

Abstract
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability and is associated with up to 5% of autism cases. Several promising drugs are in preclinical testing for FXS; however, bench-to-bedside plans for the clinic are severely limited due to lack of validated biomarkers and outcome measures. Published work from our laboratories has demonstrated altered levels of amyloid-beta (Aβ) protein precursor (APP) and its metabolites in FXS and idiopathic autism. Westmark and colleagues have focused on β-secretase (amyloidogenic) processing and the accumulation of Aβ peptides in adult FXS models while Lahiri and Sokol have studied α-secretase (nonamyloidogenic or anabolic) processing and altered levels of sAPPα and Aβ in pediatric autism and FXS. Thus, our groups have hypothesized a pivotal role for these Alzheimer’s disease (AD)-related proteins in the neurodevelopmental disorders of FXS and autism. In this review, we discuss the contribution of APP metabolites to FXS and autism pathogenesis as well as the potential use of these metabolites as blood-based biomarkers and therapeutic targets. Our future focus is to identify key underlying mechanisms through which APP metabolites contribute to FXS and autism condition-to-disease pathology. Positive outcomes will support utilizing APP metabolites as blood-based biomarkers in clinical trials as well as testing drugs that modulate APP processing as potential disease therapeutics. Our studies to understand the role of APP metabolites in developmental conditions such as FXS and autism are a quantum leap for the neuroscience field, which has traditionally restricted any role of APP to AD and aging.
Keywords: APP, fragile X syndrome, autism, biomarker, seizure, macrocephaly
1. Introduction
No validated blood-based biomarkers or behavioral outcome measures currently exist for fragile X syndrome (FXS), which is impeding bench-to-bedside clinical efforts. Several recent mega-sequencing studies suggest a strong molecular link between FXS and autism.1–3 Elucidation of shared and contrasting neurobiologies of these neurodevelopmental conditions is expected to identify novel disease biomarkers and therapeutic targets, which are critical needs for the field. Research from our laboratories has demonstrated amyloid-β (Aβ) protein precursor (APP) and its metabolites are dysregulated in FXS and autism. APP is a large membrane-spanning glycoprotein that undergoes proteolytic cleavage by secretases to form either Aβ peptides found in the extracellular cerebral amyloid plaques of Alzheimer’s disease (AD) and neurotoxic secreted APPβ (sAPPβ) or non-toxic, non-amyloidogenic p3 peptide (Aβ17–40/42) and secreted APP alpha (sAPPα) believed to have neurotrophic properties. The localization of APP at dendritic synapses and its roles in neurogenesis and cell adhesion suggest that this protein and its metabolites play pivotal roles in development. Our central hypothesis is that APP metabolite profiles are viable biomarkers for disease severity in FXS and autism. To date, there is a dearth of available data regarding APP metabolite profiles as a function of age, potential alterations in proteolytic processing with development, or associations between APP metabolite levels and FXS and autism traits. This lack of knowledge is a significant problem because it prohibits broader understanding of how APP metabolites are connected to neuronal function and behavior as well as their potential use as biomarkers for disease severity and drug efficacy testing. Our hypothesis is formulated, in a large part, based on published data from our laboratories, which addresses this lack of knowledge from opposing but complementary directions in studying amyloidogenic (β-secretase) versus non-amyloidogenic (α-secretase) APP processing and in working with samples from adult FXS versus pediatric autism subjects. Herein, we meld our findings with those from the Bagni laboratory exploring the role of sAPPα in FXS,4 the Wegiel laboratory demonstrating Aβ 17–40/42 (also known as p3 peptide, generated by the a-secretase pathway) accumulation in autistic brain,5 and the Erickson laboratory regarding the use of APP metabolites as drug responsive biomarkers6 to generate a model of how APP metabolites contribute to FXS and autism pathogenesis as well as their potential use as blood-based biomarkers and drug targets.
2. FXS and APP Metabolites
FXS is the most common form of inherited intellectual disability with a frequency of 1 in 2,500 births, and is associated with up to 5% of autism cases.7–9 It is clinically characterized by highly variable cognitive function (overall IQ<70), autistic-like behaviors, seizures, macrocephaly and macroorchidism.10 FXS results from a mutation in a single gene on the X-chromosome, FMR1, which codes for fragile X mental retardation protein (FMRP). FMRP expression is absent or greatly reduced in FXS and many disease phenotypes are manifested in Fmr1KO mice, which lack expression of FMRP. FMRP is a multi-functional RNA binding protein that is ubiquitously expressed throughout the body with significantly higher levels in young animals.11 It is involved in the transport, localization and translational regulation of mRNA ligands and is required for normal dendrite development. In aggregate, over 500 mRNA ligands for FMRP have been identified, many with the potential to influence synaptic formation and plasticity.12–14
The prevailing theory regarding the pathogenic mechanism underlying FXS is that uncontrolled metabotropic glutamate receptor 5 (mGluR5) signaling causes exaggerated protein synthesis in the absence of the translational repressor FMRP.15 Exaggerated translation at synapses underlies abnormal dendritic spine morphology, seizure activity and FXS behaviors. There has been an intense effort to identify mRNAs that are translationally repressed by FMRP as the proteins they code for are potential disease biomarkers and therapeutic targets. We identified App mRNA as a FMRP synaptic target.16 Using the UV crosslinking-immunoprecipitation (CLIP) assay developed by the Darnell laboratory, Westmark and colleagues demonstrated that FMRP binds directly to a guanine-rich region in the coding region of App mRNA;16 this important finding was reproduced by Myriam Gorospe’s laboratory.17 Stimulation of cortical synaptoneurosomes with (S)-3,5-dihydroxyphenylglycine (DHPG), a group 1 mGluR agonist, releases FMRP from App mRNA leading to APP synthesis or regulated translation. In synaptoneurosomes and primary cultured neurons prepared from Fmr1KO mice, basal APP levels are increased and do not change in response to DHPG suggesting that constitutive translation occurs in FXS.16 Elevated APP levels provide more target for secretase processing consistent with the finding that Aβ is elevated in older Fmr1KO mice16 (Figure 1).
Figure 1.
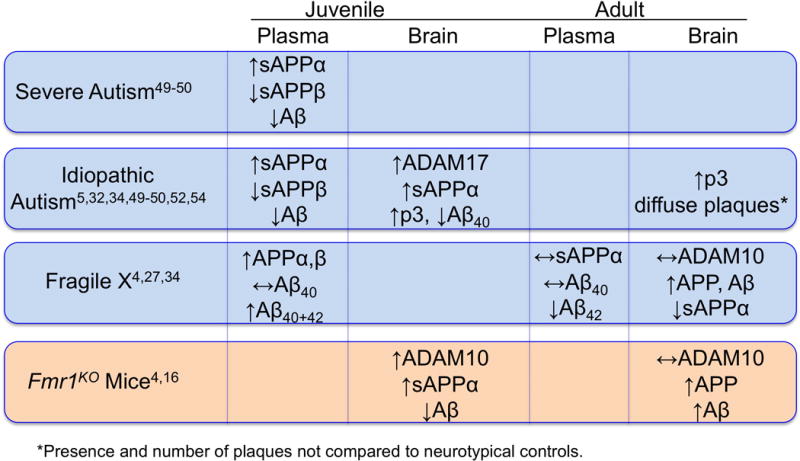
Comparison of APP metabolite levels in human autism and FXS and in Fmr1KO mice as a function of age (juvenile versus adult) and tissue (plasma and brain). APP is dysregulated in both FXS and ASD. While APP dysregulation seems to be persistent in life (young and adult/old), its processing changes during development. In young individuals and juvenile mice, the upregulated APP is processed by the a-secretase (non-amyloidogenic pathway) liberating sAPPa (human blood and mouse brain). In aged individuals and mice, the upregulated APP is processed by β-secretase liberating Aβ (human blood and mouse brain). Thus, there is a switch in the processing of APP during aging and the majority of the papers published report consistent findings in that young patients with FXS and ASD have an excess of sAPPa (possibly due to increased ADAM10 during that specific developmental window as shown in mice) while there is an increase in Aβ with age (possibly due to increased BACE1 activity).
