in Table 6 achieved Cmax values in excess of the in vitro IC90 values. Since these sulfoxides function as tight-binding inhibitors, we anticipated that they would provide substantial inhibition of 15-PGDH in vivo. Unfortunately, the excellent in vitro stability of methyl ether 12w did not translate into a longer in vivo half-life compared to (+)-1, potentially because of the very low plasma protein binding.
Table 6.
ADME properties of selected 15-PGDH inhibitors
| Compound: | (+)-1 | (R)-12q | (S)-12q | 12w | 32 | 44c |
|---|---|---|---|---|---|---|
| In Vitro | ||||||
| mouse S9 (t1/2, min) | 13 (83)a | 35 | 289 | >240 | 83 | 91 |
| human S9 (t1/2, min) | >240 | 224 | ||||
| plasma protein binding (mouse, %) | 99.98a | 78 | 56 | 99 | 90 | |
| In Vivo (mouse, 10 mg/kg IP) | ||||||
| Cmax (μM) | 1.6 (1.5)a | 3.1 | 6.2 | 13.0 | 10.6 | |
| C3h (μM)b | 0.13 (0.26)a | <0.01 | 0.06 | 4.2 | 0.06 | |
| AUC (min*μg/mL) | 43 (83)a | 47 | 116 | 959 | 172 | |
Data from racemic compound.
Plasma concentration 3h after dose.
Four of the most encouraging 15-PGDH inhibitors were evaluated in vivo (Figure 4). Mice were treated with a single dose of compound (2.5 mg/kg IP), and tissues were harvested at various time points and analyzed for PGE2 levels with an ELISA assay. Compared to untreated controls (t = 0 h), the inhibitors (+)-1, (+)-12q, and (+)-44c doubled the PGE2 levels within the bone marrow within 3 h of treatment. Levels decreased by 6 h before returning to baseline by 12h. By contrast, (+)-32 was less effective than the other 3 analogs under these conditions. Several aspects of these results are noteworthy. First, we observed similar 2-fold elevation of PGE2 levels in the colon, lung and liver (see Figure S1). Second, this magnitude of PGE2 elevation appears maximal, as tissue PGE2 levels are consistently 2-fold higher in the 15-PGDH knockout mouse compared to wild-type littermates. Third, the pharmacological effects of (+)-1, (+)-12q and (+)-44c last much longer than the pharmacokinetic half-lives might predict. For example, plasma levels of (+)-12q drop below 10 nM within 3 h after an IP dose of 2.5 mg/kg body weight. Nonetheless, tissue PGE2 levels remain elevated at 3h. Since (+)-12q is a tight binding inhibitor of 15-PGDH (Kiapp=0.06 nM), we suspect that it remains bound to the enzyme even after being cleared from the plasma. The levels of PGE2 continue to increase while the 15-PGDH is substantially inhibited, and likely return to normal as more enzyme is synthesized. In an important control experiment, the less active enantiomer (−)-12q had no impact on tissue PGE2 levels (see supporting information). Finally, while ethylene glycol 32 potently inhibited 15-PGDH in vitro and in cells and had excellent exposure and stability in vivo, it only elevated PGE2 levels modestly in vivo.
Figure 4.
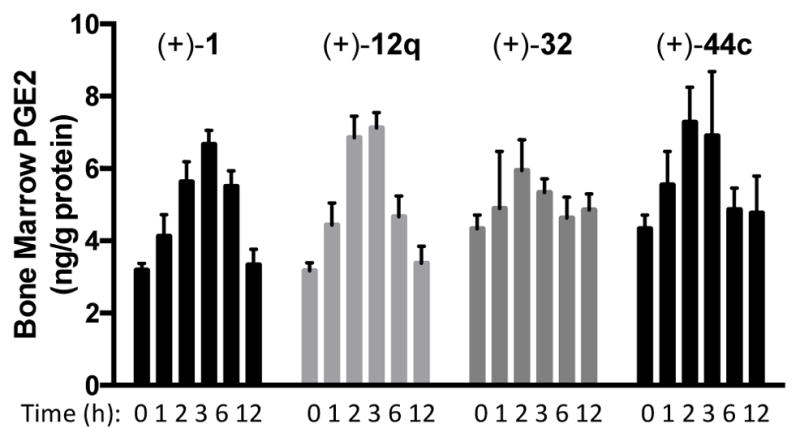
Elevation of PGE2 levels in bone marrow following a single IP dose of 2.5 mg/kg 15-PGDH inhibitor. C57BL/6J mice, n=3 per time point.
Inhibitor (+)-12q displayed IC50 values >10 μM against important cytochrome P450 enzymes (CYP 1A, 2B6, 2C8, 2C9, 2C19, 2D6, 3A). The Safety44 panel of receptors, enzymes and channels (Cerep) indicated significant binding only to the μ-opioid channel and the adenosine A2A receptor at 10 μM. Functional assays showed no activation or inhibition of the μ-opioid channel up to 10 μM and an inconsequential IC50 of 900 nM against the A2A receptor. Finally, mice treated with 25 mg/kg (+)-12q twice daily for 21 days, which is at least 10-fold higher than the dose required for maximal elevation of PGE2 levels, showed no ill effect on behavior, weight, blood count, blood chemistry or initial liver pathology.
CONCLUSION
Taken together, these results reveal that heterocyclic sulfoxides are potent inhibitors of 15-PGDH. Among these inhibitors, sulfoxides provided the most potent activity, and non-polar side chains outperformed polar groups. The 2-thienyl ring in 1 could be replaced with other aromatic and heteroaromatic rings, but removal proved detrimental. By contrast, the phenyl substituent from compound 1 could be removed or modified while retaining in vitro activity, although metabolic stability was improved with an aryl or heteroaryl ring at this position. Finally, the central ring system of the inhibitors could feature either a thienopyridine or thienopyimidine while retaining high potency.
Our results indicate that potent inhibition of 15-PGDH in vitro is necessary but not sufficient for inhibition of the enzyme in cells. Although we do not fully understand all the factors involved, cell penetration is likely a key factor in determining cellular activity. In this study, systematic modification of our initial inhibitor led to the discovery of two compounds with improved solubility and drug-like properties, (+)-12q and (+)-44c. These inhibitors retain robust in vivo activity as evidenced by their ability to elevate PGE2 levels in multiple tissues. They appear to represent attractive lead compounds towards the development of inhibitors of 15-PGDH for use in accelerating recovery following bone marrow transplant and in tissue regeneration more broadly.
EXPERIMENTAL SECTION
General
All tested compounds have purity of >95% as judged by HPLC analysis (UV detection at 210 nM). Compounds indicated as optically active were >97% ee as judged by HPLC using the Chiralpak ADH or Chiralcel ODH analytical HPLC columns. Chemical shifts δ are in ppm, and spectra were referenced using the residual solvent peak. The following abbreviations are used: singlet (s), doublet (d), triplet (t), quartet (q), double doublet (dd), quintet (quin), multiplet (m), broad signal (br s). “Mass spectra (m/z) were recorded on an Agilent LC/MS 1100 or 1290 Infinity using ESI ionization. All chemicals were used as received unless otherwise noted.
Synthetic methods. General procedure for synthesis of 2-sulfinyl-thieno[2,3-b]pyridine-3-amines as exemplified by the synthesis of 2-(butylsulfinyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (1)
Step 1
3-phenyl-1-(thiophen-2-yl)prop-2-en-1-one was prepared from benzaldehyde and 1-(thiophen-2-yl)ethanone via aldol condensation using procedure described by Azam.22 1H NMR (400 MHz, CDCl3) δ 7.88 – 7.80 (m, 2H), 7.67 (dd, J = 4.9, 1.1 Hz, 1H), 7.66 – 7.59 (m, 2H), 7.47 – 7.34 (m, 4H), 7.18 (dd, J = 5.0, 3.8 Hz, 1H). ESI-MS (m/z): 215.1 [M+H]+.
Step 2
To a solution of 3-phenyl-1-(thiophen-2-yl)prop-2-en-1-one (2.34 mmol, 500 mg) and cyanothioacetamide (7.0 mmol, 717 mg, 3.0 equiv) in ethanol (7 mL), a few drops of piperidine were added. The reaction was stirred at reflux for 3 h. The solid that formed was collected, suspended in acetic acid and heated at 80 °C. After 30 min of heating, the mixture was cooled to room temperature and filtered to give 4-phenyl-6-(thiophen-2-yl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile in 46% isolated yield. 1H NMR (400 MHz, DMSO-d6) δ 8.17 (d, J = 3.8 Hz, 1H), 7.96 (d, J = 5.0 Hz, 1H), 7.74 – 7.62 (m, 2H), 7.54 (dd, J = 5.1, 2.0 Hz, 3H), 7.31 – 7.19 (m, 1H), 7.01 (s, 1H). ESI-MS (m/z): 295.0 [M+H]+.
Step 3
A mixture of the thiopyridone from the previous step (0.34 mmol, 101 mg), butyl(chloromethyl)sulfane (0.34 mmol, 48 mg, 1.0 equiv.) and Et3N (0.51 mmol, 0.07 mL, 1.5 equiv) was stirred at reflux in dry CH3CN (0.35 mL) for 20 min. The reaction mixture was then diluted with EtOAc and water. The organic phase was separated and the aqueous layer was extracted twice with EtOAc. The combined organic phases were washed with saturated NaCl, dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography to give 124 mg of 2-(((butylthio)methyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile (92%). 1H NMR (400 MHz, CDCl3) δ 7.70 (dd, J = 3.8, 1.1 Hz, 1H), 7.64 – 7.56 (m, 1H), 7.55 – 7.47 (m, 5H), 7.40 (d, J = 1.1 Hz, 1H), 7.14 (dd, J = 5.0, 3.8 Hz, 1H), 4.53 (s, 2H), 2.74 (t, J = 8.0 Hz, 2H), 1.72 – 1.57 (m, 2H), 1.49 – 1.34 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 163.0, 154.4, 153.7, 143.2, 135.9, 130.5, 130.1, 129.0, 128.6, 128.3, 127.2, 115.6, 114.1, 114.1, 103.2, 34.5, 32.1, 31.3, 22.0, 13.7. ESI-MS (m/z): 397.0 [M+H]+.
Step 4
Acetic Acid (0.90 mL) and hydrogen peroxide (0.57 mmol, 1.5 equiv, 30% solution in water) were added to the solution of 2-(((butylthio)methyl)sulfinyl)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile (0.38 mmol, 150 mg) in chloroform (0.90 mL). The reaction mixture was stirring at 32 °C for 45 min. The reaction was then diluted with EtOAc and washed with saturated NaHCO3 solution, dried over magnesium sulfate, filtered and concentrated under reduced pressure to give 153 mg of 2-(((butylthio)methyl)sulfinyl)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile (98%). 1H NMR (400 MHz, CDCl3) δ 7.75 (dd, J = 3.8, 1.1 Hz, 1H), 7.66 – 7.57 (m, 2H), 7.58 – 7.51 (m, 4H), 7.47 (s, 1H), 7.16 (dd, J = 5.0, 3.8 Hz, 1H), 4.74 (d, J = 13.0 Hz, 1H), 4.41 (d, J = 13.0 Hz, 1H), 2.97 (dt, J = 13.0, 8.2 Hz, 1H), 2.81 (dt, J = 12.9, 7.3 Hz, 1H), 1.94 – 1.76 (m, 2H), 1.53 – 1.38 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.9, 154.7, 154.0, 142.4, 135.4, 131.0, 130.4, 129.1, 128.8, 128.3, 127.8, 115.1, 115.0, 110.0, 103.1, 51.5, 490, 24.5, 22.5, 13.7. ESI-MS (m/z): 413.1 [M+H]+.
Step 5
According to a procedure described by Kalugin,23 To the solution of 2-(((butylsulfinyl)methyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile (0.53 mmol, 220 mg) in DMF 2.10 mL/MeOH 1.05 mL was added KOH (0.32 mmol, 18 mg, 0.6 equiv, 1.7 M in water). The reaction mixture was stirred at 35 °C for 30 min. Once complete, the reaction was diluted with EtOAc and washed with 10 % aq. solution of AcOH, the organic phase was separated and the aqueous layer was extracted twice with EtOAc, dried over magnesium sulfate, filtered and concentrated under reduced pressure to give 211 mg of 2-(butylsulfinyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (1) (96 %). 1H NMR (400 MHz, CDCl3) δ 7.8 – 7.29 (m, 7H), 7.32 (s, 1H), 7.10 (dd, J = 5.0, 3.7 Hz, 1H), 4.54 (s, 2H), 3.26 (ddd, J = 12.8, 9.1, 6.0 Hz, 1H), 3.09 (ddd, J = 12.8, 9.1, 6.6 Hz, 1H), 1.83 – 1.61 (m, 2H), 1.53 – 1.38 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.4, 150.9, 146.6, 143.9, 143.6, 136.6, 129.3, 129.1, 128.8, 128.6, 128.4, 128.3, 126.0, 120.8, 117.1, 108.4, 55.3, 25.3, 21.9, 13.73. ESI-MS (m/z): 413.1 [M+H]+. Enantiomers of 1 were separated on Chiralpak IC column using EtOH/Et2NH 1000/1 with 0.5 mL/min flow rate. The 1st peak was at 12.6 min and the 2nd peak was at 14.3 min. Optical Rotation: Peak 1: [α]D23 −90.04 (c= 0.22, EtOH), Peak 2: [α]D23 +104.18 (c= 0.22, EtOH). The absolute stereochemistry was assigned by X-ray crystallography. See Figure S3.
11-(3-Amino-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-2-yl)pentan-1-one (10a)
Step 1
A mixture of 4-phenyl-6-(thiophen-2-yl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile (0.068 mmol, 20 mg), Et3N (0.11 mmol, 15 μL, 1.6 equiv) and 2-butyloxirane (0.11 mmol, 11 mg, 1.6 equiv) in MeOH (500 μL) was stirred at rt. When the reaction was completed as judged by TLC, the reaction mixture was evaporated; the crude product was dissolved in CH2Cl2 and the Dess-Martin periodinane (0.10 mmol, 1.5 equiv) was added at 0 °C. The reaction mixture was stirred at rt for 2 h and then was quenched by addition of 1:1 mixture of 20% Na2S2O3/NaHCO3 solution. The organic layer was separated, dried over magnesium sulfate and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography to afford 2-((2-oxohexyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile in 72% yield. 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.97 (d, J = 3.1 Hz, 1H), 7.71 – 7.59 (m, 2H), 7.55 (d, J = 3.2 Hz, 1H), 7.55 – 7.46 (m, 4H), 4.52 (s, 2H), 2.75 (t, J = 7.8 Hz, 2H), 1.73 – 1.54 (m, 2H), 1.51 – 1.26 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 393.1 [M+H]+.
Step 2
To the solution of the ketone from step 1 (0.13 mmol, 50 mg) in ethanol (0.5 mL) was added KOH (0.13 mmol, 2 mg, 1.0 equiv). The reaction mixture was stirred at 50 °C for 30 min. Once complete, the reaction was diluted with EtOAc and washed with 10% aq. HCl. The organic phase was separated and the aqueous layer was extracted twice with EtOAc, dried over magnesium sulfate, filtered and concentrated under reduced pressure to afford ketone 10a in 98% yield. 1H NMR (400 MHz, CDCl3) δ 7.65 (dd, J = 3.8, 1.1 Hz, 1H), 7.62 – 7.56 (m, 2H), 7.55 – 7.48 (m, 4H), 7.40 (s, 1H), 7.13 (dd, J = 5.0, 3.8 Hz, 1H), 4.13 (s, 2H), 2.72 (t, J = 7.4 Hz, 2H), 1.72 – 1.56 (m, 2H), 1.42 – 1.25 (m, 2H), 0.88 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 393.1 [M+H]+.
3-Amino-4-phenyl-N-propyl-6-(thiophen-2-yl)thieno[2,3-b]pyridine-2-carboxamide (10b)
A mixture of 4-phenyl-6-(thiophen-2-yl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile (0.12 mmol, 35 mg), 2-chloro-N-propylacetamide (0.12 mmol, 16 mg, 1.0 equiv) and EtONa (0.19 mmol, 1.6 equiv) in EtOH (1 mL) was stirred at 50 °C. When the reaction was complete as judged by TLC, the reaction was diluted with EtOAc and washed with 10 % aq. HCl. The organic phase was separated and the aqueous layer was extracted twice with EtOAc, dried over magnesium sulfate, filtered and concentrated under reduced pressure to afford 10b in 61 % yield. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (dd, J = 3.8, 1.2 Hz, 1H), 7.77 (t, J = 5.7 Hz, 1H), 7.73 (s, 1H), 7.70 (dd, J = 5.1, 1.1 Hz, 1H), 7.63 – 7.47 (m, 5H), 7.16 (dd, J = 5.1, 3.7 Hz, 1H), 5.80 (s, 2H), 3.12 (q, J = 6.7 Hz, 2H), 1.47 (h, J = 7.4 Hz, 2H), 0.83 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 165.4, 159.3, 159.2, 151.7, 147.4, 145.8, 143.9, 136.8, 129.1, 128.7, 128.6, 128.5, 128.3, 126.0, 121.5, 117.0, 41.3, 23.1, 11.7. ESI-MS (m/z): 394.1 [M+H]+
Ethyl 3-amino-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridine-2-carboxylate (10c)
A mixture of 4-phenyl-6-(thiophen-2-yl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile (0.34 mmol, 100 mg), ethyl 2-chloroacetate (0.54 mmol, 1.6 equiv) and EtONa (0.54 mmol, 1.6 equiv) in ethanol (1mL) was stirred at reflux. When the reaction was complete as judged by TLC, the reaction was diluted with EtOAc and washed with 10% aq. HCl. The organic phase was separated and aqueous layer was extracted twice with EtOAc, dried over magnesium sulfate, filtered and concentrated under reduced pressure to 10c in 79 % yield. 1H NMR (400 MHz, DMSO-d6) δ 8.01 (d, J = 3.7 Hz, 1H), 7.87 – 7.67 (m, 2H), 7.56 (d, J = 6.5 Hz, 5H), 7.36 – 6.90 (m, 1H), 5.73 (s, 2H), 4.23 (q, J = 7.1 Hz, 2H), 1.25 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 165.3, 161.6, 161.4, 152.2, 147.6, 147.6, 143.9, 136.8, 129.2, 128.9, 128.8, 128.5, 128.3, 126.2, 120.5, 116.6, 60.50, 14.27. ESI-MS (m/z): 381.1[M+H]+
3-Amino-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridine-2-carboxylic acid (10d)
To a solution of 4-phenyl-6-(thiophen-2-yl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile (0.34 mmol, 100 mg) and ethyl 2-chloroacetate (0.54 mmol, 1.6 equiv) in ethanol (1 mL), Et3N (0.54 mmol, 1.6 equiv.) was added. The reaction was refluxed for 20 min. The reaction was then diluted with EtOAc and water. The organic phase was separated and aqueous layer was extracted twice with EtOAc. The combined extractions were washed with saturated NaCl solution, dried over magnesium sulfate, filtered and concentrated under reduced pressure to afford ethyl 2-((3-cyano-4-phenyl-6-(thiophen-2-yl)pyridin-2-yl)thio)acetate, which was then dissolved in DMF and treated with 1M aq. NaOH at 50 °C to give 10d in 63% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.00 (dd, J = 3.7, 1.1 Hz, 1H), 7.79 – 7.64 (m, 2H), 7.55 (dt, J = 7.6, 3.2 Hz, 5H), 7.16 (dd, J = 5.0, 3.7 Hz, 1H), 5.72 (s, 2H). ESI-MS (m/z): 353.1[M+H]+.
