

Summary
RAS genes are frequently mutated in cancers, yet an effective treatment has not been developed. This is partly due to an incomplete understanding of signaling within Ras-related tumors. To address this, we performed a genetic screen in Drosophila, aiming to find mutations that cooperate with oncogenic Ras (RasV12) to induce tumor overgrowth and invasion. We identified fiery mountain (fmt), a regulatory subunit of the protein phosphatase 6 (PP6) complex, as a tumor suppressor that synergizes with RasV12 to drive JNK-dependent tumor growth and invasiveness. We show that Fmt negatively regulates JNK upstream of dTAK1. We further demonstrate that disruption of PpV, the catalytic subunit of PP6, mimics fmt loss of function induced tumorigenesis. Finally, Fmt synergizes with PpV to inhibit JNK-dependent tumor progression. Our data here further highlight the power of Drosophila as a model system to unravel molecular mechanisms that may be relevant to human cancer biology.
Keywords: Ras, Fmt, PpV, JNK, tumorigenesis, Drosophila
Graphical abstract
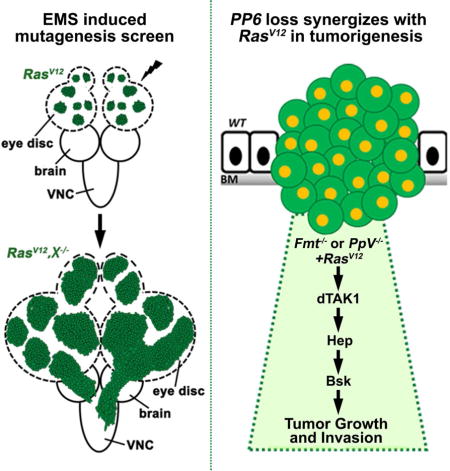
Introduction
The RAS family genes (HRAS, NRAS and KRAS) are the most frequently mutated genes in cancer (Ryan et al., 2015; Vogelstein et al., 2013). The discovery of RAS in 1982 has catapulted the pursuit of an anti-RAS therapy to the forefront of pharmaceutical cancer research (Cox et al., 2014; Der et al., 1982; Ryan et al., 2015; Samatar and Poulikakos, 2014). However, despite several decades of concentrated effort and breakthroughs, successful therapeutic methods targeting RAS-related cancers remain to be developed (Cox et al., 2014; Ryan et al., 2015; Stephen et al., 2014), largely due to the lack of a systematic understanding of the complex signaling crosstalk within RAS tumors.
A large scale screenable tool would be beneficial to comprehensively dissect genetic alternations in the RAS tumors. Given conservation of cancer-related genes and signaling pathways between humans and Drosophila (Reiter et al., 2001), and taking into account the difficulty of systematically study the mechanisms of tumor progression in patients, Drosophila has been widely used as an in vivo model to study the genetic mechanism and signaling pathways that regulate various aspects of cancer biology, using relative easy genetic manipulation (deletion/overexpression) and large scale screens (Brumby et al., 2011; Chi et al., 2010; Khoo et al., 2013; Pastor-Pareja and Xu, 2013). Indeed, over the past decade, numerous tumor growth and invasion models have been established in larvae and adult flies (Figueroa-Clarevega and Bilder, 2015; Gonzalez, 2013; Kwon et al., 2015; Pagliarini and Xu, 2003; Pastor-Pareja and Xu, 2013; Rudrapatna et al., 2012; Willoughby et al., 2013). In particular, oncogenic Ras (RasV12) has been shown to cooperate with mutants that disrupt cell polarity to drive tumor growth and invasion in fly (Brumby and Richardson, 2003; Pagliarini and Xu, 2003). The existence of these models and tools makes Drosophila a fantastic in vivo model to dissect RasV12 mediated tumorigenesis.
Here we performed a large scale EMS-induced genetic screen, aiming to unearth tumor suppressors that can synergistically enhance RasV12 induced tumor growth. We identify Fmt as a tumor suppressor that negatively regulates JNK signaling. The combination of RasV12 with loss of fmt induces dramatic tumor overgrowth and invasion behavior upstream of dTAK1. We further show that PpV, the homolog of mammalian catalytic subunit of the protein phosphatase 6 (PP6), also functions as a tumor suppressor and synergizes with Fmt in vivo to inhibit JNK mediated tumorigenesis. These findings not only shed light on the molecular mechanism of PP6-mediated tumorigenesis, but also provide a potential target for drug development for oncogenic Ras related cancer.
Results
Fmt encodes a novel tumor suppressor that synergizes with RasV12 to drive tumorigenesis and invasion
Based on the observation that oncogenic Ras (RasV12) alone can only induce mild benign tumors (Brumby and Richardson, 2003; Pagliarini and Xu, 2003), we utilized the ey-FLP based MARCM (mosaic analysis with repressible cell marker) technique to conduct an EMS-induced forward genetic screen on Drosophila chromosome 3L, aiming to identify novel tumor suppressors that can synergize with RasV12 to drive dramatic tumor overgrowth and invasion in the developing eye (Figure 1A). We screened more than 20,500 mutagenized chromosomes and successfully identified over 200 mutations that accelerate the growth of RasV12 tumors (details of the screen will be published elsewhere). Among the candidates, we isolated a recessive-lethal complementation group consisting of four alleles that exhibited invasive tumor overgrowth (Figures S1A–B). Subsequent deficiency mapping revealed that each one disrupted the gene CG10289, which encodes a highly conserved 991 amino acid protein, homologous to human PPP6R1, PPP6R2 and PPP6R3 (protein phosphatase 6 regulatory subunits) (Figure S1C). We named CG10289 fiery mountain (fmt), after a famous Chinese mountain in Xinjiang province.
