

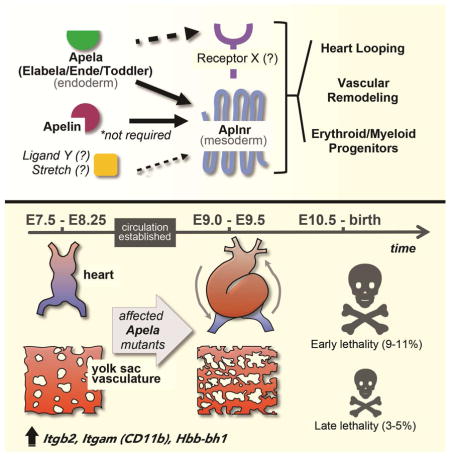
SUMMARY
Apela (also known as Elabela, Ende and Toddler) is a small signaling peptide that activates the G protein-coupled receptor Aplnr to stimulate cell migration during zebrafish gastrulation. Here, using CRISPR/Cas9 to generate a null, reporter-expressing allele, we study the role of Apela in the developing mouse embryo. We found that loss of Apela results in low penetrance cardiovascular defects that manifest after the onset of circulation. Three-dimensional micro computed tomography revealed a higher penetrance of vascular remodeling defects from which some mutants recover, and identified extraembryonic anomalies as the earliest morphological distinction in Apela mutant embryos. Transcriptomics at late gastrulation identified aberrant upregulation of erythroid and myeloid markers in mutant embryos prior to appearance of physical malformations. Double mutant analyses showed that loss of Apela signaling impacts early Aplnr-expressing mesodermal populations independently of the alternative ligand Apelin, leading to lethal cardiac defects in some Apela null embryos.
Keywords: Apela, Elabela, Ende, Toddler, Aplnr, APJ, Apelin, gastrulation, cardiovascular development, micro computed tomography (microCT), macrophages, vascular remodeling
Graphical Abstract
INTRODUCTION
Apela encodes a small, secreted peptide whose sequence is highly conserved among vertebrates. Genes that encode small peptides have been overlooked due to difficulties in annotating their short open reading frames, and study of their functions may provide insights into mechanisms that control cell behaviors that cannot be fully explained by canonical signaling pathways (Pauli et al., 2015). Loss of Apela, or its receptor Aplnr (also known as APJ and Agtrl1), affects lateral cardiac mesoderm progenitors and leads to variable and severe heart and posterior malformations in zebrafish (Chng et al., 2013, Pauli et al., 2014). During gastrulation, Apela signaling increases cell velocity in a non-directional manner to advance mesendoderm internalization (Pauli et al., 2014). However, it also participates in guided cell migration by driving angioblast migration to the midline in dorsal aorta formation (Helker et al., 2015).
Apela-Aplnr signaling also impacts erythroid and myeloid genes, which are ectopically expressed and mislocalized in apela mutants (Chng et al., 2013, Pauli et al., 2014), whereas aplnra/b morphants have defects in hematopoietic progenitor migration to the anterior lateral plate mesoderm (Paskaradevan and Scott, 2012). In Xenopus, overstimulation of Xmsr (homolog of Aplnr) expands the distribution of hematopoietic precursors (Inui et al., 2006).
The functional requirement for Apela in mammals is unknown. Approximately 20% of Aplnr null mice have defects in the dorsal aorta and yolk sac (YS) vasculature at embryonic day (E)10.5, while an additional 20% exhibit structural heart defects and vascular smooth muscle deficiencies in late gestation (Kang et al., 2013). However the developmental events leading to these phenotypes have not been characterized. Apelin is the canonical ligand for Aplnr, yet Apelin null mice survive in expected ratios (Kuba et al., 2007), raising the possibility that Apela may be the critical ligand for Aplnr during embryogenesis.
In this study, we investigate the role of Apela in mammalian development. Apela is expressed in the definitive endoderm (DE) in mice (Hassan et al., 2010). However, its loss-of-function mainly impacted mesodermal derivatives that express Aplnr (Devic et al., 1999). A low penetrance of yolk sac (YS) vascular remodeling defects, global structural malformations, and cardiac failure were evident as early as E9.5. These embryonic lethal defects were not compensated for by Apelin, and relied in part on signaling through Aplnr. Since embryos appeared morphologically normal prior to this stage, these phenotypes are likely linked to the onset of circulation, and possibly to earlier extraembryonic abnormalities including aberrant regulation of erythromyeloid progenitor progenitors.
RESULTS
Apela null mice have reduced survival
To disrupt the coding function of Apela in mice, we knocked an H2B-GFP-pA reporter cassette into the endogenous translational start site, thereby deleting the CDS of Exon 2 (Figure 1A). The Neomycin selection cassette was removed by crossing ApelaH2B-GFP-NEO/+ mice (referred to as ‘NEO-IN’) with CAG-Cre mice (Katsunaga Sakai, 1997), thereby generating ApelaH2B-GFP/+ mice (referred to as ‘NEO-OUT’). H2B-GFP protein was only detected after Neomycin removal. At E7.75, H2B-GFP was expressed in the presumptive floorplate overlying the midline, and colocalized with FOXA2 in the anterior endoderm (Figure 1B), consistent with reported Apela expression (Hassan et al., 2010).
FIGURE 1. Apela null mice generated by H2B-GFP-pA knock-in have reduced survival.

(A) Targeting of the Apela locus. Untranslated (white boxes) and coding (black boxes) sequence. Homology directed repair deleted 145bp. Elements are to scale except where indicated (double hash). Modification of Apela protein sequence by gene targeting (signal peptide cleavage site, red). E, Exon. (B) Immunostaining for GFP and FOXA2 (LHF, late headfold). 3D MIP frontal views (left) and 2D sections (a–d). Nuclei are stained with Hoechst. (C) Amplification of Apela transcript fragments (orange lines) from embryos by RT-PCR. Spliced (blue asterisk) and unspliced (red asterisk) 5′ UTR products. H2B-GFP transcripts were amplified from NEO-IN embryos, but may be unstable or insufficient for protein detection. (D) Expression of Apela by qRT-PCR. Data are represented as mean ± SEM. (E) Chi squared analysis of recovered Apela genotypes from homozygous null males crossed to heterozygous females.
To examine transcriptional activity at targeted Apela locus, we performed RT-PCR and observed differential amplification of untranslated Apela sequences (Figure 1C). The Apela 5′ UTR could not be detected in NEO-IN mutants, while the 3′ UTR was amplified at very low levels. Conversely, the Apela 3′ UTR was not amplified in NEO-OUT mutants, though both spliced and unspliced 5′ UTR RNA species, confirmed by sequencing, were detected from the NEO-OUT allele. Nonetheless, Apela was not significantly expressed in either mutant strain when compared to heterozygotes (Figure 1D). We cannot rule out the possibility of residual untranslated activity of Apela (Li et al., 2015), but the disruption of sequences in both targeted alleles abolishes the coding functions of Apela.
