

Abstract
Fear, which can be expressed innately or after conditioning, is triggered when a danger or a stimulus predicting immediate danger is perceived. Its role is to prepare the body to face this danger. However, dysfunction in fear processing can lead to psychiatric disorders in which fear outweighs the danger or possibility of harm. Although recognized as highly debilitating, pathological fear remains insufficiently treated, indicating the importance of research on fear processing. The neurobiological basis of normal and pathological fear reactions is reviewed in this article. Innate and learned fear mechanisms, particularly those involving the amygdala, are considered. These fear mechanisms are also distinguished in specific phobias, which can indeed be nonexperiential (implicating innate, learning-independent mechanisms) or experiential (implicating learning-dependent mechanisms). Poor habituation and poor extinction are presented as dysfunctional mechanisms contributing to persistence of nonexperiential and experiential phobias, respectively.
Fear is an emotion of anticipation that is triggered when a situation that is at risk for our safety and/or the safety of others is perceived, through either exteroceptive inputs or the endocrine and autonomic nervous systems (interoceptive inputs). To prepare the body to face this danger, these stimuli can evoke “freeze, flight, fight, fright” reactions (Bracha 2004) or “tend-and-befriend” responses (such as turning to others for help or social support, or making a situation less tense, dangerous, or uncomfortable in some way) (Taylor et al. 2000). At the neurobiological level, significant advances have been made in identifying fear circuits and mechanisms; dysfunctions in these circuits/mechanisms can lead to chronic psychiatric disorders, including post-traumatic stress disorder (PTSD) and various types of phobia, including specific phobias. Indeed, available treatments that aim to reduce pathological fear are associated with decreased symptom severity, but up to 40% of patients show only partial long-term benefit, while most of them fail to achieve complete remission (Singewald et al. 2015).
Specific phobias are extreme and persistent fears of certain objects, situations, or activities, or persons. Additionally, people who suffer from specific phobias work hard to avoid their phobia stimuli even though they know there is no threat or danger, but they feel powerless to stop their irrational fear. Dogs, heights, tunnels, darkness, water, flying, and injuries involving blood are a few of the more common irrational fears. While PTSD is always caused by a traumatic event, specific phobia can result from either a traumatic event (experiential-specific phobia) or not (nonexperiential-specific phobia).
Nonexperiential or nonassociative specific phobia is caused and activated by stimuli that arouse fear without previous direct or indirect associative learning. Genetic, familial, environmental, or developmental factors play an important role in the development of this type of specific phobia. For instance, many children experience fear of darkness, which, in certain cases, may be sensitized. Sensitization is a form of nonassociative learning manifested by exaggerated emotional reactions to specific stimuli (e.g., nyctophobia, fear of darkness, is characterized by sensitized fear during exposure to darkness or during anticipation to darkness exposure). Sensitization may serve the functional purpose of detecting threats. At brain level, it is a stimulus-specific increase of neuronal responses. In nonexperiential phobia, it is suggested to be supported by dysfunction in “learning-independent” fear circuits (i.e., circuits that include the amygdala and drive defensive behavior without prior learning) (Rosen et al. 2015). For example, in most children, darkness may activate the amygdala. However, this amygdala activation may be exaggerated (sensitized) in children who develop nyctophobia, due to the pathological changes in the excitability threshold in fear circuits. Another feature is the lack of habituation. Habituation is also a form of nonassociative learning, which is manifested by reduced emotional reactions to repeatedly presented stimuli. It may serve the functional purpose of protecting the brain from flooding with sensory information considered, with time, as irrelevant. At brain level, it is a stimulus-specific decrement of neuronal responses to repeatedly presented stimuli. For example, fear of the dark is often lost over time through repeated exposure to darkness without any harm. This may be characterized by a decrement of amygdala activation in response to repeatedly darkness exposure. A deficiency in this mechanism (i.e., amygdala habituation) may therefore contribute to the persistence of nonexperiential phobia.