Figure 1. Loss of Fmt synergizes with RasV12 to induce tumorigenesis and invasion.
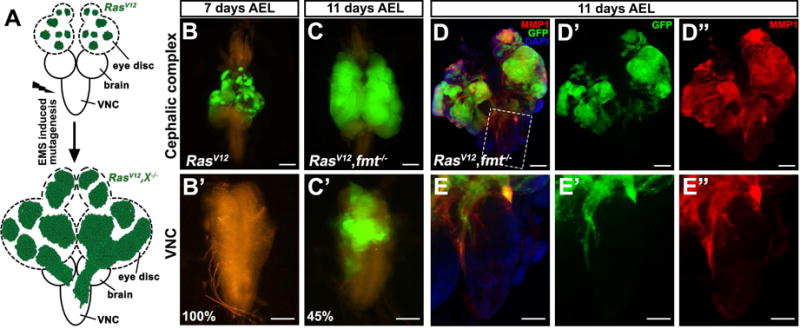
(A) Strategy of an EMS-induced forward genetic screen to identify novel RasV12 collaborating tumor suppressors on chromosome 3L.
(B–E) Fluorescence micrographs of GFP-labeled clones of dissected eye-antennal disc and ventral nerve cord (VNC) are shown. 7 days after egg laying (AEL), ectopic RasV12 expression caused benign tumor growth (B), but not invasion (B′). 11 days AEL, RasV12/Fmt−/− tumor bearing animals showed massive growth and invasion into the VNC (C and C′), as indicated by intensive MMP1 staining in both eye disc (D–D″) and VNC (E–E″). Scale bars, 200 μm in (B, C, D–D″), 100 μm in (B′, C′, E–E″). Genotypes: (B) ey-Flp1/+; Act>y+>Gal4, UAS-GFP/UAS-RasV12; tub-Gal80, FRT79E/FRT79E (C–D) ey-Flp1/+; Act>y+>Gal4, UAS-GFP/UAS-RasV12; tub-Gal80, FRT79E/fmt1, FRT79E. See also Figure S1.
7 days after egg laying (AEL), the RasV12/fmt−/− clones hyper-proliferate extensively, compared with RasV12 expression alone (Figures 1 B and S1D), whereas no GFP positive cells were observed in the ventral nerve cord (VNC) (Figures 1B′ and S1D′), a well-known organ for tumor cell invasion observation in Drosophila (Igaki et al., 2006; Pagliarini and Xu, 2003). At 11 days AEL, with continuous tumor progression, 45% of RasV12/fmt−/− animals displayed invasive behavior (Figures 1C and C′), along with intensive MMP1 activation, a protein essential for basement membrane degradation and EMT progression (Srivastava et al., 2007; Uhlirova and Bohmann, 2006), in both primary tumor and invasive leading edge (Figures 1D″, E″ and S1M–P). In agreement with this, we also observed dramatically increased autonomous mitosis and enhanced epithelial integrity in RasV12/fmt−/− tumors (Figures S1E–L). Conversely, we did not detect significant changes in apoptosis (Figures S1Q–S). Taken together, these findings identify Fmt as a novel tumor suppressor that can synergize with RasV12 to induce tumor growth and invasion.
Fmt negatively regulates JNK signaling
MMP1 serves as a transcriptional target of JNK signaling in tumor cell invasion (Srivastava et al., 2007; Uhlirova and Bohmann, 2006), suggesting that RasV12/fmt−/− may promote tumorigenesis via JNK activation. Consistent with this prediction, a canonical JNK pathway target, puc, was strongly activated in RasV12/fmt−/− tumors (Figures S2A–B). Interestingly, we found that depletion of fmt alone was sufficient to induce mild JNK activation autonomously (Figures 2B′–C′), indicating that Fmt is a negative regulator of the JNK pathway. To further test this, we first asked whether Fmt is essential for the small eye phenotype caused by ectopic expression of Eiger (Egr), the sole TNF-α ligand which activates JNK in Drosophila (Igaki et al., 2002; Moreno et al., 2002). We found that GMR>Egr induced small eye phenotype was significantly enhanced by reducing fmt expression, while expression of fmt-IR itself caused no obvious phenotype (Figures 2D–G). We have previously shown that scribbled (scirb) deficient cells undergo JNK-dependent elimination (Igaki et al., 2009), so we tested whether Fmt is required for this process. As expected, the survival defect of scrib mutant clones was significantly rescued by ectopic expression of Fmt (Figures 2H–K). Furthermore, consistent with our hypothesis, we found that inhibition of JNK activity by expression of a dominant negative form of Drosophila JNK homolog Basket (BskDN) completely abolished RasV12/fmt−/− induced tumor growth, invasive phenotype and JNK activation (Figures 2L, M and S2C–G). Together, these data indicate that Fmt negatively regulates JNK signaling in vivo.
Figure 2. Fmt negatively regulates JNK signaling.