To determine if Apela is required for survival, we performed heterozygous intercrosses and recovered fertile, adult, homozygous Apela KO animals. We then crossed homozygous KO males with heterozygous females and noted that Apela KO adults (>3 weeks of age) were not represented at Mendelian ratios (Figure 1E). Reduced survival of adult KO mutants was significant, and penetrance was equivalent for both NEO-IN and NEO-OUT alleles. Reduced numbers of Apela null adults were not accounted for by dead pups found prior to weaning. Therefore, while the majority of Apela null mutant mice are indistinguishable from non-mutants as adults, a small but significant number of animals are embryonic lethal in the absence of Apela.
Apela, Apelin, and Aplnr have differential expression patterns in mice
To assess the spatial relationships between Apela, Apelin, and Aplnr, we analyzed expression patterns in wild-type embryos from E6.5–E9.5. Apelin was expressed in extraembryonic visceral endoderm and the primitive streak at E6.5 (Figure S1A). Apela expression was first detected at E7.0 in the distal epiblast, then shortly thereafter in the DE, as previously described (Hassan et al., 2010). Thus, in contrast to zebrafish, Apela is not the first Aplnr ligand to be expressed in mice. Aplnr was expressed in cells emerging from the primitive streak that will give rise to both extraembryonic and embryonic mesoderm (Figure S1A). Apela was expressed in extraembryonic tissues as well as in the chorion at E8.25, and in trophoblast cells at the periphery of the placenta at E9.5 (Figure S1B–C). Apelin was not readily detectable in the embryo at E8.25, but was expressed in the anterior visceral YS. Punctate staining for Aplnr was observed in the allantois and in the vasculature invading the placenta at E9.5 (Figure S1B–C). Therefore, Apela-Apelin-Aplnr signaling may function in extraembryonic, as well as embryonic tissues, to impact formation of mesoderm derivatives such as YS vasculature, hematopoietic progenitors, the chorion, and allantois.
Apela mutants have variable expressivity of embryonic lethal phenotypes
We compared Apela KO embryos at E9.5 and E10.5 with heterozygotes in both NEO-IN and NEO-OUT mutant lines (Figure 2A–B). The majority of Apela mutants (90%) were unaffected at E9.5. However, affected mutants (10%) displayed unremodeled YS vasculature with variable cardiac and cranial malformations; in the most severe embryos, cardiac failure resulted in pericardial edema (Figure S2A). Affected mutants also exhibited perturbed fetal circulation with pooling of blood in the YS and posterior embryo, despite the beating heart. Approximately 1% of null embryos had severe global morphological defects and failed to turn to the fetal position. There was a low incidence of axial abnormalities that were unique to NEO-OUT mutants (Figure S2A). However, the penetrance and variable expressivity of cardiovascular defects was comparable between both strains (Figure 2C). We did not observe gross differences in Apela KO placentae at E9.5, consistent with findings from a concurrent study (Ho et al., 2017). The incidence of affected Apela null phenotypes was statistically significant, and the penetrance and severity of the phenotype remained unchanged when the mother was homozygous null for Apela (Figure 2C). Apela KO NEO-IN and NEO-OUT mutants were recovered at Mendelian ratios at E9.5 (Figure 2D), with no apparent sex bias among affected embryos (data not shown). By E10.5, affected Apela null mutants were severely dysmorphic with advanced pericardial edema, hemorrhaging, and tissue degeneration (Figure 2A–B). These mutants were no longer viable and were not recovered at E15.5.
FIGURE 2. Reduced survival and variable expressivity of Apela mutants.

(A–B) Affected Apela NEO-IN (A) and NEO-OUT (B) null phenotypes at E9.5 and E10.5. Pooling of blood in the YS and embryo in affected mutants (green arrowheads). (C) Penetrance of affected Apela null phenotypes (scored according to YS remodeling) was 9–11% at E9.5. Genotype of the mother did not impact penetrance of affected Apela null phenotypes. (D) Recovery of Apela null embryos at E9.5, E10.5, and E15.5. FB, forebrain; PE, pericardial edema.
Apelin does not compensate for Apela during mouse development
Apelin and Apela may compensate for one another despite different sites of expression since they could act at a distance from their site of production. To address this, we crossed Apelin mutants with Apela NEO-IN mutants to generate Apelin;Apela double KO (dKO) animals. At E9.5, Apelin;Apela dKO embryos exhibited the same phenotypic characteristics as Apela single KO NEO-IN embryos (Figure S2B). The majority of dKO embryos were phenotypically normal, whereas 11% had anterior, posterior, and heart malformations, sometimes with pericardial edema, and accompanied by unremodeled YS vasculature. Power analysis demonstrated no significant increase in the incidence of affected phenotypes among Apelin;Apela dKO. Overall, the penetrance and expressivity of the Apela phenotype was equivalent between Apelin;Apela dKO embryos and Apela single KO embryos. Furthermore, genetically altering the dosage of Apelin had no significant effect on the recovery of Apela KO NEO-IN adult animals (Figure S2C). We did not observe compensatory upregulation of either ligand (Figure S2D). These results demonstrate that Apelin does not compensate for loss of Apela in the mouse embryo.
Apela signals through Aplnr in early mouse development
We analyzed Aplnr KO embryos to determine whether they resemble affected Apela KO mutants at E9.5 on the same genetic background (Figure 3A). Only 2% of Aplnr KO embryos failed to turn and were severely dysmorphic with unremodeled YS. The remaining 98% did not exhibit YS remodeling defects, pericardial edema or posterior pooling of blood. Instead, 13% of Aplnr KO embryos had abnormal tail bending (Figure 3A). Aplnr mutant embryos were present in Mendelian ratios at E9.5, but far fewer adult Aplnr KO mice were recovered (Figure 3B), as expected (Kang et al., 2013).
FIGURE 3. Analysis of Aplnr KO and Aplnr;Apela double mutant embryos.

(A) Aplnr KO embryos at E9.5 isolated from homozygous KO males crossed to heterozygous or homozygous KO females. (B) Aplnr KO embryos were recovered from heterozygous intercrosses in expected Mendelian ratios at E9.5 (n=75). Survival of adult Aplnr KO mice was significantly reduced. (C) Recovery of Aplnr;Apela double mutant genotypes at E9.5 from double heterozygous intercrosses. (D–E) Aplnr;Apela (NEO-IN) double mutants. The Apela null phenotype was observed among double heterozygous embryos (D). Aplnr;Apela double KO embryos not shown were morphologically normal.
Discrepancies in the affected phenotypes of Aplnr KO versus Apela KO embryos at E9.5 raised the possibility that Apela may signal independently of Aplnr in early mouse development, and/or that Aplnr may receive additional unidentified signals. We analyzed the gross morphology of Aplnr;Apela double homozygous and double heterozygous mutant embryos at E9.5 (Figure 3C–E). Genetically reducing Apela-Aplnr signaling by heterozygous mutation of both genes sometimes resulted in a severe embryonic lethal phenotype typical of affected Apela single KO embryos, supporting the notion that Apela signals through Aplnr at these early stages. However, embryonic lethal phenotypes remained at a low penetrance in Aplnr;Apela double KO embryos, leaving open the possibility for alternative receptor:ligand partners in the pathway.