Fmr1KO mice exhibit features shared with FXS patients such as increased susceptibility to seizures, altered anxiety, and dendritic spine phenotypes.10,18–26 In order to establish if APP directly contributed to FXS pathogenesis, Westmark and colleagues modulated APP expression in Fmr1KO mice by creating mice that express only one App allele (Fmr1KO/APPHET).27 Western blot analyses confirmed APP levels were reduced by 50% in Fmr1KO/APPHET. Animals were evaluated for seizure susceptibility (audiogenic-induced seizures, AGS), repetitive behavior (marble burying), anxiety (open field), mGluR-LTD, and dendritic spine phenotypes. All of these phenotypes were attenuated in Fmr1KO/APPHET (Table 1).27
Table 1.
Summary of FXS and Autism Phenotypes Rescued by Manipulation of APP Metabolites
| Genetic Reduction of App in Fmr1KO Mice (Westmark et al27) | ||
|---|---|---|
| Phenotype | Rescue | Rescue with Fmr1KO/APPHET |
| 1. APP expression (western blot) | YES | ↓50% (equal to WT levels; n=3 mice per cohort, ANOVA P<0.0001) |
| 2. Seizures (AGS) | PARTIAL | ↓54% (intermediate between WT and Fmr1KO; n=23, Fisher Exact P<0.05) |
| 3. Perseverant behavior – marble assay | YES | 100% (equal to WT levels; n=8–10 mice per cohort, ANOVA P=0.03) |
| 4. Anxiety (open field) | YES | 100% (equal to WT levels; n=14–18 mice per cohort, ANOVA P<0.0001) |
| 5. Percent mature spines (diI labeling) | YES | 100% (equal to WT levels; n=2 individual neuronal cell preps, neurons from 2–6 coverslips per prep analyzed, 2–12 dendrites analyzed per coverslip, minimum of 746 spines analyzed per cohort) |
| 6. Dendritic spine length (diI labeling) | PARTIAL | 11% (intermediated between WT and Fmr1KO; n=2 individual neuronal cell preps, neurons from 2–6 coverslips per prep analyzed, 2–12 dendrites analyzed per coverslip, minimum of 746 spines analyzed per cohort, ANOVA P<0.0001) |
| 7. mGluR-LTD (field recordings) | YES | 100+% (decreased LTD compared to WT; n=3 mice per cohort, n=10–13 slices per cohort, ANOVA P<0.0002) |
| Reduction of ADAM10 in Fmr1KO mice (Pasciuto et al4) | ||
| Phenotype | Rescue | Rescue with TAT-Pro Peptide |
| 1. APP α-cleavage (western blot) | YES | 100% (equal to WT levels; n=5 mice per cohort, ANOVA P<0.01) |
| 2. mGluR-LTD (field recordings) | YES | 100% (equal to WT; n=4–6 mice per cohort, n=8–9 slices per cohort, ANOVA P<0.0002) |
| 3. Biomarker expression (ARC, APP, ADAM10, STEP) (western blot) | YES | 100% (equal to WT levels; n=5 per cohort, ANOVA P<0.05) |
| 4. Distance & speed (open field) | YES | 100% (equal to WT; n=11–17 mice per cohort, ANOVA P<0.05) |
| 5. Preference test for novel arm (T-maze) | NO | 100+% (increased preference for novel arm compared to WT; n=9–12 mice per cohort, Chi square P<0.001) |
| 6. Nest building | YES | 100% (equal to WT; n=5–8 mice per cohort, ANOVA P<0.001) |
| Pharmacological Rescue of APP Metabolites in FXS and Autism (Erickson et al6) | ||
| Phenotype | Rescue | Rescue with Acamprosate |
| 1. APP expression (ELISA) | YES | Significant rescue in plasma sAPP (total) and sAPPα levels (n=9 FXS/ASD, n=6 ASD, Hedge’s g, P<0.05) |
| 2. Aβ expression (ELISA) | NO | No change in Aβ 40 or Aβ 42 levels (Hedge’s g) |
Differential expression and processing of APP throughout development is expected to affect APP metabolite profiles and FXS phenotypes. Both amyloidogenic and non-amyloidogenic secretases are expressed during embryonic, postnatal and adult development.28 Recent work from Claudia Bagni’s laboratory elegantly demonstrates the role of sAPPα in mediating immature spine, mRNA translation and mGluR-LTD phenotypes in postnatal Fmr1KO mice.4 Others have shown that sAPPα, but not sAPPβ, rescues LTP and dendritic spine density in adult AppKO.29–30 Thus, sAPPα plays important roles in synaptic plasticity at multiple stages of development. These data strongly support the hypotheses that modest over-expression of APP in the context of the Fmr1KO contributes to many pathological phenotypes observed in FXS.
The aforementioned mouse studies suggest that APP metabolites are a potential disease biomarker for FXS. Westmark and colleagues evaluated APP/sAPPα and Aβ levels in blood plasma from full-mutation adult FXS subjects and found Aβ 42 was significantly lower in the FXS group (2.1–fold decrease, P<0.004).27 APP/sAPPα and Aβ 40 levels were equivalent between FXS males and controls. A reduced Aβ42/Aβ40 blood plasma ratio as seen for FXS is also a putative biomarker for AD31, autism32, and Down syndrome.33 The prevailing theory in AD and Down syndrome is that the brain acts as a sink for Aβ42 and a lower blood plasma level indicates increased brain deposition. APP/sAPPα and Aβ were measured in a limited number of adult control and FXS autopsy brain samples and there was a trend toward increased Aβ with a reciprocal decrease in sAPPα levels.27 These studies are in agreement with those in Fmr1KO mice, which exhibit elevated Aβ in the brain,16 and support a model of increased β-secretase processing in adult FXS associated with Aβ accumulation in the brain and clearance from the blood consistent with the AD “brain sink model”. Lahiri’s group recently found increased Aβ42 (P<0.001) and equivalent Aβ40 levels in FXS pediatric plasma versus control samples. sAPPα (P=0.015) and total sAPP (P<0.001) were both increased in FXS.34 These data suggest increased APP expression possibly accompanied by increases in α- and β-secretase processing in pediatric FXS.
