

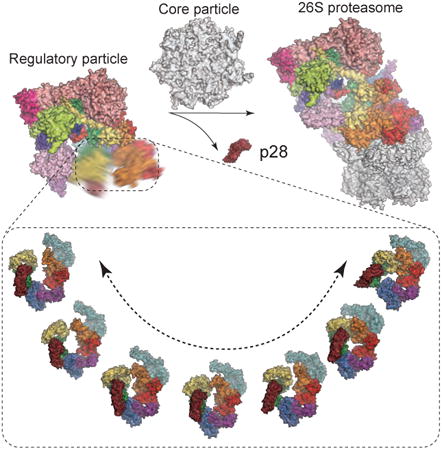
Summary
The proteasome holoenzyme is activated by its regulatory particle (RP) consisting of two subcomplexes, the lid and the base. A key event in base assembly is the formation of a heterohexameric ring of AAA-ATPases, which is guided by at least four RP assembly chaperones in mammals: PAAF1, p28/gankyrin, p27/PSMD9 and S5b. Using cryogenic electron microscopy, we analyzed the non-AAA structure of the p28-bound human RP at 4.5-Å resolution, and determined seven distinct conformations of the Rpn1-p28-AAA subcomplex within the p28-bound RP at subnanometer resolutions. Remarkably, the p28-bound AAA ring does not form a channel in the free RP and spontaneously samples multiple ‘open’ and ‘closed’ topologies at the Rpt2-Rpt6 and Rpt3-Rpt4 interfaces. Our analysis suggests that p28 assists the proteolytic core particle to select a specific conformation of the ATPase ring for RP engagement, and is released in a shoehorn-like fashion in the last step of the chaperone-mediated proteasome assembly.
Graphical abstract
At least four chaperones guide the assembly of the proteasome regulatory particle. Lu, Wu, et al. present the cryo-EM structures and dynamics of the p28 chaperone bound to the human proteasome regulatory particle and reveal how it guides conformational selection upon association of the regulatory and core particles.
Introduction
Ubiquitin-dependent protein degradation, mediated by the proteasome, regulates numerous biological processes in all eukaryotes. The complete assembly of the 26S proteasome holoenzyme consists of a 20S proteolytic core particle (CP) and two 19S regulatory particles (RP) (Chen et al., 2016; Finley, 2009; Park et al., 2010; Tomko and Hochstrasser, 2013). Peptide cleavage is carried out inside the barrel-shaped CP chamber. The RP associates with the CP to activate its ubiquitin-dependent functions. The RP consists of two subcomplexes, the lid and the base (Glickman et al., 1998). The lid contains six subunits featuring Proteasome-Cyclosome-Initiation factor (PCI) domains (Rpn3, Rpn5-7, Rpn9, Rpn12), two subunits featuring MPR1/PAD1 amino-terminal (MPN) domains (Rpn8 and Rpn11), and a small tethering protein Sem1 (Finley, 2009). The six PCI proteins are assembled into a horseshoe-like shape encircling the Rpn8-Rpn11 heterodimer (Beck et al., 2012; Chen et al., 2016; da Fonseca et al., 2012; Dambacher et al., 2016; Lander et al., 2012; Lasker et al., 2012; Luan et al., 2016; Sledz et al., 2013; Unverdorben et al., 2014). Rpn11, the only essential deubiquitylating enzyme in the proteasome, resides at the entrance to the substrate-translocation channel, and removes ubiquitin chains en bloc from a substrate during degradation (Pathare et al., 2014; Verma et al., 2002; Worden et al., 2014; Yao and Cohen, 2002). The rest of the RP subunits, apart from Rpn10, form a 9-subunit subcomplex named the base (Chen et al., 2016; Lander et al., 2012; Lasker et al., 2012). A critical component in the base is a peptide-unfolding channel made of heterohexameric AAA-ATPase ring, Rpt1-6. Each Rpt subunit features three domains, namely, coiled-coil (CC), oligonucleotide/oligosaccharide-binding (OB) and ATPase associated with diverse cellular activities (AAA) domains (Chen et al., 2016; Huang et al., 2016; Schweitzer et al., 2016; Zhang et al., 2009a; Zhang et al., 2009b). In the 26S holoenzyme, the AAA domains assemble into a helical staircase architecture and harvest the free energy from ATP hydrolysis to power substrate unfolding and translocation into the CP chamber, where the substrate is degraded (Beck et al., 2012; da Fonseca et al., 2012; Lander et al., 2012).
A key event in base assembly is the formation of the heterohexameric ring of AAA-ATPases, which is guided by at least four RP assembly chaperones in mammals: PAAF1 (proteasomal ATPase-associated factor 1), p28/gankyrin (Krzywda et al., 2004; Nakamura et al., 2007a), p27/PSMD9 and S5b (Kaneko et al., 2009; Roelofs et al., 2009). Each RP assembly chaperone binds a distinct base subunit (Kaneko et al., 2009; Park et al., 2009; Roelofs et al., 2009; Saeki et al., 2009; Thompson et al., 2009; Tomko and Hochstrasser, 2013), forming three RP assembly precursors, Rpt3-Rpt6-p28-PAAF1, Rpt4-Rpt5-p27, and Rpt1-Rpt2-S5b complexes. Release of chaperones p28, PAAF1, and S5b upon holoenzyme assembly is important for proteasome activation. The yeast ortholog of p28 is Nas6 (Funakoshi et al., 2009; Roelofs et al., 2009; Saeki et al., 2009). Modeling of the Rpt3-Nas6 structure (Nakamura et al., 2007a; Nakamura et al., 2007b) into the ATPase ring of the proteasome holoenzyme suggests that Nas6 physically occludes the formation of proper RP-CP contacts (Park et al., 2013; Roelofs et al., 2009). To date, no structural information is available for a complete RP in complex with any assembly chaperones. It remains unclear how RP chaperones interact with the RP to ensure proper complex assembly, and how the chaperones are released to activate the proteasome. To address these problems, we attempted to analyze the structures of the free RP in complex with its chaperone p28 by single-particle cryo-electron microscopy (cryo-EM).
Results
Characterization of the p28-bound RP
We purified the p28-bound RP from human embryonic kidney (HEK) 293 cells, using biotin-tagged Rpn11, followed by size-exclusion chromatography (Figure S1A-C) (Guerrero et al., 2006). The purified p28-RP complex was then examined by several biochemical assays. First, we used mass spectrometry to examine the contents of the purified RP complex and verified the presence of p28 and the absence of PAAF1 in the complex in addition to the RP subunits (Figure S1D). The absence of PAAF1 was further verified through Western blot using an anti-PAAF1 antibody (Figure S1E). Second, we examined the ATPase activity of the p28-bound RP. The result showed that it shares a similar ATPase activity as the yeast RP in the absence of p28 (Figure S1F) (Hoffman and Rechsteiner, 1996). Third, we tested the ATP-independent deubiquitylation activity of the p28-bound RP, which was reduced relative to that of the 26S proteasome holoenzyme (Figures S1G-S1M). Fourth, to test whether the p28-bound RP can unfold substrates, we employed a fluorescence-based, ubiquitin-dependent substrate-unfolding assay (see STAR Methods). As expected, we did not observe any substrate unfolding activity in the p28-bound RP (Figure S1P). Finally, we successfully reconstituted functional 26S holoenzymes in vitro by assembling the purified p28-bound RP with the purified CP (Figures S1Q and S1R). Western blot using an anti-p28 antibody confirmed that p28 was dissociated from the assembled proteasome holoenzyme after in vitro reconstitution (Figure S1S). Taken together, these results support that our purified p28-bound RP is likely an active intermediate on the pathway of proteasome holoenzyme assembly.
Structural Analysis by Cryo-EM
We collected single-particle cryo-EM data of the p28-RP complex on a Gatan K2 Summit direct electron detector mounted on the FEI Tecnai Arctica electron microscope operating at 200 kV. Through exhaustive unsupervised 2D and 3D classifications of single-particle cryo-EM data (Figures S2 and S3), we identified a dataset of the free RP, which allowed the refinement of the stable part of the structure, corresponding to the non-AAA subcomplex, to a nominal resolution of 4.5 Å by a gold-standard procedure, in which two half-datasets were refined separately (Figure 1A). The high-resolution components include all the RP components but Rpn1, Rpn13, and the AAA domains of the ATPases. An atomic model was built and refined based on this density map, which includes the complete lid, Rpn2, Rpn10, and the CC (coiled coil) and OB (oligonucleotide/oligosaccharide-binding) domains of Rpt1-Rpt6 (Figures 1B and S2; Table 1).
Figure 1. Cryo-EM Reconstructions of the p28-RP Complex.
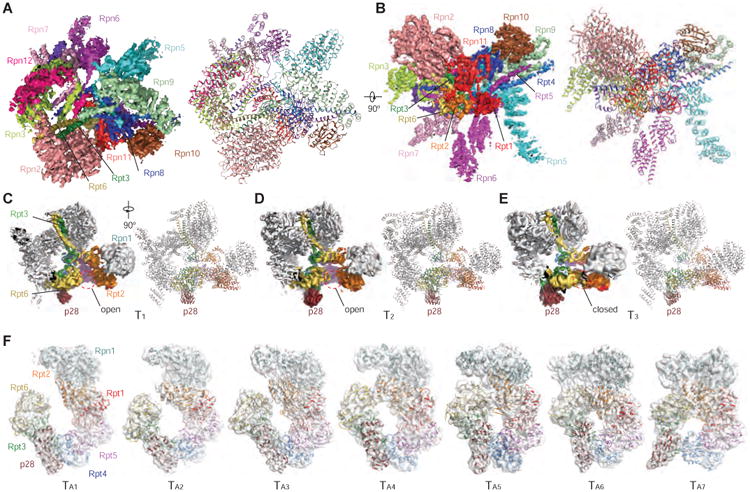
(A) High-resolution cryo-EM density map of the free RP, viewed from the top of the lid subcomplex on the left, and the atomic model refined against the cryo-EM density is shown on the right.
