

Abstract
The use of noncanonical amino acids (ncAAs) to control an organism's viability provides a strategy for the development of conditional ‘kill switches’ for live vaccines or engineered human cells. Here we report an approach inspired by the posttranslational acetylation/deacetylation of lysine residues, in which a protein encoded by a gene with an in-frame nonsense codon at an essential lysine can be expressed in its native state only upon genetic incorporation of AcK, and subsequent enzymatic deacetylation in the host cell. We have applied this strategy to two essential E. coli enzymes, the branched chain aminotransferase BCAT and the DNA replication initiator protein DnaA. We also devised a barnase-based conditional suicide switch to further lower the escape frequency of the host cells. This strategy offers a number of attractive features for controlling host viability, including a single small molecule-based kill switch, low escape frequency and unaffected protein function.
Keywords: noncanonical amino acid, deacetylation, post-translational modification, live vaccine, lysine
Graphical abstract
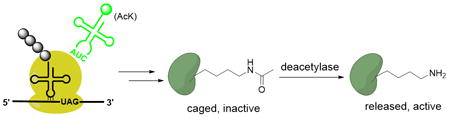
A PTM-inspired strategy for creating noncanonical amino acid dependent organisms was developed in which a protein encoded by a gene with an in-frame nonsense codon at an essential lysine can be expressed in its native state only upon genetic incorporation of AcK, and subsequent enzymatic deacetylation in the host cell. We also devised a barnase-based conditional suicide switch to further lower the escape frequency. This strategy offers a number of attractive features for controlling host viability, including a single small molecule-based kill switch, low escape frequency and unaffected protein function.
Genetically encoded noncanonical amino acids (ncAAs) have been used as probes of protein structure and function both in vitro and in living cells, and to generate proteins with improved or novel activities.[1] Recently, this technology has been used to create genetically modified organisms that are dependent on ncAAs for viability. In the presence of the ncAA (which is not naturally occurring in the host), an essential protein that contains the ncAA is translated and the organism grows at the normal rate. When the ncAA is absent, full length protein is not made and the organism cannot survive. This strategy has been applied to the development of live-attenuated vaccines,[2] synthetic auxotrophic strains[3] and bacteria with ncAA dependent antibiotic resistance.[4] However, the identification of sites in a single target protein that have a strict requirement for ncAA incorporation and where the ncAA does not impair wild type protein activity remains a challenge. Mutations at permissive sites typically suffer from high escape frequencies,[5] while mutations at active sites can negatively affect protein function, requiring computational design or in vitro evolution to achieve both strict ncAA dependence and the required level of activity for cell growth.[3-4] Therefore, additional approaches are desirable that are both simple to implement and generalizable.
The acetylation of lysine ε-amino groups is critical for a vast array of cellular processes in all kingdoms of life.[6] In bacteria, this posttranslational modification (PTM) derives from the acyl group of either acetyl-coenzyme A (acCoA) catalyzed by acetyltransferase PatZ or acetyl phosphate only. Deacetylation is carried out by the NAD+-dependent deacetylase cobB which belongs to a universally conserved sirtuin family and possesses broad substrate scope (Fig 1 A).[7] On the basis of this naturally occurring PTM, we devised a general strategy to create proteins whose activity is strictly dependent on the presence of an ncAA. We reasoned that when an essential lysine in a protein is replaced with genetically encoded N-ε-acetyl-L-Lys (AcK) in response to the amber nonsense codon, an endogenous deacetylase may be able to remove the acetyl moiety to afford the target protein in its native state (Fig 1B). When AcK is depleted, wild type protein is not biosynthesized and the cell cannot survive. Because deacetylases, including sirtuins and histone deacetylases (HDACs), are present in both prokaryotic and eukaryotic cells,[8] we expect that this approach will be generalizable to a number of host organisms. Moreover, an orthogonal amber suppressor pyrrolysyl-tRNA synthetase (AcKRS)/tRNAPyl pair for AcK was previously developed that selectively incorporates AcK into proteins in both prokaryotic and eukaryotic cells.[9]
Fig 1.