3. Autism and APP Metabolites
Autism is a cluster of complex neurobiological symptoms, autism spectrum disorder (ASD), that normally present in the second or third years of life. The core features include impairments in social interaction and communication, and repetitive stereotyped behavior. Many autistic children exhibit intellectual disability and marked delay in motor milestones. ASD are estimated to occur in 1 in 68 children with prevalence 4.5-fold higher in males via conventional epidemiological estimates,35 with total-population sample studies proposing rates as high as 1 in 38.36 The etiology of autism is unknown but genetic, epigenetic and environmental factors likely affect symptom severity.37–39 FXS shows high comorbidity with autism (although not vice versa).40–41 Recent mega-sequencing studies suggest a strong molecular link between these disorders. In particular, many of the protein-interacting partners of FMRP harbor autism-associated common variants,1 there is an enrichment of autism rare de novo variations in FMRP targets,2 and there is an enrichment of FMRP targets in neuronal gene expression modules in autism brain.3 Autism is, nevertheless, distinct from FXS in several neuroanatomical and behavioral aspects.42–48 Seminal work from the Sokol and Lahiri laboratory demonstrated significantly elevated sAPPα with reduction of sAPPβ and Aβ peptides in plasma from severely autistic children.49–51 Most of these subjects exhibited intellectual disability and seizures. Two subjects were comorbid for FXS with autism and exhibited the highest levels of sAPPα49 Importantly, they observed a negative correlation between sAPPα levels and age. In a larger study, they repeated the finding of reduced Aβ in plasma and brains of idiopathic autism subjects, independent of severity.34 No significant correlation between sAPPa levels and age was found, but they did report a significant positive relationship between α-secretase and age in non-FXS autism brains samples. Two independent laboratories have replicated portions of this work with different patient cohorts.32,52 Bailey and colleagues found significantly increased sAPPα in 60% of known autistic children.52 Al-Ayadhi and colleagues found significantly lower Aβ40 and Aβ42 and a lower Aβ40/42 ratio in Saudi autistic children.32 Fatemi and colleagues found increased mGluR5 in the superior frontal cortex of children with autism versus healthy controls53 and a corresponding increase in APP.54 They found decreased APP in the vermis of adult subjects with autism. Overall, these findings support abnormal levels of mGluR5 and APP in autism and support a model of increased α-secretase processing in severe childhood autism. It remains to be determined how extensively APP processing changes with aging and how altered metabolite levels contribute to ongoing disease pathology (Figure 2), as there are currently no reported studies of sAPPα levels in adults with autism, although a significant positive relationship has been reported between age and ADAM17 (the primary α-secretase) in idiopathic autistic brain versus controls (P=0.011).34 There have been limited studies to examine APP metabolite levels or function in mouse models of autism other than in Fmr1KO and TgsAPPα mice. The TgsAPPα mice developed by Bailey and colleagues exhibit hypoactivity, impaired sociability, increased brain glial fibrillary protein (GFAP), and altered Notch1 and IL-6 levels.55–56 Longitudinal studies in Tg-sAPPa mice could identify age-related effects of sAPPa.
Figure 2.
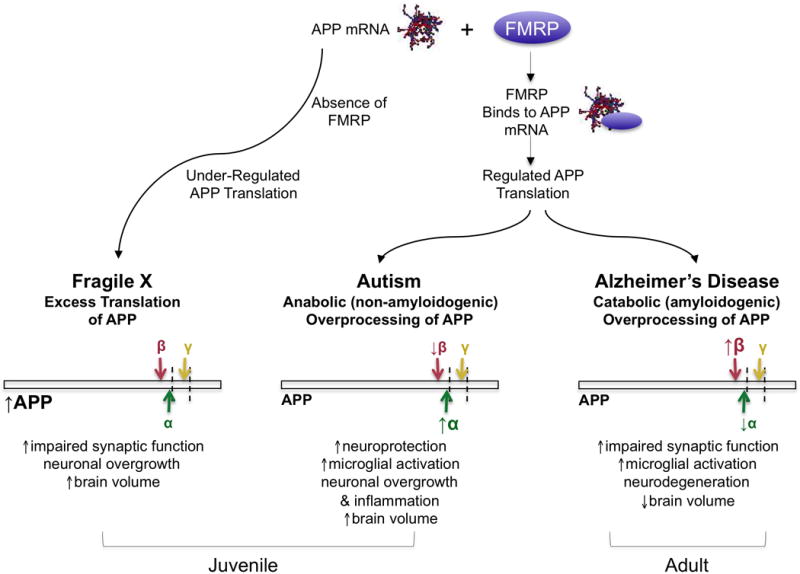
APP expression and processing contrasted among FXS, autism, and Alzheimer’s disease. At the level of mRNA translation, with a normal FMR1 gene, FMRP binds to (among other targets) APP mRNA and inhibits translation resulting in regulated APP synthesis. In FXS, loss of the translational repressor FMRP leads to exaggerated protein synthesis resulting in elevated APP levels. At the level of protein processing, excess APP provides more target for both anabolic and catabolic secretase processing. In the case of FXS, APP processing may change with age such that exaggerated anabolic processing in childhood leads to neuronal overgrowth followed by increased catabolic processing in adulthood both accompanied by associated outcomes. In the case of autism, α-secretase processing is increased resulting in increased levels of anabolic/neurotrophic sAPPα. By comparison, relative levels of catabolic products (e.g., Aβ) are insufficient to compensate, resulting in neuronal overgrowth and associated outcomes. In Alzheimer’s disease (normally a geriatric condition), excess catabolic processing by β-secretase, possibly accompanied by insufficient anabolic processing, results in inflammation, neurodegeneration, and loss of brain volume.
Ferreira and Klein elegantly reviewed the numerous synaptic activities of Aβ-derived diffusible ligands (ADDLs) and proposed that the molecular and synaptic similarities between AD, FXS and autism may be sufficient to regard AD, in principle, as a type of ASD that shows very late onset and to view FXS as an early manifestation of Aβ oligomer-induced disease.57 Indeed, microarray analysis of cerebellar samples demonstrated altered expression of 40% of AD-related genes in autistic subjects.58 Validation of this theory awaits further experimentation including a thorough analysis of APP metabolite profiles throughout the lifespan and determination of their contribution to the behavioral and cognitive phenotypes of the aforementioned disorders. Notably, certain APP metabolites are implicated in seizure propensity and macrocephaly.
Epileptic seizures may drive the development of autism in neurodevelopmental disorders.59–60 Epilepsy is highly comorbid in autism with a 21.4% prevalence in autistic subjects with intellectual disability and 8% in subjects without intellectual disability.61 EEG abnormalities were found in 31% of children with ASD.62 Seizures and EEG abnormalities are also found in FXS63 and AD.64–65 These disorders are all characterized by abnormal levels of APP metabolites, albeit each with potentially distinct profiles.66 Genetic suppression of transgenic APP in mice rescued epileptiform activity suggesting that abnormal APP levels are a potential cause of seizure activity.67 Abnormal EEG discharges in the Born study were independent of plaque load and reduction of Aβ levels implicating full-length APP or another metabolite in seizure propensity.67 Other studies indicate that Aβ oligomers induce dynamic redistribution of mGluR5 receptors to synapses,68 facilitate mGluR-LTD,69 and induce intrinsic excitability in CA1 pyramidal neurons.70 Thus, multiple APP fragments or the ratio of various fragments may contribute to seizure activity.