(B) High-resolution cryo-EM density map of the free RP viewed from the lateral side with the OB ring exposed, low-resolution density from the AAA domains of ATPase is not shown. The atomic model refined against the cryo-EM density is shown on the right.
(C)-(E) The cryo-EM density maps and their corresponding pseudo-atomic models of the conformational states T1 (C), T2 (D) and T3 (E). The opening at the Rpt2-Rpt6 interface is marked by a red dashed circle.
(F) The corresponding cryo-EM density in a transparent surface representation superimposed with pseudo-atomic models of Rpn1-p28-AAA components in a cartoon representation. The map-superposed models correspond to the seven states TA1-7 and are aligned into a sequence following the decrease of the Rpt2-Rpt6 gap from TA1 to TA7, and from the left to the right.
Table 1. Cryo-EM Data Collection and Structure Determination Statistics.
| Data collection | |||||||
|
| |||||||
| Electron energy (kV) | 200 | ||||||
| Electron dose (e-/Å2) | 50 | ||||||
| Pixel size corresponding to the physical detector sensor (Å) | 0.98 | ||||||
| Defocus range (μm) | -3.0 to -1.0 | ||||||
| Number of micrographs | 16,111 | ||||||
|
| |||||||
| Structure determination of the non-AAA subcomplex | |||||||
|
| |||||||
| Cryo-EM refinement | |||||||
| Number of particles used in refinement | 117,471 | ||||||
| Final Resolution of RP complex (Å) | 4.5 | ||||||
| Isotropic B-factor of RP complex (Å2) | -90 | ||||||
| Pseudo-crystallographic refinement of atomic model | |||||||
| Cell dimension a, b, c(Å) | 250.9,250.9,250.9 | ||||||
| Cell angle α, β, γ (Å) | 90,90,90 | ||||||
| Space group | P1 | ||||||
| Resolution range | 250.88-4.5 | ||||||
| Number of reflections | 652,468 | ||||||
| Number of atoms | 36,739 | ||||||
| Rwork factor (%) | 31.2 | ||||||
| Geometric Parameters (RMSD) | |||||||
| Bond length (Å) | 0.0043 | ||||||
| Bond angle (°) | 0.88 | ||||||
| Ramachandran plot statistics | |||||||
| Preferred (%) | 91.0 | ||||||
| Allowed (%) | 8.9 | ||||||
| Outlier (%) | 0.13 | ||||||
| MolProbity validation | |||||||
| Rotamer outlier (%) | 0.37 | ||||||
| MolProbity score | 2.3 | ||||||
| Clashscore | 15.78 | ||||||
| C-beta outliers | 0 | ||||||
|
| |||||||
| Conformational states of the p28-bound RP | |||||||
|
| |||||||
| State | Particle number | Resolution (Å) | B-factor (Å2) | Ramachandran plot (%) | Rotamer outlier (%) | ||
| Favored | Allowed | Outlier | |||||
| T1 | 39,520 | 5.7 | -80 | 87.8 | 12.0 | 0.27 | 0.99 |
| T2 | 27,629 | 6.1 | -120 | 87.4 | 12.4 | 0.28 | 1.21 |
| T3 | 10,891 | 6.8 | -120 | 88.9 | 11.0 | 0.11 | 0.52 |
| TA1 | 12,223 | 8.5 | -350 | 81.2 | 18.1 | 0.67 | 0.79 |
| TA2 | 11,082 | 8.3 | -400 | 79.5 | 19.9 | 0.61 | 2.1 |
| TA3 | 21,031 | 7.3 | -350 | 83.6 | 16.0 | 0.41 | 0.72 |
| TA4 | 9,004 | 8.3 | -400 | 79.6 | 19.7 | 0.7 | 1.15 |
| TA5 | 11,767 | 7.9 | -450 | 83.0 | 16.5 | 0.48 | 0.62 |
| TA6 | 11,610 | 8.9 | -450 | 82.3 | 17.2 | 0.46 | 0.67 |
| TA7 | 14,464 | 7.7 | -450 | 82.0 | 17.6 | 0.36 | 1.05 |
Through 3D classification, we obtained three conformational states of the complete p28-bound free RP, designated T1, T2 and T3, differing mainly in their AAA domains of the ATPase at 5.2 Å, 6.1 and 6.8 resolution, respectively (Figures 1C-1E and S2E). The AAA and CC-OB domains of each Rpt subunit are connected through a flexible loop that allows inter-domain motions. In the cryo-EM image data, the movement of the entire hexameric AAA ring relative to the OB ring is coupled with the inter-domain motion between adjacent Rpt AAA domains in the free RP. As a result, Rpn1, p28, and the AAA domains of Rpt3-Rpt6 exhibit limited resolutions and diffuse densities in the cryo-EM map. To de-couple the two dynamic modes, we employed a ‘density-subtraction’ strategy in which the density of the non-AAA subcomplex is subtracted from each single-particle image. Maximum-likelihood-based classification (Scheres, 2012a, b) of the density-subtracted images allowed us to obtain seven conformational states of the Rpn1-p28-AAA subcomplex, designated TA1-7, each refined separately to 7-9 Å resolution (Figures 1F, S2F and S3). The cryo-EM maps of these Rpn1-p28-AAA conformations significantly improved the local resolution in Rpn1, p28/gankyrin, and the AAA domains of Rpt3-Rpt6 (Figures S2K and S2L). Based on the atomic structure of the human proteasome holoenzyme (Chen et al., 2016), we built pseudo-atomic models for the Rpn1-p28-AAA subcomplex in the TA1-7 states.
Overall Structure and Dynamics of the p28-RP Complex
The cryo-EM densities of the non-AAA components in the T1, T2 and T3 states show virtually the same conformation as that in the 4.5-Å map of the non-AAA subcomplex (Figures 2A-2C). The T1, T2 and T3 structures contain six Rpt subunits. The local resolutions are lower in the AAA domains than in the OB domains. While the structure of OB ring in all three states are well formed and identical to their counterpart in the fully assembled proteasome holoenzyme (Chen et al., 2016), the AAA rings exhibit a prominent opening between Rpt2 and Rpt6 in both T1 and T2 states (Figures 2A and 2B), and an opening between Rpt3 and Rpt4 in the T3 state. However, the AAA domains of Rpt2 and Rpt6 are in direct contact in the T3 state (Figure 2C). The N-terminal elements of Rpn2, Rpn3 and Rpn12 that form a trimeric interface at the apex of RP also suffer diffuse cryo-EM densities in the T1 state indicative of local conformational dynamics (Figure 1C), whereas corresponding local densities in the T2 and T3 states are substantially stronger (Figures 1D and 1E).
Figure 2. Transitions between RP Assemblies in Different Conformations.
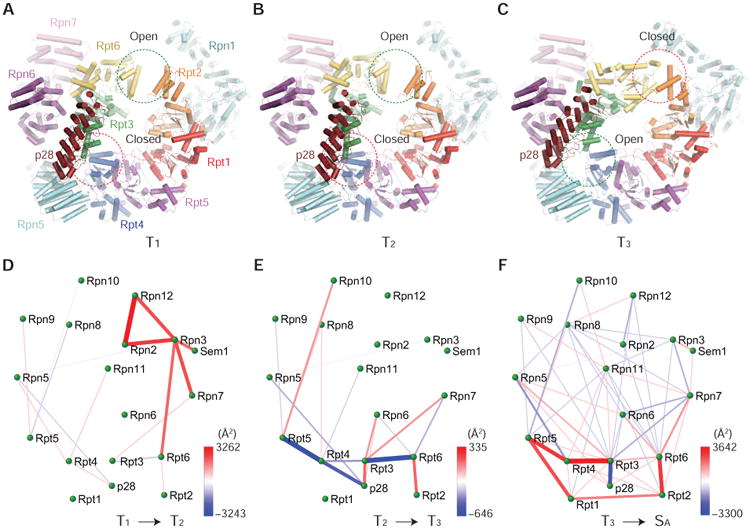
(A)-(C) Side-by-side comparison of the three RP structures in the T1 (A), T2 (B) and T3 (C) states viewed from the side of p28-bound ATPase ring. The opening and closing at Rpt2-Rpt6 and Rpt3-Rpt4 interfaces are marked by green and red circles, respectively.
(D)-(F) The microinteractome networks visualize the changes of inter-subunit interface areas during the four transitions, from T1 to T2 (D), from T2 to T3 (E), from T3 to SA (F) (Chen et al., 2016).
To capture the ‘hotspots’ of inter-subunit interface changes upon structural transitions, we calculated the differences of inter-subunit interfaces and mapped them into differential networks of inter-subunit interactions, termed ‘microinteractome networks’ (Figures 2D-2F). The T1-to-T2 transition features a ‘hotspot’ around Rpn3 (Figure 2D). In the T2-to-T3 transition, an increased Rpt2-Rpt6 interaction trades off for a decreased interaction between p28 and Rpt4 (Figure 2E). The closure of Rpt2-Rpt6 gap is accompanied with increased Rpt3 interactions with Rpn6 and Rpn7. The fully assembled human proteasome holoenzyme was recently found to adopt four co-existing conformations: the substrate-accepting SA state, the substrate-commitment SB state, the gate-priming SC state and the open-gate SD state (Chen et al., 2016). The T3-to-SA transition network suggests subtle inter-subunit rearrangement in the non-ATPase components, in contrast to dramatic strengthening of inter-Rpt interactions upon RP-CP association (Figure 2F).
Conformational Changes of the Non-AAA Subcomplex upon RP-CP Association
Comparison of the free RP structure with that of proteasome holoenzyme in the SA state reveals defined conformational changes of the lid upon RP-CP association (Figures 3A-3D). When the free RP structure is aligned with the holoenzyme structure based on their lid subcomplexes (Figure 3A), the N-terminal PCI domain of Rpn7 is translated ∼15 Å towards the CP, due to rearrangement of Rpt3 and Rpt6 during RP-CP association. The most prominent conformational change is observed in Rpn6. The N-terminal PCI domain of Rpn6 exhibits a dramatic rotation of 40°, allowing Rpn6 to interact with α2 subunit of the CP (Figure 3A). When the subunit structures are aligned separately on their own, there is a ∼40° rotation of the C-terminal helix around a pivot point at residue Pro317 in Rpn6 (Figure 4B), whereas a ∼20° rotation of the C-terminal helix is seen in Rpn7 around a pivot point at residue Pro359 (Figure 3C). The rest of the lid subunits, as well as Rpn2 and Rpn10, are nearly identical in both structures (Figure 3D).