2-(Methylsulfinyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (12a) was prepared using the synthetic procedures described for the preparation of 1. 1H NMR (500 MHz, CDCl3) δ 7.67 – 7.50 (m, 5H), 7.50 – 7.36 (m, 3H), 7.16 – 7.09 (m, 1H), 4.58 (s, 2H), 2.99 (s, 3H). ESI-MS (m/z): 371.1 [M+H]+.
4-Phenyl-2-(propylsulfinyl)-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (12b) was prepared using synthetic procedures described for the preparation of analog 1. 1H NMR (400 MHz, CDCl3) δ 7.65 (dd, J = 3.8, 1.1 Hz, 1H), 7.61 - 7.49 (m, 4H), 7.49 - 7.41 (m, 3H), 7.12 (dd, J = 5.0, 3.7 Hz, 1H), 3.28 (ddd, J = 12.7, 8.4, 6.3 Hz, 1H), 3.07 (ddd, J = 12.7, 8.6, 7.0 Hz, 1H), 1.91 - 1.65 (m, 2H), 1.08 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 161.5, 150.9, 146.8, 143.8, 143.3, 136.8, 129.2, 128.8, 128.7, 128.6, 128.3, 126.1, 121.3, 117.1, 107.9, 57.1, 26.2, 16.8, 13.1. APCI-MS (m/z): 399.1 [M+H]+.
4-Phenyl-2-(isopropylsulfinyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (12c). was prepared using synthetic procedures described for the preparation of analog 1. 1H NMR (400 MHz, CDCl3) δ 7.64 (dd, J = 3.7, 1.1 Hz, 1H), 7.58 - 7.47 (m, 5H), 7.47 -7.39 (m, 2H), 7.10 (dd, J = 5.0, 3.7 Hz, 1H), 4.59 (s, 2H), 3.38 (p, J = 6.8 Hz, 1H), 1.43 (d, J = 6.9 Hz, 3H), 1.25 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 161.8, 150.8, 146.7, 143.8, 143.7, 136.8, 129.2, 128.8, 128.7, 128.6, 128.3, 126.0, 121.5, 117.0, 105.9, 55.0, 15.9, 15.5. ESI-MS (m/z): 399.1 [M+H]+.
2-(Pentylsulfinyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (12d)
Hydrogen chloride gas was bubbled through 1-pentanethiol (10 ml, 8.1 mmol) cooled in a dry ice/acetone bath. At a stabilized internal temperature of −74.2 °C, paraformaldehyde (3.02 g, 10.1 mmol) was added slowly via a solid addition funnel. Minimal methylene chloride was used to rinse the clumped paraformaldehyde in the funnel. Hydrogen chloride gas was bubbled for 3 hours into the cold stirring reaction, and then the reaction mixture was allowed to warm to ambient temperature overnight. The aqueous layer was removed and the organic layer was washed with brine, dried over Na2SO4, filtered and condensed to give (chloromethyl)(pentyl)sulfane with less than 2 % dimer according to 1H NMR analysis. The crude product was used without purification. 1H NMR (500 MHz, CDCl3) δ 4.76 (d, J = 1.6 Hz, 2H), 2.75 (t, J = 7.5 Hz, 2H), 1.67 (p, J = 7.3 Hz, 2H), 1.47 – 1.26 (m, 4H), 0.92 (dd, J = 7.7, 6.2 Hz, 3H). Pentyl sulfoxide 12d was prepared using (chloromethyl)(pentyl)sulfane and the synthetic procedures described for the preparation of analog 1. 1H NMR (500 MHz, CD2Cl2) δ 7.98 – 7.36 (m, 8H), 7.33 – 6.85 (m, 1H), 4.47 (s, 2H), 3.28 – 3.15 (m, 1H), 3.09 – 2.99 (m, 1H), 1.81 – 1.59 (m, 2H), 1.50 – 1.25 (m, 4H), 0.88 (t, J = 7.2 Hz, 3H). ESI-MS (m/z): 427.1 [M+H]+.
2-(Hexylsulfinyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (12e)
(Chloromethyl)(hexyl)sulfane (1H NMR (400 MHz, CDCl3,) δ 4.76 (s, 2H), 2.76 (dd, J = 7.9, 6.9 Hz, 2H), 1.73 – 1.59 (m, 2H), 1.51 – 1.20 (m, 6H), 0.97 – 0.82 (m, 3H)) was prepared analogously to (chloromethyl)(pentyl)sulfane. It was used in the synthesis of 12e using synthetic procedures described for the preparation of analog 1. 1H NMR (500 MHz, CD2Cl2) δ 7.78 – 7.66 (m, 1H), 7.63 – 7.46 (m, 7H), 7.27 – 7.02 (m, 1H), 4.11 (s, 2H), 3.43 – 3.20 (m, 1H), 3.11 (ddd, J = 13.8, 9.4, 6.4 Hz, 1H), 1.89 – 1.63 (m, 2H), 1.58 – 1.39 (m, 4H), 1.40 – 1.21 (m, 2H), 0.91 (d, J = 6.8 Hz, 3H). ESI-MS (m/z): 441.1 [M+H]+.
2-((3-Methoxypropyl)sulfinyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (12f)
Step 1
4-phenyl-6-(thiophen-2-yl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile was alkylated with 4-((chloromethyl)thio)butyl acetate as described for the synthesis of 1. The crude mixture was condensed and purified by flash chromatography over SiO2 (0–30% EtOAc/hexanes to give 3-((((3-cyano-4-phenyl-6-(thiophen-2-yl)pyridin-2-yl)thio)methyl)thio)propyl acetate in 26% yield. 1H NMR (400 MHz, CHCl3) δ 7.72 (dd, J = 3.8, 1.1 Hz, 1H), 7.66 – 7.58 (m, 2H), 7.54 (dd, J = 4.2, 2.9 Hz, 4H), 7.44 (d, J = 1.3 Hz, 1H), 7.17 (dd, J = 5.0, 3.8 Hz, 1H), 4.55 (s, 2H), 4.18 (t, J = 6.3 Hz, 2H), 2.84 (t, J = 7.3 Hz, 2H), 2.05 (s, 3H), 2.05 – 1.97 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 171.2, 162.3, 154.7, 153.9, 143.3, 136.0, 130.7, 130.3, 129.2, 128.7, 128.5, 127.4, 115.7, 114.4, 103.5, 63.1, 34.5, 28.9, 28.4, 21.1. ESI-MS (m/z): 441.0 [M+H]+.
Step 2
K2CO3 (152 mg, 1.10 mmol) was added to a solution of the acetate from step 1 (240.5 mg, 0.55 mmol) in methanol (4.0 ml) and water (1.0 ml), and the reaction was stirred for 2.5 hours. The mixture was dried then diluted with EtOAc and washed twice with water and then brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduce pressure to give 2-((((3-hydroxypropyl)thio)methyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile in 98% yield, which was used without further purification. 1H NMR (400 MHz, CHCl3) δ 7.70 (dd, J = 3.8, 1.1 Hz, 1H), 7.63 – 7.58 (m, 2H), 7.53 (dd, J = 5.3, 1.7 Hz, 4H), 7.41 (s, 1H), 7.15 (dd, J = 5.0, 3.8 Hz, 1H), 4.54 (s, 2H), 3.76 (t, J = 6.0 Hz, 2H), 2.88 (t, J = 7.1 Hz, 2H), 1.93 (ddd, J = 13.2, 7.2, 6.1 Hz, 2H), 1.86 – 1.81 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 162.8, 154.5, 153.8, 143.2, 135.9, 130.6, 130.2, 129.1, 128.7, 128.4, 127.3, 115.7, 114.3, 103.3, 61.5, 34.6, 31.8, 29.1 ESI-MS (m/z): 399.1 [M+H]+.
Step 3
A solution of the alcohol from step 2 (41.6 mg, 0.10 mmol) in DMF (1.0 ml) was cooled in an ice bath before NaH (microspatula tip full) was added. The suspension was stirred at room temperature for 15 minutes before the addition of MeI (34 μl, 0.55 mmol). The reaction was stirred overnight, then diluted with EtOAc and washed several times with water and then brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduce pressure. Purification by flash chromatography on SiO2 (0–60% EtOAc/hexanes) provided 2-((((3-methoxypropyl)thio)methyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile in 56% yield. 1H NMR (400 MHz, CHCl3) δ 7.72 (dd, J = 3.8, 1.1 Hz, 1H), 7.66 – 7.59 (m, 2H), 7.54 (h, J = 4.0, 3.6 Hz, 4H), 7.43 (s, 1H), 7.17 (dd, J = 5.0, 3.8 Hz, 1H), 4.55 (s, 2H), 3.49 (t, J = 6.1 Hz, 2H), 3.34 (s, 3H), 2.85 (t, J = 7.3 Hz, 2H), 2.02 – 1.88 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 163.0, 154.6, 153.9, 143.3, 130.6, 130.3, 129.2, 128.7, 128.4, 127.3, 115.7, 114.3, 103.4, 71.1, 58.8, 34.7, 29.5, 29.3. ESI-MS (m/z): 413.1 [M+H]+.
Step 4
The sulfide from step 3 was oxidized with H2O2 and cyclized with KOtBu as described for the synthesis of 1 to provide 12f. 1H NMR (400 MHz, CHCl3) δ 7.64 (ddd, J = 7.2, 3.8, 1.7 Hz, 2H), 7.58 – 7.52 (m, 4H), 7.48 – 7.42 (m, 2H), 7.12 (qd, J = 3.7, 1.8 Hz, 1H), 4.57 (s, 2H), 3.50 (td, J = 6.1, 1.6 Hz, 2H), 3.40 – 3.29 (m, 4H), 3.26 – 3.13 (m, 1H), 2.09 – 1.95 (m, 2H). ESI-MS (m/z): 429.1 [M+H]+.
2-(Butylsulfinyl)-4-phenyl-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12g)
2-bromo-1-(thiazol-2-yl)ethan-1-one24 was converted to the corresponding Wittig reagent25 and then used to prepare thiazole 12g using procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.92 (d, J = 3.2 Hz, 1H), 7.65 – 7.39 (m, 6H), 4.63 (s, 2H), 3.28 (ddd, J = 12.8, 9.0, 6.2 Hz, 1H), 3.11 (ddd, J = 12.8, 9.0, 6.8 Hz, 1H), 1.85 – 1.63 (m, 2H), 1.56 – 1.42 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 168.1, 161.2, 149.6, 147.3, 144.3, 142.9, 136.5, 129.3, 128.7, 128.6, 123.5, 122.2, 117.6, 108.9, 54.8, 25.0, 21.9, 13.5. ESI-MS (m/z): 414.1 [M+H]+.
2-(Butylsulfinyl)-6-(oxazol-2-yl)-4-phenylthieno[2,3-b]pyridin-3-amine (12h)
(E)-1-(Oxazol-2-yl)-3-phenylprop-2-en-1-one26 was used in the synthesis of oxazole 12h using procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.84 (d, J = 0.8 Hz, 1H), 7.58 - 7.41 (m, 5H), 7.33 (d, J = 0.8 Hz, 1H), 4.65 (s, 2H), 3.30 (ddd, J = 12.9, 8.8, 6.2 Hz, 1H), 3.10 (ddd, J = 12.8, 8.9, 6.9 Hz, 1H), 1.86 - 1.64 (m, 2H), 1.42 – 1.54 (m, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.9, 160.2, 147.24, 143.8, 142.8, 140.3, 136.1, 129.5, 129.2, 128.8, 128.6, 123.5, 119.6, 109.1, 54.5, 25.0, 21.9, 13.7. ESI-MS (m/z): 398.1 [M+H]+.
2-(Butylsulfinyl)-6-(1-methyl-1H-imidazol-2-yl)-4-phenylthieno[2,3-b]pyridin-3-amine (12i)
(E)-1-(1-Methyl-1H-imidazol-2-yl)-3-phenylprop-2-en-1-one27 was used in the synthesis of imidazole 12i using procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.58 – 7.32 (m, 5H), 7.11 (d, J = 1.1 Hz, 1H), 7.00 (d, J = 1.1 Hz, 1H), 4.58 (s, 2H), 4.19 (s, 3H), 3.27 (ddd, J = 12.7, 9.0, 6.0 Hz, 1H), 3.08 (ddd, J = 12.8, 9.1, 6.6 Hz, 1H), 1.79 – 1.60 (m, 2H), 1.56 – 1.37 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 160.7, 148.7, 146.8, 143.8, 143.2, 136.6, 129.2, 128.7, 128.6, 125.2, 121.5, 120.4, 107.3, 54.6, 36.8, 25.1, 21.9, 13.7. ESI-MS (m/z): 411.1 [M+H]+.
2-(Butylsulfinyl)-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (12j)
6-(Thiophen-2-yl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile28 was used in the synthesis of the 4-unsubstituted analog 12j using procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 8.5, 1H), 7.65 – 7.49 (m, 2H), 7.39 (dt, J = 5.1, 0.7 Hz, 1H), 7.06 (dd, J = 5.0, 3.7, Hz, 1H), 5.20 (s, 2H), 3.26 (ddd, J = 12.8, 9.0, 6.2 Hz, 1H), 3.10 (ddd, J = 12.8, 9.1, 6.6 Hz, 1H), 1.78 – 1.60 (m, 2H), 1.55 – 1.39 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 337.1 [M+H]+.
Ethyl 3-amino-2-(propylsulfinyl)-6-(thiophen-2-yl)thieno[2,3-b]pyridine-4-carboxylate (12k)
Ethyl 2,4-dioxo-4-(thiophen-2-yl)butanoate29 was used in the synthesis of ester 12k using procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.69 (dd, J = 3.8, 1.1 Hz, 1H), 7.46 (dd, J = 5.0, 1.1 Hz, 1H), 7.11 (dd, J = 5.0, 3.8 Hz, 1H), 6.09 (s, 2H), 4.49 (q, J = 7.1 Hz, 2H), 3.36 – 3.22 (m, 1H), 3.15 – 3.00 (m, 1H), 1.87 – 1.68 (m, 2H), 1.47 (t, J = 7.1 Hz, 3H), 1.07 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.5, 163.4, 151.5, 143.5, 142.9, 135.1, 129.4, 128.5, 126.6, 121.1, 116.8, 109.2, 63.1, 56.6, 16.8, 14.1, 13.3. ESI-MS (m/z): 394.9 [M+H]+.
3-Amino-2-(propylsulfinyl)-6-(thiophen-2-yl)thieno[2,3-b]pyridine-4-carboxylic acid (12l)
To a solution of ester 12k (20 mg, 0.051 mmol) in THF:MeOH:H2O (0.11 mL: 0.11 mL: 0.036 mL) was added LiOH (3.6 mg, 0.15 mmol, 3 equiv) at rt for 3h to provide acid 12l in 40% yield following extraction into EtOAc and chromatographic purification with SiO2. 1H NMR (400 MHz, C3D7NO) δ 8.56 (s, 1H), 8.29 (d, J = 3.7 Hz, 1H), 8.19 (s, 1H), 7.99 (d, J = 5.0 Hz, 1H), 7.47 – 7.41 (m, 1H), 3.36 (ddd, J = 12.8, 8.4, 6.1 Hz, 1H), 3.24 (ddd, J = 12.8, 8.6, 6.8 Hz, 1H), 1.98 – 1.85 (m, 2H), 1.23 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMF) δ 168.8, 162.6, 151.5, 143.3, 142.3, 137.7, 130.2, 129.1, 127.9, 121.3, 116.9, 111.7, 57.3, 16.7, 12.9. ESI-MS (m/z): 366.8 [M+H]+.
3-Amino-2-(butylsulfinyl)-N,N-dimethyl-6-(thiophen-2-yl)thieno[2,3-b]pyridine-4-carboxamide (12m)
Step 1
Ethyl 2-(((butylthio)methyl)thio)-3-cyano-6-(thiophen-2-yl)isonicotinate was analogously to the corresponding intermediate thioether used in the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 7.87 (s, 1H), 7.75 (dd, J = 3.8, 1.1 Hz, 1H), 7.55 (dd, J = 5.1, 1.1 Hz, 1H), 7.16 (dd, J = 5.0, 3.8 Hz, 1H), 4.50 (q, J = 7.2 Hz, 2H), 4.47 (s, 2H), 2.72 (t, J = 7.2 Hz, 2H), 1.70 – 1.56 (m, 2H), 1.46 (t, J = 7.1 Hz, 3H), 1.43 – 1.33 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 393.1 [M+H]+
Step 2
The ester from step 1 was hydrolyzed to 2-(((butylthio)methyl)thio)-3-cyano-6-(thiophen-2-yl)isonicotinic acid in 94% isolated yield using conditions analogous to those described previously for compound 12l. 1H NMR (400 MHz, CDCl3) δ 10.65 (s, 1H), 7.95 (s, 1H), 7.76 (dd, J = 3.8, 1.1 Hz, 1H), 7.57 (dd, J = 5.0, 1.0 Hz, 1H), 7.17 (dd, J = 5.0, 3.7 Hz, 1H), 4.46 (s, 2H), 2.72 (t, J = 7.2 Hz, 2H), 1.67 – 1.55 (m, 2H), 1.40 (h, J = 7.4 Hz, 2H), 0.90 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.1, 164.7, 154.8, 142.3, 140.1, 131.7, 128.9, 128.4, 114.3, 114.1, 102.8, 34.8, 32.1, 31.21, 22.0, 13.7. ESI-MS (m/z): 365.0 [M+H]+.
Step 3
Dimethylamine hydrochloride (14.8 mg, 0.18 mmol, 1.1 equiv) was added to a solution of the acid from step 2 (60 mg, 0.16 mmol), HATU (0.18 mmol, 69 mg, 1.1 equiv), and DMF (0.425 mL followed by DIPEA (0.33 mmol, 0.057 mL, 2.0 equiv). The solution was stirred at room temperature for 3 hours, then diluted with EtOAc and washed with water. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to give 2-(((butylthio)methyl)thio)-3-cyano-N,N-dimethyl-6-(thiophen-2-yl)isonicotinamide in 53% yield following flash chromatography on silica gel (20% EtOAc/hexanes). 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 3.8 Hz, 1H), 7.54 (d, J = 5.0 Hz, 1H), 7.34 (s, 1H), 7.15 (dd, J = 4.8, 3.9, 1H), 4.49 (s, 2H), 3.16 (s, 3H), 2.98 (s, 3H), 2.72 (t, J = 7.2 Hz, 2H), 1.62 (p, J = 7.7 Hz, 2H), 1.41 (h, J = 7.3 Hz, 2H), 0.90 (t, J = 7.3, 3H). 13C NMR (101 MHz, CDCl3) δ 165.4, 162.7, 154.7, 149.4, 142.6, 131.2, 128.7, 127.9, 113.9, 111.2, 100.9, 38.3, 35.0, 34.5, 32.1, 31.2, 21.9, 13.6. ESI-MS (m/z): 392.1 [M+H]+.