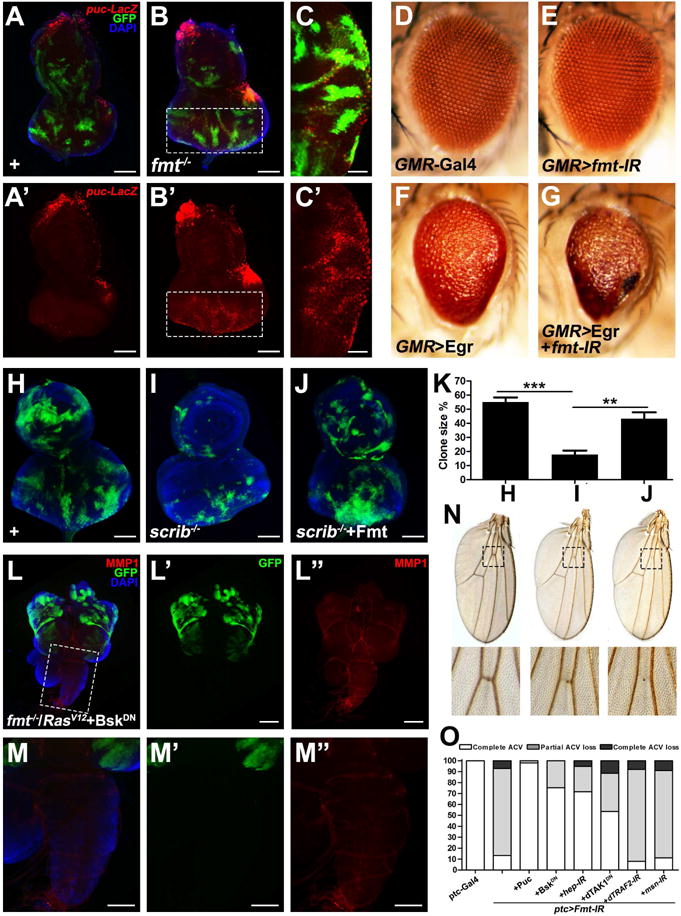
(A–C) Fluorescence micrographs of eye discs are shown. Compared with wild type (A), loss of Fmt induces mild JNK activation (B and C). (D–G) Light micrographs of Drosophila adult eyes of indicated genotypes are shown. Loss of Fmt synergistically enhanced GMR>Egr-induced small eye phenotype (F and G), whereas expression of Fmt-IR alone gave no obvious phenotype (E). (H–K) Compared with wild type (H), scrib depletion induced cell elimination (I) was rescued by expression of Fmt (J). (K) Quantification of cell elimination phenotype in H–J. **P<0.01, ***P<0.001. (mean + s.d., n=5). (L–M) Inhibition of JNK signaling completely abolished RasV12/Fmt−/− induced MMP1 activation and invasion behavior. (N) Light micrographs of Drosophila adult wings are shown. Loss of Fmt under ptc-Gal4 caused partial (middle lane) or complete loss of anterior cross vein (right lane). (O) Quantification of vein loss phenotype of indicated genotypes. Scale bars, 100 μm in (A, B, H–J, M–M″), 50 μm in (C, C′), 200 μm in (L–L″). Genotypes: (A) ey-Flp1/+; Act>y+>Gal4, UAS-GFP/+; tub-Gal80, FRT79E, pucE69/FRT79E (B–C) ey-Flp1/+; Act>y+>Gal4, UAS-GFP/+; tub-Gal80, FRT79E, pucE69/fmt1, FRT79E (D) GMR-Gal4/+ (E) GMR-Gal4/+; UAS-Fmt-IR/+ (F) UAS-Egr/+; GMR-Gal4/+ (G) UAS-Egr/UAS-Fmt-IR; GMR-Gal4/+ (H) eyFlp1/+; Act>y+>Gal4, UAS-GFP.S65T/+; FRT82B, tub-Gal80/FRT82B (I) eyFlp1/+; Act>y+>Gal4, UAS-GFP.S65T/+; FRT82B, tub-Gal80/FRT82B, scrib1 (J) eyFlp1/+; Act>y+>Gal4, UAS-GFP.S65T/UAS-Fmt; FRT82B, tub-Gal80/FRT82B, scrib1 (L–M) ey-Flp1/UAS-BskDN; Act>y+>Gal4, UAS-GFP/UAS-RasV12; tub-Gal80, FRT79E/fmt1, FRT79E (O) Left to right, ptc-Gal4, UAS-GFP/+. ptc-Gal4, UAS-GFP/UAS-Fmt-IR. ptc-Gal4, UAS-GFP/UAS-Fmt-IR; UAS-Puc/+. ptc-Gal4, UAS-GFP/UAS-Fmt-IR; UAS-BskDN/+. ptc-Gal4, UAS-GFP/UAS-Fmt-IR; UAS-hep-IR/+. ptc-Gal4, UAS-GFP/UAS-Fmt-IR; UAS-dTAK1DN/+. ptc-Gal4, UAS-GFP/UAS-Fmt-IR; UAS-dTRAF2-IR/+. ptc-Gal4, UAS-GFP/UAS-Fmt-IR; UAS-msn-IR/+. See also Figure S2.