Figure 3. Conformational Transitions of the Non-AAA Subcomplex upon 26S Assembly.
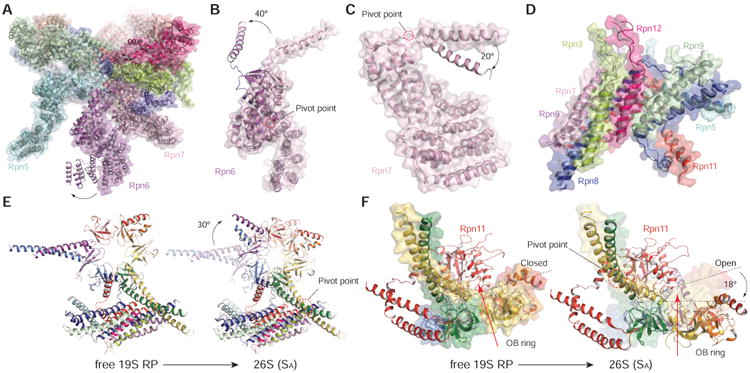
(A) Superposition of the structures of the free RP with the RP in 26S in the Sa state, after aligning them over the non-AAA subcomplex (Chen et al., 2016).
(B)-(D) Superposition of the structures of Rpn6 (B), Rpn7 (C), and the lid central helical bundle (D), by alignment their own structure in the free RP with that in the 26S.
(E) Side-by-side comparison of the RP components including CC-OB and lid helical bundle, showing a 30° in-plane rotation between the free RP in the T3 state and the 26S in the SA sate (Chen et al., 2016). On the right, the T3 structure is rendered in transparent cartoon for convenience of comparison.
(F) Side-by-side comparison of the RP components including CC-OB and lid helical bundle, showing a 18° out-of-plane rotation between the free RP in the T3 state and the 26S in the SA sate (Chen et al., 2016). On the right, the T3 structure is rendered in transparent surface for convenience of comparison.
Figure 4. Conformational States of the Rpn1-p28-AAA Components in the p28-Bound RP.
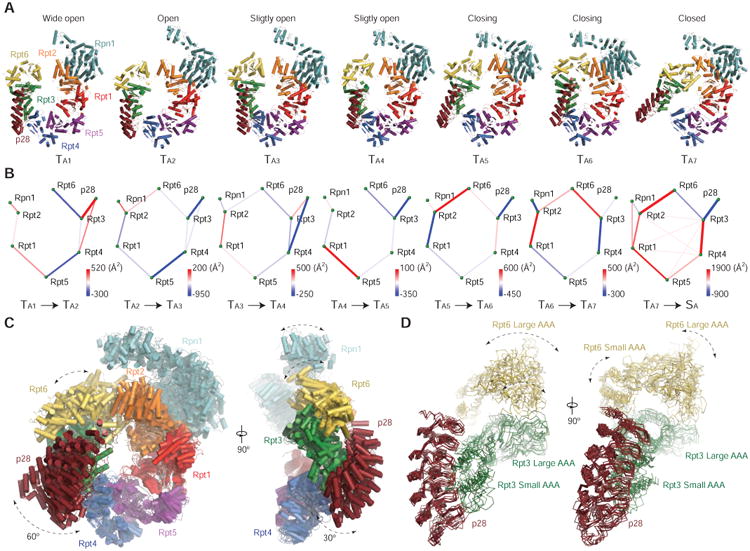
(A) The seven pseudo-atomic models of Rpn1-p28-AAA subcomplex from a viewing perspective that shows a large in-plane rotation of the p28/gankyrin.
(B) The seven differential networks show the changes of inter-subunit interactions during the state transitions.
(C) The seven pseudo-atomic models of the Rpn1-p28-AAA subcomplex are superimposed. On the right, a viewing perspective orthogonal to the left is shown.
(D) The seven states are aligned relative to the p28-Rpt3 structure and superimposed with ribbon representations.
Although the structure of the AAA ring in the free RP is significantly different from that in the assembled proteasome, the OB ring in the free RP is nearly identical to that in the holoenzyme (Chen et al., 2016). Upon RP-CP assembly, the OB ring rotates in-plane by 30° and tilts out-of-plane by 18° (Figures 3E and 3F). The gap between Rpn11 and the OB ring is slightly narrower in the free RP than in the SB, SC and SD states of the proteasome holoenzyme (Chen et al., 2016), suggesting that Rpn11 tightly blocks the substrate entry port in the free RP (Figures 3F and S4). Indeed, the unfolding assay used in this study to characterize the free RP suggests that it essentially lacks substrate unfolding activity toward a fluorescent model substrate (Figure 1D). The free RP exhibits certain ATP-independent deubiquitylating activity that is lower than that of the proteasome holoenzyme (Figure S1F). In line with this observation, docking of the ubiquitin structure to the surface of Rpn11 in the free RP indicates that the active site for deubiquitylation is exposed on the molecular surface (Figures S4E and S4F). Conformation of the Rpn11-OB interface in a SB, SC or SD-like state has been suggested to enhance Rpn11 activity (Chen et al., 2016; Pathare et al., 2014; Worden et al., 2014), which may also account for the deubiquitylating activity of the free RP.
Microstates of Rpn1-p28-AAA Subcomplex
A proper hexamerization of the AAA domains of Rpt1-6 is crucial for activating the substrate unfolding activity of the RP in an assembled proteasome holoenzyme. The cryo-EM structures of the Rpn1-p28-AAA subcomplex in seven states TA1-7 exhibit dramatic motions in their p28-Rpt3-Rpt6 and Rpn1 components (Figure 4A). The AAA domains form a C-like shape with an opening between Rpt2 and Rpt6 in all but the TA6 and TA7 states. The width of the opening is different in each state, and we used this feature to order the states from the widest to the tightest (Figure 4A; Table S1), revealing a sequence of motion of the AAA ring through consecutive intermediates in an open-to-closed transition. Consistent with our assignment of the state sequence, the microinteractome networks of inter-subunit interactions exhibit a gradually increased connectivity from TA1 to TA7 (Figure 4B; Table S2). Remarkably, Rpn1 rotates as the Rpt2-Rpt6 gap narrows. Rpn1-Rpt2 interaction is established starting from the TA5 state, suggesting that Rpn1 assists the open-to-closed transition. Correspondingly, Rpt3-bound p28 rotates in-plane by ∼60° and out-of-plane by ∼30° (Figure 4C). Accompanying closure of the Rpt2-Rpt6 gap, the Rpt3-Rpt4 interface is opened in the TA7 state, whereas in other states p28 appears to bridge the opening between Rpt3 and Rpt4.
The elements that interact with p28 seem to undergo multiple modes of conformational dynamics (Figures 4C and 4D). The p28 movement orthogonal to the hexameric axis of the ATPase concurs with its wobbling along the axis (Figure 4C). Similar multi-modal remodeling at the Rpt2-Rpt6 interface is also observed (Figure 4D). Aligning the structures of p28-Rpt3 in the seven states reveals that the p28-Rpt3 interface is much more rigid than the Rpt3-Rpt6 interface (Figure 4D). Each AAA domain contains a large N-terminal α/β subdomain and a small C-terminal α-helical subdomain, separated by a short flexible linker. The small and large AAA subdomains exhibit different amplitudes of rotational motion (Figure 4D). In summary, the multi-modal dynamics of the AAA domains is characteristic of the p28-bound RP intermediate assembly.
RP Remodeling upon RP-CP Association
To understand how the p28-RP approaches the CP for proteasome activation, we docked the pseudo-atomic model of the p28-RP complex in the TA7 state onto the CP, by aligning the AAA-ATPase in the p28-RP structure with that in the holoenzyme structure in the SA state. Notably, the p28 protein shows minimal clashes with the α-ring of CP, Rpn6 or Rpn5 (Figure 5A), suggesting that p28 does not necessarily block the RP-CP association in its first-encounter state when the CP engages with a p28-bound RP in the TA7 state (Figures 5A-5C). By contrast, there are prominent clashes between α2-subunit and p28 in the other six states, TA1-6 (Figure S5A). This suggests that p28 helps the CP to select the TA7 conformation to facilitate the RP-CP association by increasing interface contact area while introducing no significant occlusion (Figure 5D). Comparison of the docked p28-RP-CP intermediate complex with the holoenzyme structure reveals that the lid subcomplex undergoes a 40° rotation along an axis that is tilted ∼30° from the heptameric axis of the CP (Figure 5E).
Figure 5. RP Remodeling upon RP-CP Association.
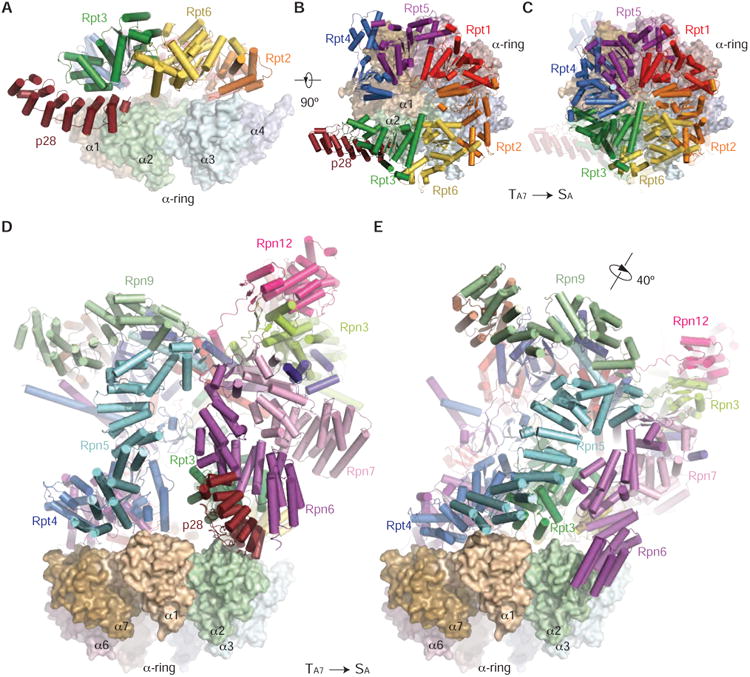
(A) The side view of the p28-AAA subcomplex in the TA7 state docking onto the α-ring surface, by aligning the AAA domains in the free RP against those in the 26S in the SA state. The model predicts that p28 is subject to little or no clashes with the α-ring surface when the free RP first encounters the CP.