Steps 4 and 5
The amide from step 3 was oxidized and cyclized using procedures described for the synthesis of compound 1 to provide 12m in 17% isolated yield over 2 steps. 1H NMR (400 MHz, CDCl3) δ 7.66 (dd, J = 3.8, 1.1 Hz, 1H), 7.52 – 7.42 (m, 2H), 7.13 (dd, J = 5.0, 3.7 Hz, 1H), 3.34 – 3.23 (m, 1H), 3.21 (s, 3H), 3.15 – 3.02 (m, 1H), 2.96 (s, 3H), 1.79 – 1.62 (m, 2H), 1.55 – 1.36 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.0, 162.4, 151.4, 143.3, 141.6, 139.1, 129.3, 128.3, 126.4, 120.6, 112.7, 110.4, 55.3, 39.5, 35.3, 24.9, 21.9, 13.7. ESI-MS (m/z): 408.1 [M+H]+.
2-(Butylsulfinyl)-4,6-di(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12n)
(E)-1,3-di(thiazol-2-yl)prop-2-en-1-one25 was converted to bis-thiazole 12n using procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.01 (d, J = 3.1 Hz, 1H), 7.96 (d, J = 3.2 Hz, 1H), 7.61 (d, J = 3.3 Hz, 1H), 7.52 (d, J = 3.1 Hz, 1H), 6.69 (s, 2H), 3.30 (ddd, J = 12.8, 9.2, 6.0 Hz, 1H), 3.14 (ddd, J = 12.8, 9.2, 6.4 Hz, 1H), 1.83 – 1.60 (m, 2H), 1.43 – 1.53 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.4, 166.0, 163.2, 149.8, 144.7, 144.3, 143.6, 138.0, 122.6, 122.5, 122.1, 117.5, 108.9, 54.5, 25.0, 21.9, 13.7. ESI-MS (m/z): 421.0 [M+H]+.
2-(Butylsulfinyl)-4-(pyridin-3-yl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12o)
Pyridine 12o was prepared using the procedures described for the preparation of analog 1. 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1 H), 8.78 (dd, J = 4.9, 1.7 Hz, 1H), 8.04 (s, 1H), 7.91 (d, J = 3.2 Hz, 1H), 7.87 – 7.85 (m, 1H), 7.51 (d, J = 3.1 Hz, 1H), 7.47 (dd, J = 7.8, 4.8 Hz, 1H), 4.53 (s, 2H), 3.28 (ddd, J = 12.8, 8.8, 6.3 Hz, 1H), 3.11 (ddd, J = 12.8, 8.9, 6.9 Hz, 1H), 1.86 – 1.70 (m, 2H), 1.57 – 1.38 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 415.0 [M+H]+.
2-(Butylsulfinyl)-4-(1-methyl-1H-imidazol-2-yl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12p)
Imidazole 12p was prepared using the procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.91 (d, J = 3.2 Hz, 1H), 7.50 (d, J = 3.1 Hz, 1H), 7.24 (d, J = 1.2 Hz, 1H), 7.13 (d, J = 1.2 Hz, 1H), 5.78 (s, 2H), 3.80 (s, 3H), 3.26 (ddd, J = 12.8, 9.1, 6.0 Hz, 1H), 3.10 (ddd, J = 12.8, 9.2, 6.5 Hz, 1H), 1.82 – 1.57 (m, 2H), 1.56 – 1.35 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.6, 162.6, 149.4, 144.3, 143.8, 142.8, 134.4, 128.8, 123.9, 123.9, 122.5, 117.1, 110.3, 54.9, 35.0, 24.9, 21.9, 13.7. ESI-MS (m/z): 418.1 [M+H]+.
2-(Butylsulfinyl)-4-(1,2-dimethyl-1H-imidazol-5-yl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12q)
Imidazole 12q was prepared using the procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.03 (s, 1H), 7.90 (d, J = 3.1 Hz, 1H), 7.50 (d, J = 3.2 Hz, 1H), 7.11 (s, 1H), 4.76 (s, 2H), 3.39 (s, 3H), 3.27 (ddd, J = 12.9, 8.7, 6.4 Hz, 1H), 3.09 (ddd, J = 12.8, 8.8, 6.9 Hz, 1H), 2.47 (s, 3H), 1.83 – 1.62 (m, 2H), 1.57 – 1.38 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.6, 161.4, 149.9, 147.6, 144.3, 142.8, 134.7, 128.1, 126.6, 124.3, 122.4, 110.0, 118.7, 54.9, 31.3, 24.9, 21.9, 13.7, 13.6. ESI-MS (m/z): 432.1 [M+H]+. Enantiomers of 12q were separated on a 1 cm Chiralpak AD column using 100% MeOH with 2.5 mL/min flow rate. The (−)-(S)-enantiomer eluted at 13.4 min and the (+)-(R)-enantiomer eluted at 19.9 min. Peak 1: [α]D23 −63.86 (c = 0.106, EtOH), Peak 2: [α]D23 +64.75 (c = 0.162, EtOH). The absolute stereochemistry was assigned by X-ray crystallography. See Figure S4.
2-(Butylsulfinyl)-4-(2-isopropyl-1-methyl-1H-imidazol-5-yl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12r)
Imidazole 12r was prepared using the procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.92 (d, J = 3.1 Hz, 1H), 7.51 (d, J = 3.2 Hz, 1H), 7.15 (s, 1H), 4.71 (s, 2H), 3.41 (s, 3H), 3.27 (ddd, J = 13.0, 8.5, 6.5 Hz, 1H), 3.19 – 2.98 (m, 2H), 1.83 – 1.59 (m, 2H), 1.58 – 1.41 (m, 2H), 1.39 (d, J = 6.7 Hz, 6H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.7, 161.4, 155.7, 149.9, 144.3, 142.8, 134.8, 127.9, 126.4, 124.6, 122.5, 118.8, 109.7, 45.8, 30.0, 26.5, 24.9, 21.9, 21.2, 13.7. ESI-MS (m/z): 460.1 [M+H]+.
2-(Butylsulfinyl)-4-(2-cyclopropyl-1-methyl-1H-imidazol-5-yl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12s)
Imidazole 12s was prepared using the procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.91 (d, J = 3.1 Hz, 1H), 7.50 (d, J = 3.1 Hz, 1H), 7.07 (s, 1H), 4.77 (s, 2H), 3.51 (s, 3H), 3.27 (ddd, J = 12.9, 8.7, 6.4 Hz, 1H), 3.10 (ddd, J = 12.9, 8.8, 6.9 Hz, 1H), 1.95 – 1.78 (m, 1H), 1.81 – 1.62 (m, 2H), 1.58 – 1.37 (m, 2H), 1.17 – 0.98 (m, 4H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.7, 161.5, 152.4, 149.9, 144.3, 142.7, 134.6, 127.8, 126.6, 124.4, 122.5, 118.8, 109.6, 54.8, 30.9, 24.9, 21.9, 13.7, 7.5, 3.6. ESI-MS (m/z): 458.1 [M+H]+.
2-(Butylsulfinyl)-4-(2-chloro-1-methyl-1H-imidazol-5-yl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12t)
Imidazole 12t was prepared using the procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CD2Cl2) δ 8.09 (s, 1H), 7.94 (d, J = 3.2 Hz, 1H), 7.56 (d, J = 3.2 Hz, 1H), 7.16 (s, 1H), 4.70 (s, 2H), 3.45 (s, 3H), 3.33 – 3.18 (m, 1H), 3.18 – 2.98 (m, 1H), 1.84 – 1.63 (m, 2H), 1.56 – 1.40 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 167.5, 161.4, 150.0, 144.4, 142.2, 135.1, 133.5, 128.7, 128.5, 124.5, 122.6, 118.9, 110.0, 55.2, 32.0, 24.8, 21.9, 13.5. ESI-MS (m/z): 453.1 [M+H]+.
2-(Butylsulfinyl)-4-(2-methyl-1H-imidazol-5-yl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12u)
Imidazole 12u was prepared using the procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 10.51 (s, 1H), 8.10 (s, 1H), 7.89 (d, J = 3.2 Hz, 1H), 7.46 (d, J = 3.2 Hz, 1H), 7.40 (s, 1H), 3.31 (ddd, J = 12.8, 9.3, 5.8 Hz, 1H), 3.15 (ddd, J = 12.8, 9.3, 6.2 Hz, 1H), 2.42 (s, 3H), 1.79 – 1.58 (m, 2H), 1.57 – 1.38 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 418.1 [M+H]+.
2-(Butylsulfinyl)-4-(1,2-dimethyl-1H-imidazol-5-yl)-6-(4-methylthiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12v)
Thiazole 12v was prepared using the procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CD2Cl2) δ 8.04 (s, 1H), 7.12 (s, 1H), 7.08 (s, 1H), 4.75 (s, 2H), 3.40 (s, 3H), 3.26 (ddd, J = 13.1, 8.9, 6.2 Hz, 1H), 3.11 (ddd, J = 12.9, 8.9, 6.5 Hz, 1H), 2.50 (s, 3H), 2.47 (s, 3H), 1.79 – 1.64 (m, 2H), 1.57 – 1.42 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 166.6, 161.4, 154.7, 150.0, 147.5, 142.7, 135.0, 128.0, 126.7, 124.4, 118.7, 117.2, 109.8, 55.1, 31.2, 24.9, 21.9, 17.0, 13.5, 13.3. ESI-MS (m/z): 446.1 [M+H]+.
4-(1,2-Dimethyl-1H-imidazol-5-yl)-2-((2-methoxyethyl) sulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12w)
Methyl ether 12w was prepared using the procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.92 (d, J = 3.2 Hz, 1H), 7.51 (d, J = 3.2 Hz, 1H), 7.11 (s, 1H), 4.73 (s, 2H), 3.88 – 3.82 (m, 1H), 3.75 – 3.62 (m, 1H), 3.57 (ddd, J = 13.1, 6.0, 3.9 Hz, 1H), 3.40 (s, 3H), 3.37 (s, 3H), 3.25 (ddd, J = 12.8, 8.0, 4.4 Hz, 1H), 2.48 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 167.6, 161.6, 150.0, 147.6, 144.3, 142.6, 134.8, 128.1, 126.6, 124.3, 122.5, 118.8, 110.0, 65.9, 59.1, 55.4, 31.3, 13.6. ESI-MS (m/z): 434.1 [M+H]+.
4-(1,2-Dimethyl-1H-imidazol-5-yl)-2-((3-methoxypropyl) sulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (12x)
Methyl ether 12x was prepared using the procedures described for the synthesis of compound 1. 1H NMR (400 MHz, Acetone-d6) δ 8.03 (s, 1H), 7.99 (d, J = 3.2 Hz, 1H), 7.82 (d, J = 3.2 Hz, 1H), 7.09 (s, 1H), 5.06 (s, 2H), 3.51 (s, 3H), 3.48 (t, J = 6.1 Hz, 2H), 3.26 (s, 3H), 3.26 – 3.18 (m, 1H), 3.18 – 3.12 (m, 1H), 2.43 (s, 3H), 2.00 – 1.89 (m, 2H). ESI-MS (m/z): 448.1 [M+H]+.
2-(Butylthio)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (13a)
A flask was charged with Zn dust (63 mg, 0.97 mmol, 8.0 equiv) and purged with nitrogen for 10 min. Dry THF (6 mL) was then added, the grey suspension was cooled to 0 °C, and TiCl4 (0.48 mmol, 91 mg, 4.0 equiv) was added. After 10 min of stirring at 0 °C sulfoxide 1 (50 mg, 0.12 mmol) in 2 mL of dry THF was added, and the reaction mixture was stirred at room temperature for 2h. The reaction was quenched with 5 mL of 3N NaOH and 5 mL of water. The aqueous layer was extracted witch EtOAc. The organic layer was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography. The purified product was found to slowly oxidize back to sulfoxide 1 upon standing exposed to air. 1H NMR (400 MHz, CD2Cl2) δ 7.63 (dd, J = 3.7, 1.2 Hz, 1H), 7.57 – 7.50 (m, 3H), 7.50 – 7.46 (m, 2H), 7.43 (s, 1H), 7.41 (dd, J = 5.0, 1.1 Hz, 1H), 7.10 (dd, J = 5.0, 3.7 Hz, 1H) 3.98 (s, 2H), 2.76 (t, J = 7.3 Hz, 2H), 1.66 – 1.57 (m, 2H), 1.42 (h, J = 7.3 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.5, 149.5, 145.2, 144.3, 141.8, 137.5, 128.9, 128.8, 128.5, 128.1, 128.0, 125.3, 121.5, 116.6, 104.1, 36.9, 31.8, 21.7, 13.7. ESI-MS (m/z): 397.1 [M+H]+.
2-(Butylsulfonyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (14a)
Acetic Acid (48.5 μL) and hydrogen peroxide (0.031 mmol, 31.5 μL, 50% w/w) were added to the solution of sulfoxide 1 (0.024 mmol, 10 mg) in chloroform (48.5 μL). The reaction mixture was stirred at 32 °C for 4 h. After cooling it was quenched with saturated bicarbonate solution, diluted with chloroform and washed with water, and dried over Na2SO4. The crude mixture was purified on a short pipette column in 1% MeOH/DCM to provide sulfone 14a in 23% yield. 1H NMR (400 MHz, CDCl3) δ 7.71 (dd, J = 3.8 1.2 Hz, 1H), 7.64 – 7.54 (m, 3H), 7.53 – 7.42 (m, 4H), 7.15 (dd, J = 5.0, 3.7 Hz, 1H), 5.09 (s, 2H), 3.38 – 3.02 (m, 2H), 1.92 – 1.67 (m, 2H), 1.52 – 1.28 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). ESI-MS (m/z): 428.8 [M+H]+.
2-(Butylsulfonyl)-4-(1,2-dimethyl-1H-imidazol-5-yl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (14b)
Sulfoxide 14b was formed as a byproduct in the final cyclization step in the synthesis of analog 12q. 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 7.95 (d, J = 3.2 Hz, 1H), 7.55 (d, J = 3.2 Hz, 1H), 7.16 (s, 1H), 5.31 (s, 2H), 3.41 (s, 3H), 3.24 – 3.18 (m, 2H), 2.51 (s, 3H), 1.87 – 1.73 (m, 2H), 1.49 – 1.36 (m, 2H), 0.95 – 0.85 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 167.3, 161.2, 151.5, 148.0, 144.5, 144.2, 136.4, 127.9, 126.1, 123.4, 122.9, 119.0, 111.8, 56.6, 31.2, 24.6, 21.4, 13.3, 13.2. ESI-MS (m/z): 448.1 [M+H]+.
3-((3-Amino-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-2-yl)sulfinyl)propan-1-ol (16a)
Step 1
Porcine lipase (5.52 g) was added to a solution of 3-mercapto-1-propanol (5.03 g, 54.6 mmol) in ethyl acetate (70 ml).30 The reaction was heated at 28 °C for 12 days. Despite incomplete conversion the mixture was filtered and condensed. Purification was carried out on an automated flash chromatography system in 100% DCM to give 3-mercaptopropyl acetate as an oil in 66% yield. 1H NMR (400 MHz, CHCl3) δ 4.17 (t, J = 6.2 Hz, 2H), 2.60 (q, J = 7.4 Hz, 2H), 2.05 (s, 3H), 1.93 (p, J = 6.6 Hz, 2H), 1.39 (t, J = 8.1 Hz, 2H).
Step 2
Hydrogen chloride gas was bubbled for 60 minutes into 3-((chloromethyl)thio)propane-1-thiol (4.80 g, 35.7 mmol) in a dry ice/acetone bath until the internal temperature stabilized before paraformaldehyde (1.59 g, 53.3 mmol) was slowly added using a solid addition funnel. The reaction was stirred at −78 °C for 1.5 h during which hydrogen chloride bubbling was continued. HCl addition was terminated and the reaction was allowed to warm to ambient temperature with stirring overnight. The crude mixture was diluted with minimal DCM. The aqueous phase was removed and the organic layer was washed with brine and dried over Na2SO4, filtered and condensed to give 3-((chloromethyl)thio)propane-1-thiol as an oil in 80% yield of a 2.4:1 mixture of desired chloromethyl thioether and dithiane dimer. 1H NMR (400 MHz, CHCl3) δ 4.74 (s, 2H), 4.17 (t, J = 6.4 Hz, 2H), 2.91 – 2.77 (m, 2H), 2.06 (d, J = 1.0 Hz, 3H), 2.03 – 1.94 (m, 2H).
Step 3
4-Phenyl-6-(thiophen-2-yl)-2-thioxo-1,2-dihydropyridine-3-carbonitrile (352.2 mg, 1.2 mmol) was alkylated with the chloromethyl thioether from step 2 (602.3 mg, 3.1 mmol) using triethylamine (250 ml, 1.8 mmol) in acetonitrile (1.2 ml) as described for the synthesis of compound 1. 3-((((3-Cyano-4-phenyl-6-(thiophen-2-yl)pyridin-2-yl)thio)methyl)thio)propyl acetate (15) was isolated in 26% yield following flash chromatography over SiO2 (0–30% EtOAc/hexanes). 1H NMR (400 MHz, CHCl3) δ 7.72 (dd, J = 3.8, 1.1 Hz, 1H), 7.66 – 7.58 (m, 2H), 7.54 (dd, J = 4.2, 2.9 Hz, 4H), 7.44 (d, J = 1.3 Hz, 1H), 7.17 (dd, J = 5.0, 3.8 Hz, 1H), 4.55 (s, 2H), 4.18 (t, J = 6.3 Hz, 2H), 2.84 (t, J = 7.3 Hz, 2H), 2.05 (s, 3H), 2.05 – 1.97 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 171.2, 162.3, 154.7, 153.9, 143.3, 136.0, 130.7, 130.3, 129.2, 128.7, 128.5, 127.4, 115.7, 114.4, 103.5, 63.1, 34.5, 28.9, 28.4, 21.1. ESI-MS (m/z): 441.0 [M+H]+.
Step 4
The thioether from step 4 was oxidized using procedures described for the synthesis of compound 1 to provide 3-((((3-cyano-4-phenyl-6-(thiophen-2-yl)pyridin-2-yl)thio)methyl)sulfinyl)propyl acetate in 90% yield. 1H NMR (400 MHz, CDCl3) δ 7.77 (dd, J = 3.8, 1.1 Hz, 1H), 7.67 – 7.61 (m, 2H), 7.57 (ddd, J = 6.9, 4.4, 2.1 Hz, 4H), 7.51 (s, 1H), 7.19 (dd, J = 5.0, 3.8 Hz, 1H), 4.81 (d, J = 13.1 Hz, 1H), 4.44 (d, J = 13.1 Hz, 1H), 4.22 (td, J = 6.4, 1.3 Hz, 2H), 3.09 (dt, J = 13.0, 8.1 Hz, 1H), 2.96 – 2.83 (m, 1H), 2.23 (tt, J = 7.3, 6.2 Hz, 2H), 2.04 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.0, 159.8, 155.1, 154.2, 142.6, 135.6, 131.2, 130.6, 129.3, 129.0, 128.4, 128.0, 115.3, 115.2, 103.4, 62.9, 49.2, 48.4, 22.3, 21.0. ESI-MS (m/z): 457.1 [M+H]+
Step 5
The sulfoxide from step 5 was cyclized using procedures described for the synthesis of compound 1 to provide 3-((3-amino-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-2-yl)sulfinyl)propyl acetate in 37% yield. 1H NMR (400 MHz, CDCl3) δ 7.58 (m, 5H), 7.45 (dd, J = 5.0, 1.1 Hz, 2H), 7.41 (s, 1H), 7.10 (dd, J = 5.0, 3.7 Hz, 1H), 4.61 (s, 2H), 4.26 – 4.14 (m, 2H), 3.38 – 3.27 (m, 1H), 3.15 (dt, J = 13.0, 7.7 Hz, 1H), 2.16 – 2.06 (m, 2H), 2.05 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 171.3, 171.0, 161.9, 151.3, 146.9, 143.82, 143.80, 136.8, 129.5, 129.1, 128.9, 128.83, 128.75, 128.4, 126.3, 121.3, 117.3, 106.4, 62.8, 60.5, 51.8, 22.8, 21.0, 14.3. ESI-MS (m/z): 457.1 [M+H]+.