Affected Apela mutants exhibit defects in cardiac looping
We further analyzed the cardiovascular defects in affected Apela mutants. At E8.25, when the embryonic circulation is not fully established, the cardiac morphology of Apela KO NEO-IN and KO NEO-OUT embryos was indistinguishable from heterozygous littermates. Mutants expressed Pitx2 normally in the left lateral plate mesoderm (Figure 4A) (Ryan et al., 1998, Piedra et al., 1998, Yoshioka et al., 1998, Campione et al., 1999). However, by E9.5, NEO-IN and KO-NEO-OUT mutants exhibited inadequate bulbous cordis (BC) formation resulting in opening of the outflow tract into a common ventricle, highlighted by abnormal Hand1 expression (Figure 4B).
FIGURE 4. Affected Apela KO embryos have insufficient heart looping and vascular defects.

(A) Expression of Pitx2 in the left lateral plate mesoderm at E8.25. (B) Expression of Hand1 in the LV at E9.5. Apela KO (NEO-IN and NEO-OUT) embryos fail to fully form the BC (red arrowheads) due to insufficient heart looping. (C) Optical sections from microCT imaging at E9.5. 3D reconstruction showing embryo curvature within the deciduum. (D) Expression of CBF:H2B-Venus in Apela KO (NEO-IN) mutants at E9.5. Planes of section (a–h) are indicated on wholemounts. Sections were stained for F-ACTIN (Phalloidin) and nuclei (Hoechst). VENUS+ presumptive endothelial progenitors (yellow arrows). (E) YS tissue sections stained for F-ACTIN (Phalloidin) and nuclei (Hoechst). (F) MIPs of PECAM-1 (CD31) expression in whole flatmounted YS. Regions of higher magnification (yellow boxes). LV, left ventricle; OFT, outflow tract; BC, bulbous cordis; CV, common ventricle.
We also performed phenotypic analysis with three-dimensional micro computed tomography (3D microCT) of KO NEO-IN embryos at E8.25 and E9.5 (Hsu et al., 2016). Digital sections confirmed the heart looping defect at E9.5 (Figure 4C). Affected mutants also had a wider angle of juxtaposition of the embryo torso to the tail, indicative of differences in embryonic turning (Figure 4C). MicroCT did not reveal abnormalities in linear heart tube formation in Apela KO NEO-IN embryos at E8.25, therefore embryonic morphological defects are only apparent after the onset of circulation.
Normal endothelial cell specification, but aberrant vessel remodeling in affected Apela mutants
We assessed the development of the embryonic vasculature in Apela KO NEO-IN embryos using a CBF:H2B-Venus Notch signaling reporter expressed in endothelial cells (Nowotschin et al., 2013). The parallel bilateral dorsal aortae evident at E8.5, converge and fuse along the midline to form a single large vessel at E9.5 (Figure 4D). However, in affected Apela KO NEO-IN embryos, the dorsal aortae remained bilateral and engorged with blood. The nephric ducts could not be discerned in affected Apela KO NEO-IN embryos, and the dorsal aorta was not distinguishable despite the presence of VENUS+ presumptive endothelial cells in the proper location. This is in agreement with the role of Apela signaling in zebrafish, in which angioblast specification is unaffected, but rather migration and vasculogenesis is perturbed (Helker et al., 2015). Furthermore, VENUS+ cells of the intersomitic vasculature were highly disorganized and the sclerotome portion of the somites was small compared to the adjacent dermomyotome (Figure 4D).
We examined the YS phenotype in these mutants, and confirmed that Apela was not required to form endoderm and mesoderm layers of the visceral YS (Figure 4E). Endothelial cells in affected YS expressed the vascular cell surface marker PECAM-1 (CD31), and were organized into a network associated with the vitelline vessels. However, the YS vasculature failed to form a hierarchy of large- and small-caliber vessels (Figure 4F). Instead, affected vessels had primary capillary plexus geometry throughout the YS, but with enlarged lumens compared with capillary regions of normal YS. Our findings suggest that Apela is not required for the specification of endothelial cells, but rather their organization and remodeling during vasculogenesis.
Viable Apela mutants survive mild defects in vascular remodeling
Since the majority of mice survive in the absence of Apela, we used 3D microCT to determine if Apela null mutants retained within the deciduum had non-lethal vascular phenotypes (Figure 5A). While embryonic lethal phenotypes (such as pericardial edema and/or unlooped heart) were present in only 10% of Apela KO embryos, we observed over twice as many embryos with unremodeled YS and/or vitelline vessels that often coincided with enlarged left ventricles (Figure 5B). This was prevalent in 16–22ss KO embryos, but not in 23–27ss KO embryos, suggesting that Apela KO mutants may recover from mild vascular remodeling defects, which were not observed in stage-matched heterozygous embryos (n=21, data not shown).
FIGURE 5. Vitelline vessel remodeling defects and extraembryonic anomalies in affected Apela KO embryos.

(A) 3D reconstructions of microCT images at E9.5. Vitelline vessels are pseudocolored: remodeled vitelline vein (blue) and vitelline artery (pink), unremodeled vitelline vessels (yellow). (B) Summary matrix of microCT results at E9.5 from 5 litters (n=46 KO NEO-IN decidua, 3 reabsorbed embryos). X-axis lists individuals by somite stage (ss; ND, not determined). Y-axis lists normal (black boxes) and abnormal (orange boxes) classification of phenotypes. (C) 3D reconstructions of microCT at E8.25 with pseudocolored structures: normal allantois (dark green), cylindrical allantois (light green), ectopic chorion protrusion (orange), normal blood island (BI; black arrowheads), abnormal BI protrusions (purple, with red arrowheads). (D) Posterior views of microCT images used to measure allantois width at E8.25. Data are represented as mean ± SD.
Extraembryonic anomalies precede embryonic malformations in Apela mutants
Morphological defects in affected Apela KO embryos were not evident until after circulation. To characterize defects in extraembryonic tissues, where both Apela and Aplnr are expressed at earlier stages, microCT analysis of Apela KO NEO-IN embryos was used. Results from E8.25 revealed ectopic protrusions from the anterior chorion (Figure 5C), which was not observed in stage-matched Apela heterozygous embryos (n=24, data not shown). There were also instances of ectopic protrusions from the YS mesoderm where the blood islands form (Figure 5C). Measurements of the proximal and mid-length regions of the allantois were comparable between affected and unaffected Apela KO embryos. However, the distal portion of the allantois was significantly thinner in 12% of affected Apela KO embryos, resulting in a smaller contact area with the chorion (Figure 5D).
Erythroid and myeloid genes are upregulated in affected Apela mutants
Apela is expressed in the embryo at late gastrulation, shortly after the emergence of extraembryonic mesoderm. We analyzed Apela KO NEO-IN embryos at E7.5 with lineage-specific markers to examine mesoderm (FLK-1, BRACHYURY) and endoderm (SOX17, FOXA2) populations, and we observed dispersal of the visceral endoderm by intercalation of DE using the Afp-GFP transgene (Kwon et al., 2008) (Figure S3A). Apela KO NEO-IN embryos appeared morphologically normal without qualitative defects in endoderm or mesoderm. However, one-fifth of Apela KO NEO-OUT mutants had asymmetric or kinked neuroectoderm at E7.5, despite correct patterning (Figure S3B). The reason for this remains unclear, but may be related to differential expression of untranslated regions of the Apela transcript.