(B) The top view of the p28-AAA subcomplex docking onto the α-ring surface.
(C) The top view of the AAA hexamer on the α-ring surface in the 26S structure for comparison. The structure of p28-AAA subcomplex is superposed as a transparent cartoon.
(D) The side view of the pseudo-atomic model of the free RP in the TA7 state docking onto the α-ring surface, with the same alignment as used in panel (A).
(E) The side view of the 26S in the SA state, in the same orientation of the CP, showing that a large RP rotation accompanying the release of p28.
The assembly chaperone p28 must be released upon the RP-CP assembly and activation of proteasome (Roelofs et al., 2009). Our structural analysis suggests a shoehorn-like mechanism for p28 release. Although p28 can be an integral part of the RP-CP first-encounter complex, RP-CP association has to be reinforced with considerable remodeling of the AAA-ATPase and lid subcomplexes as well as the RP-CP interface. The stronger affinity of the α-ring with the ATPases, the closure of the gap between Rpt3 and Rpt4, and a tighter arrangement of the ATPase hexamer, may all help to drive p28 dissociation from Rpt3. Importantly, Rpt3 is translated ∼15 Å upon the completion of proteasome assembly to close its gap with Rpt4, which dramatically reorients its p28 binding site toward the α1-α2 surface (Figures 5B and 5C). Consistent with this picture, a yeast proteasome mutant Rpt3-Δ1, in which the C-terminal residue from Rpt3 was deleted, was observed to be unable to release Nas6, the yeast ortholog of p28 (Park et al., 2009), perhaps because the mutated Rpt3 loses the affinity of its C-terminal tail with α-ring and is unable to complete the Rpt3 remodeling that ejects the assembly chaperone.
Conformation of p28-Free RP Resembles the TA7 State of p28-Bound RP
To test the shoehorn-like mechanism for p28 release upon RP-CP assembly, we determined a cryo-EM structure of p28-free RP. To this end, we attempted to purify p28-free RPs by washing the RP off the CP in the purified proteasome holoenzymes through high ionic strength in the buffer. However, we failed to yield enough p28-free RPs for structural determination due to excessive protein aggregation. Fortunately, we were able to overcome this hurdle and analyze the structure of p28-free RP from a set of cryo-EM micrographs taken on the purified human proteasome holoenzymes (Chen et al., 2016). Although the proteasome holoenzymes were purified by size-exclusion chromatography to remove the free RPs from cytosol, we still observed many single-particles of free RPs coexisting with the purified holoenzymes in these cryo-EM micrographs (Figure S6A). We speculate that these free RPs could be dissociated from the purified holoenzymes during the cryo-plunging procedure (see STAR Methods) and be free of p28. Indeed, after analyzing this dataset by focusing on the free RP, 3D classification identified two major classes, both refined to 8.8-Å resolution, one with no clear p28 density and the other showing the p28 density (Figures 6A and S6B-S6D); the particle abundance of the two classes were comparable. This sharply contrasts with the observation that all 3D classes of the free RP purified from cytosols include strong p28 density. Comparison of the p28-free RP structure with the p28-RP models suggests that it is compatible with the T3 or TA7 state of the p28-bound RP, but differing from other states, T1, 2 and TA1-6, of the p28-RP complex (Figure 6A, 6B, and S6C). In both the p28-free RP conformation and the T3 or TA7 conformation of the p28-bound RP, the Rpt2-Rpt6 interface is closed, whereas the Rpt3-Rpt4 interface is open. Thus, the discontinuity of the AAA ring in the free RP is not disrupted by the presence of p28, but manifests its intrinsic metastability. These observations support that the TA7 state is an intermediate conformation on the RP assembly pathway prior to RP-CP association (Figure 6C), and that p28 is released in a shoehorn-like fashion.
Figure 6. p28-Free RP Reconstruction and Insights into RP Assembly Pathway.
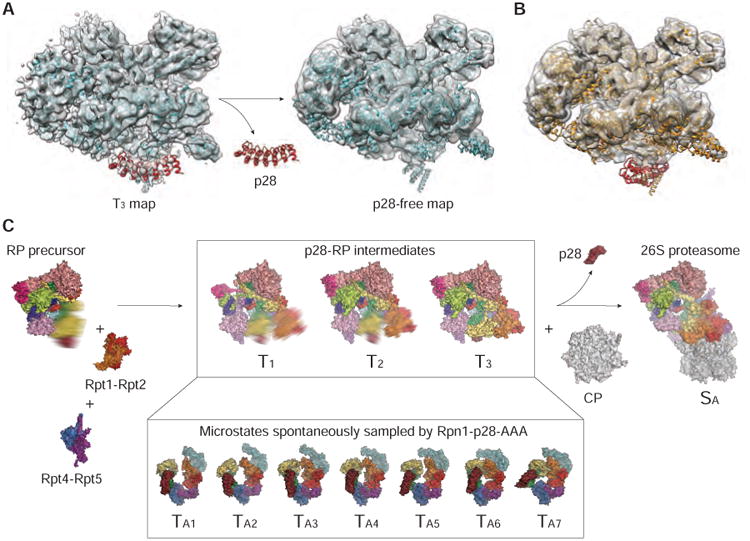
(A) Side-by-side comparison of the T3 density map of the p28-RP complex (on the left) with the cryo-EM map of the p28-free RP (on the right). Both maps, shown in a transparent surface representation, are superimposed with the pseudo-atomic model of the T3 state shown in cyan cartoon ribbons.
(B) The pseudo-atomic model of the p28-bound RP in the TA7 state fits well into the p28-free RP density except for the p28 highlighted in red. The p28 model is completely out of the cryo-EM density.
(C) A schematic illustrating the hypothetical CP-independent assembly pathway of RP prior to the 26S formation, which is compatible with our structural data. However, this hypothesis is subject to further tests and does not exclude alternative RP assembly pathways (Park et al., 2009; Roelofs et al., 2009).
Discussion
To ensure a correct assembly of the heterohexameric AAA-ATPase, several assembly chaperones are involved in the RP assembly process to regulate the assembly sequence or rectify mis-assembled complexes (Funakoshi et al., 2009; Hanssum et al., 2014; Kaneko et al., 2009; Li et al., 2017; Park et al., 2013; Park et al., 2009; Roelofs et al., 2009; Saeki et al., 2009; Thompson et al., 2009). The structures of the T1-3 subassemblies characterize an overall conformational landscape along the proteasome assembly pathway, hypothetically representing states of the ATPase hexamerization during its last step prior to the completion of holoenzyme assembly. In the absence of interactions with CP, the ATPase ring is unable to assume a stable rigid structure but instead samples a variety of conformations. Its conformational landscape was captured by seven microstates TA1-7 in the Rpn1-p28-AAA subcomplex. The approximately equal population of the seven states of the Rpn1-p28-AAA subcomplex suggests that each state may be transiently sampled by the free RP along the trajectory traversing the frustrated energy landscape of the complex (Wensley et al., 2010). This is consistent with the fact that they assemble into consecutive snapshots recapitulating an open-to-closed transition of the AAA ring. Our analysis suggests that the TA7 state of the AAA ring may facilitate RP-CP association and subsequent p28 release in a shoehorn-like fashion. This is supported by the structural similarity between the p28-bound RP in the TA7 state and the p28-free RP. By contrast, the TA1-6 states all demonstrated prominent clashes between p28 and the α-ring in the CP, suggesting that p28 inhibited the assembly of RP with CP through the TA1-6 states (Figure S5). Thus, p28 reconciles the remarkable structural differences in the RP before and after proteasome assembly, with guiding the CP to choose a specific conformation of the ATPase ring for RP engagement, which completes the last step in the chaperone-mediated proteasome assembly. This picture is also consistent with a recent study showing the nucleotide-dependent switch in yeast proteasome assembly mediated by Nas6 (Li et al., 2017).
A prominent feature of the free RP is the blocking of substrate-entry port by Rpn11, which renders the OB ring incompetent in accepting substrates. Moreover, the AAA channel is not yet formed in the free RP and undergoes large conformational changes that spontaneously sample both open and closed ring topologies (Figure 4). The ATP-independent deubiquitylating activity of the free RP is detected in the absence of complete substrate unfolding in a fluorescent unfolding assay (Figure 6). Unfolded proteins, in the absence of degradation, could form aggregates impairing numerous cellular functions. The ‘closed’ OB-ring and ‘open’ AAA-ring in the free RP minimizes this risk prior to its incorporation into the 26S proteasome holoenzyme.