Step 6
K2CO3 (14.9 mg, 0.108 mmol) was added to a solution of 3-((3-amino-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-2-yl)sulfinyl)propan-1-ol (21.2 mg, 0.046 mmol) in methanol (0.4 ml) and water (0.1 ml) and the reaction was stirred for 1 hour. The methanol was removed under reduced pressure, then the reaction mixture was extracted with EtOAc and washed twice with water and then brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduce pressure to give acetate 16a in 84% yield. 1H NMR (400 MHz, CHCl3) δ 7.63 (dd, J = 3.8, 1.1 Hz, 1H), 7.55 (s, 4H), 7.46 (dd, J = 5.0, 1.1 Hz, 2H), 7.43 (s, 1H), 7.11 (dd, J = 5.0, 3.7 Hz, 1H), 4.60 (s, 2H), 3.77 (t, J = 5.8 Hz, 2H), 3.49 – 3.33 (m, 1H), 3.21 (dt, J = 13.4, 6.9 Hz, 1H), 2.17 – 1.96 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 162.0, 151.3, 146.9, 143.8, 143.5, 136.8, 129.5, 129.0, 128.8, 128.4, 126.3, 121.4, 117.3, 106.4, 61.3, 52.8, 27.1, 14.3. ESI-MS (m/z): 415.1 [M+H]+.
2-((3-Chloropropyl)sulfinyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (16b)
Step 1
K2CO3 (152 mg, 1.10 mmol) was added to a solution of acetate 15 (240.5 mg, 0.55 mmol) in methanol (4.0 ml) and water (1.0 ml), and the reaction was stirred for 2.5 h. The mixture was dried then diluted with EtOAc and washed twice with water and then brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduce pressure to give 2-((((3-hydroxypropyl)thio)methyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile in 98% yield, which was used without further purification. 1H NMR (400 MHz, CHCl3) δ 7.70 (dd, J = 3.8, 1.1 Hz, 1H), 7.63 – 7.58 (m, 2H), 7.53 (dd, J = 5.3, 1.7 Hz, 4H), 7.41 (s, 1H), 7.15 (dd, J = 5.0, 3.8 Hz, 1H), 4.54 (s, 2H), 3.76 (t, J = 6.0 Hz, 2H), 2.88 (t, J = 7.1 Hz, 2H), 1.93 (ddd, J = 13.2, 7.2, 6.1 Hz, 2H), 1.86 – 1.81 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 162.8, 154.5, 153.8, 143.2, 135.9, 130.6, 130.2, 129.1, 128.7, 128.4, 127.3, 115.7, 114.3, 103.3, 61.5, 34.6, 31.8, 29.1 ESI-MS (m/z): 399.1 [M+H]+.
Step 2
Triethylamine (180 μl, 1.32 mmol) was added to an ice-cooled solution of the primary alcohol fro step 1 (180.4 mg, 0.45 mmol) in CH2Cl2 (4.5 ml), followed by the dropwise addition of methanesulfonyl chloride (81 ml, 1.0 mmol). The reaction mixture was stirred in the ice batch for 30 minutes, after which all starting material had been consumed. The reaction mixture was washed with brine and the organic layer was dried over Na2SO4, filtered and concentrated under reduce pressure to give 3-((((3-cyano-4-phenyl-6-(thiophen-2-yl)pyridin-2-yl)thio)methyl)thio)propyl methanesulfonate in quantitative yield. 1H NMR (400 MHz, CHCl3) δ 7.73 (dd, J = 3.6, 1.1 Hz, 1H), 7.62 (dd, J = 6.6, 2.9 Hz, 2H), 7.55 (ddd, J = 5.6, 4.2, 1.8 Hz, 4H), 7.45 (s, 1H), 7.18 (dd, J = 5.0, 3.8 Hz, 1H), 4.56 (s, 2H), 4.36 (t, J = 6.1 Hz, 2H), 3.03 (s, 3H), 2.90 (t, J = 7.0 Hz, 2H), 2.13 (p, J = 6.6 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 162.3, 154.47, 154.45, 153.8, 142.9, 135.7, 130.7, 130.2, 129.0, 128.7, 128.3, 127.4, 115.5, 114.32, 114.26, 68.19, 68.16, 68.1, 46.1, 46.09, 46.07, 37.3, 34.3, 28.6, 28.2. ESI-MS (m/z): 477.0 [M+H]+.
Step 3
The mesylate from step 2 (48.2 mg, 0.10 mmol) was dissolved in DMF (1.0 ml), and lithium chloride (67.3 mg, 1.59 mmol) was added. The reaction mixture was stirred for 5 hours, diluted with EtOAc, and washed several times with water and then brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduce pressure. The crude reaction mixture was purified via flash chromatography over silica gel (0–50% EtOAc/hexanes) to provide 2-((((3-chloropropyl)thio)methyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile in 69% yield. 1H NMR (400 MHz, CHCl3) δ 7.71 (dd, J = 3.8, 1.1 Hz, 1H), 7.64 – 7.58 (m, 2H), 7.56 – 7.52 (m, 4H), 7.43 (s, 1H), 7.16 (dd, J = 5.0, 3.8 Hz, 1H), 4.54 (s, 2H), 3.67 (t, J = 6.3 Hz, 2H), 2.92 (t, J = 7.0 Hz, 2H), 2.22 – 2.06 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 162.7, 154.6, 153.9, 143.2, 136.0, 130.7, 130.3, 129.2, 128.7, 128.4, 127.4, 115.6, 114.4, 103.4, 43.5, 34.5, 32.0, 29.6. ESI-MS (m/z): 417.0 [M+H]+.
Step 4
The thioether from step 3 was oxidized to 2-((((3-chloropropyl)sulfinyl)methyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile using procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CHCl3) δ 7.76 (dd, J = 3.9, 1.1 Hz, 1H), 7.62 (ddd, J = 5.1, 3.8, 2.1 Hz, 2H), 7.59 – 7.54 (m, 4H), 7.51 (s, 1H), 7.18 (dd, J = 5.0, 3.8 Hz, 1H), 4.74 (d, J = 13.1 Hz, 1H), 4.51 (d, J = 13.1 Hz, 1H), 3.80 – 3.63 (m, 2H), 3.29 – 3.16 (m, 1H), 2.96 (ddd, J = 13.0, 7.5, 6.4 Hz, 1H), 2.45 – 2.31 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 159.8, 155.0, 154.2, 142.6, 135.6, 131.2, 130.6, 129.3, 129.0, 128.4, 127.9, 115.3, 115.2, 103.4, 49.6, 48.9, 43.6, 25.9. ESI-MS (m/z): 433.0 [M+H]+.
Step 5
The sulfoxide from step 4 was cyclized using procedures described for the synthesis of compound 1 to provide chloride 16b in 88% yield. 1H NMR (400 MHz, CHCl3) δ 7.56 (m, 5H), 7.45 (m, 2H), 7.40 (s, 1H), 7.09 (t, J = 4.4 Hz, 1H), 4.61 (s, 2H), 3.66 (tt, J = 8.0, 4.0 Hz, 2H), 3.40 (dt, J = 14.1, 7.3 Hz, 1H), 3.24 (dt, J = 13.1, 7.6 Hz, 1H), 2.25 (p, J = 7.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ. 161.9, 151.4, 147.0, 143.9, 143.7, 136.8, 129.5, 129.0, 128.8, 128.4, 126.3, 121.3, 117.3, 106.2, 52.1, 43.4, 26.4. ESI-MS (m/z): 433.0 [M+H]+.
2-((3-Fluoropropyl)sulfinyl)-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-3-amine (16c)
Kryptofix 222 (44.4 mg, 0.012 mmol), KF (6.1 mg, 0.10 mmol) and K2CO3 (3.0 mg, 0.022 mmol) were added to a vial containing the mesylate from step 2 in the synthesis of 16b (54.2 mg, 0.11 mmol). DMF (1.1 ml) was added and the reaction was heated at 85 °C for 65 minutes. The cooled mixture was diluted with EtOAc and washed several times with water and then brine. The organic layer was dried over Na2SO4, filtered and condensed to provide 2-((((3-fluoropropyl)thio)methyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile in 96%, which was carried forward without additional purification. Oxidation and cyclization using procedures described for the synthesis of compound 1 provided fluoride 16c in 32% yield. 1H NMR (400 MHz, CHCl3) δ 7.68 (dd, J = 3.8, 1.1 Hz, 1H), 7.56 (q, J = 2.7 Hz, 4H), 7.53 – 7.44 (m, 3H), 7.14 (dd, J = 5.1, 3.7 Hz, 1H), 4.65 (td, J = 5.8, 3.2 Hz, 1H), 4.60 (s, 2H), 4.53 (td, J = 5.8, 3.1 Hz, 1H), 3.40 (dt, J = 13.0, 7.3 Hz, 1H), 3.24 (dt, J = 13.1, 7.6 Hz, 1H), 2.19 (dtt, J = 26.4, 7.5, 5.7 Hz, 2H). ESI-MS (m/z): 417.1 [M+H]+.
4-((3-Amino-4-phenyl-6-(thiophen-2-yl)thieno[2,3-b]pyridin-2-yl)sulfinyl)butanenitrile (16d)
DMF (1.1 ml) was added to a vial containing the mesylate from step 2 of the synthesis of 16b (56.4 mg, 0.12 mmol) and potassium cyanate (76.9 mg, 1.18 mmol). The reaction was heated to 85 °C for 4 hours. The reaction mixture was cooled, diluted with EtOAc and washed several times with water, then brine. The organic layer was dried over Na2SO4, filtered and concentrated to provide 2-((((3-cyanopropyl)thio)methyl)thio)-4-phenyl-6-(thiophen-2-yl)nicotinonitrile in 89% yield, which was carried forward without additional purification. 1H NMR (400 MHz, CHCl3) δ 7.71 (dd, J = 3.4, 1.4 Hz, 1H), 7.60 (dt, J = 6.4, 1.9 Hz, 2H), 7.54 (ddt, J = 7.1, 4.2, 1.6 Hz, 4H), 7.44 (d, J = 1.4 Hz, 1H), 7.16 (ddd, J = 5.2, 3.8, 1.5 Hz, 1H), 4.53 (s, 2H), 2.92 – 2.84 (m, 2H), 2.52 (td, J = 7.1, 1.5 Hz, 2H), 2.08 – 2.00 (m, 2H). ESI-MS (m/z): 408.1 [M+H]+. Oxidation and cyclization were performed as described for the synthesis of analog 1 to provide nitrile 16d in 45% yield. 1H NMR (400 MHz, CDCl3 δ 7.65 (d, J = 3.7 Hz, 1H), 7.56 (d, J = 6.5 Hz, 4H), 7.52 – 7.43 (m, 3H), 7.13 (t, J = 4.4 Hz, 1H), 4.64 (s, 2H), 3.40 (dt, J = 13.2, 6.6, 5.9 Hz, 1H), 3.19 (dt, J = 13.0, 7.5 Hz, 1H), 2.59 (t, J = 7.1 Hz, 2H), 2.20 (p, J = 7.2 Hz, 2H). ESI-MS (m/z): 424.0 [M+H]+.
N-(4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)benzyl)acetamide (21a)
Step 1
2-Cyanothioacetamide (275mg, 2.74 mmol) and methyl (E)-4-(3-oxo-3-(thiazol-2-yl)prop-1-en-1-yl)benzoate (250 mg, 0.915 mmol) were combined in a vial that was evacuated and backfilled with O2. Ethanol (2.75 mL) and piperdine (2 drops) were added. The solution was sparged with O2 for a few minutes then stirred at 80 °C for 4 hours. The solvent was evaporated under reduced pressure, and the product was alkylated directly. Thus, butyl(chloromethyl)sulfane (252.5 mg, 1.83 mmol) in acetonitrile (2 mL), was added to the thiopyridone followed by Et3N (278 mg, 2.75 mmol). The solution was stirred at 80 °C for 20 minutes. The reaction mixture was diluted with EtOAc and washed with water, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude solid was purified using automated flash chromatography (20% EtOAc/hexanes) to provide methyl 4-(2-(((butylthio)methyl)thio)-3-cyano-6-(thiazol-2-yl)pyridin-4-yl)benzoate (17a) as a solid in 24% yield. 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.4 Hz, 2H), 8.02 (s, 1H), 7.98 (d, J = 3.1 Hz, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 3.2 Hz, 1H), 4.52 (s, 2H), 3.95 (s, 3H), 2.76 (t, J = 7.3 Hz, 2H), 1.64 (tt, J = 7.7, 6.3 Hz, 2H), 1.42 (h, J = 7.3 Hz, 2H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.9, 166.2, 163.6, 153.9, 152.4, 144.9, 139.6, 131.7, 130.3, 128.5, 123.7, 114.9, 105.7, 52.4, 34.8, 32.2, 31.2, 22.0, 13.7. ESI-MS (m/z): 456.1 [M+H]+
Step 2
To the solution of the ester from step 1 (336 mg, 0.74 mmol) in THF (8.41 mL), LiBH4 (96.3 mg, 4.42 mmol) was added at 0 °C. The reaction was stirred at room temperature for 36 h and then diluted with EtOAc and water. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure, to give 2-(((butylthio)methyl)thio)-4-(4-(hydroxymethyl)phenyl)-6-(thiazol-2-yl)nicotinonitrile (18) in 96% yield, which was used without additional purification. 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.98 (d, J = 3.1 Hz, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.56 (d, J = 3.1 Hz, 1H), 7.52 (d, J = 8.0 Hz, 2H), 4.79 (d, J = 4.3 Hz, 2H), 4.52 (s, 2H), 2.75 (t, J = 7.4 Hz, 2H), 1.71 – 1.58 (m, 2H), 1.49 – 1.33 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.3, 163.3, 154.8, 152.2, 144.8, 143.3, 134.6, 128.6, 127.3, 123.4, 115.3, 114.9, 105.6, 64.6, 34.7, 32.1, 31.2, 22.1, 13.6. ESI-MS (m/z): 428.1 [M+H]+
Step 3
To a solution of alcohol 18 (70 mg, 0.16 mmol) in toluene was added diphenyl phosphoryl azide (55 mg, 0.20 mmol, 1.2 equiv) and 1,8-diazabicyclo[5.4.0]undec-7-ene (39 mg, 0.26 mmol, 1.3 equiv), and the reaction was stirred overnight at room temperature. Once complete as judged by LC/MS, the reaction was diluted with EtOAc and water. The organic phase was separated, and the aqueous layer was extracted twice with EtOAc. The organic layer was dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography to give 59 mg of 4-(4-(azidomethyl)phenyl)-2-(((butylthio)methyl)thio)-6-(thiazol-2-yl)nicotinonitrile (19, 79% yield). 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.98 (d, J = 3.1 Hz, 1H), 7.67 (d, J = 7.5 Hz, 2H), 7.57 (d, J = 3.1 Hz, 1H), 7.48 (d, J = 8.0 Hz, 2H), 4.52 (s, 2H), 4.44 (s, 2H), 2.75 (t, J = 7.4 Hz, 2H), 1.69 – 1.58 (m, 2H), 1.50 – 1.35 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.1, 163.4, 154.3, 152.3, 144.9, 137.9, 135.3, 129.8, 128.9, 128.6, 123.5, 120.1, 115.2, 114.9, 110.0, 105.6, 54.2, 34.8, 32.15, 31.2, 21.9, 13.6 ESI-MS (m/z): 453.1 [M+H]+.
Step 4
To a solution of azide 19 (407 mg, 0.89 mmol) in THF (8.5 mL) was added PPh3 (5.4 mmol, 1.41 g, 6.0 equiv), and the reaction mixture was stirred overnight at room temperature. Once complete, water was added and reaction was stirred for additional 5 h at room temperature and diluted with EtOAc. The organic phase was separated, and the aqueous layer was extracted twice with EtOAc. The organic layer was dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography to give 4-(4-(aminomethyl)phenyl)-2-(((butylthio)methyl)thio)-6-(thiazol-2-yl)nicotinonitrile (20) in 62% yield. 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.97 (d, J = 3.1 Hz, 1H), 7.63 (d, J = 8.2 Hz, 2H), 7.55 (d, J = 3.2 Hz, 1H), 7.47 (d, J = 8.2 Hz, 2H), 4.51 (s, 2H), 3.96 (s, 2H), 2.75 (t, J = 7.3 Hz, 2H), 1.72 – 1.52 (m, 2H), 1.42 (h, J = 7.3 Hz, 2H), 0.90 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 163.3, 154.8, 152.2, 145.2, 144.8, 134.0, 128.7, 127.8, 123.41, 115.3, 114.9, 110.0, 105.6, 45.9, 34.7, 32.1, 31.2, 23.0, 13.6. ESI-MS (m/z): 427.1 [M+H]+.
Step 5
To a solution of amine 20 (30 mg, 0.07 mmol) in THF was added acetic anhydride (21.4 mg, 0.21mmol, 3.0 equiv) and pyridine (16.6 mg, 0.21 mmol, 3.0 equiv), and the reaction was stirred at 50 °C overnight. Upon completion, the reaction was diluted with EtOAc and water. The organic phase was separated and aqueous layer was extracted twice with EtOAc. The organic layer was dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography to give N-(4-(2-(((butylthio)methyl)thio)-3-cyano-6-(thiazol-2-yl)pyridin-4-yl)benzyl)acetamide in quantitative yield. 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.97 (d, J = 3.1 Hz, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 3.2 Hz, 1H), 7.43 (d, J = 8.1 Hz, 2H), 5.95 (t, J = 7.5 Hz, 1H), 4.51 (s, 2H), 4.49 (d, J = 7.4 Hz, 2H), 2.74 (t, J = 7.3 Hz, 2H), 2.04 (s, 3H), 1.72 – 1.54 (m, 2H), 1.49 – 1.32 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 469.1 [M+H]+.
Step 6
N-(4-(2-(((Butylsulfinyl)methyl)thio)-3-cyano-6-(thiazol-2-yl)pyridin-4-yl)benzyl)acetamide was prepared in 78% yield via oxidation with hydrogen peroxide as described previously for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.07 (s, 1H), 7.98 (d, J = 3.1 Hz, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 3.1 Hz, 1H), 7.43 (d, J = 8.1 Hz, 2H), 6.04 (t, J = 6.3 Hz, 1H) 4.70 (d, J = 13.1 Hz, 1H), 4.50 (d, J = 5.9 Hz, 2H), 4.39 (d, J = 13.1 Hz, 1H), 2.96 (dt, J = 13.0, 8.1 Hz, 1H), 2.81 (dt, J = 12.9, 7.3 Hz, 1H), 2.05 (s, 3H), 1.91 – 1.73 (m, 2H), 1.62 – 1.37 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). ESI-MS (m/z): 485.1 [M+H]+.