To investigate the earliest regulatory events leading to abnormalities in Apela mutants, we performed RNA-Seq on whole and morphologically normal E7.5 embryos (3 wild-type, 6 Apela KO NEO-IN, and 6 Apela KO NEO-OUT individuals) (Figure S4A). Mapping of reads to Apela loci confirmed differential expression of 5′ UTR and 3′ UTR transcripts at very low levels in Apela mutants (Figure S4B), correlating with RT-PCR results (Figure 1C). Of note, modification of the Apela locus did not influence expression of neighboring genes (Figure S4C).
Initially, we highlighted DEGs that were altered across all Apela KO samples (Figure S4D, Supplemental File 1). Downregulated genes included mitochondrial genes, Ceacam2 and Ulk4 which play roles in cell adhesion and motility (Lang et al., 2014, Salaheldeen et al., 2012), and Mov10l1 (also known as CHAMP) which is an RNA helicase downstream of Mef2C, Nkx2.5, and ERK5 in the developing heart (Liu et al., 2001, Regan et al., 2002, Ueyama et al., 2003). Upregulated genes included the vascular endothelial growth factor Vegfc that is required for lymphatic endothelial cell migration (Karkkainen et al., 2004). Gene ontology (GO) analysis showed enrichment for receptor binding, receptor activity, immune system response, angiogenesis, and metabolism (Supplemental File 1).
Due to the variability in the Apela null phenotype, we explored intragroup variation among individuals by principal component (PC) analysis (Figure 6A). Samples aligned along the PC1 axis according to genotype. However, KO9 and KO1 were spread further along the PC2 axis. KO9 exhibited the most variability, with 116 DEGs uniquely misexpressed compared to others of the same genotype (Figure S5A). KO1 clustered with KO9, as predicted by their relationship in the PC analysis (Figure 6B), and both were characterized by downregulation of mesoderm genes (Figure S5A, Supplemental File 1). Genes upregulated in KO9 were primarily clusters of erythroid genes (Group 1; Kingsley et al., 2013) and myeloid genes (Group 2; Figure 6C, Supplemental File 1). The most upregulated were macrophage markers Integrin beta 2 (Itgb2), Lysozyme 2 (Lyz2), C4b, and Cfp (Figure 6D, Supplemental File 1), as well as the fetal hemoglobin gene Hbb-bh1. The early myeloid lineage marker MPO, and the differentiated macrophage marker CX3CR1 were not upregulated (Figure S5B), suggesting an enrichment for maturing but not fully differentiated myeloid cells.
FIGURE 6. Upregulation of erythroid and myeloid markers in Apela KO embryos.

(A) PC analysis separated samples according to genotype along PC1. KO9 exhibited unique, yet similar, variance to KO1 according to PC2. (B) Heat map of sample-to-sample distances between KO9 and other samples based on 116 DEGs identified in Figure S5A. Color scale indicates rlog transformed expression values. (C) Upregulated DEGs identified in (B) were classified by expression of erythroid (Group 1) and myeloid (Group 2) genes. (D) Boxplots of normalized expression values for genes with the highest upregulation in KO9. (E) Itgb2 expression by wholemount in situ hybridization in Apela KO (NEO-IN) embryos shows ectopic clusters of cells localized to extraembryonic tissue (blue arrows). (F–G) qRT-PCR of individual Apela (NEO-IN) and (NEO-OUT) embryos at E7.5. Data are represented as mean ± SEM.
We confirmed the upregulation of erythroid and myeloid genes in additional Apela null individuals at E7.5 (Figure 6E). We observed Apela mutants with ectopic and clustered Itgb2 expression in the proximal extraembryonic tissue. We speculate that these clusters of cells may lead to extraembryonic blood island protrusions that were observed one day later by microCT (Figure 5C). In addition, we performed qRT-PCR for myeloid (Lyz2, Itgb2, Itgam/CD11b, Adgre1/F4/80), erythroid (Hbb-bh1, Hbb-B1), and endothelial (VE-cadherin/VEC, Kdr/Flk-1) transcripts in an additional cohort of embryos (Figure 6F–G). Outliers were identified (KO20, KO23, KO33, and KO38, Figure S5C), and remaining samples were analyzed collectively (Figure 6F–G). There were no significant changes in expression among samples that were analyzed collectively. However, outliers expressed upregulation of erythroid and myeloid markers, as well as elevation of endothelial genes, suggesting that Apela signaling may affect EMP progenitors (Figure S5D). Taken together, our data showed that loss of Apela signaling impacted expression of hematopoietic genes in 13% of late gastrulation stage embryos (n=5/39 Apela KO embryos).
Surviving Apela null embryos have defects in vasculature and CD11b-expressing populations
To examine the effects of loss of Apela at later stages of development, we analyzed embryos at E10.5 using an antibody to PECAM-1 to label and quantify the somitic and cranial vasculature (Figure 7A–B). Apela KO NEO-IN embryos had smaller somites and significant differences in the total length of somitic vessel edges and number of branchpoints (Figure 7B). While the overall vascular density was comparable to controls, the complexity of somitic vasculature was reduced in Apela KO NEO-IN embryos. This was not observed in the cranial vasculature (Figure 7B).
FIGURE 7. Defects in somitic vasculature, CD11b-expressing cells, and coronary vasculature in surviving Apela KO embryos.

(A) MIPs of PECAM-1 (CD31) localization at E10.5. Regions of somitic (blue outline) and cranial (white dotted lines) vasculature quantified in (B). HB, hindbrain; Tel, telencephalon. (B) Quantification of vasculature in 35–37ss Apela (NEO-IN) heterozygous (n=5) and KO (n=5) embryos. Data are represented as mean ± SD. *p<0.05, **p<0.01, ***p<0.001. (C) FLK-1 and ITGAM (CD11b) localization at E10.5. Accumulation of ITGAM-expressing cells in the hindbrain (white arrowheads) and ectopic clustering (yellow arrowheads) below branchial arches (BA1, BA2). (D) FLK-1 and APLNR localization in the heart at E15.5. Nuclei are stained with Hoechst. Normal (white arrowheads) and truncated (yellow arrowheads) trajectory of the right coronary vein (RCV) expressing APLNR.
Itgam and Itgb2 were upregulated in Apela KO embryos at E7.5 (Figure 6C–G). CD11b (Integrin alpha M/ITGAM) is the heterodimeric partner of ITGB2, together constituting the macrophage-1 antigen receptor MAC-1. At E10.5, approximately half of Apela KO NEO-IN embryos had increased ITGAM expression in the hindbrain, along with accumulations of ITGAM-expressing clusters along the trajectory of the neural tube, extending from the branchial arches to the forelimb (Figure 7C). These results suggest that Apela signaling may regulate myeloid progenitors at E10.5, but it is possible that these abnormalities occur in response to earlier alterations in gene expression.