The overall conformation of the non-AAA components is virtually identical to that in the SB, SC and SD states of the human proteasome holoenzyme (Figures S4A and S4B) and resembles the yeast counterpart in the s2, s3 and substrate-engaged states (Chen et al., 2016; Luan et al., 2016; Matyskiela et al., 2013; Sledz et al., 2013; Unverdorben et al., 2014). This suggests that the non-AAA structure in the free RP could represent a ground state of this subcomplex. Indeed, the total buried interfacial area of the non-AAA subcomplex is about 900-Å2 larger in the free RP than in the proteasome holoenzyme in the SA state (Chen et al., 2016). The non-AAA conformation of the proteasome in the SA state is of higher potential energy and is stabilized by an additional interaction between Rpn5/Rpn6 and the α-ring of the CP, which buries ∼600-Å2 of interfacial area (Chen et al., 2016; Huang et al., 2016; Schweitzer et al., 2016). Taken together, our data suggest that the non-AAA subcomplex conformation switches mainly between its ground state and its higher-energy state during and after proteasome assembly (Chen et al., 2016; Luan et al., 2016; Matyskiela et al., 2013; Sledz et al., 2013; Unverdorben et al., 2014). The energetic proximity of the two states in the non-AAA subcomplex poises the RP adjacent to a ‘critical’ point that facilitates allosteric regulation of proteasome activation and function (Chen et al., 2016).
The purified human p28-bound RP can assemble with the purified human CP in vitro to reconstitute functional 26S holoenzymes. This observation verifies that the presence of p28 in the free RP does not only serve to antagonize the assembly of RP with CP to form the holoenzyme. Rather, p28 serves as a conformation-selection chaperone to guide RP-CP association along a specific pathway (Figure 6C). In the absence of p28, the free RP exhibited a high propensity of physical aggregation, which would impair the intracellular assembly of proteasome holoenzymes. The presence of p28 might have prevented the free RP from detrimental off-pathway aggregation. Thus, p28 might help maintain a reservoir of free RP assembly in cells, which could be essential for dynamically regulating the intracellular abundance level of fully assembled proteasome holoenzyme (Hanssum et al., 2014). Finally, our data suggest the possibility of a CP-independent RP assembly pathway that awaits further investigation (Figure 6C).
STAR*METHODS
Contact For Reagent And Resource Sharing
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Youdong Mao (youdong_mao@dfci.harvard.edu).
Experimental Model And Subject Details
Cell lines
HEK293 cells stably expressing RPN11-HTBH (His-Tev-Biotinylation-His) (Wang et al., 2007) were cultured in DMEM containing 10% FBS (fetal bovine serum) and 1% Penicillin-Streptomycin under 37 °C with 5% CO2. At least 1 L monolayer cells cultured to a full confluence were harvested for proteasome RP purification with ∼30 mL medium of 150 × 25 mm plates.
Method Details
Protein expression and purification
Human RP was affinity-purified on a large scale from a stable HEK293 cell line harboring HTBH tagged hRPN11 (a gift from L. Huang, Departments of Physiology and Biophysics and of Developmental and Cell Biology, University of California, Irvine, California 92697) (Wang et al., 2007). The cells were Dounce-homogenized in a lysis buffer (50 mM NaH2PO4 [pH7.5], 100 mM NaCl, 10% glycerol, 5 mM MgCl2, 0.5% NP-40, 5 mM ATP and 1 mM DTT) containing protease inhibitors. Lysates were clarified, then incubated with the NeutrAvidin agarose resin (Thermo Scientific) overnight at 4°C. The beads were then washed with excess lysis buffer followed by the wash buffer (50 mM Tris-HCl [pH7.5], 1 mM MgCl2 and 1 mM ATP). Usp14 was removed from the proteasomes using the wash buffer including 150 mM NaCl for 30 min (Lee et al., 2010). Human RP was eluted from the beads by cleavage, using TEV protease (Invitrogen), and was further purified by gel filtration, using a Superose 6 10/300 GL column at a flow-rate of 0.15 ml/minute in buffer (30 mM Hepes pH7.5, 60 mM NaCl, 1 mM MgCl2, 10% Glycerol, 0.5 mM DTT, 0.8 mM ATP). Gel-filtration fractions were concentrated to about 2 mg/ml. Right before cyro-EM sample preparation, the RP sample was buffer-exchanged into 50 mM Tris-HCl [pH7.5], 1 mM MgCl2, 3 mM ATP, 0.5 mM TCEP, to remove glycerol, and was supplemented with 0.005% NP-40.
Yeast RP and 26S proteasome were purified as previously described from a ubp6Δ strain (Leggett et al., 2005). Briefly, cells were grown to an OD600 of ∼7, then harvested. The cell pellet was washed in PBS once, resuspended in the lysis buffer (50 mM Tris-HCl [pH 8.0], 5 mM MgCl2, 1 mM EDTA, 1 mM ATP), and supplemented with a protease inhibitor cocktail. Cells were then lysed by passing the cell suspension through a French press at a pressure of 20,000 psi. Clarified yeast lysate was incubated with IgG resin for one hour at 4 °C. After incubation, the resin was washed with 100 bed volumes of the wash buffer (50 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 100 mM NaCl, 1 mM EDTA, 1 mM ATP), followed by an equilibration with 20 bed volumes of elution buffer (50 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 1 mM EDTA, 1 mM ATP, 10% glycerol). TEV cleavage was carried out by incubating resin with 2 bed volumes of elution buffer supplemented with 3 units of AcTEV protease, at 30 °C for 1 hour. The cleaved fraction was then collected and the resin was further washed with 5 bed volumes of elution buffer. All eluted fractions were pooled and passed through equilibrated Ni-NTA resin to remove the AcTEV protease. Proteasomes were then concentrated by ultrafiltration using devices with a 30-kDa molecular weight cut-off. Yeast RP was purified similarly, except that 500 mM NaCl was used during the washes.
Deubiquitylation and degradation assays
About 500 nM ubiquitylated pySic1-iRFP was mixed with purified RP or 26S proteasome in a buffer (50 mM Tris-HCL [pH 7.5], 50 mM NaCl, 5 mM MgCl2, 0.5 mM DTT, 0.2 mg/ml BSA) supplemented with 3 mM ATP or other nucleotides, incubated at room temperature. pySic1-iRFP was affinity purified using a Ni-NTA column after ubiquitylation.
ATPase activity assay
ATP hydrolysis was measured through the detection of phosphate by Malachite Green. 500 μM ATP (in 20 mM HEPES, pH 7.3, 5 mM MgCl2, 1 mM DTT) was incubated with 3 μg of 26S or 2 μg of RP at 37 °C in total volume of 50 μL for 2 h and 1 h, respectively. Then hydrolysis solution was mixed with 400 μL Malachite Green solution (of 0.034% Malachite Green, 1.1% ammonium molybdate, 1 M HCl, and 0.04% Triton X-100) for a 1 min and then 50 μL 34% citric acid was added. After 5 min at room temperature, absorbance was measured at 660 nm.
In vitro 26S assembly assay
The in vitro assembly of purified human RP and CP was conducted as described. 0.5-1.5 μg RP and 0.5-2.5 μg CP were mixed in a buffer containing 50 mM Tris (pH 8), 10 mM DTT, 500 μM ATP and 10 mM MgCl2. The final volume was 10μL. The mixture was incubated under 37 °C for 45 min for RP-CP in vitro assembly and then were characterized by native gel electrophoresis. As shown in Figure S1Q-S1S, after the reconstitution, the band corresponding to RP disappeared; and new bands were observed, which correspond to singly capped and doubly capped 26S proteasome. The dissociation of p28 during the holoenzyme reconstitution was monitored by Western blot of native gel using an anti-p28 antibody.
Fluorescent unfolding assay
We used iRFP as a fluorescence reporter to investigate whether translocation occurs during the RP-catalyzed deubiquitylation (Shcherbakova and Verkhusha, 2013). If the Sic1-fused iRFP is unfolded and threaded through the ATPase channel, a loss of fluorescent signal will ensue. To prevent refolding of iRFP, we employed a mutated protein-folding chaperone, GroEL, to entrap unfolded peptides (Weber-Ban et al., 1999). 500 nM purified, ubiquitylated pySic1-iRFP substrate was mixed with 20 nM RP or recombinant ClpA in a buffer (50 mM Hepes [pH7.5], 20 mM MgCl2, 10 mM ATP, 150 mM NaCl, 10% glycerol, 0.5 mM DTT), in the presence or absence of GroEL trap at 500 nM, continuously monitored in a Cary Eclipse fluorescence spectrophotometer (ex680/em715) at room temperature. In the fluorescent degradation assay, 20 nM human 26S proteasome was employed instead of RP. The signal was normalized by the initial fluorescence intensity.
Cryo-EM data collection
A 2.5-μl drop of 3 mg/ml RP solution was applied to a glow-discharged C-flat grid (R 1/1, 400 Mesh, Protochips, CA, USA), blotted for 2 s at 4 °C and 100% humidity, then plunged into liquid ethane and flash frozen using the FEI Vitrobot Mark IV. The cryo-grid was imaged in an FEI Tecnai Arctica microscope, equipped with an Autoloader, at a nominal magnification of 54,000 times and an acceleration voltage of 200 kV. Coma-free alignment was manually conducted prior to data collection. Cryo-EM data were collected semi-automatically by the Leginon version 3.1 (Suloway et al., 2005) on the Gatan K2 Summit direct detector camera (Gatan Inc., CA, USA) in a counting mode, with 9.0 s of total exposure time and 250 ms per frame. This resulted in movies of 36 frames per exposure and an accumulated dose of 50 electrons/Å2. The calibrated physical pixel size is 0.98 Å. The defocus in data collection was set in the range of from -1.0 to -3.0 μm. A total of 20,000 movies were collected, among which 16,111 movies were selected for further data analysis.
Cryo-EM data processing and reconstruction
The raw 36 frames in each movie file were first corrected for their gain reference and was used to generate a micrograph corrected for sample movement and drift (Li et al., 2013). These drift-corrected micrographs were used for the determination of actual defocus with the CTFFind3 program (Mindell and Grigorieff, 2003). Initial candidate particles were automatically picked in SPIDER by a template-matching approach (Shaikh et al., 2008). 250,251 particles were manually checked from 16,111 micrographs in EMAN2 (Tang et al., 2007) before extracting them for structure determination.