To further dissect the mechanism of how Fmt modulates JNK signaling, we performed epistasis analysis between Fmt and known JNK pathway components in the developing wing. Similar to elevated JNK activation (Ma et al., 2015; Ma et al., 2013), expression of fmt-IR under patched (ptc) promoter resulted in partial or complete loss of the anterior cross vein (Figures 2N and S2H–I), which can be significantly suppressed by co-expression of the JNK phosphatase Puckered (Puc), expression of BskDN, reducing activity of JNK kinase Hemipterous (Hep), or inhibition of dTAK1, whereas it remained unaffected by blocking dTRAF2 or Msn (Figure 2O). In agreement with this genetic evidence, we found inhibition of dTAK1 activity also significantly impeded RasV12/fmt−/− induced tumor growth and completely suppressed the invasive behavior (Figures S2J–K). Together, these data demonstrate that Fmt negatively regulates JNK signaling upstream of dTAK1.
PpV depletion synergizes with RasV12 to induce dTAK1-JNK dependent tumorigenesis
Fmt contains an evolutionarily conserved SAPS (SIT4 phosphatase-associated proteins) domain (Figure S1C), encoding a regulatory subunit, which interacts with another catalytic subunit (PPP6C, PpV in Drosophila) to form a functional PP6 holoenzyme (Douglas et al., 2010; Hosing et al., 2012). Interestingly, PPP6C has been recently recognized as a potential tumor suppressor, since it is significantly mutated in melanoma, although the underlying mechanism remains poorly understood (Hodis et al., 2012; Krauthammer et al., 2012). Paradoxically, studies in fly indicate that PpV depletion results in a growth defect (Friedman et al., 2011), suggesting a growth promoting role for PPP6C. To resolve this contradiction and clarify PpV’s in vivo role during tumor progression, we generated a null allele of PpV that deletes its entire coding region by imprecise P element excision (Figure S3A). Similar to that of fmt disruption, we observed mild JNK activation in PpV mutant clones (Figures 3A–B). In line with this, we found loss of PpV significantly enhanced GMR>Egr induced small eye phenotype (Figures 3C–D and S3C–D), illustrating the role of PpV as a negative regulator of JNK signaling. More importantly, loss of PpV synergizes with RasV12 to drive massive MMP1 activation, tumor overgrowth, invasion and metastasis into other organs (Figures 3E–F, I–J and S3B, E–F). Consistent with the epistasis analysis of Fmt, we found PpV−/−/RasV12 induced tumor progression was completely or dramatically impeded blocking JNK activity (Figures 3G, H, K and S3G), or inhibition of dTAK1 (Figure S3H). Collectively, these data indicate that PpV encodes a tumor suppressor that collaborates with RasV12 to drive JNK-dependent tumor growth and invasion through dTAK1.
Figure 3. Loss of PpV collaborates with RasV12 to drive JNK-dependent tumor growth and invasion.
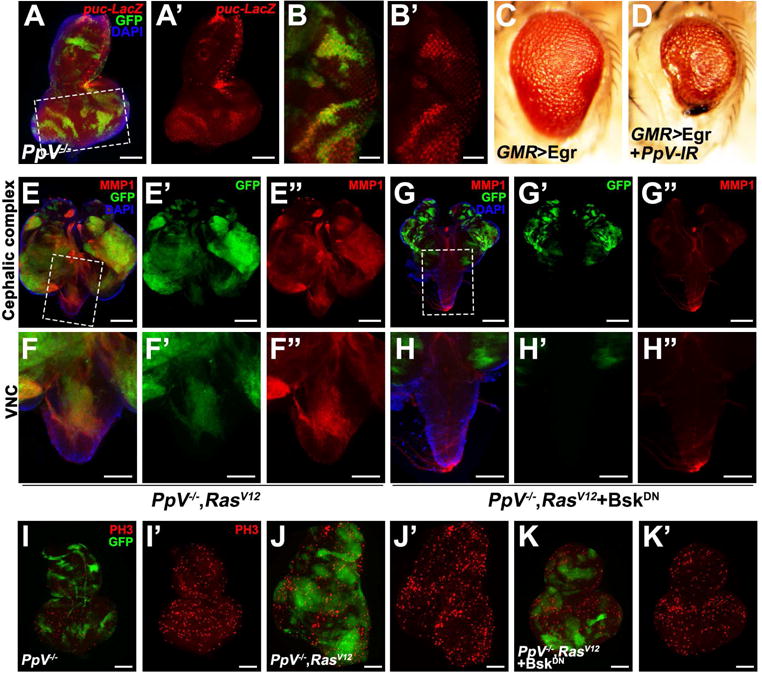
(A–B) Loss of PpV induces mild JNK activation. (C–D) Light micrographs of Drosophila adult eyes of indicated genotypes are shown. Loss of PpV synergistically enhances GMR>Egr induced small eye. (E–H) Fluorescence micrographs of GFP-labeled clones of eye-antennal disc and VNC are shown. PpV−/−/RasV12 induced MMP1 activation, tumorigenesis and invasion behavior (E and F) were all completely suppressed by blocking JNK signaling (G and H). (I–K) Compared with loss of PpV alone (I′), PpV−/−/RasV12 clones displayed strongly increased mitosis (J′), which was dramatically suppressed by reducing JNK activity (K′). Scale bars, 100 μm in (A, F–F″, H–H″, I–K′), 50 μm in (B–B′), 200 μm in (E–E″, G–G″). Genotypes: (A–B) PpVΔ1, FRT19A/tub-Gal80, FRT19A; ey-Flp5, Act>y+>Gal4, UAS-GFP/+; pucE69/+ (C) UAS-Egr/+; GMR-Gal4/+ (D) UAS-Egr/+; GMR-Gal4/UAS-PpV-IR (E, F, J) PpVΔ1, FRT19A/tub-Gal80, FRT19A; ey-Flp5, Act>y+>Gal4, UAS-GFP/UAS-RasV12 (G, H, K) PpVΔ1, FRT19A/tub-Gal80, FRT19A; ey-Flp5, Act>y+>Gal4, UAS-GFP/UAS-RasV12; UAS-BskDN/+ (I) PpVΔ1, FRT19A/tub-Gal80, FRT19A; ey-Flp5, Act>y+>Gal4, UAS-GFP/+. See also Figure S3.