Experiential-specific phobia results from an unfortunate experience. It has been suggested that its acquisition is due to classical fear conditioning, while its maintenance is due to operant fear conditioning, which, in turn, reinforces the avoidance behavior (Tillfors 2004). Experimental studies show that classical fear conditioning can occur by associating the presentation of a neutral cue, like a sound, with an aversive event, like an electric shock. Following training, the presentation of the cue (or conditioned stimulus), in the absence of the unconditioned stimulus, causes fear behaviors. Fear can also be acquired through other ways, including an observational conditioning, as shown in primates (Cook et al. 1985; Mineka and Cook 1993) and in rodents (Chen et al. 2009; Jeon et al. 2010). In observational conditioning, a subject A observes a conspecific B experiencing classical fear conditioning. Following training, the subject A displays fear behaviors when later exposed to the conditioned stimulus alone. These findings demonstrate the social transmission of fear. Interestingly, mechanisms involved in observational conditioning and those of direct classical conditioning are similar (Mineka and Cook 1993). However, only studies using direct classical conditioning paradigm have largely contributed to the characterization of the circuits and mechanisms underlying fear conditioning (LeDoux 2014; Maren 2015). Behavioral abnormalities relative to experiential-specific phobia may therefore be supported by dysfunctions in these “learning-dependent” fear circuits and mechanisms. In this case, one possible dysfunction is deficiency in extinction (i.e., a failure to acquire a reduction of conditioned response through repeated presentations of a conditioned stimulus), explaining maintenance of experiential-specific phobia.
This review focuses mainly on the involvement of the amygdala in normal innate fear and dysfunction of innate fear in nonexperiential phobia and amygdala mechanisms of classical fear conditioning and their potential involvement in experiential phobia.
Rodent and primate studies on substrates of innate fear
Evidence accumulated from animal studies and human lesion and neuroimaging data indicate that the amygdala plays a pivotal role in innate fear (Table 1). For example, odors from predators are ecologically relevant stimuli used by prey animals as warnings for the presence of danger. These odors are not painful, but they have innate threat-like properties. This is also true in laboratory rodents that have been bred in animal colonies for generations without exposure to predators. Studies with adolescent (Chan et al. 2011) and adult (Dielenberg et al. 2001; McGregor et al. 2004) rats and adult mice (Janitzky et al. 2015) show that exposure to a cat odor induces fear responses, which are associated with increases of the number of Fos-positive cells in the amygdala, periaqueductal gray, dorsomedial prefrontal cortex, bed nucleus of the stria terminalis, paraventricular nucleus of the hypothalamus, and the locus coeruleus. These substrates are interconnected with projections from the amygdala, dorsomedial prefrontal cortex, and bed nucleus of the stria terminalis to the periaqueductal gray controlling responses such as freezing (Vianna and Brandão 2003; Chan et al. 2011). Particularly, inactivation of the prelimbic area decreases freezing and abolishes Fos expression in the periaqueductal gray (Chan et al. 2011). In monkeys, fear can be assessed in laboratory by placing the animal alone in an isolated cage. An unfamiliar human enters the room, avoids any eye contact with the animal and leaves the room, and enters again, remains motionless while staring at the animal. In this second condition, animal expresses innate fear behaviors, including freezing. Based on this human intruder paradigm, it has been found that bilateral lesions of the central amygdala abolish animal fear responses (Kalin et al. 2004). In a human study, it has been reported that bilateral calcification and atrophy of the amygdala is associated with impairment of the recognition of fear in facial expressions (Adolphs et al. 1995). Functional magnetic resonance imaging in humans has also confirmed the involvement of the amygdala in fear, by demonstrating that viewing fearful faces activates this specific brain region (Wright et al. 2006). In another study (Mobbs et al. 2010), participants were placed in a neuroimaging apparatus and asked to position their foot with the shoe removed into an open-topped imminence box (they believed that they could observe, via a camera feed, the experimenter moving a live tarantula closer or further away from their foot in real time). As the tarantula was placed closer to a subject's foot, there was an increased activity in the amygdala, periaqueductal gray, and bed nucleus of the stria terminalis. Interestingly projections of the amygdala to the bed nucleus of the stria terminalis control stress reactions involving the pituitary axis (Crestani et al. 2013).