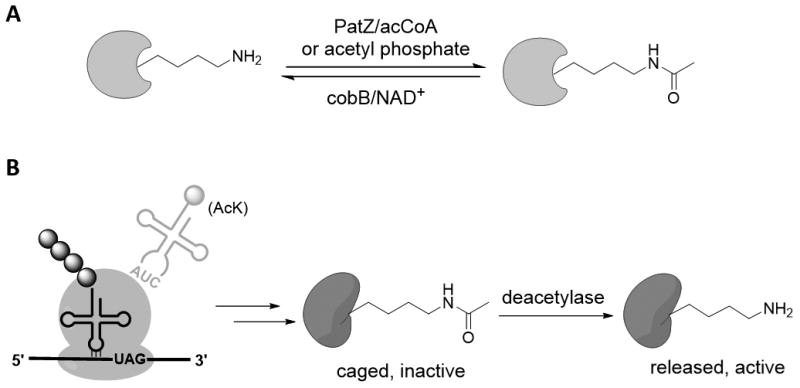
A) Protein lysine acetylation/deacetylation in E. coli cellular metabolism; B) The replacement of an essential lysine by genetically encoded AcK that is inserted in response to an amber (TAG) nonsense codon, followed by deacetylation by an endogenous deacetylase affords protein in the native state.
To test this approach, we initially targeted the essential branched chain aminotransferase BCAT in E. coli which catalyzes the synthesis of Leu, Ile and Val and has an active site K159 which covalently binds pyridoxal 5′-phosphate (PLP). When K159 in E. coli BCAT (encoded by the ilvE gene) was mutated to any of the other 19 amino acids, none of the variants had sufficient activity to allow host cell growth on M9-glucose plates (Fig S1). To genetically incorporate AcK, an E. coli ΔilvE strain was co-transformed with a plasmid containing ilvE-K159TAG and a second plasmid containing AcKRS and its cognate tRNAPyl. As shown in Fig 2A, this strain showed robust growth in the presence of 2 mM AcK, while no growth was observed in the absence of AcK after 24 hours (indicating that there is no background amber suppression with Lys in the absence of the ncAA; Fig. S6). Also, the presence of 20 mM NAM (nicotinamide, a noncompetitive inhibitor of the sirtuin deacetylation reaction) blocked AcK-dependent growth, indicating that the endogenous deacetylase is required to uncage the active site lysine. To confirm the genetic incorporation of AcK and subsequent deacetylation reaction, a C-terminal His-tagged K159AcK mutant of BCAT was expressed in E. coli ΔilvE, purified by Ni-NTA column and analyzed by LC-ESI-QTOF mass spectrometry. As shown in Fig 2B, the observed mass (35070.55 Da) of the BCAT-K159AcK mutant expressed in the absence of NAM corresponded to that of the wild type enzyme (35070.66 Da); a mass corresponding to that of BCAT-K159AcK (35112.67 Da) was observed (35112.37 Da) only when protein was expressed in the presence of 20 mM NAM (Fig 2B). Notably, the desired deacetylation reaction went to the completion. Also, no deacetylation was observed when BCAT-K159AcK was expressed in an E. coli ΔcobB strain, indicating that cobB is the endogenous deacetylase responsible for the observed deacetylation reaction (Fig S2). Direct deacetylation was also observed by incubating BCAT-K159AcK, cobB and NAD+ in 20 mM, pH 8, HEPES buffer at 37 °C (Fig S3). As a control, we inserted TAG at a permissive site, K21, on the protein surface. This mutant also showed AcK-dependent growth on M9-glucose plates, but addition of NAM no longer inhibited cell growth on agar plates, consistent with the permissive nature of this site (Fig S5). In M9-glucose liquid medium, NAM reduced the growth of BCAT-K159AcK mutant much more than that of BCAT-K21 AcK mutant (Fig S6).