Fmt synergizes with PpV in vivo
To gain further insights into the in vivo function(s) of Fmt and PpV in regulating the JNK pathway, we knocked down or ectopically expressed Fmt and PpV in different tissues. We found that simultaneous reduction of Fmt and PpV under nubbin (nub) promoter synergistically reduced wing size, phenocopying elevated JNK activity (Lee et al., 2006), an effect that is not observed under expression of Fmt-IR or PpV-IR alone (Figures 4H–L). Similarly, removing one copy each of fmt and PpV synergistically enhance GMR>Eiger eye phenotype and result in a complete loss of the eye tissue (Figures 4A–D). Conversely, ectopic expression of Fmt and PpV suppress GMR>Egr-induced small eye in a synergistic way (Figures 4E–G). We previously showed that the combination of cell polarity disruption and RasV12 activation induces JNK dependent tumor growth and invasive behavior (Igaki et al., 2006; Pagliarini and Xu, 2003). In accordance with the observation that Fmt and PpV negatively regulate the JNK pathway (Figures 2 and 3), we found that over-expression of Fmt or PpV alone is sufficient to partially or completely suppress the tumor invasion and rescued the lgl−/−/RasV12 bearing animals to pupae stage (Figures 4F–H, F″–H″), while the tumor size remained relatively unaffected (Figures 4F′–H′). Strikingly, when Fmt and PpV were co-expressed, lgl−/−/RasV12 induced tumor growth, invasion and MMP1 activation were dramatically suppressed (Figures 4I–I″ and S4C–D), indicating a synergistic role of Fmt and PpV in tumor inhibition. It is noteworthy that residual MMP1 activation still detected, in line with our observation that co-expression of Fmt and PpV cannot fully suppress GMR>Egr small eye phenotype (Figure 4G). As tumor progression is frequently accompanied by apoptosis (Menendez et al., 2010; Vidal et al., 2006), to test if Fmt and PpV block tumorigenesis by inducing apoptosis, we monitored caspase 3 activation and found that co-expression of Fmt and PpV did not induce massive apoptosis in RasV12/lgl−/−clones (Figure S4), suggesting that the tumor suppressing role of Fmt and PpV is uncoupled from apoptosis. Finally, we found that co-expression of RasV12 and PpV-IR induced mild tumor overgrowth and MMP1 activation was dramatically enhanced by deleting one copy of fmt (Figures S4E–G), further confirming the in vivo synergistic effect between Fmt and PpV in tumorigenesis.
Figure 4. PpV synergizes with Fmt in vivo.
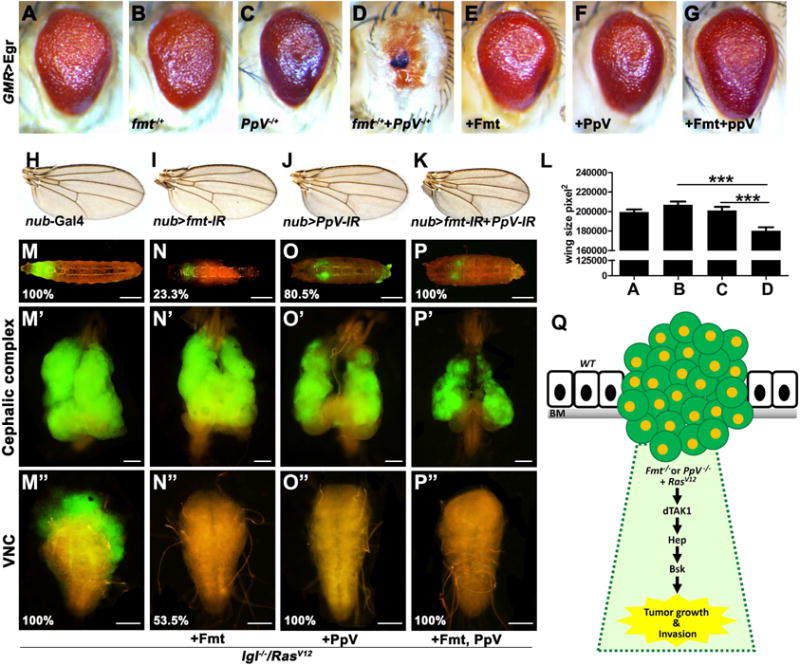
(A–G) Light micrographs of Drosophila adult eyes are shown. Removing one copy of fmt or PpV slightly enhances GMR>Egr small eye, while simultaneously remove one copy of each results in complete eye loss. Co-expression of Fmt and PpV synergistically suppress GMR>Egr eye phenotype.