The initial model was generated by EMAN2. A subset of 19,182 particles from one of the data sets was used for reference-free classification performed by e2refine2d.py, generating 80 classes. 42 classes were selected for the generation of an initial model using e2initialmodel.py. In the following steps, reference-free 2D classification was performed in ROME (Wu et al., 2016), whereas unsupervised 3D classification was performed in RELION 1.3 (Scheres, 2012b). After the first round of reference-free 2D classification using all 250,251 particles binned to a pixel size of 4.00 Å, bad particles and non-RP class averages were rejected as a whole class upon inspection of class average quality. We selected class averages for free RP and manually removed non-RP class averages. 211,417 particles were selected. The initial model, low-pass filtered to 60 Å, was used as an input reference to conduct unsupervised 3D classification into six classes without assumption of any symmetry, using particles binned to a pixel size of 2.00 Å separately, with an angular sampling of 7.5 and a regularization parameter T of 4.0. This resulted in several classes with different conformations. After two rounds of 3D classification, we got 3 classes containing 13,620, 39,520 and 64,331 particles with different conformations, which had a common rigid part of the non-AAA subcomplex but diverse conformations in Rpn1 and AAA domains of the ATPase subunits. We named 39,520 particles dataset as the T1 dataset, which gave rise to reduced densities in the N-terminal regions of Rpn12 and Rpn3. Two rounds of 3D classification were applied on the 64,331-particle dataset, and yielded two classes named the T2 dataset (27,629 particles) and the T3 dataset (10,891 particles). The T1-T3 datasets were then subject to the refinement separately, and yielded the gold-standard FSC resolution of 5.7, 6.1 and 6.8 Å. To improve the resolution of the non-AAA component of the p28-RP structure, we performed a refinement by combining the three 3D classes of 13,620, 39,520 and 64,331 particles (in total of 117,471 particles), which all showed the similar conformations in the non-AAA structure. Based on the refinement of the combined 117,471 particles, we re-centered the particles and decreased the box size of particles from 3202 to 2562. These particles were subsequently used for particle-based motion correction in RELION (hereafter referred to as “particle polishing”). For these calculations, we only used the 2nd to 20th frames of each micrograph with running averages of five frames and a standard deviation of 1 pixel for translational alignment. These polished particles gave a reconstruction with a nominal resolution of 4.5 Å in the non-AAA subcomplex of the p28-RP.
When the single-particle images were classified in 3D based on the whole RP conformation, p28-Rpt3-Rpt6 densities always appeared to be averaged out to a weaker level of densities regardless how each 3D classes were sorted, most likely because the motion of p28-Rpt3-Rpt6 heterotrimer is convoluted with the overall motion of AAA hexameric ring. To improve the resolution of Rpt3 and Rpt6, we used a density subtraction strategy following a projection-matching procedure. A local mask (including the non-AAA subcomplex) was added on auto-refinement of 103,851 polished particles (combining 64,331 and 39,520 particles), which converged at 4.5 Å resolution measured by a gold-standard FSC of 0.143 cutoff. In the last iteration of auto-refinement, we extracted the maximum probability orientation for each particle. The refined 4.5-Å map was used for density subtraction. First, we used SPIDER command “mmc” to apply a mask of the rigid part to two half unfiltered maps. We segmented the rigid part from these two unfiltered maps that correspond to the non-AAA subcomplex and used each as the input map for density subtraction. With this input map and corresponding STAR file, we executed RELION 1.3 command relion_project to subtract projections of the non-AAA subcomplex structure out of the raw particle images, leaving only flexible parts (AAA domains of Rpt1, Rpt2, Rpt3, Rpt4, Rpt5 and Rpt6; p28; and Rpn1). We segmented the Rpt AAA domains, Rpn1 and p28 components out of the 4.5-Å map and used it for the initial model in the following 3D classification. After two rounds of 3D classification, we got seven classes with 12,223, 11,082, 21,031, 9,004, 11,767, 11,610 and 14,464 particles, named TA1, TA2, TA3, TA4, TA5, TA6, and TA7, respectively. Auto-refinement of each class was performed separately, resulting in reconstructions of Rpn1-p28-AAA subcomplex in TA1, TA2, TA3, TA4, TA5, TA6, and TA7 at 8.5, 8.3, 7.3, 8.3, 7.9, 8.9, and 7.7 Å, respectively. Reported resolutions were based on the gold-standard FSC 0.143 criterion, and FSC curves were corrected for the effects of a soft mask. Prior to visualization, all density maps were corrected for the modulation transfer function (MTF) of the detector, and then sharpened by applying a negative B-factor that was estimated using automated procedures (Tang et al., 2007). Local resolution variations were estimated using ResMap (Kucukelbir et al., 2014).
Cryo-EM analysis of the p28-free RP
To determine a structure of the free RP without p28, a total of 5,645 movies collected on purified human 26S proteasome holoenzyme were analyzed. This was a subset of data used for determining the 26S proteasome structures reported recently (Chen et al., 2016). Cryo-EM data were collected semi-automatically by the Leginon version 3.1 on the Gatan K2 Summit direct detector camera (Gatan Inc., CA) in a super-resolution counting mode, with 6.0 s of total exposure time and 200 ms per frame, on a 200 kV FEI Tecnai Arctica at a nominal magnification of 21,000 times. This resulted in movies of 30 frames per exposure and an accumulated dose of 30 electrons/Å2. The calibrated physical pixel size and the super-resolution pixel size are 1.72 Å and 0.86 Å, respectively. The defocus in data collection was set in the range of -1.0 to -3.0 μm.
The raw movie frames were first corrected for their gain reference. Each movie was used to generate a micrograph that was corrected for sample movement and drift with the MotionCorr program (Li et al., 2013). These drift-corrected micrographs were used for the determination of actual defocus of each micrograph with the CTFFind3 program (Mindell and Grigorieff, 2003). 259,459 particles were manually checked from 5,645 micrographs in EMAN2 before extracting them for structure determination. After using all 259,459 particles binned to a pixel size of 3.44 Å for the first round of reference-free 2D classification in ROME, bad particles and non-RP class averages were rejected based on a whole class upon inspection of class average quality. We selected class averages for free RP and manually removed non-RP from the free RP class averages. 191,643 particles were selected. The initial model, low-pass filtered to 60 Å, was used as the input reference to conduct unsupervised 3D classification into four classes without assumption of any symmetry, with the particles binned to a pixel size of 1.72Å, using an angular sampling of 7.5 and a regularization parameter T of 4.0. After 3D classification, followed by 2D classification, were repeated twice, we got a dataset of 84,121 particles whose reconstruction highly resembled the T1 state. Based on the auto-refinement of the 84,121 particles, we re-centered the particles and decreased the box size of particles from 3602 to 3002. These particles gave a reconstruction with an overall resolution of 6.8 Å, in which the p28 density is very weak as compared to other subunits. Thus, we combined the TA1 and TA7 states, along with the T1 state, which served as two new initial models. The two initial models, low-pass filtered to 20 Å, were used as the input references to conduct supervised 3D classification into two classes without assumption of any symmetry, using an angular sampling of 7.5 and a regularization parameter T of 4.0. 3D classification applied to these 84,121 particles was repeated twice, and two classes including 21,885 and 19,661 particles were obtained. The two datasets then were subjected to refinement separately, and both yielded the gold-standard FSC resolution of 8.8 Å. The 21,885-particle dataset exhibits a p28-free RP structure and 19,661-particle dataset exhibits a p28-bound RP structure (see Figure S6).