Table 1.
Summary of studies on the amygdala

Under resting conditions, the amygdala is inhibited by an extensive γ-aminobutyric acid (GABA) network and amygdala neurons exhibit low firing rates (Quirk and Gehlert 2003). The reduction of GABAergic activity is therefore required to lower the threshold for amygdala activation and facilitates amygdala activation, necessary to expression of fear. This idea is, in part, supported by a study demonstrating that injections of muscimol, a GABAA receptor agonist that increases GABAergic inhibition, into the basolateral amygdala blocks predator odor-induced fear behavior (Müller and Fendt 2006). Dopamine and norepinephrine may play key roles in amygdala activation in response to predator scent. First, predator odor causes increased dopamine metabolism in the amygdala (Morrow et al. 2000), which, in turn, reduces GABAergic inhibitory control (Marowsky et al. 2005). Second, absorption of reboxetine, a selective norepinephrine reuptake inhibitor that increases the levels of norepinephrine, has been reported to increase basolateral amygdala responses to fearful faces (Onur et al. 2009); the norepinephrine signaling being involved in facilitation of GABAergic inhibition in the same area (Skelly et al. 2017). In addition, a human pharmacological study with propranolol, a compound blocking the action of epinephrine and norepinephrine, shows that this treatment reduces basolateral amygdala reactivity (Hurlemann et al. 2010), while, in rodents, it impairs unlearned fear to predatory threats (Do Monte et al. 2008). Increased amygdala activity enhances, in turn, activity of the paraventricular nucleus of the hypothalamus through direct projections (Gray et al. 1989). The pituitary gland is therefore activated from corticotrophin releasing hormone released by the paraventricular nucleus of the hypothalamus. The pituitary gland increases, in turn, the levels of peripheral adrenocorticotropic hormone, which stimulates the adrenal glands to secrete glucocorticoids (mainly cortisol in primates and corticosterone in rodents). This is supported by several fear studies, both in primates and rodents. A rodent study (Muñoz-Abellán et al. 2011) showed that the odor of a predator increases plasma levels of peripheral adrenocorticotropic hormone and corticosterone. In monkeys, the human intruder paradigm also induces increases in plasma levels of adrenocorticotropic hormone and corticosterone and central levels of corticotrophin releasing hormone. Lesions of the central amygdala, inducing reduction in innate fear, are also accompanied by a significant decrease in levels of these hormones (Kalin et al. 2004). The peripheral hormonal and autonomic responses mediated by the hypothalamic–pituitary–adrenal axis play particularly adaptive roles in responding to threat from fear processing in the amygdala (and extending amygdala: the bed nucleus of the stria terminalis). The adaptive roles initiated by activation of this axis include increased cardiovascular tone, respiratory rate, and intermediate metabolism, along with inhibition of general vegetative functions such as feeding, digestion, growth, reproduction, and immunity (Sapolsky et al. 2000). In addition, the hypothalamic–pituitary–adrenal axis is closely linked to another axis, the hypothalamic–pituitary–gonadal axis (Viau 2002), with the ability to inhibit each other (Fenchel et al. 2015). For example, as shown recently in a rodent study (Fenchel et al. 2015), testosterone (a product of the hypothalamic–pituitary–gonadal axis) and corticosterone levels are lower and higher, respectively, in animals exposed to predator scent, while there are higher and lower, respectively, in unexposed animals. However, although testosterone has repeatedly been shown to have anxiolytic properties in rodents, findings in primates are more mixed. In monkeys, Suarez-Jimenez et al. (2013) reported that animals exposed to an unfamiliar human staring directly at them (i.e., the human intruder paradigm) exhibited less anxious behaviors when given a treatment lowering testosterone levels.