(H–K) Light micrographs of Drosophila adult wings are shown. Compared with controls (H), loss of either Fmt (I) or PpV (J) driven by nub-Gal4 produced no obvious phenotype, while co-expression of Fmt-IR and PpV-IR resulted in smaller wings (K). (L) Quantification data of wing size in A–D. (M–P) Fluorescence micrographs of GFP-labeled clones of eye-antennal disc and VNC are shown. lgl−/−/RasV12 induced tumor overgrowth (M′), invasion (M″), and eventually death as giant larvae (M). Ectopic expression of Fmt partially restored the pupation (N), significantly suppressed tumor invasion (N″), but not tumor growth (N′). Expression of PpV alone is sufficient to completely suppressed invasive phenotype (O″) and dramatically restored pupation (O), whereas tumor size was only slightly suppressed, if there is any (O′). Co-expression of Fmt and PpV dramatically suppressed tumor progression, including tumorigenesis and invasion (P′ and P″), and rescued all the animals to pupal stage (P). % indicates phenotype penetrance ratio. (Q) Model of PpV and Fmt in RasV12-induced tumorigenesis. Scale bars, 400 μm in (M–P), 200 μm in (M′–P′), 100 μm in (M″–P″). Genotypes: (A) UAS-Egr/+; GMR-Gal4/+ (B) UAS-Egr/+; GMR-Gal4/fmt1, FRT79E (C) PpVΔ1, FRT19A/+; UAS-Egr/+; GMR-Gal4/+ (D) PpVΔ1, FRT19A/+; UAS-Egr/+; GMR-Gal4/fmt1, FRT79E (E) UAS-Egr/UAS-Fmt; GMR-Gal4/+ (F) UAS-Egr/+; GMR-Gal4/UAS-PpV (G) UAS-Egr/UAS-Fmt; GMR-Gal4/UAS-PpV (H) nub-Gal4/+ (I) nub-Gal4/UAS-Fmt-IR (J) nub-Gal4/+; UAS-PpV-IR/+ (K) nub-Gal4/UAS-Fmt-IR; UAS-PpV-IR/+ (M) ey-Flp1/+; tub-Gal80, FRT40A/lgl4, FRT40A, UAS-RasV12; Act>y+>Gal4, UAS-GFP/+ (N) ey-Flp1/+; tub-Gal80, FRT40A/lgl4, FRT40A, UAS-RasV12; Act>y+>Gal4, UAS-GFP/UAS-Fmt (O) ey-Flp1/+; tub-Gal80, FRT40A/lgl4, FRT40A, UAS-RasV12; Act>y+>Gal4, UAS-GFP/UAS-PpV (P) ey-Flp1/+; tub-Gal80, FRT40A/lgl4, FRT40A, UAS-RasV12; Act>y+>Gal4, UAS-GFP/UAS-Fmt, UAS-PpV. See also Figure S4.
Discussion
In this study, we have conducted an unbiased, EMS-based forward genetic screen and identified Fmt and PpV as tumor suppressors that cooperate with RasV12 to induce massive tumor overgrowth and invasion. Our genetic epistasis analysis establish Fmt and PpV as essential negative regulators of the JNK pathway, acting upstream of dTAK1 (Figure 4J). Moreover, we found that Fmt/PpV synergistically inhibits JNK-mediated tumorigenesis in vivo. Interestingly, the human homolog of PpV, the catalytic subunit of the PP6 holoenzyme (PPP6C), was recently identified as a driver mutation during melanoma progression. Approximately 10% of patients were found to harbor PPP6C somatic mutations (Hodis et al., 2012; Krauthammer et al., 2012), and surprisingly, all of them had BRAF or RAS mutations as well (Krauthammer et al., 2012). Apart from the known roles of PP6 in regulating cell cycle and mitosis (Stefansson and Brautigan, 2007; Zeng et al., 2010), little is understood about the genetic mechanism by which it modulates tumor growth. Here, we have uncovered the underlying mechanism of PP6−/−/RasV12 induced tumor overgrowth, and identified dTAK1-JNK signaling as the essential molecular link, which also further demonstrates the value of the Drosophila model system for gaining insight into human cancer biology. Interestingly, human PP6 is known to directly bind and dephosphorylate TAK1 at Thr-187 (Kajino et al., 2006), suggesting a conserved role of PP6-TAK1 module. Given the conservation of signaling pathways between Drosophila and humans, similar mechanisms could be involved in human PP6-RasV12 related cancer progression. Further investigation in mammal and human may provide potential therapeutic targets for cancer treatment, especially melanoma.