Macrocephaly in autism is a disputed topic.71–81 An NIH study has questioned early brain overgrowth in autism as detected by head circumference measurements, which may represent norm bias rather than an autism-specific biomarker.71 Despite the controversy regarding dramatic brain overgrowth in autism during the first year of life, there is a possible subtle divergence in head circumference during the second year.71 Brain overgrowth is a feature of several genetic syndromes that are comorbid with autism including FXS;82 however, autism can also be comorbid with several microcephalic syndromes such as Down syndrome, which is trisomic for the APP gene. In a sample of children ages four and older, Wegiel and colleagues conducted a stereological study of nuclear and cytoplasmic volumes of neurons in 16 brain structures of autistic and control subjects.83 They found significant deficits of neuronal soma and nuclear volumes in 13 of the 16 brain regions examined in pediatric autism samples and increased nuclear volumes in 8 of 16 structures in autistic teenagers and young adults. Their findings suggest global abnormalities in brain development in autism. Enlargement in white, rather than gray matter, likely accounts for macrocephaly.74 White matter abnormalities have also been observed in FXS brain and implicated in the Fmr1KO.84–86 The secreted sAPPα fragment of APP, which has known neurotrophic properties, is elevated in both brain and plasma of autistic patients34, particularly those with severe autism.49–51 It remains to be determined if sAPPα localizes to white matter and contributes to brain overgrowth and how this may relate to autism severity, particularly since brain overgrowth, while less common than previously thought, may be predictive of more severe autism.87
How could APP metabolites, in particular sAPPα, facilitate white matter brain overgrowth? Evidence associating sAPPα with brain overgrowth was found by Bailey and colleagues in sAPPα-overexpressing transgenic mice, which produce an abundance of astrocytes, GFAP and brain CD8 T cells.55–56 Convergent, albeit indirect evidence for the role of sAPPα in aberrant brain growth was reported by Zeidan-Chulia et al who found anabolic upregulation of GRIN1, NMDA glutamate receptors, and MAP3K1, known activators of ERK pathways, in cerebellar samples from autistic individuals.88 They hypothesized that deregulated glutamatergic synaptic transmission/plasticity, caused by increased GRIN1 with resultant increased density of NMDA receptors, favors the nonamyloidogenic pathway and production of sAPPα through ERK mediated α-secretase activity. sAPPα, then may facilitate proliferation by activating the P13K/Akt/mTOR pathway. They also showed altered expression of genes in the AD and WNT pathways in autism. The WNT–β -catenin adhesion pathway is abnormal in autism.89 Disruption of brain cell adhesion would favor brain overgrowth; further, APP has been associated with downregulation of α-catenin.90
Unlike AD individuals who show confusion and visual memory disturbance, autism subjects exhibit hypervigilance and often excellent visual memory.91 Could a derangement in APP metabolites within white matter account for this? Although AD is considered a gray matter disease, there is evidence that oligodendrocytes and myelin are targeted via Aβ peptides before signs of amyloid deposition and Tau aggregates.92 Desai et al speculate that early derangement in myelin might make the affected axon vulnerable to inflammation or oxidative stress. No studies to date of sAPPα in white matter exist; however, tumor necrosis factor alpha (TNF) converting enzyme (TACE or ADAM17) was found essential for oligodendroglia development and CNS myelination in a nonpathological mouse model.93 Therefore, by proxy sAPPα may localize to oligodendrocytes. In contrast, Wegiel et al5 reported signs of elevated a-secretase activity, specifically increased Aβ 17–40/42, in the presence of microcephaly in individuals with chromosome 15q11.2–q13 duplications (Dup(15)) suggesting that α-secretase is not associated with brain overgrowth. Up to 80% of individuals with Dup(15) acquire microcephaly by age 2,94 unlike congenital microcephaly that exists at birth and in opposition to the prevalence of macrocephaly in other syndromic forms of autism (e.g., FXS with autism) and in up to 20% of idiopathic autism.95 The Dup(15) ubiquitin protein ligase E3A (UBE3A), associated with Angelman’s syndrome, interacts with the primary microcephaly protein ASPM.96 This association may override an effect of sAPPa in contributing to macrocephaly. In any case, APP has been associated with the ubiquitin proteosome machinery that degrades and clears proteins, and underlies UBE3A.97 Furthermore, APP has been shown to modulate β-catenin degradation which occurs via ubiquitinylation and proteosome degradation.90
4. The APP Metabolite Profile as a Blood-Based Biomarker
Currently, FXS and autism therapy development efforts range from early translational studies to human clinical trials. As promising candidate therapies move forward, there is a critical need for sensitive, reliable and valid biomarkers in model organisms, particularly measures that can predict efficacy in human trials and can be used in titrating and monitoring therapies. The recent discontinuation of clinical trials by Novartis and Roche of promising mGluR5 antagonists in FXS subjects highlights the compelling need to identify and validate physiologic measures that successfully bridge pre-clinical and clinical studies in this disorder.98–99
Published data from our laboratories present a complex picture that may show significant age-related changes in the molecular neurobiology of FXS and autism. Our groups demonstrated reduced Aβ in blood plasma and brains from autistic children, but increased levels in FXS children.34,50 Full-mutation FXS adults, on the other hand, had reduced blood Aβ versus controls.27 Our results differ in showing elevated sAPPα in blood plasma from autistic and FXS children34,49–50 but no difference in sAPPα in adult FXS.27 These observations create important gaps in the literature regarding: (1) differential APP processing as a function of tissue, age and disease status; (2) methodological differences in blood collection protocols that could contribute to APP processing post-sample collection; and (3) utility of using APP metabolites as disease biomarkers and drug targets for FXS and autism. Toward addressing the first aforementioned gap, Lahiri and colleagues measured APP metabolites in brain tissue of autistic and FXS children compared to age-matched controls. In left temporal lobe samples from the Autism Tissue Program, they found a similar APP profile as in plasma, i.e. non-significant elevation in sAPPα for autism (P > 0.05) (n=7) compared to controls but significant elevation in FXS vs. controls (P = 0.01) and decreased Aβ40 (P = 0.001) in autism but not FXS vs. controls (P > 0.05).34 Studies from the Wegiel laboratory demonstrate abnormal intracellular accumulation and extracellular deposition of Aβ17–40/42 in idiopathic and Dup(15) autism brains in both adults and children.5 A recent study showed high levels of sAPPα in children with autism (n=6) in insular cortex gray matter,56 consistent with the Lahiri laboratory temporal lobe findings. In total, these data suggest a preponderance of α-secretase processing in idiopathic autism. The Lahiri laboratory also evaluated APP/sAPP and Aβ levels in blood plasma from FXS children (n=18) and found elevated total sAPP, sAPPα, sAPPβ, Aβ40 and Aβ42.34 These data in conjunction with the adult FXS and pediatric autism studies suggest both elevated catabolic and anabolic APP processing in pediatric FXS with a shift toward decreased α-secretase activity with aging.
The major difference in blood collection protocols was the choice of anticoagulant. The adult blood samples were collected in heparin and the pediatric samples in EDTA. To determine effects of anticoagulant on APP metabolite profiles, Westmark et al collected blood samples from 5 adult control subjects splitting the blood into various anticoagulant tubes (lithium heparin, sodium heparin, EDTA, sodium citrate) and assessed APP metabolite levels.100 With EDTA or sodium citrate, sAPPα measurements were significantly higher compared to sodium or lithium heparin. There was a significant inverse effect on Aβ42 levels with no change in Aβ40. These data suggest that anticoagulant affects sAPP stability post-blood collection, which could account for varied results concerning sAPPα levels, but does not account for varied results with Aβ, which may be due to subject age and disease severity. The possibility exists that EDTA chelation of calcium or other divalent ions may explain some differences in Aβ detectability.101 Overall, these data demonstrate that FXS is associated with dysregulated post-transcriptional synthesis and processing of APP and prompt studies to assess APP metabolite levels as a function of age and disease severity under standardized blood collection protocols.
Regarding the third aforementioned gap in the literature, the utility of using APP metabolites as disease biomarkers for FXS and autism, Bailey and colleagues developed a sensitive ELISA to specifically measure sAPPα in human plasma and umbilical cord blood. They found significantly elevated levels of plasma sAPPα in 60% of autistic children and 10 of 150 human umbilical cord blood samples.52 It remains to be determined which, if any, of the newborn babies from which cord blood was analyzed develop autism, which would support using sAPPα measurements as an early diagnostic tool. Erickson and colleagues are developing APP metabolites as blood-based biomarkers that are sensitive to drug treatment in autism and FXS clinical trials. Their team found acamprosate was associated with a significant reduction in plasma sAPP and sAPPα with no change in Aβ40 or Aβ42 in ASD youth6 (Table 1). Furthermore, they showed that youth with FXS-associated ASD showed increased sAPPα processing compared to age-, gender- and IQ-matched youth with idiopathic ASD. Thus, APP metabolite profiles are a potential blood-based biomarker for disease severity and drug efficacy in FXS and autism.
Based on our results, we suggest that the APP biochemical pathway is dysregulated in both FXS and autism, but these two disorders differ in specific metabolite profiles (for example, low Aβ in pediatric ASD and high Aβ in pediatric FXS). Altered APP metabolite profiles could explain neuroanatomical differences between ASD and FXS. Interestingly, this observation is consistent with the amygdala being enlarged in ASD (low Aβ) and reduced in FXS (high Aβ causing atrophy). Differential Ab expression in FXS and ASD is not contradictory to the commonality of high sAPP in both disorders. How is that possible mechanistically? APP cleavage by β-site APP-cleaving enzyme (BACE1, aka β-secretase) depends on the availability of APP in the endosome where cleavage occurs102. That is, unless both APP and BACE1 meet in the endosomal compartment, there is no direct interaction and no Aβ formation. Circumstantial evidence suggests that clathrin-coated endosomes are deficient in ASD, which might lead to reduced cleavage of APP by BACE1 and thus decreased Aβ formation in ASD but not in FXS. What is interesting is that an endosomal mechanism can disrupt APP processing toward the anabolic pathway even when levels of APP, BACE1, and ADAM proteins are normal. This is a testable hypothesis, which can be verified by analyzing endosomal structure in ASD cases versus controls.