Fig 2.
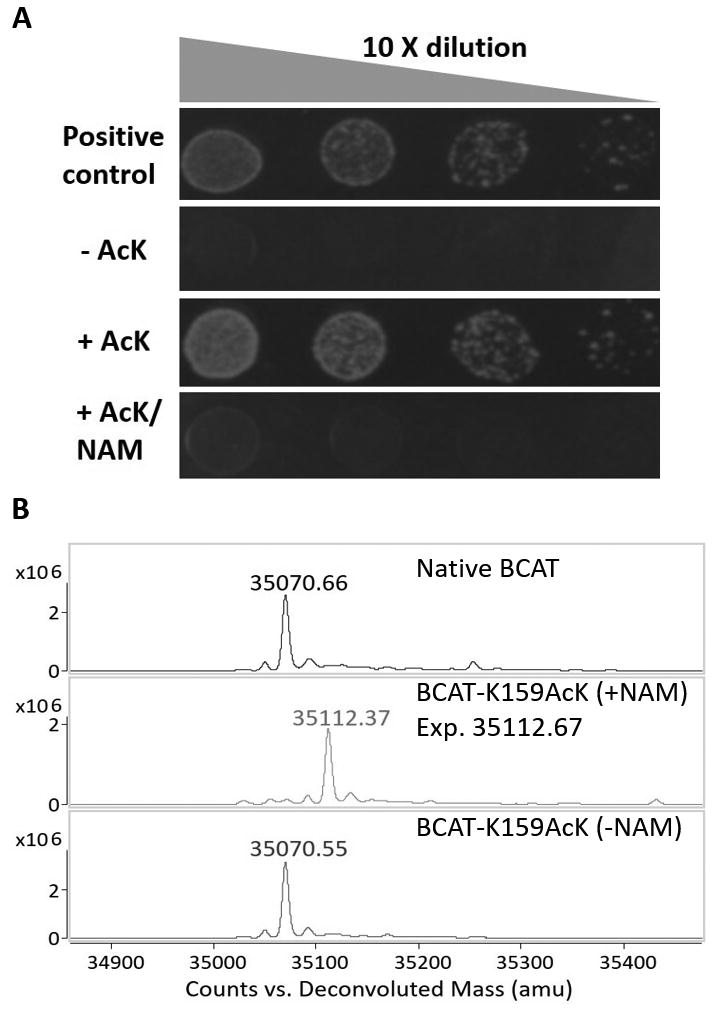
ncAA dependence of E. coli ΔilvE strain. A) Viability of E. coli ΔilvE strain harboring ilvE-K159TAG and AcKRS/tRNAPyl on M9-glucose plates at 37 °C after 24 hours. The positive control contained the wild type ilvE gene; 2 mM AcK and 20 mM NAM were used; B) LC-ESI-QTOF analysis of BCAT-K159AcK expressed in the presence or absence of NAM.
To determine the stringency of AcK dependent cell growth, cultures containing these two ilvE mutants (K159TAG and K21TAG) were passaged for over 100 generations in M9-glucose medium. The escape frequencies were determined to be 1.9 × 10−8 and 1.4 × 10−3 for the K159TAG and K21TAG mutants, respectively, underscoring the strict requirement for substitution of AcK in the active site. To understand the escape mechanism, the ilvE escape mutant was identified and analyzed. No mutation in the plasmid DNA was observed. However, sequencing of the six copies of lysyl-tRNA in the genome revealed an U to A mutation in the third nucleotide of lysyl-tRNA anticodon for the LysT gene generating a 5′-UUA-3′ anticodon.[10] This mutation affords a nonsense suppressor tRNA that can insert Lys at the TAG codon by G/U wobble pair formation. However, one solution is to simply delete the LysT gene in the genome.[3a[ As an alternative, we developed a conditional suicide switch. Barnase is a bacterial ribonuclease, and is lethal to the cell when expressed without its inhibitor barstar. We hypothesized that a barnase variant with in-frame ochre mutations can serve as a conditional suicide switch that will only be turned on by the presence of the mutant LysT suppressor tRNA. As a result, expression of this barnase variant should further reduce the escape frequency from the K159AcK BCAT mutant, since escape by generating ochre suppressors is lethal.