Step 7
The sulfoxide from step 6 was cyclized using the procedure described for the synthesis of compound 1 to provide acetate 21a in 52% yield. 1H NMR (400 MHz, CD2Cl2) δ 8.01 (s, 1H), 7.89 (d, J = 3.1 Hz, 1H), 7.53 (d, J = 3.2 Hz, 1H), 7.47 – 7.41 (m, 4H), 6.25 (s, 1H), 4.59 (s, 2H), 4.50 (d, J = 6.1 Hz, 2H), 3.23 (ddd, J = 13.0, 9.2, 6.1 Hz, 1H), 3.08 (ddd, J = 12.9, 9.2, 6.5 Hz, 1H), 2.03 (s, 3H), 1.57 – 1.86 (m, 2H), 1.57 – 1.35 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, acetone) δ 169.1, 167.6, 161.2, 149.5, 147.4, 144.5, 142.1, 141.5, 135.1, 128.7, 127.7, 123.5, 122.7, 117.4, 110.9, 54.9, 42.5, 24.8, 22.1, 21.6, 13.1. ESI-MS (m/z): 485.1 [M+H]+.
1-(4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)benzyl)-3-ethylurea (21b)
To the solution amine 20 (50 mg, 0.117 mmol) in THF was added ethyl isocyanate (13 mg, 0.23 mmol, 19 μl) at 0 °C. The reaction was stirred at room temperature for 1h. During this time a solid formed, which was filtered, washed with small amount of EtOAc, and dried under reduced pressure to give 40 mg of 1-(4-(2-(((butylthio)methyl)thio)-3-cyano-6-(thiazol-2-yl)pyridin-4-yl)benzyl)-3-ethylurea (69% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.13 – 8.02 (m, 2H), 7.91 (s, 1H), 7.73 – 7.61 (m, 2H), 7.49 – 7.37 (m, 2H), 6.40 (t, J = 6.1 Hz, 1H), 5.94 (t, J = 5.6 Hz, 1H), 4.62 (s, 2H), 4.27 (d, J = 6.0 Hz, 2H), 3.02 (q, J = 6.8 Hz, 2H), 2.68 (t, J = 7.5 Hz, 2H), 1.66 – 1.45 (m, 2H), 1.41 – 1.20 (m, 2H), 0.98 (t, J = 6.9 Hz, 3H), 0.82 (t, J = 7.4 Hz, 3H). ESI-MS (m/z): 498.1 [M+H]+. The resulting thioether was oxidized and cyclized using procedures described for the synthesis of compound 1 to provide urea 21b in 37% yield over 2 steps. 1H NMR (400 MHz, CD2Cl2) 7.95 (s, 1H), 7.86 (d, J = 3.1 Hz, 1H), 7.50 (d, J = 3.1 Hz, 1H), 7.46 – 7.30 (m, 4H), 5.43 (s, 1H), 4.98 (s, 1H), 4.59 (s, 2H), 4.41 (d, J = 6.1 Hz, 2H), 3.32 – 3.13 (m, 3H), 3.14 – 3.00 (m, 1H), 1.92 – 1.57 (m, 2H), 1.57 – 1.37 (m, 2H), 1.11 (t, J = 7.2 Hz, 1H), 0.93 (t, J = 7.3 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 167.9, 161.5, 159.6, 149.7, 148.1, 143.9, 143.5, 142.1, 134.7, 128.5, 127.2, 122.8, 122.6, 117.2, 108.5, 54.5, 42.9, 34.5, 24.9, 21.4, 14.4, 12.6. ESI-MS (m/z): 514.1 [M+H]+.
4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)benzyl ethylcarbamate (22a)
Carbamate 22a was prepared from alcohol 18 using synthetic procedures described for the preparation of analog 21b. 1H NMR (400 MHz, CD3OD) δ 7.97 – 7.83 (m, 2H), 7.75 – 7.64 (m, 1H), 7.62 – 7.41 (m, 4H), 5.17 (s, 2H), 3.24 (ddd, J = 12.1, 5.9, 3.1 Hz, 1H), 3.15 (q, J = 7.5 Hz, 2H), 3.07 (ddd, J = 12.4, 6.5, 3.0 Hz, 1H), 1.78 – 1.55 (m, 2H), 1.56 – 1.38 (m, 2H), 1.11 (t, J = 7.2 Hz, 3H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 167.9, 167.1, 1615, 149.7, 147.8, 143.9, 143.4, 138.9, 135.7, 128.6, 127.5, 122.7, 122.7, 117.2, 108.7, 65.1, 54.5, 35.2, 24.9, 21.4, 13.9, 12.6. ESI-MS (m/z): 515.1 [M+H]+.
(4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)phenyl)methanol (23a)
Thioether 18 was oxidized and cyclized to provide alcohol 23a in 16% isolated yield using procedures described for the synthesis of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.90 (d, J = 3.2 Hz, 1H), 7.59 – 7.40 (m, 5H), 4.80 (s, 2H), 4.63 (s, 2H), 3.27 (ddd, J = 12.8, 9.0, 6.1 Hz, 1H), 3.10 (ddd, J = 12.8, 9.1, 6.6 Hz, 1H), 1.78 – 1.61 (m, 2H), 1.55 – 1.40 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.2, 161.4, 149.6, 147.4, 144.2, 143.1, 142.3, 135.5, 128.8, 127.2, 123.5, 122.2, 117.8, 108.3, 64.5, 54.5, 25.0, 21.9, 13.7. ESI-MS (m/z): 444.1 [M+H]+.
4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)benzyl dimethylglycinate (23b)
N,N-Dimethylglycine (3.5 mg, 0.034 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (6.5 mg, 0.034 mmol), and DMAP (4.1 mg, 0.0334 mmol) were combined with (4-(3-amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)phenyl)methanol 23a (10 mg, 0.023 mmol) and dissolved in DMF (270 μL). The reaction mixture stirred at room temperature overnight, then neutralized with 1M NaOH, washed with H2O and extracted with EtOAc. The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified using flash chromatography (7 % MeOH, 93 % DCM) to give a quantitative yield of green solid product 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.90 (d, J = 3.2 Hz, 1H), 7.56 – 7.43 (m, 5H), 5.25 (s, 2H), 4.61 (s, 2H), 3.35 – 3.27 (m, 1H), 3.26 (s, 2H), 3.16 – 3.04 (m, 1H), 2.37 (s, 6H), 1.78 – 1.63 (m, 2H), 1.55 – 1.39 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 529.1 [M+H]+.
(3-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)phenyl)methanol (23c)
Following the procedure for the preparation of analog 23a, (E)-3-(3-oxo-3-(thiazol-2-yl)prop-1-en-1-yl)benzoate was converted to alcohol 23c. 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.88 (d, J = 3.1 Hz, 1H), 7.55 – 7.30 (m, 5H), 4.75 (s, 2H), 4.62 (s, 2H), 3.26 (ddd, J = 12.8, 9.1, 6.0 Hz, 1H), 3.09 (ddd, J = 12.8, 9.2, 6.5 Hz, 1H), 1.76 – 1.61 (m, 2H), 1.51 – 1.38 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H) 13C NMR (101 MHz, CDCl3) δ 171.1, 168.1, 161.4, 149.6, 147.2, 144.2, 143.1, 136.5, 128.9, 128.8, 127.7, 127.1, 126.9, 123.2, 122.3, 117.7, 64.5, 54.5, 25.0, 21.9, 13.7. ESI-MS (m/z): 444.1 [M+H]+.
4-(4-(Aminomethyl)phenyl)-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (24)
Azide 19 was oxidized and cyclized as described for the synthesis of compound 1. To a solution of the resultant 4-(4-(azidomethyl)phenyl)-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (10 mg, 0.02 mmol) in THF was added PPh3 (6 equiv), and the reaction mixture was stirred overnight at room temperature. Once complete, water was added and reaction was stirred for additional 5 h at room temperature and diluted with EtOAc. The organic phase was separated, and the aqueous layer was extracted twice with EtOAc. The organic layer was dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography to give 7 mg of amine 24 (79% yield). 1H NMR (400 MHz, Acetone-d6) δ 7.98 (s, 1H), 7.94 (d, J = 3.2 Hz, 1H), 7.78 (d, J = 3.2 Hz, 1H), 7.65 – 7.44 (m, 4H), 4.89 (s, 2H), 4.52 (s, 2H), 3.17 (ddd, J = 12.7, 8.9, 6.0 Hz, 1H), 3.11 – 2.99 (m, 1H), 1.78 – 1.59 (m, 2H), 1.60 – 1.36 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 443.1 [M+H]+
N-(4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)benzyl)methanesulfonamide (25)
Amine 24 (10 mg, 0.023 mmol) was suspended in DCM (300 μL) and Et3N (3.5 μL, 0.025) and cooled to 0 °C under N2. Methanesulfonyl chloride (1.8μL, 0.023 mmol) was added to the reaction mixture, which was stirred at room temperature for four hours before an additional methanesulfonyl chloride (1 equiv). After stirring overnight the reaction mixture was diluted with EtOAc, washed with water and brine, and the organic layer was separated. The combined organic layers were dried over sodium sulfate, concentrated under reduced pressure and purified using automated chromatography on silica gel (3% MeOH, 97% CH2Cl2) and then preparative thin layer chromatography (100% EtOAc) to give 24 in 3.3% isolated yield. 1H NMR (400 MHz, acetone-d6) δ 8.02 (s, 1H), 7.99 (d, J = 3.1 Hz, 1H), 7.82 (d, J = 3.2 Hz, 1H), 7.70 – 7.58 (m, 4H), 4.84 (s, 1H), 4.46 (s, 2H), 3.23 – 3.13 (m, 1H), 3.12 – 3.02 (m, 1H), 2.93 (s, 3H), 1.76 – 1.63 (m, 2H), 1.55 – 1.41 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 521.1 [M+H]+.
Methyl 4-(3-amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)benzoate (26a)
Methyl ester 26a was synthesized from intermediate 17a according to the procedures described for compound 1. 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 7.5 Hz, 2H), 8.04 (s, 1H), 7.91 (d, J = 3.2 Hz, 1H), 7.67 – 7.54 (m, 2H), 7.50 (d, J = 3.2 Hz, 1H), 3.97 (s, 3H), 3.27 (ddd, J = 12.8, 8.9, 6.2 Hz, 1H), 3.10 (ddd, J = 12.8, 9.0, 6.8 Hz, 1H), 1.81 – 1.63 (m, 2H), 1.54 – 1.39 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.8, 166.2, 161.4, 149.6, 145.9, 144.2, 142.8, 140.9, 131.0, 129.9, 128.9, 122.9, 122.4, 117.4, 109.4, 54.7, 52.5, 25.0, 21.9, 13.7. ESI-MS (m/z): 472.1 [M+H]+.
Methyl 3-(3-amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)benzoate (26b)
Methyl ester 26b was synthesized from intermediate 17b according to the procedures described for compound 1. 1H NMR (400 MHz, CDCl3) δ 8.26 – 8.11 (m, 2H), 8.02 (s, 1H), 7.89 (d, J = 3.2 Hz, 1H), 7.76 – 7.56 (m, 2H), 7.49 (d, J = 3.1 Hz, 1H), 4.54 (s, 2H), 3.93 (s, 3H), 3.27 (ddd, J = 12.8, 9.0, 6.2 Hz, 1H), 3.09 (ddd, J = 12.8, 9.0, 6.7 Hz, 1H), 1.79 – 1.61 (m, 2H), 1.55 – 1.39 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.9, 166.1, 162.5, 161.4, 149.7, 145.9, 144.2, 142.7, 136.7, 133.0, 130.7, 130.4, 129.7, 128.8, 123.2, 122.4, 117.7, 54.7, 52.5, 25.1, 21.9, 13.7. ESI-MS (m/z): 472.1 [M+H]+.
4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)benzoic acid (27a)
To a solution of ester 26a (29 mg, 0.06 mmol) in THF (278 μL), MeOH (278 μL), and H2O (93 μL), LiOH (4.4 mg, 0.18 mmol) was added. After 3 h of stirring at room temperature the reaction mixture was diluted with EtOAc and washed with 1M HCl. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to give bright green solid in 84% isolated yield. 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.4 Hz, 2H), 8.05 (s, 1H), 7.95 (d, J = 3.2 Hz, 1H), 7.68 – 7.55 (m, 2H), 7.52 (d, J = 3.2 Hz, 1H), 3.40 – 3.24 (m, 1H), 3.24 – 3.04 (m, 1H), 1.83 – 1.65 (m, 2H), 1.55 – 1.37 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.7, 168.3, 161.6, 149.3, 146.2, 143.9, 143.3, 141.0, 131.1, 130.3, 129.0, 122.8, 122.6, 117.8, 108.9, 54.3, 25.1, 21.9, 13.7. ESI-MS (m/z): 458.1 [M+H]+
4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)-N,N-dimethylbenzamide (28a)
Dimethylamine hydrochloride (4.82 mg, 0.059 mmol) was added to a solution of acid 27a (24.6 mg, 0.05 mmol), HATU (22.5 mg, 0.06 mmol), and DMF (140 μL) followed by DIPEA (19 μL, 0.10 mmol). The solution was stirred at room temperature for 3 h then diluted with EtOAc and washed with water. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to give amide 28a in 59% yield. 1H NMR (400 MHz, CDCl3) δ 8.03 (s, 1H), 7.91 (d, J = 3.2 Hz, 1H), 7.66 – 7.44 (m, 5H), 3.36 – 3.21 (m, 1H), 3.14 (s, 3H), 3.13 – 3.06 (m, 1H), 3.02 (s, 3H), 1.81 – 1.64 (m, 2H), 1.55 – 1.41 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 485.1 [M+H]+.
3-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)-N,N-dimethylbenzamide (28b)
Amide 28b was synthesized from ester 17b following the procedures described for the synthesis of 28a. 1H NMR (400 MHz, Chloroform-d) δ 8.02 (s, 1H), 7.90 (d, J = 3.1 Hz, 1H), 7.62 – 7.51 (m, 4H), 7.49 (d, J = 3.1 Hz, 1H), 4.59 (s, 2H), 3.27 (ddd, J = 12.8, 9.0, 6.1 Hz, 1H), 3.15 – 2.97 (m, 7H), 1.78 – 1.64 (m, 2H), 1.55 – 1.39 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 485.1 [M+H]+.
3-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)-N-(2-hydroxyethyl)benzamide (28c)
Amide 28c was synthesized from ester 17b following the procedures described for the synthesis of 28a. 1H NMR (400 MHz, CDCl3) δ 8.04 – 7.92 (m, 3H), 7.90 (d, J = 3.1 Hz, 1H), 7.64 – 7.51 (m, 3H), 7.50 (d, J = 3.2 Hz, 1H), 4.54 (s, 2H), 3.81 (t, J = 4.9 Hz, 2H), 3.62 (q, J = 4.7 Hz, 2H), 3.30 – 3.15 (m, 1H), 3.15 – 2.96 (m, 1H), 1.72 – 1.54 (m, 2H), 1.53 – 1.33 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 501.1 [M+H]+.
2-(4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)phenyl)propan-2-ol (30)
To the solution of methyl ester 26a (15 mg, 0.031 mmol) in THF (500 μL) −78 °C, CH3Li·LiBr (1.5 M in ether, 104μL, 0.156 mmol) was added, and the reaction mixture was stirred for 3 h before an additional 2.0 equiv of CH3Li·LiBr was added. After an additional hour of stirring the reaction mixture was quenched with water and diluted with EtOAc. The organic layer was separated, dried over sodium sulfate, and concentrated under reduced pressure. The crude product was purified using automated chromatography over silica gel (4% MeOH in CH2Cl2) to give tertiary alcohol 30 in 25% isolated yield. 1H NMR (400 MHz, (CD3)2CO) δ 8.03 (s, 1H), 7.98 (d, J = 3.2 Hz, 1H), 7.81 (d, J = 3.2 Hz, 1H), 7.77 (d, J = 8.6 Hz, 2H), 7.57 (d, J = 8.1 Hz, 2H), 4.87 (s, 1H), 4.84 (s, 1H), 3.25 – 3.13 (m, 1H), 3.11 – 3.01 (m, 1H), 1.76 – 1.63 (m, 2H), 1.59 (s, 6H), 1.53 – 1.40 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 472.1 [M+H]+.
2-(4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)phenyl)ethan-1-ol (31)
Step 1
2-(((butylthio)methyl)thio)-4-(4-iodophenyl)-6-(thiazol-2-yl)nicotinonitrile was synthesized from 1-(thiazol-2-yl)-2-(triphenyl-l5-phosphanylidene)ethan-1-one (1.67 g, 4.31 mmol) and 4-iodobenzaldehyde (1.0 g, 4.3 mmol) using the procedures described for the preparation of compound 1. 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 3.0 Hz, 1H), 7.94 (d, J = 16.1 Hz, 1H), 7.89 (d, J = 16.1 Hz, 1H), 7.75 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 3.0 Hz, 1H), 7.41 (d, J = 8.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 181.4, 144.7, 144.4, 138.2, 134.1, 130.2, 126.6, 121.2, 97.5. ESI-MS (m/z): 341.9 [M+H]+.
Step 2
The aryl iodide from step 1 (114 mg, 0.218 mmol), Pd2(dba)3 (52 mg, 0.057 mmol), tri-(2-furyl)-phosphine (46 mg, 0.20 mmol), and 4 Å molecular sieve (700 mg) were combined in a 4 ml vial, and this vial was charged with argon. Dry and degased diisopropylamine (1.5 ml) and t-butoxylacetylene (1.6 ml, 0.8 M in diethyl ether) were added to the reaction vial sequentially at room temperature.31 The iodide was consumed as judged by TLC analysis after stirring overnight. The reaction mixture was filtered through an aluminum oxide column (Brockmann I, basic, activated), and the desired fraction was eluted with 300 ml EtOAc. The crude reaction product was concentrated under reduced pressure, dissolved in hexanes:MeOH (1:10, 4 mL), and stirred at 70 °C overnight. The reaction solvents were removed under reduced pressure, and automated chromatography over silica gel (20 % EtOAc, 80 % Hex) provided methyl 2-(4-(2-(((butylthio)methyl)thio)-3-cyano-6-(thiazol-2-yl)pyridin-4-yl)phenyl)acetate in 36% yield. 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.97 (d, J = 3.1 Hz, 1H), 7.62 (d, J = 7.5 Hz, 2H), 7.55 (d, J = 3.1 Hz, 1H), 7.44 (d, J = 8.0 Hz, 2H), 4.51 (s, 2H), 3.71 (s, 3H), 3.70 (s, 2H), 2.74 (t, J = 7.5 Hz, 2H), 1.69 – 1.57 (m, 2H), 1.49 – 1.34 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.3, 167.2, 163.4, 154.6, 152.2, 144.9, 136.5, 134.2, 130.1, 128.7, 123.4, 115.3, 115.0, 105.6, 52.2, 40.9, 34.7, 32.1, 31.2, 22.0, 13.6, 11.7.
Step 3
The thioether from step 2 was oxidized and cyclized as described for the synthesis of compound 1 to provide methyl 2-(4-(3-amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)phenyl)acetate in 68% yield. 1H NMR (400 MHz, CD3)2CO) δ 8.00 (s, 1H), 7.96 (d, J = 3.2 Hz, 1H), 7.79 (d, J = 3.2 Hz, 1H), 7.61 – 7.49 (m, 4H), 3.80 (s, 2H), 3.68 (s, 3H), 3.22 – 3.13 (m, 1H), 3.11 – 3.01 (m, 1H), 1.76 – 1.61 (m, 2H), 1.54 – 1.37 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 486.1 [M+H]+.