5. APP Metabolites as Drug Targets for FXS and Autism
Therapies directed at modulating APP metabolites, as studied for AD, may be applicable to FXS and autism. For example, drugs that modulate secretase activity could be repurposed.103–104 β- and γ-secretase inhibitors are currently in the preclinical stage of investigation and could provide a means to reduce the production of Aβ. BACE1 cleaves at the amino-terminus of Aβ and is the rate-limiting step in Aβ peptide generation. BACE1 inhibitor design has proven difficult due to the large size of the enzyme catalytic pocket.105 It is also difficult for BACE1 inhibitors to reach the central nervous system. Nevertheless, GRL-8234 is a blood-brain barrier permeable compound that potently blocks BACE1 activity106. A single dose of GRL-8234 reduced plasma levels of Aβ by 50–65%106–107 and brain interstitial fluid levels by 50% within 3 hr in Tg2576 AD mice107. Fmr1KO mice exhibit elevated brain Aβ expression but not to the same extent as Tg2576. Thus, optimized pharmacokinetic treatment conditions could be determined to normalize Aβ levels in Fmr1KO. Another option to reduce production of carboxyl-terminal fragments (CTF) of APP is γ-secretase inhibitors, which should decrease Aβ and g-CTF but increase b-CTF, which may also be neurotoxic. The problem associated with the use of γ-secretase inhibitors is that these drugs inhibit proteolytic processing of other proteins such as Notch that are critical for cellular function.108 Supplementary Table 1 summarizes the numerous Aβ-modulating drugs in clinical trials for AD.109–110 In addition, new approaches that modulate the expression rather than the catalytic activity of BACE1 are actively being sought. The Puglielli laboratory has identified novel biochemical inhibitors of acetyltransferases that significantly reduce the levels of BACE1 and the generation of Aβ in cellular systems.111–113 Alternatively, the disintegrins proteins such as ADAM10 and ADAM17 possess α-secretase activity.114 Cleavage by α-secretase increases sAPPα as well as carboxyl-terminal metabolites of APP. sAPPα has neuroprotective properties, but as previously discussed, elevated levels in autism and FXS may be contributing to macrocephaly. Data from the Bagni laboratory suggest that pharmacological inhibition of ADAM10 may be a viable therapeutic option for FXS (Table 1). Considering the complexity of APP processing and the potential feedback mechanisms associated with various APP metabolites, it may be necessary to concurrently modulate α- and β-secretase processing as well as APP synthesis to attain APP homeostatic conditions.
Despite decades of research, the exact roles of APP and Aβ in AD are still surrounded by inconsistencies and controversies.115 Our work complicates matters by adding FXS and autism to the list of disorders that display abnormal APP metabolite profiles. We found altered levels of APP and Aβ in FXS mice as well as altered levels of Aβ42 in plasma samples from adult FXS males. We found higher levels of total APP, sAPPα and lower levels of Aβ peptide in plasma from children with autism with the same pattern in brain tissue specimens from autistic patients. In part, these results have been replicated by independent laboratories. Furthermore, transgenic mouse models link the overexpression of varied APP metabolites with FXS phenotypes, autistic features, increased abundance of brain astrocytes and seizures. Recently, APP metabolites were reported as potentially useful biomarkers in a FXS drug study. These studies highlight the importance of future endeavors to understand the role of various APP metabolites in the development and pathology of FXS and autism and to develop these proteins as disease biomarkers and therapeutic targets. The goal is to repurpose decades of AD drug-related research to provide therapeutics for FXS and autism.
Supplementary Material
Acknowledgments
CJW has received funding from FRAXA Research Foundation, NIH (R21AG044714 and R03HD075881), the University of Wisconsin (UW)-Madison Alzheimer’s Disease Research Center (NIA P50AG033514), the UW-Madison Clinical and Translational Science Award (CTSA) program (NCATS UL1TR000427), Lundbeck USA, Merz Pharmaceuticals and Pierre Fabre.
DKS has received funding from the Riley Memorial Foundation, Mental Health Association for the Advancement of Mental Health Research and Education, Clarian Health Partners, Indiana University Collaborative Research Grant (22-140-29).
DKL has received funding from NIH (R01AG051086, R21AG4687100, P30AG010133), Alzheimer’s Association, Indianan Clinical and Translational Sciences Institute (ICTSI), and ISDH Spinal Cord and Brain Injury Board.
Funding agencies played no role in the preparation of this publication. We thank the reviewers for their careful and insightful critiques, which greatly improved the manuscript.
Footnotes
Financial Disclosures
CJW: none
DKS: none
DKL: reports grants as a Principal Investigator from the National Institute on Aging, National Institute of Health, USA; Member, Scientific Advisory Board, QR Pharma, Inc., Berwyn, PA, USA; Member, Scientific Advisory Board, Yuma Therapeutics, Boston, MA; Member, Scientific Advisory Board, Entia Biosciences, Inc., Sherwood, OR; Member, International Advisory Board of the Drug Discovery and Therapy World Congress, Boston, MA; and Editor-in-Chief, “Current Alzheimer Research”, Bentham Sciences Publishers.