To test this notion, we constructed barnase variants with in-frame TAA mutations under control of the araBAD promoter. In order to minimize unwanted toxicity to normal strains, we introduced a single TAA codon at K27 which is essential for ribonuclease activity.[11] As expected, the expression of this plasmid-encoded barnase variant was lethal to the escape strain, but obvious toxicity was also observed in the original ΔilvE strain. When a second TAA at K62 was introduced into this barnase variant, the resulting toxicity was negligible in the wild type strain, but remained high for the escape strain (Fig S7). Therefore, we incorporated this barnase variant into the plasmid with ilvE-K159TAG, and found that it could effectively kill the escape strain (Fig S8), and had minimal effect on the growth rate of the corresponding strains (Fig S9). The escape frequency was examined as described above after 100 generations of iterative growth in the presence of AcK. No escape strain was observed in experiment performed with the improved complementation plasmid containing the conditional suicide switch (Fig 3B). This result suggests that the generation of a lysyl-tRNA derived ochre suppressor is the major challenge to ncAA dependence, and the barnase-based conditional suicide switch can reduce the escape frequency to an undetectable level.
Fig 3.
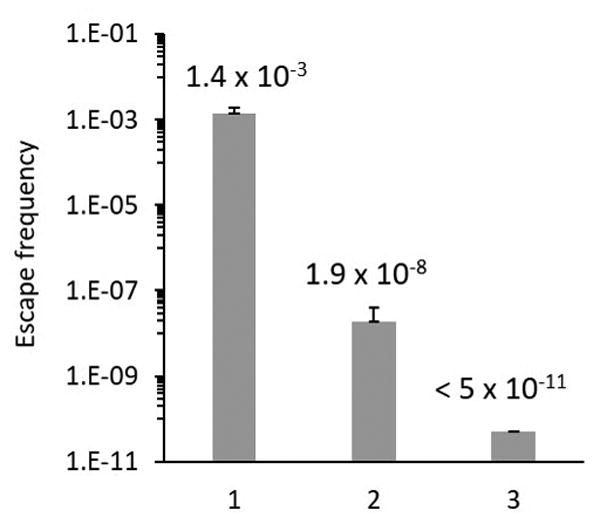
Escape frequency of ΔilvE strain harboring different ilvE variants and AcKRS/tRNAPyl. 1. K21TAG; 2. K159TAG; 3. K159TAG-barnase (2TAA). Positive error bar indicates the standard deviation.
As a further demonstration of this strategy, we attempted to replace K178 in the E. coli chromosome replication initiator protein DnaA with AcK. In E. coli, chromosome replication is initiated in each cell division cycle by DnaA, in which the conserved K178 is essential for ATP binding, and regulated by reversible acetylation.[12] We employed an E. coli strain BLR(DE3) [a ΔRecA strain (TetR) derived from BL21(DE3)],[13] and integrated a copy of the conditional suicide switch and a selectable maker gene (KanR) into the DnaA locus in the presence of an IPTG inducible complementation plasmid containing AcKRS/tRNAPyl and DnaA-K178TAG (Fig 4A). Gene insertion was verifed by colony PCR (Fig S10) and further confirmed by DNA sequencing. This mutant strain showed both AcK and IPTG dependent growth on LB agar plates, and the growth was inhibited in the presence of NAM (Fig 4B). The growth rate was slightly slower than that of the original strain, and strictly controlled by the concentration of AcK, ranging from 0.05 – 5 mM (Fig S11). The escape frequency was examined after 100 generations of iterative growth in the presence of 4 mM AcK, and no escape was observed after screening 2 × 1010 colonies on LB agar plates.