At E15.5, we observed some Apela NEO-IN KO embryos that were paler than their heterozygous and KO littermates (Figure 7D). Since Aplnr is expressed in the coronary vasculature (Arita et al., 2014), we analyzed expression of FLK-1 and APLNR in the heart. In Apela heterozygous embryos, the right coronary vein (RCV) expressed APLNR and extended to the posterior tip of the ventricle. However, in some Apela KO NEO-IN mutants, the RCV appeared truncated with smaller branches terminating more anteriorly. Taken together, our data suggest that Apela signaling impacts endothelial and hematopoietic cell populations during multiple stages of mouse embryonic development.
DISCUSSION
We investigated the role of Apela, a small peptide hormone, during mouse embryonic development. Affected Apela KO embryos resemble mutants with defective heart morphogenesis resulting from poor circulation (Shalaby et al., 1995, Huang et al, 2003. Xie et al., 2009). Establishment of circulation is a critical developmental event requiring flow of YS-derived hematopoietic cells to the embryo, as well as fetal-maternal exchange. This generates hemodynamic forces that are key for YS remodeling and cardiac looping (Huang et al., 2003; Koushik et al., 2001; Lucitti et al., 2007). Prior to these events, Apela null embryos form a linear heart tube, suggesting adequate heart field formation (Saga et al., 1999). Although, we cannot rule out secondary heart field insufficiencies that may lead to later structural heart defects.
In mice, Aplnr is expressed in nascent mesoderm, and Apela in the DE. In contrast to zebrafish, Apela KO mice did not exhibit endoderm defects (Chng et al., 2013, Pauli et al. 2014). This suggests non-conserved roles of Apela in vertebrate gastrulation, perhaps due to species-specific mechanisms of mesendoderm migration. Differences in the Apela and Aplnr mutant phenotypes at E9.5 led us to question the fidelity of these signaling partners. We showed that Apela-Aplnr signaling could be impaired by double heterozygous mutation, thereby leading to the same embryonic lethal defects observed in Apela KO embryos. This supports the notion of Apela-Aplnr signaling. However, the inability to increase the penetrance of the phenotype in Aplnr;Apela double KO mutants suggests that each ligand and receptor may have alternative partners that participate in the same developmental processes. An alternative receptor for Apela has been speculated from work in human ESCs (Li et al., 2015). Our Apelin;Apela double KO analysis does not support a role for Apelin as a critical ligand for Aplnr, but Aplnr may be activated by unidentified ligand(s) or ligand-independent mechanisms such as mechanosensory stretch (Paskaradevan and Scott, 2012, Scimia et al., 2012).
Erythroid and myeloid genes were unregulated in a subset of Apela mutants prior to blood flow. Primitive hematopoietic cells are present in the YS as early as E7.0 (Isern et al., 2011, Palis et al., 1999), whereas erythro-myeloid progenitors (EMPs) emerge from hemogenic endothelium in the YS at E8.5 (Kasaai et al., 2017). Distribution of EMPs from the YS is facilitated by circulation (Kasaai et al., 2017), yet there is also evidence for active cell mobilization (Tanaka et al., 2014). Overrepresentation of YS-derived myeloid cells has been linked to remodeling of the venous plexus of the YS (Kasaai et al., 2017), leading to embryonic lethal phenotypes resembling Apela mutants (Cortegano et al., 2014, Venkatesh et al., 2008). Whether erythromyeloid defects in late gastrulation stage Apela null embryos can be directly linked to lethal cardiovascular defects observed after circulation remains an open question. Evidence exists that Aplnr activation targets myeloid cells in other biological settings such as arterial-venous alignment (Kidoya et al., 2015).
A contemporaneous study of Apela function in the mouse reports both mother and fetus as sources of circulating Apela, the loss of which provides a model for preeclampsia in pregnant females (Ho et al., 2017). Systemic administration of Apela peptide to pregnant Apela mutant mothers rescued maternal pathology (hypertension and proteinuria) and KO embryo birth weights. Apelin was upregulated in Apela KO placentae, a compensatory mechanism that we did not observe in the embryo, however this was insufficient to rescue placental defects. Ho and colleagues reported low penetrance embryonic lethal defects in 43% of Apela KO embryos, in addition to placental defects from E10.5 onwards (Ho et al., 2017), whereas our study reports a 10% penetrance of embryonic lethal defects at E9.5. Furthermore, our data did not reveal a maternal effect, consistent with results from zebrafish mutants (Chng et al., 2013, Pauli et al, 2014). One possible explanation for the differences in the penetrance and maternal effects of Apela null mutation may be due to differences in the mouse strain background. Another possibility, alluded to by Ho and colleagues, could be differences in Apela mouse husbandry that may lead to transgenerational adaptation. Since our study focused on events leading up to cardiovascular defects at E9.5, we did not examine placental morphology at E10.5. However, both studies have noted expression of Apela in the placenta, where vascular remodeling of the fetal-maternal interface relies on factors secreted by macrophages in maternal decidual tissue (Hanna et al., 2006, Hazan et al., 2010). Considering the results from our study, along with the effect of Aplnr mutation on myeloid gene expression (Kidoya et al., 2015), one might speculate that misregulation of maternal and/or fetal macrophages contributes to the placental defects observed at later stages of development (Ho et al, 2017).
The cause of variable expressivity and low penetrance of the Apela null phenotype remains an open question. One possibility is alternative signaling options (either at the ligand or receptor level) and/or redundancy in parallel mechanisms that feed into the same developmental pathways. Another possible explanation is the inherent robustness of the circulatory system. For example, vascular remodeling from E9–E10 requires hemodynamic forces, yet tolerates natural embryo-to-embryo variabilities in circulation (Lucitti et al., 2007, McGrath et al., 2003). Furthermore, recovery from mild or impartial YS remodeling defects has been previously reported for mutants of other signaling pathways (Byrd et al., 2002).
Further understanding the effects of Apela peptide signaling on target tissues may provide insights into mechanisms that regulate the proliferative and migratory behavior of early hematopoietic and endothelial progenitors, and how this impacts the development of the mammalian cardiovascular system. Finally, if mesodermal derivatives were to be affected during development but not result in embryonic lethality, it raises the question of whether surviving adult mutant mice might exhibit reduced tolerance to physiological or pathological challenges.
EXPERIMENTAL PROCEDURES
Mice
ApelaH2B-GFP-NEO mice (JAX Stock No. 030845) were generated using a CRISPR/Cas9-nickase gene targeting strategy in C57BL/6-Tyrc-2J albino mESCs. Details of the targeting cnstruct design and screening of targeted mESC clones are provided in Supplemental Experimental Procedures. Two correctly targeted Apela heterozygous mESC clones were injected into C57BL/6 blastocysts. Male chimeras were crossed to CD-1 (Charles River Laboratories) females for two generations. Apelin (Kuba et al., 2007) and APJ (Ishida et al., 2004) mice were imported on a C57BL/6 background and crossed to CD-1 for two generations. Mice were housed under a 12-hour light/dark cycle and maintained in accordance with the guidelines of the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee. The date of vaginal plug was considered E0.5. Embryos were dissected in DMEM/F-12 (1:1) (GIBCO 11320) with 5% newborn calf serum, and stages defined using morphological landmarks (Downs and Davies, 1993) and pairs of somites (somite stage, ss). See Supplemental Experimental Procedures for details on mouse strains, genotyping, and statistical analysis.