Serotonin, or 5-hydroxotryptamine (5-HT), has also been examined given its involvement in a range of functions including regulation of emotional states such as mood and anxiety. The amygdala receives dense serotonergic projections from the dorsal raphe nucleus and expresses multiple subtypes of 5-HT receptors (Sadikot and Parent 1990). Studies with 5-HT knockout mice have shown a reduction in binding density and/or function of 5-HT1A receptors in several brain areas, including the amygdala (e.g., Li et al. 2000) and facilitation of anxiety-like behaviors (for review, see Adamec et al. 2008). Importantly, administration of vilazodone, an agonist of these 5-HT receptors, following predator stress interferes with the development of anxiety-related changes (Adamec et al. 2004), supporting the involvement of reduced 5-HT activity in the amygdala in mechanisms of innate fear. It has been suggested that 5-HT inhibits fear circuits in the amygdala likely through local action on GABAergic interneurons (Lee et al. 2013).
Potential mechanism of persistent fear in nonexperiential-specific phobia
Brain circuits of fear are also activated in phobic patients, but to significantly greater levels than in healthy individuals. For example, as seen above, the amygdala and bed nucleus of the stria terminalis display increases in activity in response to a threat stimulus. In a recent study, it has been reported that phobic patients have significantly higher activation of these areas than controls in response to threat (Münsterkötter et al. 2015). Moreover, the same authors found enhancement of functional connectivity between the two neuroanatomical substrates in phobic subjects (Münsterkötter et al. 2015). Other neuroanatomical substrates that show an exaggerated anxiety response to threat, as shown recently with spider pictures in spider phobics, are the mid insula, dorsal anterior cingulate, and ventrolateral prefrontal cortex (Zilverstand et al. 2017). Another recent study has also shown a decrease in parietal processing in specific phobia, suggesting attentional avoidance of affective stimuli (Klahn et al. 2017).
Studies on selective serotonin reuptake inhibitors (SSRI), a class of drugs that enhance 5-HT, have indicated that these drugs are effective for phobias (Cassano et al. 2002). Interestingly, improvement of specific phobias with SSRI is accompanied by decreased regional cerebral blood flow in the amygdala (Fredrikson and Furmark 2003), revealing that low levels of 5-HT in the amygdala may be involved in the pathophysiology of phobia. This is confirmed by a positron emission tomography study showing a significantly lower 5-HT1A receptor binding in the amygdala and insula (Lanzenberger et al. 2007).
As seen for normal fear, phobias are also characterized by changes affecting the dopaminergic and GABAergic systems. This is supported, in part, by the fact that patients with Parkinson's disease, which is associated with dopamine dysfunction, seem to have increased risk for developing phobia (Richard 2005). It has also been found that monoamine oxidase inhibitors, a potentially dopamine-enhancing class of drugs, are effective in treating phobia (Ipser et al. 2008). Regarding GABAergic changes, treatment with GABA uptake inhibitor tiagabine, a potentially GABA-enhancing drug, is also effective in treating phobia (Dunlop et al. 2007), indicating reduction of GABAergic tonic inhibition in the amygdala of phobic patients. Animal studies have shown that the ventromedial prefrontal cortex inhibits the amygdala by activating intra-amygdala GABAergic interneurons (Paré et al. 2004). Decreased GABAergic activity may, in addition, result from reduction of this down-regulation of amygdala activity. Indeed, phobic patients are characterized by diminished responses in the ventromedial prefrontal cortex during provocation of phobic reactions (Hermann et al. 2007, 2009). In another study, the authors (Kerr et al. 2012) found that phobic patients exhibit greater activity in the same cortical area during the anticipation of phobic stimuli under conditions of control over stimulus presentation when compared with conditions without any control. In addition, this study also confirms the inhibition of the amygdala by the ventromedial prefrontal cortex.