Experimental Procedures
Drosophila stocks and genetics
All crosses were raised on standard Drosophila media at 25°C unless otherwise indicated. Fluorescently labeled clones were produced in the eye discs as previously described (Pagliarini and Xu, 2003) using the following strains: tub-Gal80, FRT19A; ey-Flp5, Act>y+>Gal4, UAS-GFP (19A tester); ey-Flp1; tub-Gal80, FRT40A; Act>y+>Gal4, UAS-GFP (40A tester); ey-Flp1; Act>y+>Gal4, UAS-GFP; tub-Gal80, FRT79E (79E tester); eyFlp1; Act>y+>Gal4, UAS-GFP.S65T; FRT82B, tub-Gal80 (82B tester). Additional strains, including KG09672, GMR-Gal4, ptc-Gal4, nub-Gal4, UAS-GFP, UAS-msn-IR, UAS-PpV (#53770) were obtained from Bloomington Drosophila Stock Center; UAS-dTAK1DN, UAS-dTRAF2-IR (Xue et al., 2007), pucE69, UAS-Egr, lgl4, UAS-RasV12, UAS-Puc (Ma et al., 2014), UAS-BskDN, UAS-hep-IR (Ma et al., 2015), scrib1 (Igaki et al., 2009) were previously described. UAS-PpV-IR (V31690) and UAS-Fmt-IR (V16005) were obtained from the Vienna Drosophila RNAi Center; UAS-PpVHA (F000874) was obtained from FlyORF.
PpV mutants were generated by imprecise excision of the the P element insertion line KG09672. Genomic DNA of isolated candidate mutants were isolated and analyzed by PCR. Sequence analysis indicated that the PpVΔ1 allele deletes the entire coding region of PpV, suggesting that PpVΔ1 is a null allele.
UAS-Fmt transgenic flies were generated by standard P element-mediated transformation. Two independent lines (on second and third chromosomes) were produced and examined for each transgene, and gene expression was verified by RT-PCR.
EMS mutagenesis and genetic screen
We focus on the left arm of chromosome 3, which covers around 20% of the fly genome. Male flies carrying a FRT79E (Sp/CyO-GFP; FRT79E) were starved for 8 hr and subsequently fed a 25 mM ethyl methanesulphonate (EMS) solution overnight at room temperature. The mutagenized males were mated to females of the genotype UAS-RasV12; sb/TM6B. Single F1 males of the genotype UAS-RasV12/CyO-GFP; *FRT79E/TM6B were crossed to Sp/CyO; sb/TM6B first and then crossed to a 79E tester line. The larvae are transparent and can be easily scored for overgrowth of GFP-labeled tumor caused by enhancer mutations.
Immunostaining
Third instar larvae eye-antennal discs were dissected in 1× PBS, fixed in freshly made 4% paraformaldehyde and stained as described previously (Ma et al., 2013), using mouse anti-MMP1 (1:200), mouse anti-β-Gal (1:1000, DSHB, Developmental Studies Hybridoma Bank), rabbit anti-phospho histone 3 (PH3) (1:200), rabbit anti-active Caspase 3 (1: 400) and Alexa Fluor®555 (1:100, Cell Signaling Technology). Secondary antibodies were anti-rabbit-Alexa (1:400), and anti-mouse-Cy3 (1:400, Thermo Fisher Scientific). Tumor growth and invasion images in Figureure 1 and 4 were taken with a Leica MZ FLIII fluorescence stereomicroscope with an Optronics Magnafire S99802 digital camera.
Statistical analysis
Clone and wing size were measured with Image J and Photoshop, respectively. Quantification of the data was presented in bar graphs created with Graphpad Prism 5. Data represents mean values + SD. We used a one-way ANOVA with Bonferroni correction for multiple comparisons to calculate statistical significance (**P<0.01; ***P<0.001).
Supplementary Material
Highlights.
An EMS-based genetic screen in fly identifies Fmt as a tumor suppressor
Loss of Fmt synergizes with RasV12 to induce JNK-dependent tumorigenesis
PpV depletion phenocopies Fmt disruption-induced tumorigenesis
Fmt synergizes with PpV in vivo
Acknowledgments
We thank Bloomington and VDRC stock centers for providing fly stocks, C. Chabu, B. Dunn for discussion and comments, J. Kouptsova for critical reading of the manuscript. This research was supported by a NIH/NCI grant to T.X, T.X. is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
X.M., J-Y L. and T.X. conceived the study. X.M. and J-Y L. performed experiments and analyzed data. Y.D. and D.L. assisted in immunofluorescence staining. J.M. assisted with model drawing. X.M., and T.X. wrote the manuscript.