Fig 4.
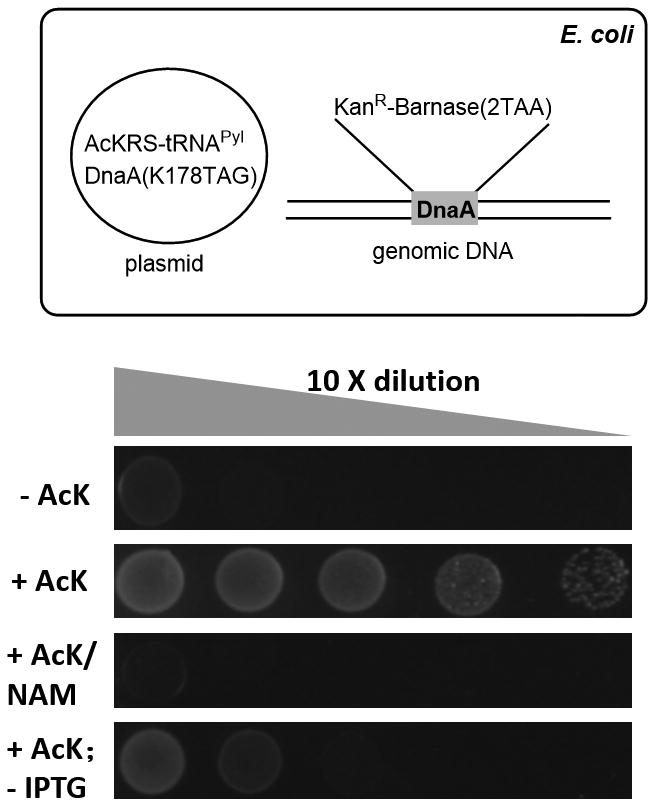
A) Wild type DnaA in E. coli BLR(DE3) genome was detected by integrating the conditional suicide switch and selectable maker gene (KanR) in the presence of complementation plasmid pUltra-AckRS/tRNAPyl-DnaA(K178TAG). B) Viability of the obtained E. coli BLR(DE3) mutant on LB plates at 37°C for 20 hours in the presence of 12.5 μg/mL tetracycline and 25 μg/mL kanamycin. 4 mM AcK, 20 mM NAM and 1 mM IPTG were used.
In summary, we have developed a novel strategy to create ncAA dependent organisms in which a native protein encoded by a gene with an in-frame TAG mutation at an essential lysine is expressed by the genetic incorporation of AcK, and subsequent enzyme-catalyzed deacetylation. This strategy leads to stringent ncAA dependence without affecting protein activity, and in the presence of the barnase-based suicide switch has a very low escape frequency. This simple strategy requires minimal genomic manipulation, and is easily adaptable to other organisms since the PylRS/tRNAPyl pair functions in both prokaryotic and eukaryotic organisms, and many organisms have lysine deacetylases. In addition we are exploring other PTMs such as Ser/Tyr phosphorylation with the corresponding phosphatase, to target other essential residues. Finally we are beginning to explore the utility of this approach in two settings. We are testing whether ncAA dependent hosts cultured in the presence of the ncAA, and then used as immunogens in vivo, induce robust immune responses and then die as the ncAA is depleted through rounds of cell division. We are also testing this strategy as a molecular kill switch for CAR-T cells.
Supplementary Material
Acknowledgments
We acknowledge Dr. Sean Reed and Dr. Travis Young for productive discussions and Kristen Williams for her assistance in manuscript preparation. This work is supported by NIH grant R01 GM062159 (P.G.S). This is manuscript 29434 of The Scripps Research Institute.