Sensitization-associated increased amygdala activity is a key amygdala mechanism contributing to fear sensitization in nonexperiential phobia. Direct support of this idea comes from animal studies. For example, predator stress can induce long-lasting enhancement of right amygdala afferent and efferent neural transmission, which likely mediates behavioral effects (Adamec et al. 2005). Repeated predator exposure (in a procedure allowing animals to see, hear, and smell each other, but without any physical contact) induces long-lasting sensitization of basolateral amygdala norepinephrine receptors via a corticotropin-releasing factor receptor 1-dependent mechanism, as revealed by double-immunofluorescence labeling on days 11 and 28 after stress (Rajbhandari et al. 2016). In other words, norepinephrine sensitization may provoke reduced GABAergic inhibition control, which may result in both reduction in amygdala activation threshold and hyperexcitability of the amygdala by threat stimuli. This dysfunction may be part of mechanisms that provoke persistent fear in nonexperiential phobia (Table 2).
Table 2.
Summary of potential amygdala mechanisms of fear alteration in specific phobias

Innate fear of stimuli such as animals, darkness, and heights is frequent in children. These innate fears gradually diminish with time, with repeated, nontraumatic exposure to the feared object or situation, a phenomenon known as habituation. However, children with poor habituation are at high risk of developing nonexperiential-specific phobias. In laboratory, freezing habituation can be observed in rats exposed, over a period of days, to a cat odor (e.g., Dielenberg and McGregor 1999), but there are also contradictory data (e.g., Wallace and Rosen 2000). Studies in healthy humans (Fischer et al. 2003; Herry et al. 2007; Denny et al. 2014) and mice (Herry et al. 2007) indicate that habituation is associated with decreased activity in the amygdala. In other word, persistence of amygdala activity characterizes poor habituation. Nonexperiential phobia may therefore be characterized by reduced amygdala habituation, responsible of fear persistence in patients with this type of phobia (Table 2).
Animal studies on mechanisms of fear conditioning
One of the most significant properties of the nervous system is its ability to modify its structure and function in response to experience. At the synaptic level, this ability is characterized by synaptic plasticity (i.e., activity-dependent changes of the strength of synaptic transmission). Synaptic plasticity, such as long-term potentiation (LTP), which is an increase in synaptic efficacy, at appropriate synapses during memory formation is believed to be both necessary and sufficient for storage of information (Garcia 2001; Lynch 2004), including fear conditioning (LeDoux 2014; Maren 2015).
In fear conditioning, innate fear response (e.g., freezing) can be converted into learned response when fear is evoked (for instance with a footshock application) during or immediately after the presentation of a neutral stimulus (e.g., a sound). The neutral stimulus becomes a conditioned stimulus (an aversive stimulus) because it is capable of evoking by itself the fear response (freezing). This example of classical fear conditioning corresponds to auditory fear conditioning. It is the only example that is reviewed below to examine mechanisms of fear conditioning.
The amygdala, which is critical for innate fear, is also critical for auditory fear conditioning, as revealed in rats with lesions targeting the lateral nucleus (LeDoux et al. 1990; Sheng et al. 2015). It is now well established that during training, information from the auditory stimulus and information from the nociceptive stimulus (footshock application) converge onto the basolateral amygdala complex (for a recent review, see Silva et al. 2016). Excitatory auditory inputs from the medial geniculate body are directly and indirectly, through the auditory cortex, conveyed to the amygdala. Regarding excitatory nociceptive inputs from the spinal and trigeminal dorsal horn, they seem to reach the amygdala through two pathways: via a spino-thalamic tract (Bienvenu et al. 2015) and via a spino-periaqueductal–thalamic pathway (Johansen et al. 2010). In the search of mechanisms at the basis of fear conditioning, it is therefore possible to place a stimulation electrode in the auditory thalamus or in the bundle of fibers connecting the auditory thalamus to the amygdala and a recording electrode in the basolateral complex of the amygdala, and more specifically in the lateral nucleus. Two major discoveries derive from this approach. First, LTP is induced in this pathway by means of high-frequency stimulation (Humeau