Imaging
Embryos were imaged on a Leica M165FC microscope with Axiocam MRc camera, and raw data was processed with ZEN Blue software. Immunostained tissue sections and whole embryos placed on MatTek dishes were imaged on a Zeiss LSM880 confocal microscope, and raw data was processed using ZEN Black software to generate maximum intensity projections (MIPs).
PCR
RNA was isolated from whole embryos using the RNeasy Plus Mini Kit (Qiagen 74134). Embryos were obtained from Apela KO males crossed to heterozygous females and genotyped according to Apela expression. cDNA was made using Superscript III First-Strand Synthesis and polydT oligos (Invitrogen 18080-051). Reverse transcription PCR (RT-PCR) was carried out using standard protocols. Apela 5′ UTR fragments were cloned using TOPO TA (Invitrogen) for sequencing. Quantitative RT-PCR (qRT-PCR) was carried out using SYBR Select Master Mix for CFX (Applied Biosystems 4472942) with a Bio-Rad CFX96 thermal cycler. CT values were averaged from technical triplicates and normalized to 18S. Biological replicates were analyzed by comparative CT relative to expression from E7.5 late bud heterozygotes. SEM was calculated using mean variance and Gaussian error propagation. Primer sequences are provided in Supplemental Experimental Procedures.
Immunofluorescence
Fixed embryos were blocked in 5% horse serum and incubated with primary and Alexa Fluor conjugated secondary antibodies (Life Technologies, 1:500) overnight (O/N) at 4°C. Hoechst 33342 (Life Technologies, 1:500) and A lexa Fluor 568 Phalloidin (Life Technologies A12380, 1:500) were used. Tissue sections and YS wholemounts were coverslipped with Fluoromount G (Southern Biotech). CBF:H2B-Venus (Nowotschin et al., 2013) and Afp-GFP (Kwon et al., 2006) were imaged directly. See Supplemental Experimental Procedures for details on embryo processing, primary antibodies, and vascular quantification.
RNA in situ Hybridization
Fixed embryos were dehydrated in a methanol series. Riboprobes were detected with anti-Digoxigenin-AP, Fab fragments (Roche 11093274910, 1:10,000) and BM purple (Roche 11442074001). See Supplemental Experimental Procedures for details on hybridization, blocking, and riboprobes.
microCT Imaging
Whole decidua were fixed O/N in 4% PFA at 4°C, wash ed 3 × 10 min, and stored in 0.1% sodium azide in PBS. Iodine contrast was achieved by immersion in 0.1N (v/v) iodine solution (Sigma) O/N at room temperature. Images were captured with a SKYSCAN 1272 microCT scanner (Bruker) and digitally segmented on CTAn (Hsu et al., 2016). Decidua were isolated from Apela KO NEO-IN intercrosses (for mutant analysis) or Apela KO NEO-IN males crossed to CD-1 females (for controls).
RNA-Seq
RNA was isolated from whole embryos using the RNeasy Plus Mini Kit (Qiagen 74134). Mutant embryos were obtained from Apela KO intercrosses, and controls were isolated separately. New York Genome Center performed quality control (RSeQC; Picard), TruSeq mRNA Library Prep (Illumina), paired end sequencing (Illumina HiSeq2500; 50bp reads; 30–50MB), trimming and alignment of reads to the mouse genome (mm10), gene quantification (FeatureCounts), gene annotation (Gencode), transcript quantification (Kallisto), normalization and differential expression (DESeq2). GOSeq and GOStat were used for gene ontology analysis. Boxplots were generated in R.
Supplementary Material
All DEGs with non-zero log2 fold change and adjusted p-value <0.01 are listed (DEG_NEOINvsControl and DEG_NEOOUTvsControl). GO analysis was performed using GOStat and GOSeq (GO_Analysis). DEGs are listed with log2 fold change <−0.05 (Common_DOWNreg_DEG) and >0.05 (Common_UPreg_DEG) with adjusted p-value <0.01. DEGs identified when comparing KO9 to all other Apela KO NEO-OUT samples with FDR 1% are listed (DEGs_KO9_vs_otherKO).
A complete list of all DEGs with adjusted p-value <0.01 that are common in Apela KO NEO-IN and Apela KO NEO-OUT mutants before applying log2 fold change cutoff (Common_DEG). All other DEGs that were unique to each mutant strains are listed separately (NEO-IN_non-shared and NEO-OUT_non-shared).
Acknowledgments
We thank members of the Hadjantonakis lab and Nicolas Porchet for comments and suggestions on the manuscript; MSKCC’s Mouse Genetics Core Facility for production of germline transmitting chimeras; Rockefeller University’s Gene Targeting Facility for assistance in the generation of targeted ES cells; the New York Genome Center for sequence data generation and analysis; the Optical Imaging and Vital Microscopy core at BCM for microCT data. This work was supported by a Ruth L. Kirschstein fellowship from the National Institutes of Health F32GM115089 (LF), R01DK084391, P30CA008748 (AKH), and U54HG006348, R01HL128064 (MED) and the Natural Sciences and Engineering Research Council of Canada RGPIN418298-12 (PAH). The ApelaH2B-GFP (Apelaem1.1Hadj/J) strain of mice is available from the Jackson Labs Mouse Mutant Resource as JAX Stock No.030845.