References
- 1.a) Liu CC, Schultz PG. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]; b) Xiao H, Schultz PG. Cold Spring Harb Perspect Biol. 2016;8 doi: 10.1101/cshperspect.a023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Si LL, Xu H, Zhou XY, Zhang ZW, Tian ZY, Wang Y, Wu YM, Zhang B, Niu ZL, Zhang CL, Fu G, Xiao SL, Xia Q, Zhang LH, Zhou DM. Science. 2016;354:1170–1173. doi: 10.1126/science.aah5869. [DOI] [PubMed] [Google Scholar]; b) Wang N, Li Y, Niu W, Sun M, Cerny R, Li Q, Guo J. Angew Chem Int Ed. 2014;53:4867–4871. doi: 10.1002/anie.201402092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Rovner AJ, Haimovich AD, Katz SR, Li Z, Grome MW, Gassaway BM, Amiram M, Patel JR, Gallagher RR, Rinehart J, Isaacs FJ. Nature. 2015;518:89–93. doi: 10.1038/nature14095. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mandell DJ, Lajoie MJ, Mee MT, Takeuchi R, Kuznetsov G, Norville JE, Gregg CJ, Stoddard BL, Church GM. Nature. 2015;518:55–60. doi: 10.1038/nature14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tack DS, Ellefson JW, Thyer R, Wang B, Gollihar J, Forster MT, Ellington AD. Nat Chem Biol. 2016;12:138–140. doi: 10.1038/nchembio.2002. [DOI] [PubMed] [Google Scholar]
- 5.a) Eggertsson G, Söll D. Microbiol Rev. 1988;52:354–374. doi: 10.1128/mr.52.3.354-374.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ko Jh, Wang YS, Nakamura A, Guo LT, Söll D, Umehara T. FEBS Lett. 2013;587:3243–3248. doi: 10.1016/j.febslet.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Soppa J. ARCHAEA. 2010;2010 Article ID 820681. [Google Scholar]; b) Drazic A, Myklebust LM, Ree R, Arnesen T. Biochimica et Biophysica Acta. 2016;1864:1372–1401. doi: 10.1016/j.bbapap.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 7.a) AbouElfetouh A, Kuhn ML, Hu LI, Scholle MD, Sorensen DJ, Sahu AK, Becher D, Antelmann H, Mrksich M, Anderson WF, Gibson BW, Schilling B, Wolfe AJ. MicrobiologyOpen. 2015;4:66–83. doi: 10.1002/mbo3.223. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao K, Chai X, Marmorstein R. J Mol Biol. 2004;337:731–741. doi: 10.1016/j.jmb.2004.01.060. [DOI] [PubMed] [Google Scholar]
- 8.a) Blander G, Guarente L. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]; b) Ruijter AJMd, Gennip AHv, Caron HN, Kemp S, Kuilenburg ABPv. Biochem J. 2003;370:737. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. Mol Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Elsasser SJ, Ernst RJ, Walker OS, Chin JW. Nat Meth. 2016;13:158–164. doi: 10.1038/nmeth.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uemura H, Thorbjarnardottir S, Gamulin V, Yano J, Andresson OS, Soll D, Eggertsson G. J Bacteriol. 1985;163:1288–1289. doi: 10.1128/jb.163.3.1288-1289.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mossakowska DE, Nyberg K, Fersht AR. Biochemistry. 1989;28:3843–3850. doi: 10.1021/bi00435a033. [DOI] [PubMed] [Google Scholar]
- 12.Zhang QF, Zhou AP, Li SX, Ni JJ, Tao J, Lu J, Wan BS, Li S, Zhang J, Zhao SM, Zhao GP, Shao F, Yao YF. Scientific Reports. 2016;6 doi: 10.1038/srep30837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kogoma T. Microbiology and Molecular Biology Reviews. 1997;61:212. doi: 10.1128/mmbr.61.2.212-238.1997. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.