References
- Brumby AM, Goulding KR, Schlosser T, Loi S, Galea R, Khoo P, Bolden JE, Aigaki T, Humbert PO, Richardson HE. Identification of novel Ras-cooperating oncogenes in Drosophila melanogaster: a RhoGEF/Rho-family/JNK pathway is a central driver of tumorigenesis. Genetics. 2011;188:105–125. doi: 10.1534/genetics.111.127910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. The EMBO journal. 2003;22:5769–5779. doi: 10.1093/emboj/cdg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi C, Zhu H, Han M, Zhuang Y, Wu X, Xu T. Disruption of lysosome function promotes tumor growth and metastasis in Drosophila. The Journal of biological chemistry. 2010;285:21817–21823. doi: 10.1074/jbc.M110.131714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nature reviews Drug discovery. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:3637–3640. doi: 10.1073/pnas.79.11.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Zhong J, Ye R, Moorhead GB, Xu X, Lees-Miller SP. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Molecular and cellular biology. 2010;30:1368–1381. doi: 10.1128/MCB.00741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Clarevega A, Bilder D. Malignant Drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Developmental cell. 2015;33:47–55. doi: 10.1016/j.devcel.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AA, Tucker G, Singh R, Yan D, Vinayagam A, Hu Y, Binari R, Hong P, Sun X, Porto M, et al. Proteomic and functional genomic landscape of receptor tyrosine kinase and ras to extracellular signal-regulated kinase signaling. Science signaling. 2011;4:rs10. doi: 10.1126/scisignal.2002029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat Rev Cancer. 2013;13:172–183. doi: 10.1038/nrc3461. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosing AS, Valerie NC, Dziegielewski J, Brautigan DL, Larner JM. PP6 regulatory subunit R1 is bidentate anchor for targeting protein phosphatase-6 to DNA-dependent protein kinase. The Journal of biological chemistry. 2012;287:9230–9239. doi: 10.1074/jbc.M111.333708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, Miura M. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. The EMBO journal. 2002;21:3009–3018. doi: 10.1093/emboj/cdf306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Current biology: CB. 2006;16:1139–1146. doi: 10.1016/j.cub.2006.04.042. [DOI] [PubMed] [Google Scholar]
- Igaki T, Pastor-Pareja JC, Aonuma H, Miura M, Xu T. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Developmental cell. 2009;16:458–465. doi: 10.1016/j.devcel.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajino T, Ren H, Iemura S, Natsume T, Stefansson B, Brautigan DL, Matsumoto K, Ninomiya-Tsuji J. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. The Journal of biological chemistry. 2006;281:39891–39896. doi: 10.1074/jbc.M608155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo P, Allan K, Willoughby L, Brumby AM, Richardson HE. In Drosophila, RhoGEF2 cooperates with activated Ras in tumorigenesis through a pathway involving Rho1-Rok-Myosin-II and JNK signalling. Disease models & mechanisms. 2013;6:661–678. doi: 10.1242/dmm.010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nature genetics. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Song W, Droujinine IA, Hu Y, Asara JM, Perrimon N. Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Developmental cell. 2015;33:36–46. doi: 10.1016/j.devcel.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Koh H, Kim M, Park J, Lee SY, Lee S, Chung J. JNK pathway mediates apoptotic cell death induced by tumor suppressor LKB1 in Drosophila. Cell death and differentiation. 2006;13:1110–1122. doi: 10.1038/sj.cdd.4401790. [DOI] [PubMed] [Google Scholar]
- Ma X, Li W, Yu H, Yang Y, Li M, Xue L, Xu T. Bendless modulates JNK-mediated cell death and migration in Drosophila. Cell death and differentiation. 2014;21:407–415. doi: 10.1038/cdd.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Xu W, Zhang D, Yang Y, Li W, Xue L. Wallenda regulates JNK-mediated cell death in Drosophila. Cell death & disease. 2015;6:e1737. doi: 10.1038/cddis.2015.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Yang L, Yang Y, Li M, Li W, Xue L. dUev1a modulates TNF-JNK mediated tumor progression and cell death in Drosophila. Developmental biology. 2013;380:211–221. doi: 10.1016/j.ydbio.2013.05.013. [DOI] [PubMed] [Google Scholar]
- Menendez J, Perez-Garijo A, Calleja M, Morata G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14651–14656. doi: 10.1073/pnas.1009376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E, Yan M, Basler K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Current biology: CB. 2002;12:1263–1268. doi: 10.1016/s0960-9822(02)00954-5. [DOI] [PubMed] [Google Scholar]
- Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science. 2003;302:1227–1231. doi: 10.1126/science.1088474. [DOI] [PubMed] [Google Scholar]
- Pastor-Pareja JC, Xu T. Dissecting social cell biology and tumors using Drosophila genetics. Annual review of genetics. 2013;47:51–74. doi: 10.1146/annurev-genet-110711-155414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome research. 2001;11:1114–1125. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrapatna VA, Cagan RL, Das TK. Drosophila cancer models. Dev Dyn. 2012;241:107–118. doi: 10.1002/dvdy.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MB, Der CJ, Wang-Gillam A, Cox AD. Targeting RAS-mutant cancers: is ERK the key? Trends in cancer. 2015;1:183–198. doi: 10.1016/j.trecan.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nature reviews Drug discovery. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Pastor-Pareja JC, Igaki T, Pagliarini R, Xu T. Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc Natl Acad Sci U S A. 2007;104:2721–2726. doi: 10.1073/pnas.0611666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson B, Brautigan DL. Protein phosphatase PP6 N terminal domain restricts G1 to S phase progression in human cancer cells. Cell cycle. 2007;6:1386–1392. doi: 10.4161/cc.6.11.4276. [DOI] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Uhlirova M, Bohmann D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. The EMBO journal. 2006;25:5294–5304. doi: 10.1038/sj.emboj.7601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Larson DE, Cagan RL. Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Developmental cell. 2006;10:33–44. doi: 10.1016/j.devcel.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby LF, Schlosser T, Manning SA, Parisot JP, Street IP, Richardson HE, Humbert PO, Brumby AM. An in vivo large-scale chemical screening platform using Drosophila for anti-cancer drug discovery. Disease models & mechanisms. 2013;6:521–529. doi: 10.1242/dmm.009985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Igaki T, Kuranaga E, Kanda H, Miura M, Xu T. Tumor suppressor CYLD regulates JNK-induced cell death in Drosophila. Developmental cell. 2007;13:446–454. doi: 10.1016/j.devcel.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Zeng K, Bastos RN, Barr FA, Gruneberg U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. The Journal of cell biology. 2010;191:1315–1332. doi: 10.1083/jcb.201008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.