Atomic model building and refinement
Direct rigid-body fitting of crystal structures of yeast Rpn2, Rpn6, Rpn9, Rpn10 and Rpn12 monomers and Rpn11-Rpn8 dimer or into our cryo-EM map suggests an excellent agreement in the secondary structural elements between our cryo-EM structure and the crystal structures. The initial atomic modelling of non-AAA subcomplex in Coot (Emsley and Cowtan, 2004) was based on the atomic structure of human 26S proteasome (Chen et al., 2016). Refinement was first carried out on the Rpn subunits and six Rpt subunits in real space using Phenix (Adams et al., 2010) with secondary structure and geometry restraints to prevent over-fitting. Each part of cryo-EM map was segmented in Chimera. The cryo-EM map and the atomic model were placed into a pseudo-unit cell and the refinement was performed in Phenix in Fourier space using both amplitudes and phases. The final refinement statistics are shown in Table 1. The FSC curve between the map and the final refined atomic model gave an estimated resolution of 4.7 Å at FSC = 0.5. Overfitting of the overall model was monitored by refining the model in one of the two independent maps from the gold-standard refinement approach, and testing the refined model against the other map.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-PAAF1 rabbit polyclonal antibody | Abcam | Cat#ab103566 |
| Anti-p28 rabbit monoclonal antibody | Abcam | Cat#ab182576 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NaH2PO4 | Sigma-Aldrich | Cat#S8282; CAS: 7558-80-7 |
| Na2HPO4 | Sigma-Aldrich | Cat#431478; CAS: 7782-85-6 |
| NaCl | Sigma-Aldrich | Cat#S7653; CAS: 7647-14-5 |
| MgCl2 | Sigma-Aldrich | Cat#M8266; CAS: 7786-30-3 |
| IGEPAL® CA-630 (NP-40) | Sigma-Aldrich | Cat#I3021; CAS: 9002-93-1 |
| ATP disodium salt | Sigma-Aldrich | Cat#A2383; CAS: 34369-07-8 |
| DTT | Sigma-Aldrich | Cat#D0632; CAS: 3483-12-3 |
| TEV protease | Sigma-Aldrich | Cat#T4455 |
| HCl | Sigma-Aldrich | Cat#H1758; CAS: 7647-01-0 |
| Tris base | Sigma-Aldrich | Cat#T1503; CAS:77-86-1 |
| HEPES | Sigma-Aldrich | Cat#H3375; CAS: 7365-45-9 |
| TCEP | Sigma-Aldrich | Cat#C4706; CAS: 51805-45-9 |
| EDTA disodium salt dihydrate | Sigma-Aldrich | Cat#E4884; CAS: 6381-92-6 |
| Glycerol | Sigma-Aldrich | Cat#G9012; CAS: 56-81-5 |
| Malachite Green | Sigma-Aldrich | Cat#HT8028; CAS: |
| Ammonium molybdate | Sigma-Aldrich | Cat#A1343; CAS: 12054-85-2 |
| TWEEN® 20 | Sigma-Aldrich | Cat#P9416; CAS: 9005-64-5 |
| Triton X-100 | Sigma-Aldrich | Cat# X100; CAS: 9002-93-1 |
| AcTEV protease | Thermo Fisher Scientific | Cat# 12575015 |
| DMEM | Thermo Fisher Scientific | Cat# 11965084 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15070063 |
| PBS | Corning | Cat#21-040-CV |
| Fetal Bovine Serum | TaKaRa | Cat#631106 |
| pySic1-iRFP | This paper | N/A |
| ClpA | TWeber-Ban et al., 1999 | N/A |
| GroEL | TWeber-Ban et al., 1999 | N/A |
| Critical Commercial Assays | ||
| Pierce™ NeutrAvidin™ Agarose | Thermo Fisher Scientific | Cat# 29200 |
| Superose 6 10/300 GL column | GE Life Science | Cat# 17517201 |
| NI-NTA AGAROSE | QIAGEN | Cat#30210 |
| IgG resin | GE Life Science | Cat#17-0969-01 |
| Deposited Data | ||
| CryoEM map of the non-AAA structure of free RP | This paper | EMBD: EMD-8672 |
| Atomic model of the non-AAA structure of free RP | This paper | PDB: 5VGZ |
| CryoEM map of the p28-bound RP structure in T1 | This paper | EMBD: EMD-8674 |
| Pseudo-atomic model of the p28-bound RP structure in T1 | This paper | PDB: 5VHF |
| CryoEM map of the p28-bound RP structure in T2 | This paper | EMBD: EMD-8675 |
| Pseudo-atomic model of the p28-bound RP structure in T2 | This paper | PDB: 5VHH |
| CryoEM map of the p28-bound structure of RP in T3 | This paper | EMBD: EMD-8676 |
| Pseudo-atomic model of the p28-bound RP structure in T3 | This paper | PDB: 5VHI |
| CryoEM map of the Rpn1-p28-AAA subcomplex in TA1 | This paper | EMBD: EMD-8677 |
| Pseudo-atomic model of the Rpn1-p28-AAA subcomplex in TA1 | This paper | PDB: 5VHJ |
| CryoEM map of the Rpn1-p28-AAA subcomplex in TA2 | This paper | EMBD: EMD-8678 |
| Pseudo-atomic model of the Rpn1-p28-AAA subcomplex in TA2 | This paper | PDB: 5VHM |
| CryoEM map of the Rpn1-p28-AAA subcomplex in TA3 | This paper | EMBD: EMD-8679 |
| Pseudo-atomic model of the Rpn1-p28-AAA subcomplex in TA3 | This paper | PDB: 5VHN |
| CryoEM map of the Rpn1-p28-AAA subcomplex in TA4 | This paper | EMBD: EMD-8680 |
| Pseudo-atomic model of the Rpn1-p28-AAA subcomplex in TA4 | This paper | PDB: 5VHO |
| CryoEM map of the Rpn1-p28-AAA subcomplex in TA5 | This paper | EMBD: EMD-8681 |
| Pseudo-atomic model of the Rpn1-p28-AAA subcomplex in TA5 | This paper | PDB: 5VHP |
| CryoEM map of the Rpn1-p28-AAA subcomplex in TA6 | This paper | EMBD: EMD-8682 |
| Pseudo-atomic model of the Rpn1-p28-AAA subcomplex in TA6 | This paper | PDB: 5VHQ |
| CryoEM map of the Rpn1-p28-AAA subcomplex in TA7 | This paper | EMBD: EMD-8683 |
| Pseudo-atomic model of the Rpn1-p28-AAA subcomplex in TA7 | This paper | PDB: 5VHR |
| Cryo-EM map of the p28-free RP structure | This paper | EMBD: EMD-8684 |
| Pseudo-atomic model of the p28-free RP structure | This paper | PDB: 5VHS |
| The raw micrographs and particle data | This paper | EMPIAR-10091 |
| The original gel images | This paper | http://dx.doi.org/10.17632/jzx75c6vbs.1 |
| Experimental Models: Cell Lines | ||
| HEK293 harboring HTBH tagged hRPN11 | Wang et al., 2007 | N/A |
| Software and Algorithms | ||
| Leginon | Suloway et al., 2005 | http://emg.nysbc.org/redmine/projects/leginon/wiki/Leginon_Homepage |
| MotionCor | Li et al., 2013 | http://cryoem.ucsf.edu/software/driftcorr.html |
| CTFFind3 | Mindell and Grigorieff, 2003 | http://grigoriefflab.janelia.org/ctf |
| SPIDER | Shaikh et al., 2008 | https://spider.wadsworth.org/ |
| EMAN2 | Tang et al., 2007 | http://blake.bcm.edu/emanwiki/EMAN2 |
| ROME | Wu et al., 2016 | http://ipccsb.dfci.harvard.edu/rome |
| RELION | Scheres, 2012b | http://www2.mrc-lmb.cam.ac.uk/relion |
| Bfactor | Grigorieff et al. | http://grigoriefflab.janelia.org/grigorieff/download_b.html |
| ResMap | Kucukelbir et al., 2014 | http://resmap.sourceforge.net |
| Coot | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org |
| Chimera | Pettersen et al., 2004 | http://www.cgl.ucsf.edu/chimera |
| PISA | Krissinel and Henrick, 2007 | http://www.ebi.ac.uk/pdbe/pisa/ |
| Pajek | De Nooy, et al., 2011 | http://mrvar.fdv.uni-lj.si/pajek/ |
| Matlab | The MathWorks Inc., Natick, MA, 2000 | https://www.mathworks.com |
| Other | ||
| C-flat grid | Protochips | R1/1 400 mesh |
Pseudo-atomic models of T1 –T3, except the AAA domain, Rpn1 and p28, were based on the atomic model of non-AAA subcomplex. Pseudo-atomic models of Rpn1-p28-AAA subcomplex (TA1-TA7) were based on the atomic structure of human 26S proteasome (Chen et al., 2016) and the crystal structure of human gankyrin (Krzywda et al., 2004) (PDB accession code: 1UOH). These structures were fitted into density by Chimera. The models were manually adjusted and flexibly fitted in Coot (Emsley and Cowtan, 2004), and energetically minimized using Phenix with secondary structure and geometry restraints to prevent over-fitting.
Structural analysis and visualization
Solvation energy and interaction surface for each model were calculated using PISA (Krissinel and Henrick, 2007). Total interaction surface or free energy was calculated by summing up the contributions from each interaction pair. Bias due to truncation of the termini of some subunits was compensated after the calculation. Subunit-subunit interaction networks or transition networks were visualized using Pajek (Nooy et al., 2011). Numeric processing was carried out in Matlab (The MathWorks Inc., Natick, MA). The scripts are available upon request.
Quantification and Statistical Analysis
The estimations of cryo-EM resolutions are based on the FSC cutoff at 0.143 computed between two half maps refined independently (Scheres and Chen, 2012).
Data and Software Availability
The single-particle reconstructions and atomic coordinates reported in this paper have been deposited in the Electron Microscopy Data Bank, www.emdatabank.org (accession nos. EMD-8672 for the non-AAA structure of free RP at high resolution, EMD-8674, EMD-8675, EMD-8676 for the p28-bound structure of RP in T1, T2, T3; EMD-8677, EMD-8678, EMD-8679, EMD-8680, EMD-8681, EMD-8682, EMD-8683 for the Rpn1-p28-AAA subcomplex in TA1, TA2, TA3, TA4, TA5, TA6 and TA7, respectively; and EMD-8684 for the p28-free RP structure) and Protein Data Bank, www.wwpdb.org (PDB ID codes 5VGZ the non-AAA structure of free RP at high resolution, 5VHF, 5VHH, 5VHI for the p28-bound structure of RP in T1, T2, T3; 5VHJ, 5VHM, 5VHN, 5VHO, 5VHP, 5VHQ, 5VHR for the Rpn1-p28-AAA subcomplex in TA1, TA2, TA3, TA4, TA5, TA6 and TA7, respectively; and 5VHS for the p28-free RP structure). The raw micrographs and particle data have been deposited in the Electron Microscopy Pilot Image Archive, www.ebi.ad.uk/emdb/ampiar (accession no. EMPIAR-10091). Original gel images have been deposited to Mendeley Data, http://dx.doi.org/10.17632/jzx75c6vbs.1.
Supplementary Material
Highlights.
Cryo-EM structures of p28-bound human proteasome regulatory particles (RPs)
The AAA ring in the p28-bound RPs samples seven conformations
The assembly chaperone p28 guides the core particle to select a specific RP conformation
p28 is released in a shoehorn-like fashion during proteasome assembly
Acknowledgments
The authors thank J. Jackson and T. Song for assistance in maintaining the high-performance computing system; D. Yu for assistance in drift correction; Dr. Horwich for a gift of the ClpA and GroEL constructs. This work was funded in part by a grant from NIGMS, GM02675 (M.K.), by a grant of the Thousand Talents Plan of China (Y.M.), by an Intel academic grant (Y.M.), by the research funds at Peking-Tsinghua Center for Life Science at Peking University (Y.M., Q.O.), by a grant from National Natural Science Foundation of China 91530321 (Y.M., Q.O.), and by the NIH grant R01 GM095526 (D.F.). The cryo-EM experiments were performed in part at the Center for Nanoscale Systems at Harvard University, a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award no. 1541959. The cryo-EM facility was partly funded through the NIH grant AI100645, Center for HIV/AIDS Vaccine Immunology and Immunogen Design (CHAVI-ID). The data processing was performed in part in the Sullivan supercomputer, which is funded in part by a gift from Mr. and Mrs. Daniel J. Sullivan, Jr.
Footnotes
Supplemental Information: Supplemental Information includes six figures, two tables, and can be found with this article online.