Step 4
The methyl ester from step 3 was reduced with LiBH4 following the procedure described previously for the preparation of intermediate 18 to yield primary alcohol 31 in 30% yield following chromatography on silica gel (EtOAc). 1H NMR (400 MHz, CD2Cl2) δ 8.06 (s, 1H), 7.92 (d, J = 3.2 Hz, 1H), 7.53 (d, J = 3.2 Hz, 1H), 7.47 – 7.39 (m, 4H), 4.54 (m, 2H), 3.91 (t, J = 6.6 Hz, 2H), 3.28 – 3.20 (m, 1H), 3.15 – 3.05 (m, 1H), 2.96 (t, J = 6.6 Hz, 2H), 1.75 – 1.63 (m, 2H), 1.55 – 1.42 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 458.1 [M+H]+.
2-(4-(3-Amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)phenoxy)ethan-1-ol (32)
Step 1
2-Acetylthiazole (534 μL, 5.15 mmol) was added to a solution of methyl 2-(4-formylphenoxy)acetate32 (1.0 g, 5.2 mmol) in MeOH (11mL) under N2. NaOMe (279 mg, 5.15 mmol) was added last and the reaction mixture stirred at room temperature overnight. The reaction mixture was filtered, and the precipitate was washed with small amount of MeOH then diluted with CH2Cl2 and washed with H2O. The organic layer was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure to provide solid methyl (E)-2-(4-(3-oxo-3-(thiazol-2-yl)prop-1-en-1-yl) phenoxy)acetate in 21 % isolated yield. 1H NMR (400 MHz, Chloroform-d) δ 8.03 (d, J = 3.0 Hz, 1H), 7.95 (d, J = 15.9 Hz, 1H), 7.82 (d, J = 16.0 Hz, 1H), 7.70 – 7.61 (m, 3H), 6.92 (d, J = 8.8 Hz, 2H), 4.67 (s, 2H), 3.80 (s, 3H). ESI-MS (m/z): 304.1 [M+H].
Step 2
The enone from step 1 was condensed with cyanothioacetamide, alkylated with (chloromethyl)(butyl)sulfane, oxidized and cyclized as described for the synthesis of compound 1 to provide Methyl 2-(4-(3-amino-2-(butylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-4-yl)phenoxy)acetate. 1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H), 7.89 – 7.86 (m, 1H), 7.47 (d, J = 3.1 Hz, 1H), 7.46 – 7.37 (m, 2H), 7.02 (d, J = 8.5 Hz, 2H), 4.70 (s, 2H), 4.66 (s, 2H), 3.82 (s, 3H), 3.34 – 3.18 (m, 1H), 3.16 – 3.01 (m, 1H), 1.77 – 1.64 (m, 2H), 1.51 – 1.38 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.9, 168.1, 161.4, 158.6, 149.6, 146.8, 144.2, 143.1, 130.2, 129.6, 123.4, 122.2, 117.9, 114.8, 110.0, 65.2, 54.6, 52.4, 25.0, 21.9, 13.7. ESI-MS (m/z): 502.1 [M+H]+.
Step 3
The methyl ester from step 2 was reduced with LiBH4 following the procedure described in the synthesis of intermediate 18 to provide primary alcohol 32. 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.90 (d, J = 3.2 Hz, 1H), 7.48 (d, J = 3.1 Hz, 1H), 7.46 – 7.35 (m, 2H), 7.07 – 6.99 (m, 2H), 4.69 (s, 2H), 4.18 – 4.11 (m, 2H), 4.04 – 3.96 (m, 2H), 3.34 – 3.23 (m, 1H), 3.17 – 3.04 (m, 1H), 1.80 – 1.61 (m, 2H), 1.53 – 1.40 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 168.1, 161.3, 159.6, 149.6, 147.2, 144.2, 143.4, 130.1, 128.2, 123.7, 122.2, 117.8, 114.7, 108.6, 69.6, 61.2, 54.7, 24.25.0, 21.9, 13.5. ESI-MS (m/z): 474.1 [M+H]+. Enantiomers were separated on a 1 cm Chiralpak AD column using 50% EtOH in hexanes with a 2.5 mL/min flow rate monitoring absorbance at 254 and 315 nM. The positive enantiomer eluted at 27.5–31 min and the negative enantiomer eluded at 32–36 min. Peak 1: [α]D23 = + 75.46 (c = 0.212, EtOH); peak 2: [α]D23 = − 51.24 (c = 0.263, EtOH).
4-Methyl-6-morpholino-2-(propylsulfinyl)thieno[2,3-b]pyridin-3-amine (38a)
Step 1
Anhydrous MeOH (3.97 mL) was added to 2,6-dichloro-4-methylnicotinonitrile (34, 500 mg, 2.67 mmol) under a N2 atmosphere, and the mixture was cooled to 0 °C. Morpholine (473.7 μL, 5.493 mmol) was added dropwise to the solution and the solution stirred at room temperature overnight. The reaction mixture was filtered, and the precipitate was washed with MeOH (0.5 mL) and H2O (3–4 mL). The solid was dissolved in CH2Cl2 and dried over MgSO4. The solution was filtered and concentrated under reduced pressure. The crude product was purified using automated flash chromatography to give 2-chloro-4-methyl-6-morpholinonicotinonitrile (35a) in 85 % yield. 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 3.81 – 3.73 (m, 4H), 3.68 – 3.58 (m, 4H), 2.42 (d, J = 0.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 158.55, 153.80, 115.84, 104.32, 97.92, 94.99, 66.37, 44.80, 21.02. ESI-MS (m/z): 238.1 [M+H]+.
Step 2
NaOMe (73.6 mg, 1.36 mmol) and methyl 3-mercaptopropionate (151 μL, 1.363 mmol) were added to a solution of the chloropyridine from step 1 (324 mg, 1.36 mmol) in DMF (4.10 mL), and the reaction mixture was stirred at 80 °C for 1 h. The reaction mixture allowed to cool to rt, diluted with EtOAc and washed with H2O. The organic layer was separated, dried over MgSO4, filtered, and concentrated under reduced pressure to give a crude mixture of 1:1 starting material to desired thioether (methyl 3-((3-cyano-4-methyl-6-morpholinopyridin-2-yl)thio)propanoate), which was used directly in the next step without additional purification. ESI (m/z): 322.1 [M+H]+.
Step 3
NaH (150.8 mg, 3.769 mmol, 60% in mineral oil) and THF (10 mL) were added to a flame dried flask under N2, followed by the crude reaction mixture from step 2 dissolved in THF (10 mL). The reaction was stirred at reflux for 6 h, and an additional 2.0 equiv of NaH (60% in mineral oil) was added. The reaction mixture was stirred at reflux overnight and then allowed to cool to rt. EtOH (1.5 mL) was added then volatile solvents were removed under reduced pressure. Water (8 mL) was added and the solution was adjusted to pH 6 with concentrated HCl before filtration to give a crude solid 4-methyl-6-morpholino-2-thioxo-1,2-dihydropyridine-3-carbonitrile that was used directly in the next step without purification. ESI (m/z): 236.1 [M+H]+.
Step 4
The thiopyridone from step 3 was alkylated with (chloromethyl)(butyl)sulfane, oxidized with hydrogen peroxide, and cyclized with KOtBu as described for the synthesis of compound 1 to provide morpholine 38a. 1H NMR (400 MHz, CDCl3) δ 6.36 (s, 1H), 4.91 (s, 2H), 3.85 – 3.76 (m, 4H), 3.63 – 3.58 (m, 4H), 3.32 – 3.18 (m, 1H), 3.09 – 2.99 (m, 1H), 2.65 (s, 3H), 1.81 – 1.66 (m, 2H), 1.07 (t, J = 7.4 Hz, 3H). ESI (m/z): 340.1 [M+H]+.
2-(Butylsulfinyl)thieno[2,3-b]pyridin-3-amine (38b)
3-cyano-1,2-dihydropyridine-2-thione was alkylated with (chloromethyl)(butyl)sulfane, oxidized with hydrogen peroxide, and cyclized with KOtBu as described for the synthesis of compound 1 to provide the unsubstituted analog 38b. 1H NMR (400 MHz, CDCl3) δ 8.61 (dd, J = 4.7, 1.6 Hz, 1H), 7.89 (dd, J = 8.1, 1.6 Hz, 1H), 7.33 (dd, J = 8.1, 4.6 Hz, 1H), 3.39 – 3.18 (m, 1H), 3.20 – 3.03 (m, 1H), 1.74 (p, J = 7.6 Hz, 2H), 1.63 – 1.38 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 255 [M+H]+.
4-Methyl-2-(propylsulfinyl)-6-(thiazol-2-yl)thieno[2,3-b]pyridin-3-amine (38c)
Step 1
1,2-Dibromobutane (1.64 mmol, 310 mg, 143 μL) was added to a suspension of zinc powder (33.3 mmol, 2.18 g) in THF 2 mL. The mixture was heated to reflux for 5 minutes and then chlorotrimethylsilane (1.73 mmol, 188 mg, 220 μL) was added. Next, a solution of 2-bromothiazole (11.1 mmol, 1.82 g, 1.0 mL) in THF (10 mL) was added dropwise and the mixture was stirred at 50 ° C for 1 hour to give a THF solution of 2-thiazolylzinc bromide.
Step 2
2,6-dichloro-4-methylnicotinonitrile (0.27 mmol, 50 mg) and tetrakis(triphenylphosphine)palladium (0.013 mmol, 15 mg, 5 mol %) were added sequentially to the THF solution of 2-thiazolylzinc bromide (550 μL) from step 1, and the mixture was stirred at 60 °C. for 12 hours.33 The reaction solution was poured into saturated aqueous sodium bicarbonate solution and the aqueous layer was extracted with EtOAc. The organic phases were dried with MgSO4, filtered and concentrated under reduced pressure. The crude compound was purified by flash chromatography on silica gel to give 2-chloro-4-methyl-6-(thiazol-2-yl)nicotinonitrile in 45% yield. ESI-MS (m/z): 236.0 [M+H]+.
Step 3
The chloropyridine from step 2 was subjected to substitution with methyl 3-mercaptopropionate, alkylated, oxidized and cyclized as described for compound 38a to provide thiazole 38c. 1H NMR (400 MHz, CDCl3) δ 7.95 (s, 1H), 7.93 (d, J = 3.2 Hz, 1H), 7.48 (d, J = 3.2 Hz, 1H), 5.16 (s, 2H), 3.37 – 3.23 (m, 1H), 3.16 – 3.05 (m, 1H), 2.85 (s, 3H), 1.83 (h, J = 7.5 Hz, 2H), 1.11 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.3, 161.1, 154.2, 150.1, 148.6, 144.1, 126.3, 122.1, 118.1, 108.8, 56.9, 20.3, 16.7, 13.3. ESI-MS (m/z): 338.0 [M+H]+.
General Synthesis of thieno[2,3-d]pyrimidines as exemplified by the synthesis of 6-(butylsulfinyl)-4-phenyl-2-(thiophen-2-yl)thieno[2,3-d]pyrimidin-5-amine (44a)
Step 1
According to a procedure described by Lorente,34 a mixture of NaOiPr (1.75 mmol, 1.0 equiv, prepared in situ from sodium and dry iPrOH), thiophene-2-carbothioamide (1.75 mmol, 250 mg, 1.0 equiv) and 2-(ethoxy(phenyl)methylene)malononitrile (1.75 mmol, 346 mg, 1.0 equiv) in i-PrOH (30 mL) was stirred for 5 h at rt. The reaction was then acidified with concentrated HCl and stirred overnight. The solvent was evaporated, and the resulting solid was suspended in acetic acid and heated to 80 °C for 2 h. After cooling to room temperature the mixture was filtered to give 315 mg of 4-phenyl-2-(thiophen-2-yl)-6-thioxo-1,6-dihydropyrimidine-5-carbonitrile, which was used without additional purification (60%). ESI-MS (m/z): 296.1 [M+H]+.
Step 2
A mixture of the pyrimidine from step 1 (0.68 mmol, 200 mg), butyl(chloromethyl)sulfane (0.0.68 mmol, 94 mg, 1.0 equiv) and Et3N (1.02 mmol, 1.5 equiv) was stirred at refluxed in dry CH3CN (700 μL) for 20 min. The reaction was allowed to cool to rt and diluted with EtOAc and water. The organic phase was separated, and the aqueous layer was extracted twice with EtOAc. The combined extractions were washed with saturated NaCl solution, dried over magnesium sulfate, filtered and concentrated under reduced pressure. The residue obtained was then purified by flash chromatography to give 180 mg of 4-(((butylthio)methyl)thio)-6-phenyl-2-(thiophen-2-yl)pyrimidine-5-carbonitrile (68%). 1H NMR (400 MHz, CDCl3) δ 8.15 (dd, J = 3.7, 1.2 Hz, 1H), 8.12 – 8.03 (m, 2H), 7.61 (dd, J = 4.9, 1.2 Hz, 1H), 7.59 – 7.50 (m, 3H), 7.18 (dd, J = 4.9, 3.7 Hz, 1H), 4.54 (s, 2H), 2.75 (t, J = 7.3 Hz, 2H), 1.73 – 1.56 (m, 2H), 1.48 – 1.34 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H). 398.1 [M+H]+.
Step 3
Acetic Acid (860 μL) and hydrogen peroxide (0.54 mmol, 1.5 equiv, 30% solution in water) were added to a solution of the thioether from step 2 (0.36 mmol, 140 mg) in chloroform (860 μL). The reaction mixture was stirred at 32 °C for 45 min. Once complete, the reaction was diluted with EtOAc and washed with saturated NaHCO3 solution, dried over magnesium sulfate, filtered and concentrated under reduced pressure to give 139 mg 4-(((butylsulfinyl)methyl)thio)-6-phenyl-2-(thiophen-2-yl)pyrimidine-5-carbonitrile (94 %). 1H NMR (400 MHz, CDCl3) δ 8.21 (dd, J = 3.9, 1.3 Hz, 1H), 8.09 (dt, J = 6.8, 1.5 Hz, 2H), 7.64 (dd, J = 5.0, 1.3 Hz, 1H), 7.63 – 7.49 (m, 3H), 7.20 (dd, J = 5.0, 3.8 Hz, 1H), 4.71 (d, J = 13.1 Hz, 1H), 4.45 (d, J = 13.1 Hz, 1H), 2.94 (dt, J = 12.9, 8.1 Hz, 1H), 2.83 (dt, J = 13.0, 7.3 Hz, 1H), 1.84 (p, J = 7.6 Hz, 2H), 1.54 – 1.38 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 414 [M+H]+.
Step 4
To a solution of the sulfoxide from step 3 (0.12 mmol, 50 mg) in DMF (480 μL) was added KOH (0.06 mmol, 3.4 mg in 34 μl of water, 0.5 equiv). The reaction mixture was stirred at rt. for 20 min. Once the reaction was complete as judged by TLC analysis, the mixture was diluted with EtOAc and washed with 5% aqueous acetic acid. The organic phase was separated and aqueous layer was extracted twice with EtOAc. The combined organic layers were dried over magnesium sulfate, filtered and concentrated under reduced pressure to give crude product, which was purified by flash chromatography (EtOAc) on silica gel to provide the pyrimidine analog 44a in 40% yield. 1H NMR (400 MHz, CDCl3) δ 8.10 (dd, J = 3.7, 1.3 Hz, 1H), 7.74 – 7.65 (m, 2H), 7.62 – 7.53 (m, 3H), 7.50 (dd, J = 5.0, 1.2 Hz, 1H), 7.14 (dd, J = 5.0, 3.7 Hz, 1H), 4.79 (s, 2H), 3.28 (ddd, J = 12.8, 9.0, 6.2 Hz, 1H), 3.11 (ddd, J = 12.8, 9.0, 6.7 Hz, 1H), 1.84 – 1.63 (m, 2H), 1.54 – 1.41 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 169.7, 162.5, 157.1, 142.8, 142.3, 136.8, 130.6, 130.4, 129.8, 128.9, 128.8, 128.4, 119.1, 106.9, 54.9, 24.94, 21.9, 13.5. ESI-MS (m/z): 414 [M+H]+.
6-(Butylsulfinyl)-2,4-diphenylthieno[2,3-d]pyrimidin-5-amine (44b)
Benzothioamide and 2-(ethoxy(phenyl)methylene)malononitrile were converted to diphenylpyrimidine 44b using procedures described for the synthesis of 44a. 1H NMR (400 MHz, CDCl3) δ 8.73 – 8.37 (m, 2H), 7.78 – 7.68 (m, 2H), 7.66 – 7.55 (m, 3H), 7.53 – 7.40 (m, 3H), 4.83 (s, 2H), 3.30 (ddd, J = 12.7, 8.9, 6.3 Hz, 1H), 3.21 – 3.01 (m, 1H), 1.87 – 1.66 (m, 2H), 1.57 – 1.41 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 170.0, 162.4, 160.2, 142.1, 137.2, 136.9, 131.0, 130.3, 129, 128.8, 128.6, 119.6, 107.3, 54.9, 24.9, 21.9, 13.5. ESI-MS (m/z): 408 [M+H]+.
6-(Butylsulfinyl)-4-phenyl-2-(thiazol-2-yl)thieno[2,3-d]pyrimidin-5-amine (44c)
Thiazole-2-carbothioamide and 2-(ethoxy(phenyl)methylene)malononitrile were converted to pyrimidine 44c using procedures described for the synthesis of 44a. 1H NMR (400 MHz, CDCl3) δ8.06 (dd, J = 3.1, 1H), 7.75 - 7.66 (m, 2H), 7.75 - 7.66 (m, 3H), 7.55 (dd, J = 3.1, 1H), 4.87 (s, 2H), 3.30 (ddd, J = 12.8, 8.4, 6.3 Hz, 1H), 3.12 (ddd, J = 12.8, 8.6, 6.9 Hz, 1H), 1.85 - 1.65 (m, 2H), 1.55 – 1.40 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 169.7, 166.3, 162.8, 155.0, 145.2, 141.9, 136.4, 130.6, 128.9, 128.9, 123.5, 120.6, 108.9, 54.9, 24.9, 219, 13.5. ESI-MS (m/z): 415.1 [M+H]+. Enantiomers of 44c were separated on a 1 cm Chiralpak AD HPLC column using 30% EtOH/hexanes with a 2.5 mL/min flow rate, monitoring absorption at 315 and 254 nm. Peak 1 eluted at 28–32 min and peak 2 eluted at 35–42 min. Peak 1: [α]D23 = −52.15 (c = 0.39, EtOH), Peak 2: [α]D23 = +65.36 (c = 0.33, EtOH).
6-(Butylsulfinyl)-2-(1-methyl-1H-imidazol-2-yl)-4-phenylthieno[2,3-d]pyrimidin-5-amine (44d)
1-Methyl-1H-imidazole-2-carbothioamide and 2-(ethoxy(phenyl)methylene)malononitrile were converted to pyrimidine 44d using procedures described for the synthesis of 44a. 1H NMR (400 MHz, CDCl3) δ7.75 – 7.63 (m, 2H), 7.63 – 7.49 (m, 3H), 7.29 (s, 1H), 7.07 (s, 1H), 4.85 (s, 2H), 4.18 (s, 3H), 3.29 (ddd, J = 12.8, 8.6, 6.3 Hz, 1H), 3.11 (ddd, J = 12.8, 8.7, 6.9 Hz, 1H), 1.83 – 1.65 (m, 2H), 1.59 – 1.39 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.6, 162.3, 153.2, 143.0, 142.1, 136.6, 130.5, 129.4, 129.0, 128.9, 125.8, 119.32, 107.6, 54.7, 37.1, 25.0, 21.8, 13.7. ESI-MS (m/z): 412.1 [M+H]+.