Footnotes
ACCESSION NUMBERS
GEO: GSE101748
AUTHOR CONTRIBUTIONS
Concez ptualization, L.F., S.N., A.P., A.F.S., P.H. and A.K.H.; Methodology, S.N.; Investigation, L.F. and C.H.; Resources, J.I., K.K., and A.F.; Writing - Original Draft, L.F.; Writing - Review & Editing, L.F., S.N., A.P., A.F.S., P.H., M.E.D. and A.K.H; Visualization, L.F.; Supervision, A.P., A.F.S., P.H., M.E.D., and A.K.H.; Project Administration, A.K.H. Funding Acquisition, L.F., P.A.H., M.E.D. and A.K.H.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arita Y, Nakaoka Y, Matsunaga T, Kidoya H, Yamamizu K, Arima Y, Kataoka-Hashimoto T, Ikeoka K, Yasui T, Masaki T, et al. Myocardium-derived angiopoietin-1 is essential for coronary vein formation in the developing heart. Nat Commun. 2014;5:4552. doi: 10.1038/ncomms5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY, Jalil A, Klaine M, Jung S, Cumano A, Godin I. Three pathways to mature macrophages in the early mouse yolk sac. Blood. 2005;106:3004–3011. doi: 10.1182/blood-2005-02-0461. [DOI] [PubMed] [Google Scholar]
- Byrd N, Becker S, Maye P, Narasimhaiah R, St-Jacques B, Zhang X, McMahon J, McMahon A, Grabel L. Hedgehog is required for murine yolk sac angiogenesis. Development. 2002;129:361–372. doi: 10.1242/dev.129.2.361. [DOI] [PubMed] [Google Scholar]
- Campione M, Steinbeisser H, Schweickert A, Deissler K, van Bebber F, Lowe LA, Nowotschin S, Viebahn C, Haffter P, Kuehn MR, Blum M. The homeobox gene Pitx2: mediator of asymmetric left-right signaling in vertebrates heart and gut looping. Development. 1999;126:1225–1234. doi: 10.1242/dev.126.6.1225. [DOI] [PubMed] [Google Scholar]
- Chng SC, Ho L, Tian J, Reversade B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell. 2013;27:672–680. doi: 10.1016/j.devcel.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Cortegano I, Melgar-Rojas P, Luna-Zurita L, Siguero-Alvarez M, Marcos MA, Gaspar ML, de la Pompa JL. Notch1 regulates progenitor cell proliferation and differentiation during mouse yolk sac hematopoiesis. Cell Death Differ. 2014;21:1081–1094. doi: 10.1038/cdd.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aniello C, Fiorenzano A, Iaconis S, Liguori GL, Andolfi G, Cobellis G, Fico A, Minchiotti G. The G-protein coupled receptor APJ is expressed in the second heart field and regulates Cerberus-Baf60c axis in embryonic stem cell cardiomyogenesis. Cardiovasc Res. 2013;100:95–104. doi: 10.1093/cvr/cvt166. [DOI] [PubMed] [Google Scholar]
- Devic E, Rizzoti K, Bodin S, Knibiehler B, Audigier Y. Amino acid sequence and embryonic expression of msr/apj, the mouse homolog of Xenopu X-msr and human. APJ Mech Dev. 1999;84:199–203. doi: 10.1016/s0925-4773(99)00081-7. [DOI] [PubMed] [Google Scholar]
- Downs KM, Davies T. Staging of gastrulating mouse embryos by morphological landmarks in the dissecting microscope. Development. 1993;118:1255–1266. doi: 10.1242/dev.118.4.1255. [DOI] [PubMed] [Google Scholar]
- Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. 2006;12:1065–1074. doi: 10.1038/nm1452. [DOI] [PubMed] [Google Scholar]
- Hassan AS, Hou J, Wei W, Hoodless PA. Expression of two novel transcripts in the mouse definitive endoderm. Gene Expr Patterns. 2010;10:127–134. doi: 10.1016/j.gep.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan AD, Smith SD, Jones RL, Whittle W, Lye SJ, Dunk CE. Vascular-leukocyte interactions: mechanisms of human decidual spiral artery remodeling in vitro. Am J Pathol. 2010;177:1017–1030. doi: 10.2353/ajpath.2010.091105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helker CSM, Schuermann A, Pollmann C, Chang SC, Kiefer F, Reversade B, Herzog W. The hormonal peptide Elabela guides angioblasts to the midline during vasculogenesis. ELife. 2015 doi: 10.7554/eLife.06726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Van Dijk M, Chye STJ, Messerschmidt DM, Chng SC, Ong S, Yi LK, Boussata S, Goh GH-Y, Afink GB, Lim CY, Dunn NR, Solter D, Knowles BB, Reversade B. ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science. 2017 doi: 10.1126/science.aam6607. [DOI] [PubMed] [Google Scholar]
- Hsu CW, Wong L, Rasmussen TL, Kalaga S, McElwee ML, Keith LC, Bohat R, Seavitt JR, Beaudet AL, Dickinson ME. Three-dimensional microCT imaging of mouse development from early post-implantation to early postnatal stages. Dev Biol. 2016;419(2):229–236. doi: 10.1016/j.ydbio.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Sheikh F, Hollander M, Cai C, Becker D, Chu PH, Evans S, Chen J. Embryonic atrial function is essential for mouse embryogenesis, cardiac morphogenesis and angiogenesis. Development. 2003;130:6111–6119. doi: 10.1242/dev.00831. [DOI] [PubMed] [Google Scholar]
- Inui M, Fukui A, Ito Y, Asashima M. Xapelin and Xmsr are required for cardiovascular development in Xenopus laevis. Dev Biol. 2006;298:188–200. doi: 10.1016/j.ydbio.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Isern J, He Z, Fraser ST, Nowotschin S, Ferrer-Vaquer A, Moore R, Hadjantonakis AK, Schulz V, Tuck D, Gallagher PG, et al. Single-lineage transcriptome analysis reveals key regulatory pathways in primitive erythroid progenitors in the mouse embryo. Blood. 2011;117:4924–4934. doi: 10.1182/blood-2010-10-313676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida J, Hashimoto T, Hashimoto Y, Nishiwaki S, Iguchi T, Harada S, Sugaya T, Matsuzaki H, Yamamoto R, Shiota N, et al. Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J Biol Chem. 2004;279:26274–26279. doi: 10.1074/jbc.M404149200. [DOI] [PubMed] [Google Scholar]
- Kang Y, Kim J, Anderson JP, Wu J, Gleim SR, Kundu RK, McLean DL, Kim JD, Park H, Jin SW, et al. Apelin-APJ signaling is a critical regulator of endothelial MEF2 activation in cardiovascular development. Circ Res. 2013;113:22–31. doi: 10.1161/CIRCRESAHA.113.301324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74–80. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- Kasaai B, Caolo V, Peacock HM, Lehoux S, Gomez-Perdiguero E, Luttun A, Jones EA. Erythro-myeloid progenitors can differentiate from endothelial cells and modulate embryonic vascular remodeling. Sci Rep. 2017;7:43817. doi: 10.1038/srep43817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsunaga Sakai J-iM. A Transgenic Mouse Line That Retains Cre Recombinase Activity in Mature Oocytes Irrespective of the cre Transgene Transmission. Biochemical and Biophysical Research Communications. 1997;237:318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- Kidoya H, Naito H, Muramatsu F, Yamakawa D, Jia W, Ikawa M, Sonobe T, Tsuchimochi H, Shirai M, Adams RH, et al. APJ Regulates Parallel Alignment of Arteries and Veins in the Skin. Dev Cell. 2015;33:247–259. doi: 10.1016/j.devcel.2015.02.024. [DOI] [PubMed] [Google Scholar]