References
- 1.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–61. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun. 2014;5:5748. doi: 10.1038/ncomms6748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pasciuto E, Ahmed T, Wahle T, Gardoni F, D’Andrea L, Pacini L, et al. Dysregulated ADAM10-mediated processing of APP during a critical time window leads to synaptic deficits in fragile X syndrome. Neuron. 2015;87:382–98. doi: 10.1016/j.neuron.2015.06.032. [DOI] [PubMed] [Google Scholar]
- 5.Wegiel J, Frackowiak J, Mazur-Kolecka B, Schanen NC, Cook EH, Jr, Sigman M, et al. Abnormal intracellular accumulation and extracellular Abeta deposition in idiopathic and Dup15q11.2–q13 autism spectrum disorders. PLoS One. 2012;7:e35414. doi: 10.1371/journal.pone.0035414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erickson CA, Ray B, Maloney B, Wink LK, Bowers K, Schaefer TL, et al. Impact of acamprosate on plasma amyloid-beta precursor protein in youth: A pilot analysis in fragile X syndrome-associated and idiopathic autism spectrum disorder suggests a pharmacodynamic protein marker. J Psychiatr Res. 2014;59:220–8. doi: 10.1016/j.jpsychires.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagerman PJ. The fragile X prevalence paradox. J Med Genet. 2008;45:498–9. doi: 10.1136/jmg.2008.059055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLennan Y, Polussa J, Tassone F, Hagerman R. Fragile x syndrome. Curr Genomics. 2011;12:216–24. doi: 10.2174/138920211795677886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budimirovic DB, Kaufmann WE. What can we learn about autism from studying fragile X syndrome? Dev Neurosci. 2011;33:379–94. doi: 10.1159/000330213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hagerman RJ, Hagerman PJ. Physical and behavioral phenotype. John Hopkins University Press; Baltimore: 2002. [Google Scholar]
- 11.Khandjian EW, Fortin A, Thibodeau A, Tremblay S, Cote F, Devys D, et al. A heterogeneous set of FMR1 proteins is widely distributed in mouse tissues and is modulated in cell culture. Hum Mol Genet. 1995;4:783–9. doi: 10.1093/hmg/4.5.783. [DOI] [PubMed] [Google Scholar]
- 12.Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–87. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 13.Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–99. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- 14.Ramon y Cajal S. Recollections of My Life. The MIT Press; Cambridge: p. 1989. [Google Scholar]
- 15.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–7. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007;5:e52. doi: 10.1371/journal.pbio.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee EK, Kim HH, Kuwano Y, Abdelmohsen K, Srikantan S, Subaran SS, et al. hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat Struct Mol Biol. 2010;17:732–9. doi: 10.1038/nsmb.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–50. doi: 10.1016/s0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- 19.Yan QJ, Asafo-Adjei PK, Arnold HM, Brown RE, Bauchwitz RP. A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav. 2004;3:337–59. doi: 10.1111/j.1601-183X.2004.00087.x. [DOI] [PubMed] [Google Scholar]
- 20.Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–66. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Qin M, Kang J, Smith CB. A null mutation for Fmr1 in female mice: Effects on regional cerebral metabolic rate for glucose and relationship to behavior. Neuroscience. 2005;135:999–1009. doi: 10.1016/j.neuroscience.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 22.Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, et al. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46:94–102. doi: 10.1136/jmg.2008.061796. [DOI] [PubMed] [Google Scholar]
- 23.Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 24.de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, et al. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31:127–32. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berry-Kravis E. Epilepsy in fragile X syndrome. Dev Med Child Neurol. 2002;44:724–8. doi: 10.1017/s0012162201002833. [DOI] [PubMed] [Google Scholar]
- 26.Musumeci SA, Hagerman RJ, Ferri R, Bosco P, Dalla Bernardina B, Tassinari CA, et al. Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia. 1999;40:1092–9. doi: 10.1111/j.1528-1157.1999.tb00824.x. [DOI] [PubMed] [Google Scholar]
- 27.Westmark CJ, Westmark PR, O’Riordan KJ, Ray BC, Hervey CM, Salamat MS, et al. Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1 mice. PLoS One. 2011;6:e26549. doi: 10.1371/journal.pone.0026549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcinkiewicz M, Seidah NG. Coordinated expression of beta-amyloid precursor protein and the putative beta-secretase BACE and alpha-secretase ADAM10 in mouse and human brain. J Neurochem. 2000;75:2133–43. doi: 10.1046/j.1471-4159.2000.0752133.x. [DOI] [PubMed] [Google Scholar]
- 29.Hick M, Herrmann U, Weyer SW, Mallm JP, Tschape JA, Borgers M, et al. Acute function of secreted amyloid precursor protein fragment APPsalpha in synaptic plasticity. Acta Neuropathol. 2015;129:21–37. doi: 10.1007/s00401-014-1368-x. [DOI] [PubMed] [Google Scholar]
- 30.Tyan SH, Shih AY, Walsh JJ, Maruyama H, Sarsoza F, Ku L, et al. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol Cell Neurosci. 2012;51:43–52. doi: 10.1016/j.mcn.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schupf N, Tang MX, Fukuyama H, Manly J, Andrews H, Mehta P, et al. Peripheral Abeta subspecies as risk biomarkers of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2008;105:14052–7. doi: 10.1073/pnas.0805902105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al-Ayadhi LY, Ben Bacha AG, Kotb M, El-Ansary AK. A novel study on amyloid beta peptide 40, 42 and 40/42 ratio in Saudi autistics. Behav Brain Funct. 2012;8:4. doi: 10.1186/1744-9081-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehta PD, Capone G, Jewell A, Freedland RL. Increased amyloid beta protein levels in children and adolescents with Down syndrome. J Neurol Sci. 2007;254:22–7. doi: 10.1016/j.jns.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 34.Ray B, Sokol DK, Maloney B, Lahiri DK. Finding novel distinctions between the sAPPα-mediated anabolic pathways in fragile X syndrome and idiopathic Autism plasma and brain tissue. Scientific Reports. 2016;6:26052. doi: 10.1038/srep26052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baio J. Prevalence of autism spectrum disorder among children aged 8 years — autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summaries. 2014;63:1–21. [PubMed] [Google Scholar]
- 36.Kim YS, Leventhal BL, Koh YJ, Fombonne E, Laska E, Lim EC, et al. Prevalence of autism spectrum disorders in a total population sample. Am J Psychiatry. 2011;168:904–12. doi: 10.1176/appi.ajp.2011.10101532. [DOI] [PubMed] [Google Scholar]
- 37.Newschaffer CJ, Croen LA, Daniels J, Giarelli E, Grether JK, Levy SE, et al. The epidemiology of autism spectrum disorders. Annu Rev Public Health. 2007;28:235–58. doi: 10.1146/annurev.publhealth.28.021406.144007. [DOI] [PubMed] [Google Scholar]
- 38.Hessl D, Dyer-Friedman J, Glaser B, Wisbeck J, Barajas RG, Taylor A, et al. The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics. 2001;108:E88. doi: 10.1542/peds.108.5.e88. [DOI] [PubMed] [Google Scholar]
- 39.Lahiri DK, Maloney B, Zawia NH. The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases. Mol Psychiatry. 2009;14:992–1003. doi: 10.1038/mp.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37:738–47. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- 41.Moss J, Howlin P. Autism spectrum disorders in genetic syndromes: Implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res. 2009;53:852–73. doi: 10.1111/j.1365-2788.2009.01197.x. [DOI] [PubMed] [Google Scholar]