Author Contributions: Y.L. and Y.M. conceived this study. Y.L., Y.D. and S.S. purified proteasome regulatory particles and performed biochemical experiments with advices from M.W.K., Y.M. and D.F., respectively. Y.L. prepared the specimen for data collection and developed the unfolding assay. Y.M. collected cryo-electron microscopy data. S.C. performed initial reconstruction. Y.L., J.W. and Y.M. designed computational strategy for data processing. J.W., Y.L. and Y.B.M. processed the data and refined the cryo-EM maps. Y.L. and Y.M. built the initial atomic model. J.W., S.C. and Y.M. refined the atomic model for the near-atomic resolution structure and built the pseudo-atomic models for the medium-resolution structures. J.W., Y.D. and S.C. contributed to the preparation of Supplemental Information. Y.M. supervised the structural determination and wrote the manuscript with input from Y.L., D.F., Q.O. and M.W.K.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck F, Unverdorben P, Bohn S, Schweitzer A, Pfeifer G, Sakata E, Nickell S, Plitzko JM, Villa E, Baumeister W, et al. Near-atomic resolution structural model of the yeast 26S proteasome. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14870–14875. doi: 10.1073/pnas.1213333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wu J, Lu Y, Ma YB, Lee BH, Yu Z, Ouyang Q, Finley DJ, Kirschner MW, Mao Y. Structural basis for dynamic regulation of the human 26S proteasome. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:12991–12996. doi: 10.1073/pnas.1614614113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Fonseca PC, He J, Morris EP. Molecular model of the human 26S proteasome. Mol Cell. 2012;46:54–66. doi: 10.1016/j.molcel.2012.03.026. [DOI] [PubMed] [Google Scholar]
- Dambacher CM, Worden EJ, Herzik MA, Martin A, Lander GC. Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. Elife. 2016;5 doi: 10.7554/eLife.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annual review of biochemistry. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi M, Tomko RJ, Jr, Kobayashi H, Hochstrasser M. Multiple assembly chaperones govern biogenesis of the proteasome regulatory particle base. Cell. 2009;137:887–899. doi: 10.1016/j.cell.2009.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998;94:615–623. doi: 10.1016/s0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- Guerrero C, Tagwerker C, Kaiser P, Huang L. An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Molecular & cellular proteomics: MCP. 2006;5:366–378. doi: 10.1074/mcp.M500303-MCP200. [DOI] [PubMed] [Google Scholar]
- Hanssum A, Zhong Z, Rousseau A, Krzyzosiak A, Sigurdardottir A, Bertolotti A. An inducible chaperone adapts proteasome assembly to stress. Mol Cell. 2014;55:566–577. doi: 10.1016/j.molcel.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman L, Rechsteiner M. Nucleotidase activities of the 26 S proteasome and its regulatory complex. J Biol Chem. 1996;271:32538–32545. doi: 10.1074/jbc.271.51.32538. [DOI] [PubMed] [Google Scholar]
- Huang X, Ruan B, Wu J, Shi Y. An atomic structure of the human 26S proteasome. Nature Structural & Molecular Biology. 2016 doi: 10.1038/nsmb.3273. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Hamazaki J, Iemura S, Sasaki K, Furuyama K, Natsume T, Tanaka K, Murata S. Assembly pathway of the Mammalian proteasome base subcomplex is mediated by multiple specific chaperones. Cell. 2009;137:914–925. doi: 10.1016/j.cell.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Krzywda S, Brzozowski AM, Higashitsuji H, Fujita J, Welchman R, Dawson S, Mayer RJ, Wilkinson AJ. The crystal structure of gankyrin, an oncoprotein found in complexes with cyclin-dependent kinase 4, a 19 S proteasomal ATPase regulator, and the tumor suppressors Rb and p53. J Biol Chem. 2004;279:1541–1545. doi: 10.1074/jbc.M310265200. [DOI] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett DS, Glickman MH, Finley D. Purification of proteasomes, proteasome subcomplexes, and proteasome-associated proteins from budding yeast. Methods Mol Biol. 2005;301:57–70. doi: 10.1385/1-59259-895-1:057. [DOI] [PubMed] [Google Scholar]
- Li F, Tian G, Langager D, Sokolova V, Finley D, Park S. Nucleotide-dependent switch in proteasome assembly mediated by the Nas6 chaperone. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:1548–1553. doi: 10.1073/pnas.1612922114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan B, Huang X, Wu J, Mei Z, Wang Y, Xue X, Yan C, Wang J, Finley DJ, Shi Y, et al. Structure of an endogenous yeast 26S proteasome reveals two major conformational states. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:2642–2647. doi: 10.1073/pnas.1601561113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela ME, Lander GC, Martin A. Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol. 2013;20:781–788. doi: 10.1038/nsmb.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Nakano K, Umehara T, Kimura M, Hayashizaki Y, Tanaka A, Horikoshi M, Padmanabhan B, Yokoyama S. Structure of the oncoprotein gankyrin in complex with S6 ATPase of the 26S proteasome. Structure. 2007a;15:179–189. doi: 10.1016/j.str.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Umehara T, Tanaka A, Horikoshi M, Padmanabhan B, Yokoyama S. Structural basis for the recognition between the regulatory particles Nas6 and Rpt3 of the yeast 26S proteasome. Biochem Biophys Res Commun. 2007b;359:503–509. doi: 10.1016/j.bbrc.2007.05.138. [DOI] [PubMed] [Google Scholar]
- Nooy Wd, Mrvar A, Batagelj V. Exploratory social network analysis with Pajek Rev and expanded 2nd. England; New York: Cambridge University Press; 2011. [Google Scholar]
- Park S, Li X, Kim HM, Singh CR, Tian G, Hoyt MA, Lovell S, Battaile KP, Zolkiewski M, Coffino P, et al. Reconfiguration of the proteasome during chaperone-mediated assembly. Nature. 2013;497:512–516. doi: 10.1038/nature12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Roelofs J, Kim W, Robert J, Schmidt M, Gygi SP, Finley D. Hexameric assembly of the proteasomal ATPases is templated through their C termini. Nature. 2009;459:866–870. doi: 10.1038/nature08065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Tian G, Roelofs J, Finley D. Assembly manual for the proteasome regulatory particle: the first draft. Biochem Soc Trans. 2010;38:6–13. doi: 10.1042/BST0380006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathare GR, Nagy I, Sledz P, Anderson DJ, Zhou HJ, Pardon E, Steyaert J, Forster F, Bracher A, Baumeister W. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:2984–2989. doi: 10.1073/pnas.1400546111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Roelofs J, Park S, Haas W, Tian G, McAllister FE, Huo Y, Lee BH, Zhang F, Shi Y, Gygi SP, et al. Chaperone-mediated pathway of proteasome regulatory particle assembly. Nature. 2009;459:861–865. doi: 10.1038/nature08063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki Y, Toh EA, Kudo T, Kawamura H, Tanaka K. Multiple proteasome-interacting proteins assist the assembly of the yeast 19S regulatory particle. Cell. 2009;137:900–913. doi: 10.1016/j.cell.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Scheres SH. A Bayesian view on cryo-EM structure determination. J Mol Biol. 2012a;415:406–418. doi: 10.1016/j.jmb.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012b;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nat Methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer A, Aufderheide A, Rudack T, Beck F, Pfeifer G, Plitzko JM, Sakata E, Schulten K, Forster F, Baumeister W. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:7816–7821. doi: 10.1073/pnas.1608050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TR, Gao H, Baxter WT, Asturias FJ, Boisset N, Leith A, Frank J. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat Protoc. 2008;3:1941–1974. doi: 10.1038/nprot.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova DM, Verkhusha VV. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nature methods. 2013;10:751–754. doi: 10.1038/nmeth.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz P, Unverdorben P, Beck F, Pfeifer G, Schweitzer A, Forster F, Baumeister W. Structure of the 26S proteasome with ATP-gammaS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7264–7269. doi: 10.1073/pnas.1305782110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Thompson D, Hakala K, DeMartino GN. Subcomplexes of PA700, the 19 S regulator of the 26 S proteasome, reveal relative roles of AAA subunits in 26 S proteasome assembly and activation and ATPase activity. J Biol Chem. 2009;284:24891–24903. doi: 10.1074/jbc.M109.023218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko RJ, Jr, Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annual review of biochemistry. 2013;82:415–445. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unverdorben P, Beck F, Sledz P, Schweitzer A, Pfeifer G, Plitzko JM, Baumeister W, Forster F. Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:5544–5549. doi: 10.1073/pnas.1403409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Aravind L, Oania R, McDonald WH, Yates JR, 3rd, Koonin EV, Deshaies RJ. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- Wang X, Chen CF, Baker PR, Chen PL, Kaiser P, Huang L. Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry. 2007;46:3553–3565. doi: 10.1021/bi061994u. [DOI] [PubMed] [Google Scholar]
- Weber-Ban EU, Reid BG, Miranker AD, Horwich AL. Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature. 1999;401:90–93. doi: 10.1038/43481. [DOI] [PubMed] [Google Scholar]
- Wensley BG, Batey S, Bone FA, Chan ZM, Tumelty NR, Steward A, Kwa LG, Borgia A, Clarke J. Experimental evidence for a frustrated energy landscape in a three-helix-bundle protein family. Nature. 2010;463:685–688. doi: 10.1038/nature08743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden EJ, Padovani C, Martin A. Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat Struct Mol Biol. 2014;21:220–227. doi: 10.1038/nsmb.2771. [DOI] [PubMed] [Google Scholar]
- Wu J, Ma YB, Condgon C, Brett B, Chen S, Ouyang Q, Mao Y. Unsupervised single-particle deep classification via statistical manifold learning. arXiv. 2016 doi: 10.1371/journal.pone.0182130. 1604.04539 [physics.data-an] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- Zhang F, Hu M, Tian G, Zhang P, Finley D, Jeffrey PD, Shi Y. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009a;34:473–484. doi: 10.1016/j.molcel.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wu Z, Zhang P, Tian G, Finley D, Shi Y. Mechanism of substrate unfolding and translocation by the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell. 2009b;34:485–496. doi: 10.1016/j.molcel.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.