- Kingsley PD, Greenfest-Allen E, Frame JM, Bushnell TP, Malik J, McGrath KE, Stoeckert CJ, Palis J. Ontogeny of erythroid gene expression. Blood. 2013;121:e5–e13. doi: 10.1182/blood-2012-04-422394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushik SV, Wang J, Rogers R, Moskophidis D, Lambert NA, Creazzo TL, Conway SJ. Targeted inactivation of the sodium-calcium exchanger (Ncx1) results in the lack of a heartbeat and abnormal myofibrillar organization. FASEB J. 2001;15:1209–1211. doi: 10.1096/fj.00-0696fje. [DOI] [PubMed] [Google Scholar]
- Kuba K, Zhang L, Imai Y, Arab S, Chen M, Maekawa Y, Leschnik M, Leibbrandt A, Markovic M, Schwaighofer J, et al. Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ Res. 2007;101:e32–42. doi: 10.1161/CIRCRESAHA.107.158659. [DOI] [PubMed] [Google Scholar]
- Kwon GS, Fraser ST, Eakin GS, Mangano M, Isern J, Sahr KE, Hadjantonakis AK, Baron MH. Tg(Afp-GFP) expression marks primitive and definitive endoderm lineages during mouse development. Dev Dyn. 2006;235:2549–2558. doi: 10.1002/dvdy.20843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon GS, Viotti M, Hadjantonakis AK. The endoderm of the mouse embryo arises by dynamic widespread intercalation of embryonic and extraembryonic lineages. Dev Cell. 2008;15:509–520. doi: 10.1016/j.devcel.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang B, Pu J, Hunter I, Liu M, Martin-Granados C, Reilly TJ, Gao GD, Guan ZL, Li WD, Shi YY, et al. Recurrent deletions of ULK4 in schizophrenia: a gene crucial for neuritogenesis and neuronal motility. J Cell Sci. 2014;127:630–640. doi: 10.1242/jcs.137604. [DOI] [PubMed] [Google Scholar]
- Li M, Gou H, Tripathi BK, Huang J, Jiang S, Dubois W, Waybright T, Lei M, Shi J, Zhou M, et al. An Apela RNA-Containing Negative Feedback Loop Regulates p53-Mediated Apoptosis in Embryonic Stem Cells. Cell stem cell. 2015;16:669–683. doi: 10.1016/j.stem.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZP, Nakagawa O, Nakagawa M, Yanagisawa H, Passier R, Richardson JA, Srivastava D, Olson EN. CHAMP, a novel cardiac-specific helicase regulated by MEF2C. Dev Biol. 2001;234:497–509. doi: 10.1006/dbio.2001.0277. [DOI] [PubMed] [Google Scholar]
- Lucitti JL, Jones EA, Huang C, Chen J, Fraser SE, Dickinson ME. Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development. 2007;134:3317–3326. doi: 10.1242/dev.02883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath KE, Koniski AD, Malik J, Palis J. Circulation is established in a stepwise pattern in the mammalian embryo. Blood. 2003;101:1669–1676. doi: 10.1182/blood-2002-08-2531. [DOI] [PubMed] [Google Scholar]
- Nowotschin S, Xenopoulos P, Schrode N, Hadjantonakis AK. A bright single-cell resolution live imaging reporter of Notch signaling in the mouse. BMC Dev Biol. 2013;13:15. doi: 10.1186/1471-213X-13-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palis J, Robertson S, Kennedy M, Wall C, Keller G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development. 1999;126:5073–5084. doi: 10.1242/dev.126.22.5073. [DOI] [PubMed] [Google Scholar]
- Paskaradevan S, Scott IC. The Aplnr GPCR regulates myocardial progenitor development via a novel cell-non-autonomous, Galpha(i/o) protein-independent pathway. Biol Open. 2012;1:275–285. doi: 10.1242/bio.2012380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343:1248636. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Valen E, Schier AF. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 2015;37:103–12. doi: 10.1002/bies.201400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedra ME, Icardo JM, Algajar M, Rodriguez-Rey JC, Ros MA. Pitx2 participates in the late phase of the pathway controlling left-right asymmetry. Cell. 1998;94:319–324. doi: 10.1016/s0092-8674(00)81475-0. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Blumberg B, Rodriguez-Esteban C, Yonei-Tamura S, Tamura K, Tsukui T, de la Peña J, Sabbagh W, Greenwald J, Choe S, Norris DP, Robertson EJ, Evans RM, Rosenfeld MG, Izpisúa-Belmonte JC. Pitx2 determines left-right asymmetry of internal organs in vertebrates. Nature. 1998;394:545–551. doi: 10.1038/29004. [DOI] [PubMed] [Google Scholar]
- Regan CP, Li W, Boucher DM, Spatz S, Su MS, Kuida K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc Natl Acad Sci U S A. 2002;99:9248–9253. doi: 10.1073/pnas.142293999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki JI, Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Dev. 1999;126:3437–3447. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- Salaheldeen E, Kurio H, Howida A, Iida H. Molecular cloning and localization of a CEACAM2 isoform, CEACAM2-L, expressed in spermatids in mouse testis. Mol Reprod Dev. 2012;79:843–852. doi: 10.1002/mrd.22123. [DOI] [PubMed] [Google Scholar]
- Scimia MC, Hurtado C, Ray S, Metzler S, Wei K, Wang J, Woods CE, Purcell NH, Catalucci D, Akasaka T, et al. APJ acts as a dual receptor in cardiac hypertrophy. Nature. 2012;488:394–398. doi: 10.1038/nature11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Sanchez V, Takata N, Yokomizo T, Yamanaka Y, Kataoka H, Hoppe PS, Schroeder T, Nishikawa S. Circulation-independent differentiation pathway from extraembryonic mesoderm toward hematopoietic stem cells via hemogenic angioblasts. Cell Rep. 2014;8:31–39. doi: 10.1016/j.celrep.2014.05.055. [DOI] [PubMed] [Google Scholar]
- Ueyama T, Kasahara H, Ishiwata T, Yamasaki N, Izumo S. Csm, a cardiac-specific isoform of the RNA helicase Mov10l1, is regulated by Nkx2.5 in embryonic heart. J Biol Chem. 2003;278:28750–28757. doi: 10.1074/jbc.M300014200. [DOI] [PubMed] [Google Scholar]
- Venkatesh DA, Park KS, Harrington A, Miceli-Libby L, Yoon JK, Liaw L. Cardiovascular and hematopoietic defects associated with Notch1 activation in embryonic Tie2-expressing populations. Circ Res. 2008;103:423–431. doi: 10.1161/CIRCRESAHA.108.177808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Wu T, Xu K, Huang IK, Cleaver O, Huang CL. Endothelial-specific expression of WNK1 kinase is essential for angiogenesis and heart development in mice. Am J Pathol. 2009;175:1315–1327. doi: 10.2353/ajpath.2009.090094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka H, Meno C, Koshiba K, Sugihara M, Itoh H, Ishimaru Y, Inoue T, Ohuchi H, Semina EV, Murray JC, Hamada H, Noji S. Pitx2, a bicoid-type homeobox gene, is involved in a lefty-signaling pathway in determination of left-right asymmetry. Cell. 1998;94:299–305. doi: 10.1016/s0092-8674(00)81473-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All DEGs with non-zero log2 fold change and adjusted p-value <0.01 are listed (DEG_NEOINvsControl and DEG_NEOOUTvsControl). GO analysis was performed using GOStat and GOSeq (GO_Analysis). DEGs are listed with log2 fold change <−0.05 (Common_DOWNreg_DEG) and >0.05 (Common_UPreg_DEG) with adjusted p-value <0.01. DEGs identified when comparing KO9 to all other Apela KO NEO-OUT samples with FDR 1% are listed (DEGs_KO9_vs_otherKO).
A complete list of all DEGs with adjusted p-value <0.01 that are common in Apela KO NEO-IN and Apela KO NEO-OUT mutants before applying log2 fold change cutoff (Common_DEG). All other DEGs that were unique to each mutant strains are listed separately (NEO-IN_non-shared and NEO-OUT_non-shared).