- 42.Langen M, Durston S, Staal WG, Palmen SJ, Engeland H. Caudate nucleus is enlarged in high-functioning medication-naive subjects with autism. Biol Psychiatry. 2007;62:262–6. doi: 10.1016/j.biopsych.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 43.Gothelf D, Furfaro JA, Hoeft F, Eckert Ma, Hall SS, O’Hara R, et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP) Ann Neurol. 2008;63:40–51. doi: 10.1002/ana.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hazlett HC, Poe MD, Lightbody AA, Gerig G, Macfall JR, Ross AK, et al. Teasing apart the heterogeneity of autism: Same behavior, different brains in toddlers with fragile X syndrome and autism. J Neurodev Disord. 2009;1:81–90. doi: 10.1007/s11689-009-9009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schumann CM, Barnes CC, Lord C, Courchesne E. Amygdala enlargement in toddlers with autism related to severity of social and communication impairments. Biol Psychiatry. 2009;66:942–9. doi: 10.1016/j.biopsych.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hall SS, Lightbody AA, Hirt M, Rezvani A, Reiss AL. Autism in fragile X syndrome: a category mistake? J Am Acad Child Adoles Psychiatry. 2010;9:921–33. doi: 10.1016/j.jaac.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dissanayake C, Bui Q, Bulhak-Paterson D, Huggins R, Loesch DZ. Behavioural and cognitive phenotypes in idiopathic autism versus autism associated with fragile X syndrome. J Child Psychol Psychiatry. 2009;50:290–9. doi: 10.1111/j.1469-7610.2008.01988.x. [DOI] [PubMed] [Google Scholar]
- 48.McDuffie A, Abbeduto L, Lewis P, Kover S, Kim JS, Weber A, Brown WR. Autism spectrum disorder in children and adolescents with fragile X syndrome: within-syndrome differences and age-related changes. Am J Intellect Dev Disabil. 2010;115:307–326. doi: 10.1352/1944-7558-115.4.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA, et al. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J Child Neurol. 2006;21:444–9. doi: 10.1177/08830738060210062201. [DOI] [PubMed] [Google Scholar]
- 50.Ray B, Long JM, Sokol DK, Lahiri DK. Increased secreted amyloid precursor protein-alpha (sAPPalpha) in severe autism: Proposal of a specific, anabolic pathway and putative biomarker. PLoS One. 2011;6:e20405. doi: 10.1371/journal.pone.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lahiri DK, Sokol DK, Erickson C, Ray B, Ho CY, Maloney B. Autism as early neurodevelopmental disorder: evidence for an sAPPalpha-mediated anabolic pathway. Front Cell Neurosci. 2013;7:94. doi: 10.3389/fncel.2013.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bailey AR, Giunta BN, Obregon D, Nikolic WV, Tian J, Sanberg CD, et al. Peripheral biomarkers in autism: Secreted amyloid precursor protein-alpha as a probable key player in early diagnosis. Int J Clin Exp Med. 2008;1:338–44. [PMC free article] [PubMed] [Google Scholar]
- 53.Fatemi SH, Folsom TD. Dysregulation of fragile × mental retardation protein and metabotropic glutamate receptor 5 in superior frontal cortex of individuals with autism: A postmortem brain study. Mol Autism. 2011;2:6. doi: 10.1186/2040-2392-2-6. 2392-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fatemi SH, Folsom TD, Kneeland RE, Yousefi MK, Liesch SB, Thuras PD. Impairment of fragile × mental retardation protein-metabotropic glutamate receptor 5 signaling and its downstream cognates ras-related C3 botulinum toxin substrate 1, amyloid beta A4 precursor protein, striatal-enriched protein tyrosine phosphatase, and homer 1, in autism: A postmortem study in cerebellar vermis and superior frontal cortex. Mol Autism. 2013;4:21. doi: 10.1186/2040-2392-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bailey AR, Hou H, Obregon DF, Tian J, Zhu Y, Zou Q, et al. Aberrant T-lymphocyte development and function in mice overexpressing human soluble amyloid precursor protein-alpha: Implications for autism. FASEB J. 2012;26:1040–51. doi: 10.1096/fj.11-195438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bailey AR, Hou H, Song M, Obregon DF, Portis S, Barger S, et al. GFAP expression and social deficits in transgenic mice overexpressing human sAPPalpha. Glia. 2013;61:1556–69. doi: 10.1002/glia.22544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem. 2011;96:529–43. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zeidan-Chulia F, de Oliveira BH, Salmina AB, Casanova MF, Gelain DP, Noda M, et al. Altered expression of Alzheimer’s disease-related genes in the cerebellum of autistic patients: A model for disrupted brain connectome and therapy. Cell Death Dis. 2014;5:e1250. doi: 10.1038/cddis.2014.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Eeghen AM, Pulsifer MB, Merker VL, Neumeyer AM, van Eeghen EE, Thibert RL, et al. Understanding relationships between autism, intelligence, and epilepsy: A cross-disorder approach. Dev Med Child Neurol. 2013;55:146–53. doi: 10.1111/dmcn.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hagerman RJ. Epilepsy drives autism in neurodevelopmental disorders. Dev Med Child Neurol. 2013;55:101–2. doi: 10.1111/dmcn.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amiet C, Gourfinkel-An I, Bouzamondo A, Tordjman S, Baulac M, Lechat P, et al. Epilepsy in autism is associated with intellectual disability and gender: Evidence from a meta-analysis. Biol Psychiatry. 2008;64:577–82. doi: 10.1016/j.biopsych.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 62.Hartley-McAndrew M, Weinstock A. Autism spectrum disorder: Correlation between aberrant behaviors, EEG abnormalities and seizures. Neurol Int. 2010;2:e10. doi: 10.4081/ni.2010.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heard TT, Ramgopal S, Picker J, Lincoln SA, Rotenberg A, Kothare SV. EEG abnormalities and seizures in genetically diagnosed fragile X syndrome. Int J Dev Neurosci. 2014;38:155–60. doi: 10.1016/j.ijdevneu.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 64.Robinson DJ, Merskey H, Blume WT, Fry R, Williamson PC, Hachinski VC. Electroencephalography as an aid in the exclusion of Alzheimer’s disease. Arch Neurol. 1994;51:280–4. doi: 10.1001/archneur.1994.00540150074020. [DOI] [PubMed] [Google Scholar]
- 65.Born HA. Seizures in Alzheimer’s disease. Neuroscience. 2015;286C:251–63. doi: 10.1016/j.neuroscience.2014.11.051. [DOI] [PubMed] [Google Scholar]
- 66.Westmark CJ. What’s hAPPening at synapses? the role of amyloid beta-protein precursor and beta-amyloid in neurological disorders. Mol Psychiatry. 2013;18:425–34. doi: 10.1038/mp.2012.122. [DOI] [PubMed] [Google Scholar]
- 67.Born HA, Kim JY, Savjani RR, Das P, Dabaghian YA, Guo Q, et al. Genetic suppression of transgenic APP rescues hypersynchronous network activity in a mouse model of Alzheimer’s disease. J Neurosci. 2014;34:3826–40. doi: 10.1523/JNEUROSCI.5171-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, et al. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–54. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen X, Lin R, Chang L, Xu S, Wei X, Zhang J, et al. Enhancement of long-term depression by soluble amyloid beta protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38MAPK, STEP and caspase-3. Neuroscience. 2013;253:435–43. doi: 10.1016/j.neuroscience.2013.08.054. [DOI] [PubMed] [Google Scholar]
- 70.Tamagnini F, Scullion S, Brown JT, Randall AD. Intrinsic excitability changes induced by acute treatment of hippocampal CA1 pyramidal neurons with exogenous amyloid beta peptide. Hippocampus. 2015;25:786–97. doi: 10.1002/hipo.22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raznahan A, Wallace GL, Antezana L, Greenstein D, Lenroot R, Thurm A, et al. Compared to what? early brain overgrowth in autism and the perils of population norms. Biol Psychiatry. 2013;74:563–75. doi: 10.1016/j.biopsych.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Davidovitch M, Patterson B, Gartside P. Head circumference measurements in children with autism. J Child Neurol. 1996;11:389–93. doi: 10.1177/088307389601100509. [DOI] [PubMed] [Google Scholar]
- 73.Lainhart JE, Bigler ED, Bocian M, Coon H, Dinh E, Dawson G, et al. Head circumference and height in autism: A study by the collaborative program of excellence in autism. Am J Med Genet A. 2006;140:2257–74. doi: 10.1002/ajmg.a.31465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Herbert MR, Ziegler DA, Makris N, Filipek PA, Kemper TL, Normandin JJ, et al. Localization of white matter volume increase in autism and developmental language disorder. Ann Neurol. 2004;55:530–40. doi: 10.1002/ana.20032. [DOI] [PubMed] [Google Scholar]
- 75.Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, et al. A clinicopathological study of autism. Brain. 1998;121(Pt 5):889–905. doi: 10.1093/brain/121.5.889. [DOI] [PubMed] [Google Scholar]
- 76.Courchesne E, Carper R, Akshoomoff N. Evidence of brain overgrowth in the first year of life in autism. JAMA. 2003;290:337–44. doi: 10.1001/jama.290.3.337. [DOI] [PubMed] [Google Scholar]
- 77.Courchesne E, Karns CM, Davis HR, Ziccardi R, Carper RA, Tigue ZD, et al. Unusual brain growth patterns in early life in patients with autistic disorder: An MRI study. Neurology. 2001;57:245–54. doi: 10.1212/wnl.57.2.245. [DOI] [PubMed] [Google Scholar]