6-(Butylsulfinyl)-2-(oxazol-4-yl)-4-phenylthieno[2,3-d]pyrimidin-5-amine (44e)
Oxazole-4-carbothioamide and 2-(ethoxy(phenyl)methylene)malononitrile were converted to pyrimidine 44e using procedures described for the synthesis of 44a. 1H NMR (400 MHz, CDCl3) δ 8.51 (d, J = 1.1 Hz, 1H), 8.01 (d, J = 1.1 Hz, 1H), 7.75 – 7.61 (m, 2H), 7.62 – 7.48 (m, 3H), 4.56 (s, 2H), 3.29 (ddd, J = 12.9, 8.8, 6.3 Hz, 1H), 3.09 (ddd, J = 12.9, 8.9, 6.9 Hz, 1H), 1.81 – 1.64 (m, 2H), 1.56 – 1.39 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 399.1 [M+H]+.
6-(Butylsulfinyl)-2,4-diphenylthieno[2,3-d]pyrimidine (48)
Step 1
2,4-Dichlorothieno[2,3-d]pyrimidine35 (100 mg, 0.50 mmol), phenylboronic acid (242 mg, 2.0 mmmol, 4.0 equiv), potassium carbonate (1.5 mmol, 3.0 equiv), Pd(OAc)2 (5 mol %), SPhos (10 mol%) in CH3CN:H2O (1.5:1) were heated at 100 °C overnight. After cooling to rt the reaction mixture was diluted with EtOAc and washed with water. The organic layer was dried over magnesium sulfate and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography on silica gel to afford designed product 2,4-diphenylthieno[2,3-d]pyrimidine in 89% yield. 1H NMR (400 MHz, CDCl3) δ 8.70 – 8.59 (m, 2H), 8.14 – 8.02 (m, 2H), 7.65 – 7.44 (m, 8H). ESI-MS (m/z): 289.0 [M+H]+.
Step 2
To a solution of the diphenylpyrimidine from step 1 (53 mg, 0.28 mmol) in THF (1.0 mL) was added n-BuLi (0.56 mmol, 2.0 equiv, 225 μL of 2.5 M solution in hexanes) at −78 °C. The reaction mixture was stirred for 5 min before 1,2-dibutyldisulfane (1.14 mmol, 4.0 equiv) in THF was added. The reaction mixture was stirred for 1 h at −78 °C and then quenched with water. The crude product was purified by flash chromatography on silica gel to afford 6-(Butylthio)-2,4-diphenylthieno[2,3-d]pyrimidine in 61% yield. 1H NMR (400 MHz, CDCl3) δ 8.62 – 8.56 (m, 2H), 8.06 – 7.98 (m, 2H), 7.61 – 7.41 (m, 8H), 3.01 (t, J = 7.3, 2H), 1.76 – 1.62 (m, 2H), 1.55 – 1.38 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). ESI-MS (m/z): 377.1 [M+H]+.
Step 3
Acetic acid (250 μl) and hydrogen peroxide (20 μl, 30% solution in water) were added to the solution of the thioether from step 3 (35 mg, 0.1 mmol) in chloroform (250 μl). The reaction mixture was stirred at 32 °C for 45 min. Once complete, the reaction was diluted with EtOAc and was washed with saturated NaHCO3 solution, dried over magnesium sulfate, filtered and concentrated under reduce pressure to give sulfoxide 48 in 79% yield. 1H NMR (400 MHz, CDCl3) δ 8.69 – 8.59 (m, 2H), 8.09 – 7.99 (m, 2H), 7.95 (s, 1H), 7.65 – 7.56 (m, 3H), 7.56 – 7.45 (m, 3H), 3.18 – 3.02 (m, 2H), 1.87 – 1.64 (m, 2H), 1.54 – 1.42 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.1, 161.8, 160.8, 146.8, 137.4, 137.12, 131.0, 130.8, 129.3, 129.0, 128.7, 128.6, 125.3, 122.9, 57.4, 24.3, 21.8, 13.6. ESI-MS (m/z): 393.1 [M+H]+.
6-(Butylsulfinyl)-2-phenyl-4-(piperidin-1-yl)thieno[2,3-d]pyrimidine (51)
Step 1
4,6-Dichlorothieno[2,3-b]pyrimidine35 (50 mg, 0.24 mmol) and piperidine (0.36 mmol, 1.5 equiv) were combined in EtOH and stirred at room temperature overnight. The solvent was evaporated under reduced pressure, and the mixture was purified by flash chromatography on silica gel to give 6-chloro-4-(piperidin-1-yl)thieno[2,3-b]pyrimidine in quantitative yield. 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 6.1 Hz, 1H), 7.18 (d, J = 6.2 Hz, 1H), 4.01 – 3.67 (m, 4H), 1.92 – 1.63 (m, 6H). ESI-MS (m/z): 254.0 [M+H]+.
Step 2
To the solution of the thiophene from step 1 (52 mg, 0.20 mmol) in THF was added n-BuLi (0.4 mmol, 2.0 equiv, 1.6 M solution in hexanes) at −78 °C. The reaction mixture was stirred for 5 min before 1,2-dibutyldisulfane (0.80 mmol, 4.0 equiv) in THF was added. The reaction mixture was stirred for additional 1h at −78 °C and then quenched by the addition of water. The crude product was purified by flash chromatography on silica gel to afford 2-(butylthio)-6-chloro-4-(piperidin-1-yl)thieno[2,3-b]pyrimidine in 74 % yield. 1H NMR (400 MHz, CHCl3) δ 7.24 (s, 1 H), 3.93 – 3.74 (m, 4H), 2.83 (t, J = 7.3 Hz, 2H), 1.82 – 1.66 (m, 6H), 1.66 – 1.53 (m, 2H), 1.49 – 1.33 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.6, 157.6, 155.2, 132.5, 124.9, 115.0, 48.2, 37.5, 31.5, 25.8, 24.3, 21.6, 13.6. ESI-MS (m/z): 342.1 [M+H]+.
Step 3
The chloride from step 2 (52 mg, 0.15 mmol), phenylboronic acid (27 mg, 0.22 mmol, 1.5 equiv), potassium carbonate (0.3 mmol, 2.0 equiv), and PdCl2dtbpf (10 mol%), in CH3CN (1 mL) and H2O (0.5 mL) were heated at 100 °C overnight. After cooling to rt the reaction mixture was diluted with EtOAc and washed with water. The organic layer was dried over magnesium sulfate and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography on silica gel to afford 2-(butylthio)-6-phenyl-4-(piperidin-1-yl)thieno[2,3-b]pyrimidine in 19% yield. 1H NMR (400 MHz, CHCl3) δ 8.49 – 8.36 (m, 2H), 7.51 – 7.36 (m, 3H), 7.29 (s, 1H), 3.95-3.85 (m, 4H), 2.90 (t, J = 7.4 Hz, 2H), 1.76 – 1.73 (m, 6H), 1.70 – 1.59 (m, 2H), 1.48 – 1.39 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). ESI-MS (m/z): 384.0 [M+H]+.
Step 4
Acetic acid (50 μl) and hydrogen peroxide (5.0 μl, 30 % solution in water) were added to the solution of the thioether from step 3 (10 mg, 0.026 mmol) in chloroform (50 μl). The reaction mixture was stirred at 32 °C for 45 min. Once complete, the reaction was diluted with EtOAc and was washed with saturated NaHCO3 solution, dried over magnesium sulfate, filtered and concentrated under reduce pressure to give sulfoxide 51 in 76 % yield. 1H NMR (400 MHz, CDCl3) δ 8.54 – 8.34 (m, 2H), 7.73 (s, 1H), 7.55 – 7.36 (m, 3H), 4.09 – 3.86 (m, 4H), 3.24 – 2.94 (m, 2H), 1.97 – 1.36 (m, 10H), 0.93 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 400.1 [M+H]+.
2-(Butylsulfinyl)-5-(thiophen-2-yl)thiazolo[5,4-b]pyridine (53)
Step 1
Potassium ethyl xanthate (1.9 g, 0.012 mol) and anhydrous N-methyl-2-pyrrolidone (14.1 mL) were added to 2,6-dichloropyridin-3-amine (1.0 g, 0.0061 mol) under N2. The solution was heated at 170 °C for 3.5 hours. The reaction mixture was allowed to cool to room temperature, acidified to pH 5 using AcOH, diluted in EtOAc, and washed several times with H2O. The organic layer was separated, dried over MgSO4, and concentrated under reduced pressure. This gave 5-chlorothiazolo[5,4-b]pyridine-2-thiol (52)20 as a red solid in 18% yield which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 8.4 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H). ESI-MS (m/z): 202.9.
Step 2
K2CO3 (75 mg, 0.54 mmol), 1-bromobutane (53.3 μL, 0.493 mmol), 18-crown-6 (13.2 mg, 0.0493 mmol), and DMF (3.4 mL) were added to the thiol from step 1, and the solution was heated at 80 °C for 3 hours. The solution was diluted with EtOAc, washed with H2O, and the organic layer was separated, dried over MgSO4, and concentrated under reduced pressure. The crude product was purified using flash chromatography on silica gel to give 2-(butylthio)-5-chlorothiazolo[5,4-b]pyridine in 76 % isolated yield. 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.5 Hz, 1H), 7.30 (d, J = 8.5 Hz, 1H), 3.31 (t, J = 7.3 Hz, 2H), 1.76 (p, J = 7.5 Hz, 2H), 1.52 – 1.39 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). ESI-MS (m/z): 259.0 [M+H]+.
Step 3
2-Thienylboronic acid (49.4 mg, 0.386 mmol), CsCO3 (126 mg, 0.386 mmol), Pd(dppf)Cl2 (15.8 mg, 0.0193 mmol), CuCl (19.1 mg, 0.193 mmol) and DMF (1 mL) were added to the pyridyl chloride from step 2 (50 mg, 0.19 mmol) under N2. The reaction mixture was heated to 100 °C for 30 minutes. Then the N2 was disconnected and the vial was capped and sealed with teflon tape and allowed to stir at 100 °C overnight. The reaction mixture was diluted in EtOAc, washed with H2O, and the organic layer was separated, dried over MgSO4, filtered, and concentrated under reduced pressure. Chromatography on silica gel provided 2-(butylthio)-5-(thiophen-2-yl)thiazolo[5,4-b]pyridine in 31% yield. 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.6 Hz, 1H), 7.63 (dd, J = 3.8, 1.1 Hz, 1H), 7.51 (dd, J = 5.1, 1.1 Hz, 1H), 7.23 (d, J = 8.6 Hz, 1H), 7.14 (dd, J = 5.0, 3.8 Hz, 1H), 3.24 (t, J = 7.3 Hz, 2H), 1.78 – 1.66 (m, 2H), 1.57 – 1.41 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H). ESI-MS (m/z): 307.0 [M+H]+.
Step 4
To a solution of the thioether from step 3 (18 mg. 0.059 mmol) in chloroform (142 μL) and AcOH (142 μL) was added H2O2 (12.0 μL, 0.118 mmol, 30% solution in H2O). The reaction mixture was heated at 35 °C for 2.5 hours. The reaction was diluted with EtOAc and washed with saturated NaHCO3. The organic layer was separated, dried over MgSO4, filtered, and concentrated under reduced pressure to give analog 53 in 60% yield. 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 3.8, 1.1 Hz, 1H), 7.61 (dd, J = 5.0, 1.1 Hz, 1H), 7.19 (dd, J = 5.0, 3.8 Hz, 1H), 3.15 (ddd, J = 13.3, 9.8, 6.0 Hz, 1H), 2.96 (ddd, J = 13.3, 9.9, 4.9 Hz, 1H), 1.98 – 1.80 (m, 1H), 1.60 – 1.52 (m, 1H), 1.52 – 1.37 (m, 2H), 0.93 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 163.5, 161.4, 158.1, 147.7, 136.8, 131.0, 130.6, 130.1, 128.4, 117.8, 54.6, 23.8, 21.9, 13.7. ESI-MS (m/z): 323.0 [M+H]+.
2-(Butylsulfinyl)-5-(thiophen-2-yl)-3H-imidazo[4,5-b]pyridine (57)
Step 1
2-Thiophene boronic acid (742 mg, 5.8 mmol, 2.0 equiv), 6-chloro-3-nitropyridin-2-amine (500 mg, 2.9 mmol, 1.0 equiv), cesium carbonate (5.8 mmol, 2.0 equiv), PdCl2dppf (10 mol%), CuCl (2.9 mmol, 1.0 equiv) in DMF were heated at 100 °C for 12 h. After cooling to rt the reaction mixture was diluted with EtOAc and washed with water and then brine. The organic layer was dried over magnesium sulfate and the solvent was removed under reduced pressure. The crude product was purified by column chromatography (hexanes/EtOAc: 8/2) to afford 3-nitro-6-(thiophen-2-yl)pyridin-2-amine in 63 % yield. 1H NMR (400 MHz, CDCl3) δ 8.42 (d, J = 8.7 Hz, 1H), 7.70 (dd, J = 3.8, 1.1 Hz, 1H), 7.54 (dd, J = 5.0, 1.1 Hz, 1H), 7.15 (dd, J = 5.0, 3.8 Hz, 1H), 7.09 (d, J = 8.7 Hz, 1H). ESI-MS (m/z): 222.1 [M+H]+.
Step 2
The nitroarene from step 1 (1.20 mmol, 265.4 mg) was dissolved in a 5:1 acetone/water mixture. Zinc powder (12.0 mmol, 784 mg, 10 equiv) and ammonium chloride (18 mmol, 962.5 mg, 15 equiv) were added to the solution, which was stirred at room temperature for 1 hour. The solution was then filtered through a celite pad and washed with ethyl acetate. The filtrate was washed twice with brine then the aqueous layer was back extracted with EtOAc. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. Further purification by column chromatography gave 118.2 mg of 6-(thiophen-2-yl)pyridine-2,3-diamine (52% yield). 1H NMR (400 MHz, CD3OD) δ 7.34 (dd, J = 3.6, 1.1 Hz, 1H), 7.25 (dd, J = 5.1, 1.1 Hz, 1H), 7.00 (dd, J = 5.1, 3.6 Hz, 1H), 6.94 (d, J = 7.8 Hz, 1H), 6.90 (d, J = 7.8 Hz, 1H). ESI-MS (m/z): 192.1 [M+H]+.
Step 3
Thiourea (16.97 mmol, 223.0 mg, 5 equiv) was added to the diamine from step 2. The solution was heated at 170 °C for 2 hours. The addition of ethanol at room temperature produced solid, which was filtered to give 112.5 mg of 5-(thiophen-2-yl)-1,3-dihydro-2H-imidazo[4,5-b]pyridine-2-thione (82% yield). 1H NMR (400 MHz, DMSO-d6) δ 13.12 (s, 1H), 12.75 (s, 1H), 7.69 (dd, J = 3.7, 1.1 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.56 (dd, J = 5.0, 1.1 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 7.12 (dd, J = 5.0, 3.7 Hz, 1H). ESI-MS (m/z): 234.0 [M+H]+.
Step 4
A mixture of the thiourea from step 3 (0.39 mmol, 92 mg), potassium carbonate (0.45 mmol, 61.9 mg, 1.1 equiv), 1-bromobutane (0.39 mmol, 42.8 μL, 1 equiv), 18-crown-6 (0.039 mmol, 10.5 mg, 0.1 equiv), and DMF (2.67 mL) was heated at 80 °C for 3 hours. After cooling to room temperature, the reaction mixture was diluted with EtOAc and washed with water. The organic layer was dried over magnesium sulfate, filtered, and concentrated under high pressure to give 74.4 mg of 2-(butylthio)-5-(thiophen-2-yl)-3H-imidazo[4,5-b]pyridine (65 % yield). 1H NMR (400 MHz, CDCl3) δ 7.95 – 7.83 (m, 1H), 7.61 – 7.53 (m, 2H), 7.36 (d, J = 5.1, 1H), 7.16 – 7.06 (m, 1H), 3.28 (t, J = 7.3 Hz, 2H), 1.76 – 1.62 (m, 2H), 1.48 – 1.32 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H). ESI-MS (m/z): 290.1 [M+H]+.
Step 5
Chloroform (450 μL), acetic acid (450 μL), and hydrogen peroxide (0.37 mmol, 2.0 equiv, 40 μL) were added to the thioether from step 4 (54.4 mg, 0.19 mmol) and heated at 45 °C for 2.5 hours. The solution was then diluted with EtOAc and washed with 10% acetic acid. The organic layer was separated, dried with magnesium sulfate, filtered, concentrated, and purified via chromatography on silica gel to give 16.8 mg of sulfoxide 57 (30% yield). 1H NMR (400 MHz, CDCl3) δ 12.17 (s, 1H), 8.06 (d, J = 8.5 Hz, 1H), 7.84 – 7.60 (m, 2H), 7.46 – 7.29 (m, 1H), 7.16 – 7.05 (m, 1H), 3.35 (ddd, J = 13.4, 9.8, 5.8 Hz, 1H), 3.26 (ddd, J = 13.4, 9.8, 5.4 Hz, 1H), 2.16 – 1.89 (m, 2H), 1.59 – 1.39 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.0, 149.3, 147.4, 144.4, 135.2, 128.7, 128.2, 127.9, 125.3, 115.7, 54.7, 23.6, 21.8, 13.6. ESI-MS (m/z): 306.1 [M+H]+.
Inhibition of 15-PGDH
As described previously,14 test compounds were dissolved in DMSO as 50 mM stock solutions. Reactions were assembled with 5 nM 15-PGDH enzyme, and 2-fold dilutions of inhibitor from 2500 to 0.15 nM, plus 150 μM NAD(+) and 25 μM PGE2 in reaction buffer (50 mM Tris-HCl, pH7.5, 0.01% Tween 20). The reaction mix was incubated for 15 min at 25 °C in an Envision Reader (PerkinElmer). Enzyme activity was determined by following generation of NADH as assayed by recording fluorescence at Ex/Em=340 nM/485 nM every 30s for 3 minutes, commencing immediately after addition of PGE2. IC50 values were calculated with GraphPad Prism 5 software using the sigmoidal dose-response function and plotted against inhibitor concentration.
Elevation of PGE2 levels in A549 cell culture
The A549 cell line was maintained in F12K medium supplemented with 10% fetal calf serum (FBS) and 50 μg/mL gentamicin in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were plated in duplicate in a 24-well plate (1 mL per well) at 1×105 cells per well and grown for 24 h before stimulation with IL-1β (1 ng/mL) overnight (16 h) to induce COX2 expression and PGE2 production. Cells were then treated for an additional 8 hours with fresh medium containing the indicated concentration of 15-PGDH inhibitor. Media was then collected and the level of PGE2 analyzed using a PGE2 enzyme immunoassay Kit (R&D System). Data were collected from four independent experiments. Results were tabulated graphically with error bars corresponding to standard error of the means and compared using 2-tailed t-tests.