- 78.McCaffery P, Deutsch CK. Macrocephaly and the control of brain growth in autistic disorders. Prog Neurobiol. 2005;77:38–56. doi: 10.1016/j.pneurobio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 79.Aylward EH, Minshew NJ, Goldstein G, Honeycutt NA, Augustine AM, Yates KO, et al. MRI volumes of amygdala and hippocampus in non-mentally retarded autistic adolescents and adults. Neurology. 1999;53:2145–50. doi: 10.1212/wnl.53.9.2145. [DOI] [PubMed] [Google Scholar]
- 80.Aylward EH, Minshew NJ, Field K, Sparks BF, Singh N. Effects of age on brain volume and head circumference in autism. Neurology. 2002;59:175–83. doi: 10.1212/wnl.59.2.175. [DOI] [PubMed] [Google Scholar]
- 81.Dementieva YA, Vance DD, Donnelly SL, Elston LA, Wolpert CM, Ravan SA, et al. Accelerated head growth in early development of individuals with autism. Pediatr Neurol. 2005;32:102–8. doi: 10.1016/j.pediatrneurol.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 82.Cohen MM., Jr Mental deficiency, alterations in performance, and CNS abnormalities in overgrowth syndromes. Am J Med Genet C Semin Med Genet. 2003;117C:49–56. doi: 10.1002/ajmg.c.10013. [DOI] [PubMed] [Google Scholar]
- 83.Wegiel J, Flory M, Kuchna I, Nowicki K, Ma S, Imaki H, et al. Neuronal nucleus and cytoplasm volume deficit in children with autism and volume increase in adolescents and adults. Acta Neuropathol Commun. 2015;3:2. doi: 10.1186/s40478-015-0183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hallahan BP, Craig MC, Toal F, Daly EM, Moore CJ, Ambikapathy A, et al. In vivo brain anatomy of adult males with fragile X syndrome: An MRI study. Neuroimage. 2011;54:16–24. doi: 10.1016/j.neuroimage.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 85.Hoeft F, Carter JC, Lightbody AA, Cody Hazlett H, Piven J, Reiss AL. Region-specific alterations in brain development in one- to three-year-old boys with fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107:9335–9. doi: 10.1073/pnas.1002762107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pacey LK, Xuan IC, Guan S, Sussman D, Henkelman RM, Chen Y, et al. Delayed myelination in a mouse model of fragile X syndrome. Hum Mol Genet. 2013;22:3920–30. doi: 10.1093/hmg/ddt246. [DOI] [PubMed] [Google Scholar]
- 87.Campbell DJ, Chang J, Chawarska K. Early generalized overgrowth in autism spectrum disorder: prevalence rates, gender effects, and clinical outcomes. J Am Acad Child Adolesc Psychiatry. 2014;53:1063–73. doi: 10.1016/j.jaac.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zeidan-Chulia F, de Oliveira BH, Salmina AB, Casanova MF, Gelain DP, Noda M, et al. Altered expression of Alzheimer’s disease-related genes in the cerebellum of autistic patients: A model for disrupted brain connectome and therapy. Cell Death Dis. 2014;5:e1250. doi: 10.1038/cddis.2014.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–50. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen Y, Bodles AM. Amyloid precursor protein modulates beta-catenin degradation. J Neuroinflammation. 2007;4:29. doi: 10.1186/1742-2094-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pennington BF. Diagnosing learning disorders. (2nd) 2009 [Google Scholar]
- 92.Desai MK, Mastrangelo MA, Ryan DA, Sudol KL, Narrow WC, Bowers WJ. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am J Pathol. 2010;177:1422–35. doi: 10.2353/ajpath.2010.100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Palazuelos J, Crawford HC, Klingener M, Sun B, Karelis J, Raines EW, et al. TACE/ADAM17 is essential for oligodendrocyte development and CNS myelination. J Neurosci. 2014;34:11884–96. doi: 10.1523/JNEUROSCI.1220-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dagli AI, Mueller J, Williams CA. Angelman syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R) University of Washington, Seattle; Seattle (WA): 1993. [PubMed] [Google Scholar]
- 95.Fombonne E, Roge B, Claverie J, Courty S, Fremolle J. Microcephaly and macrocephaly in autism. J Autism Dev Disord. 1999;29:113–9. doi: 10.1023/a:1023036509476. [DOI] [PubMed] [Google Scholar]
- 96.Singhmar P, Kumar A. Angelman syndrome protein UBE3A interacts with primary microcephaly protein ASPM, localizes to centrosomes and regulates chromosome segregation. PLoS One. 2011;6:e20397. doi: 10.1371/journal.pone.0020397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Recuero M, Munive VA, Sastre I, Aldudo J, Valdivieso F, Bullido MJ. A free radical-generating system regulates AbetaPP metabolism/processing: Involvement of the ubiquitin/proteasome and autophagy/lysosome pathways. J Alzheimers Dis. 2013;34:637–47. doi: 10.3233/JAD-121510. [DOI] [PubMed] [Google Scholar]
- 98.Berry-Kravis E, Hessl D, Abbeduto L, Reiss AL, Beckel-Mitchener A, Urv TK, et al. Outcome measures for clinical trials in fragile x syndrome. J Dev Behav Pediatr. 2013;34:508–22. doi: 10.1097/DBP.0b013e31829d1f20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mullard A. Fragile X disappointments upset autism ambitions. Nat Rev Drug Discov. 2015;14:151–3. doi: 10.1038/nrd4555. [DOI] [PubMed] [Google Scholar]
- 100.Westmark CJ, Hervey CM, Berry-Kravis EM, Malter JS. Effect of anticoagulants on amyloid beta-protein precursor and amyloid beta levels in plasma. Alzheimer’s Disease and Parkinsonism. 2011;1:1–3. doi: 10.4172/2161-0460.1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Isaacs AM, Senn DB, Yuan M, Shine JP, Yankner BA. Acceleration of amyloid beta-peptide aggregation by physiological concentrations of calcium. J Biol Chem. 2006;281:27916–23. doi: 10.1074/jbc.M602061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Das U, Scott DA, Ganguly A, Koo EH, Tang Y, Roy S. Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron. 2013;79:447–60. doi: 10.1016/j.neuron.2013.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lahiri DK, Farlow MR, Sambamurti K, Greig NH, Giacobini E, Schneider LS. A critical analysis of new molecular targets and strategies for drug developments in Alzheimer’s disease. Curr Drug Targets. 2003;4:97–112. doi: 10.2174/1389450033346957. [DOI] [PubMed] [Google Scholar]
- 104.Vincent B, Govitrapong P. Activation of the alpha-secretase processing of AbetaPP as a therapeutic approach in Alzheimer’s disease. J Alzheimers Dis. 2011;24(Suppl 2):75–94. doi: 10.3233/JAD-2011-110218. [DOI] [PubMed] [Google Scholar]
- 105.Gravitz L. Drugs: A tangled web of targets. Nature. 2011;475:S9–11. doi: 10.1038/475S9a. [DOI] [PubMed] [Google Scholar]
- 106.Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni S, Xu X, Miller HB, et al. Potent memapsin 2 (beta-secretase) inhibitors: Design, synthesis, protein-ligand X-ray structure, and in vivo evaluation. Bioorg Med Chem Lett. 2008;18:1031–6. doi: 10.1016/j.bmcl.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chang WP, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM, et al. Beta-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 2011;25:775–84. doi: 10.1096/fj.10-167213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li G, Percontino L, Sun Q, Qazi AS, Frederikse PH. Beta-amyloid secretases and beta-amyloid degrading enzyme expression in lens. Mol Vis. 2003;9:179–83. [PubMed] [Google Scholar]
- 109.Vassar R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimer’s Research & Therapy. 2014;6:89. doi: 10.1186/s13195-014-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jia Q, Deng Y, Qing H. Potential therapeutic strategies for Alzheimer’s disease targeting or beyond beta-amyloid: Insights from clinical trials. Biomed Res Int. 2014;2014:837157. doi: 10.1155/2014/837157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ding Y, Ko MH, Pehar M, Kotch F, Peters NR, Luo Y, et al. Biochemical inhibition of the acetyltransferases ATase1 and ATase2 reduces beta-secretase (BACE1) levels and Abeta generation. J Biol Chem. 2012;287:8424–33. doi: 10.1074/jbc.M111.310136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ko MH, Puglielli L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J Biol Chem. 2009;284:2482–92. doi: 10.1074/jbc.M804901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem J. 2007;407:383–95. doi: 10.1042/BJ20070040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang C, Saunders AJ. Therapeutic targeting of the alpha-secretase pathway to treat Alzheimer’s disease. Discov Med. 2007;7:113–7. [PubMed] [Google Scholar]
- 115.Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. 2014;2:135. doi: 10.1186/s40478-014-0135-5. 014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.