Evaluation of PGE2 levels in mouse tissue
Female CB57/L6 mice (8 to 12 weeks of age) were obtained from Jackson laboratories. The animals were housed in standard microisolator cages and maintained on a defined irradiated diet (Prolab Isopro RMH 3000) and autoclaved water. Three-five mice/group were administered a 0.2 mL dose of inhibitor corresponding to 2.5 mg/kg by IP injection formulated as follows. (+)-1: 10% EtOH, 5% Cremophor EL, and 85% D5W; (+)-12q: 10% EtOH/10% PEG400/80% 50mM citrate-phosphate buffer pH 3.6; (+)-32: 10% DMSO, 10% Cremophor, and 80% D5W pH 7.4; (+)-44c: 6% DMSO, 4% EtOH; 10% Cremophor EL, and 80% Lactic acid pH 5.5. Solid tissues were harvested, rinsed in ice-cold PBS containing indomethacin (5.6 μg/ml), and snap frozen in liquid nitrogen. Marrow was flushed in 1.5 ml ice cold PBS with indomethacin, pelleted at 2000 rpm for 5 minutes in an Eppendorf centrifuge, and snap frozen in liquid nitrogen. Frozen samples were pulverized in liquid nitrogen. The powder was transferred to an Eppendorf tube with 500 μl of cold PBS with indomethacin and then homogenized using a tissue homogenizer. The suspension was sonicated in ice water for 5 min using cycles of 20 seconds of sonication with 20 seconds of cooling, followed by centrifugation for 10 minutes at 12,000 rpm. The PGE2 level of the supernatant was measured using a PGE2 ELISA Kit (R&D Systems, cat. # KGE004B) and normalized to protein concentration measured by BCA assay (Thermo Scientific, cat. #23225) and expressed as ng PGE2/mg protein. Each sample was assayed in triplicate.
Solubility determination
1 ml of citrate-phosphate buffer (0.1 M citric acid/disodium phosphate buffer), at pH 7 unless otherwise indicated, was added to a 20 ml scintillation vial. Determination of the solubility of the salt form of s-12q was performed in dH20. Compound was slowly added to buffer until super saturated. The solution was stirred overnight (16hr) protected from light at RT. The solution was transferred to a Teflon microcentrifuge tube and spun at 16,100×g for 5 min. The supernatant was transferred to a second clean Teflon microcentrifuge tube and spun again. 0.1 ml of supernatant from the top of the centrifuge tube was carefully removed and placed in a standard microcentrifuge tube containing 0.2 ml of methanol or acetonitrile containing 0.15% formic acid and either tolbutamide or n-benzylbenzamide internal standard (organic crash). The solution was vortexed for 15 sec, incubated at RT for 10 min, centrifuged at 16,100 × g, and transferred to an HPLC vial containing an insert. As necessary, dilutions of compound were prepared prior to mixing with organic crash. Compound concentration was determined by LC-MS/MS analysis in comparison to a standard curve prepared by addition of varying amounts of compounds to 0.3 ml of a 1:2 mixture of 0.1M citrate-phosphate buffer:organic crash identical to that used for the solubility determination. Standard curve solutions were vortexed, centrifuged, and processed in an identical fashion to solubility determination tubes. Analytical methods were developed for each compound using a SCIEX (Framingham, MA) 3200 or 4000-QTrap, combination triple quadrupole/ion trap instruments. The parent ion and the two most prominent daughter ions were followed to confirm compound identity, although only the most abundant daughter was used for quantitation. A Shimadzu (Columbia, MD) Prominence LC with Agilent C18 XDB column (5 micron packing; 50 × 4.6 mm) was used for chromatography.
Liver S9 stability
Female ICR/CD-1 mouse S9 fractions were purchased from BioreclamationIVT (Chestertown, MD). 0.025 ml (0.5 mg) of S9 protein was added to a 15 ml glass screw cap tube. 0.350 ml of a 50 mM Tris, pH 7.5 solution, containing the compound of interest was added on ice. The final concentration of compound after addition of all reagents was 2 μM. 0.125 ml of an NADPH-regenerating system (1.7 mg/ml NADP, 7.8 mg/ml glucose-6-phosphate, 6 U/ml glucose-6-phosphate dehydrogenase in 2% w/v NaHCO3/10 mm MgCl2) was added for analysis of Phase I metabolism. The tube was then placed in a 37°C shaking water bath. At varying time points after addition of phase I cofactors, the reaction was stopped by the addition of 0.5 ml of methanol containing 0.2% formic acid and either tolbutamide or n-benzylbenzamide IS. The samples were incubated 10′ at RT and then spun at 16,100 × g for 5 min in a microcentrifuge. The supernatant was analyzed by LC-MS/MS as described above. The method described in McNaney et al.36 was used with modification for determination of metabolic stability half-life by substrate depletion. A “% remaining” value was used to assess metabolic stability of a compound over time. The LC-MS/MS peak area of the incubated sample at each time point was divided by the LC-MS/MS peak area of the time 0 (T0) sample and multiplied by 100. The natural Log (LN) of the % remaining of compound was then plotted versus time (in min) and a linear regression curve plotted going through y-intercept at LN(100). The metabolism of some compounds failed to show linear kinetics at later time point, so those time points were excluded. The half-life (T ½) was calculated as T ½ = 0.693/slope.
Mouse PK analysis
Female CD-1 mice (5–6 weeks of age) were obtained from Charles River. The animals were housed in standard microisolator cages and were administered inhibitor compounds at 10 mg/kg in 0.2 ml by IP injection formulated as follows. 1: 10% EtOH, 5% Cremophor EL, and 85% D5W; 12q: 10% EtOH/10% PEG400/80% 50mM citrate-phosphate buffer pH 3.6; 12w: 100% 50 mM citrate-phosphate buffer pH3.6; 32: 10% DMSO, 10% Cremophor, and 80% D5W pH 7.4; 44c: 6% DMSO, 4% EtOH; 10% Cremophor EL, and 80% Lactic acid pH 5.5. Animals were euthanized by inhalation overdose of CO2 in groups of 3 at 10, 30, 90, 180, 360, 960, and 1440 min post-dose and blood collected by cardiac puncture, using acidified citrate dextrose (ACD) as the anti-coagulant. Plasma was isolated from blood by centrifugation at 9600 × g for 10 min and stored at −80°C until analysis. 0.1 ml of plasma was precipitated with 0.2ml of an organic crash solution containing either methanol or acetonitrile, 0.15% formic acid and an internal standard (either tolbutamide or n-benzylbenzamide). Extraction conditions were optimized prior to PK analysis for efficient and reproducible recovery over a three log range of concentrations. The solution was centrifuged twice at 16,100 × g twice for 5 min. The final supernatant was analyzed by LC-MS/MS as described above and compound concentrations determined in reference to a standard curve prepared by addition of the appropriate compound to blank plasma. A value of 3x above the signal obtained in the blank plasma was designated the limit of detection (LOD). The limit of quantitation (LOQ) was defined as the lowest concentration at which back calculation yielded a concentration within 20% of the theoretical value and above the LOD signal. The LOQ values were as follows: 1-(+): 1 ng/ml; 1-(rac): 10 ng/ml; 12q: 5 ng/ml; 12w: 1 ng/ml; 32: 5 ng/ml; 44c: 5 ng/ml. Pharmacokinetic parameters were determined using the noncompartmental analysis tool in Phoenix WinNonlin (Certara, Corp. Princeton, NJ).
Supplementary Material
Figure 1.

Biological synthesis and degradation of prostaglandin E2 (PGE2)
Acknowledgments
Financial support from the NIH (R01 GM102403, R01 CA216863, JMR) the Welch Foundation (I-1612, JMR), CPRIT (RP110708-C3, NSW). The Preclinical Pharmacology Core (NSW) is supported in part by funding from the Institute for Innovations in Medicine at UTSW. Additional funding from the NIH (P50 CA150964, SDM) and a Scholar Innovator Award from the Harrington Discovery Institute (SDM).
ABBREVIATIONS
- ADME
adsorption distribution metabolism and excretion
- BMT
bone marrow transplantation
- COX
cyclooxygenase
- dmPGE2
16,16-dimethyl-PGE2
- ELISA
enzyme-linked immunosorbent assay
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- IV
intravenous
- NAD+
nicotinamide adenine dinucleotide
- 15-PGDH
15-prostaglandin dehydrogenase
- PGE2
prostaglandin E2
- PGES
prostaglandin E synthase
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Additional pharmacokinetic and pharmacodynamic data in mice. Spectral data for synthetic compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Funk CD. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 2.Kalish BT, Kieran MW, Puder M, Panigrahy D. The growing role of eicosanoids in tissue regeneration, repair, and wound healing. Prostaglandins Other Lipid Mediat. 2013;104–105:130–138. doi: 10.1016/j.prostaglandins.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Jung P, Sato T, Merlos-Suarez A, Barriga FM, Iglesias M, Rossell D, Auer H, Gallardo M, Blasco MA, Sancho E, Clevers H, Batlle E. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 2011;17:1225–1227. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 4.Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, Zon LI. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136:1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, Weber GJ, Bowman TV, Jang IH, Grosser T, FitzGerald GA, Daley GQ, Orkin SH, Zon LI. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007–1011. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goessling W, Allen RS, Guan X, Jin P, Uchida N, Dovey M, Harris JM, Metzger ME, Bonifacino AC, Stroncek D, Stegner J, Armant M, Schlaeger T, Tisdale JF, Zon LI, Donahue RE, North TE. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011;8:445–458. doi: 10.1016/j.stem.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoggatt J, Singh P, Sampath J, Pelus LM. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood. 2009;113:5444–5455. doi: 10.1182/blood-2009-01-201335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pelus LM, Hoggatt J, Singh P. Pulse exposure of haematopoietic grafts to prostaglandin E2 in vitro facilitates engraftment and recovery. Cell Proliferation. 2011;44:22–29. doi: 10.1111/j.1365-2184.2010.00726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cutler C, Multani P, Robbins D, Kim HT, Le T, Hoggatt J, Pelus LM, Desponts C, Chen Y-B, Rezner B, Armand P, Koreth J, Glotzbecker B, Ho VT, Alyea E, Isom M, Kao G, Armant M, Silberstein L, Hu P, Soiffer RJ, Scadden DT, Ritz J, Goessling W, North TE, Mendlein J, Ballen K, Zon LI, Antin JH, Shoemaker DD. Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood. 2013:3074–3081. doi: 10.1182/blood-2013-05-503177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung P, Sato T, Merlos-Suarez A, Barriga FM, Iglesias M, Rossell D, Auer H, Gallardo M, Blasco MA, Sancho E, Clevers H, Batlle E. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 2011;17:1225–1227. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 11.Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, Tsuboi K, Sugimoto Y, Kobayashi T, Miyachi Y, Ichikawa A, Narumiya S. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Tai HH, Ensor CM, Tong M, Zhou H, Yan F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002;68–69:483–493. doi: 10.1016/s0090-6980(02)00050-3. [DOI] [PubMed] [Google Scholar]; (b) Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: Structure and biological functions. Curr Pharm Des. 2006;12:955–962. doi: 10.2174/138161206776055958. [DOI] [PubMed] [Google Scholar]
- 13.Myung SJ, Rerko RM, Yan M, Platzer P, Guda K, Dotson A, Lawrence E, Dannenberg AJ, Lovgren AK, Luo G, Pretlow TP, Newman RA, Willis J, Dawson D, Markowitz SD. 15-hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci. 2006;103:12098–12102. doi: 10.1073/pnas.0603235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Zhang Y, Desai A, Yang SY, Bae KB, Antczak MI, Fink SP, Tiwari S, Willis JE, Williams NS, Dawson DM, Wald D, Chen W-D, Wang Z, Kasturi L, Larusch GA, He L, Cominelli F, Di Martino L, Djuric Z, Milne GL, Chance M, Sanabria J, Dealwis C, Mikkola D, Naidoo J, Wei S, Tai H-H, Gerson SL, Ready JM, Posner B, Willson JKV, Markowitz SD. Inhibition of the prostaglandin-degrading enzyme 15-pgdh potentiates tissue regeneration. Science. 2015;348:aaa2340. doi: 10.1126/science.aaa2340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Markowitz S, Willson JKV, Posner BA, Ready J, Zhang Y, Tai H-H, Moss M, Antczak M. WO2013158649A1. Compositions and methods of modulating 15-hydroxy-prostaglandin dehydrogenase. 2013; (c) Markowitz S, Willson JKV, Posner B, Ready J, Antczak M, Zhang Y, Desai A, Gerson S, Greenlee W. WO2015065716A1. Preparation of thienopyridines and similar bicyclic heterocyclic compounds as modulators of 15-hydroxyprostaglandin dehydrogenase type i for therapy. 2015; (c) Markowitz SD, Ready J, Zhang Y, Antczak M, Willson JKV, Posner BA, Greenlee W. WO2016168472A1. Compositions and methods of modulating short-chain dehydrogenase activity. 2016
- 15.Michelet JF, Bernard B, Rozot R, Boulle C. US20080206320 A1. Composition comprising at least one 15-PGDH inhibitor. 2008
- 16.Rozot R, Boulle C, Dalko M. US7396525 B2. Care/makeup compositions comprising a 2-alkylideneaminooxyacetamide compound for stimulating the growth of the hair or eyelashes and/or slowing loss thereof. 2008
- 17.(a) Wu Y, Karna S, Choi CH, Tong M, Tai HH, Na DH, Jang CH, Cho H. Synthesis and biological evaluation of novel thiazolidinedione analogues as 15-hydroxyprostaglandin dehydrogenase inhibitors. J Med Chem. 2011;54:5260–5264. doi: 10.1021/jm200390u. [DOI] [PubMed] [Google Scholar]; (b) Choi D, Piao YL, Wu Y, Cho H. Control of the intracellular levels of prostaglandin E2 through inhibition of the 15-hydroxyprostaglandin dehydrogenase for wound healing. Bioorg Med Chem. 2013;21:4477–4484. doi: 10.1016/j.bmc.2013.05.049. [DOI] [PubMed] [Google Scholar]
- 18.(a) Niesen FH, Schultz L, Jadhav A, Bhatia C, Guo K, Maloney DJ, Pilka ES, Wang M, Oppermann U, Heightman TD, Simeonov A. High-affinity inhibitors of human NAD+-dependent 15-hydroxyprostaglandin dehydrogenase: Mechanisms of inhibition and structure-activity relationships. PLoS One. 2010;5:e13719. doi: 10.1371/journal.pone.0013719. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Duveau DY, Yasgar A, Wang Y, Hu X, Kouznetsova J, Brimacombe KR, Jadhav A, Simeonov A, Thomas CJ, Maloney DJ. Structure–activity relationship studies and biological characterization of human NAD+-dependent 15-hydroxyprostaglandin dehydrogenase inhibitors. Bioorg Med Chem Lett. 2014;24:630–635. doi: 10.1016/j.bmcl.2013.11.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.cLogP calculations performed in Chem Bio Draw.
- 20.Layton ME, Kelly MJ. WO2011/137046 A1. Substituted 1,3-benzothiazol-2(3H)-ones and [1,3]thiazolo[5,4-b]pyridin-2(1H)-ones as positive allosteric modulators of mGlur2. 2011
- 21.The absolute stereochemistry was determined by X-ray crystallography. See Supporting information.
- 22.Parveen H, Iqbal PF, Azam A. Synthesis and characterization of a new series of hydroxy pyrazolines. Synth Commun. 2008;38:3973–3983. [Google Scholar]
- 23.Kalugin VE, Shestopalov AM, Litvinov VP. Functionalized sulfur-containing compounds. 13. Synthesis of substituted 3-amino-2-(organylsulfinyl)-and-(organylsulfonyl)thieno[2,3-b]pyridines. Russ Chem Bull. 2006;55:529–534. [Google Scholar]
- 24.Hoover DJ, Witter KG. WO2008/004117 A1. Selective azole PDE10A inhibitor compounds. 2008
- 25.Dondoni A, Marra A, Merino P. Installation of the pyruvate unit in glycidic aldehydes via a wittig olefination-michael addition sequence utilizing a thiazole-armed carbonyl ylide. A new stereoselective route to 3-deoxy-2-ulosonic acids and the total synthesis of DAH, KDN, and 4-epi-KDN. J Am Chem Soc. 1994;116:3324–3336. [Google Scholar]
- 26.Pippel DJ, Mapes CM, Mani NS. Reactions between weinreb amides and 2-magnesiated oxazoles: A simple and efficient preparation of 2-acyl oxazoles. J Org Chem. 2007;72:5828–5831. doi: 10.1021/jo070646a. [DOI] [PubMed] [Google Scholar]
- 27.Ohmiya H, Yoshida M, Sawamura M. Copper-catalyzed conjugate additions of alkylboranes to imidazolyl α,β-unsaturated ketones: Formal reductive conjugate addition of terminal alkenes. Org Lett. 2011;13:482–485. doi: 10.1021/ol102819k. [DOI] [PubMed] [Google Scholar]
- 28.Abdelhamid AO, Al-Atoom AA. Reactions with hydrazonoyl halides 45: Synthesis of some new triazolino[4,3-a]pyrimidines, pyrazolo[3,4-d]pyridazines, isoxazolo[3,4-d]pyridazines, and thieno[2,3-b]pyridines. Synth Commun. 2006;36:97–110. [Google Scholar]
- 29.Zhang J, Didierlaurent S, Fortin M, Lefrançois D, Uridat E, Vevert JP. Potent nonpeptide endothelin antagonists: Synthesis and structure–activity relationships of pyrazole-5-carboxylic acids. Bioorg Med Chem Lett. 2000;10:2575–2578. doi: 10.1016/s0960-894x(00)00513-8. [DOI] [PubMed] [Google Scholar]
- 30.Iglesias LE, Baldessari A, Gros EG. Lipase-catalyzed chemospecific o-acylation of 3-mercapto-1-propanol and 4-mercapto-1-butanol. Bioorg Med Chem Lett. 1996;6:853–856. [Google Scholar]
- 31.Zhang W, Ready JM. The ketene-surrogate coupling: Catalytic conversion of aryl iodides to aryl ketenes via ynol ethers. Angew Chem Int Ed. 2014;53:8980–8984. doi: 10.1002/anie.201405036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoogendoorn S, Blom AEM, Willems LI, van der Marel GA, Overkleeft HS. Synthesis of pH-activatable red fluorescent bodipy dyes with distinct functionalities. Org Lett. 2011;13:5656–5659. doi: 10.1021/ol202379w. [DOI] [PubMed] [Google Scholar]
- 33.Nakayama Y, Ito H, Iwata T, Nakamura J, Matsushima Y. US2006/0202197 A1. Platinum complex and light-emitting device. 2006
- 34.Lorente A, García Navío JL, Vaquero JJ, Soto JL. Synthesis of heterocyclic compounds. XXXIX. Synthesis of 5-cyano-2-phenyl-4-thioxo-3,4-dihydropyrimidines. J Heterocyclic Chem. 1985;22:49–51. [Google Scholar]
- 35.Bourke DG, Burns CJ, Cuzzupe AN, Feutrill JT, Kling MR, Nero TL. WO2009/062258. N-Containing heterocyclic compounds. 2009
- 36.McNaney CA, Drexler DM, Hnatyshyn SY, Zvyaga TA, Knipe JO, Belcastro JV, Sanders M. An automated liquid chromatography-mass spectrometry process to determine metabolic stability half-life and intrinsic clearance of drug candidates by substrate depletion. Assay Drug Dev Technol. 2008;6:121–129. doi: 10.1089/adt.2007.103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.