et al. 2007). Second, auditory fear conditioning also induces LTP-like changes in the same pathway (Sigurdsson et al. 2007). Given that high-frequency stimulation and fear conditioning induce similar long-term enhancement of synaptic efficacy in the thalamo–amygdala pathway, LTP and LTP-like changes may share similar cellular and molecular mechanisms, which may underlie the basis of fear learning and memory. The first mechanism is the increase of glutamate release with high-frequency stimulation. Fear conditioning also provokes an increase in glutamate release in the amygdala (Venton et al. 2006). The second mechanism related to the application of high-frequency stimulation is an increase of postsynaptic depolarization, which can also be seen with fear conditioning (Rogan et al. 1997). In both cases, the depolarization results from the increase of amount of glutamate-evoked current that flows into the postsynaptic cell through α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs). Among AMPARs, the subunit glutamate receptor 1 is involved in AMPAR-dependent amygdala LTP and fear conditioning, as demonstrated in a study using knockout mice (Humeau et al. 2007). The third mechanism is calcium entry into postsynaptic cells, through several channels, and mostly N-methyl-D-aspartate receptors (NMDARs) and L-type voltage-gated calcium channels (VGCCs). LTP can require NMDARs or L-type VGCCs depending on electrical stimulation paradigm (Bauer et al. 2002). Blockade of NMDARs alters both short-term and long-term fear memory, while blockade of L-type VGCCs impairs only long-term fear memory. The rise in calcium in the postsynaptic cell triggers, in turn, a cascade of intracellular processes that ultimately induce synaptic enhancement. This includes an increase of the autophosphorylated (active) form of Ca2+/calmodulin-dependent protein kinase II (CaMKII) in lateral amygdala spines (Rodrigues et al. 2004). Interestingly, intra-amygdala infusion of a CaMKII inhibitor blocks short-term fear memory during conditioning, but not already established memory (Rodrigues et al. 2004). In addition, the same treatment also impairs LTP induction (Rodrigues et al. 2004). The other intracellular cascade resulting from the rise of intracellular calcium is the production of retrograde massagers, such as nitric oxide. Disrupting nitric oxide signaling also results in marked suppression of LTP induction in the lateral amygdala and impairment of long-term fear memory (Schafe et al. 2005). Nitric oxide acts as a retrograde messenger turning on presynaptic signaling cascades through activation of soluble guanylyl cyclase and cGMP-dependent protein kinase. As for nitric oxide, disruption of cGMP-dependent protein kinase, and particularly its type I isoform, causes a marked reduction of freezing behavior to a conditioned tone and a decrease of LTP in the lateral amygdala (Paul et al. 2008). Considering that most of these studies were done with a transient form of LTP in the lateral amygdala, while fear conditioning may require enduring changes for long-term memory, it is conceivable that there is another form of LTP, which is enduring. This enduring form exists and depends on gene expression and on new protein synthesis, through stimulation of the phosphorylation of cAMP response element-binding proteins (CREB) in amygdala cells (Huang et al. 2000). By manipulating CREB function in the lateral amygdala principal neurons in mice, it has been shown that neurons with high levels of CREB have more dendritic spines, while neurons with low CREB function have relatively fewer spines compared with control neurons (Sargin et al. 2013). Poor fear memory characterizes mice with low CREB function (Sargin et al. 2013), confirming the critical role of CREB in fear conditioning.
Potential mechanisms of persistent fear in experiential-specific phobia
Studies on the conditioning of fear in phobias indicate that fear is high in phobic compared with nonphobic subjects. However, there are studies indicating exaggerated general conditionability in phobia (Lissek et al. 2008; Vriends et al. 2012), while in other studies there was either no general conditionability (Hermann et al. 2002; Schweckendiek et al. 2011) or exaggerated conditionability only with phobia-related conditioned stimuli (Schweckendiek et al. 2011). The authors also found that phobic subjects display higher amygdala activation in response to the phobia-related conditioned stimulus than to the nonphobia-related conditioned stimulus. In another study, the authors (Vriends et al. 2012) found a stronger conditioning effect in flying phobia. Particularly, compared with healthy controls, patients with flying phobia rated conditioned stimuli more frightening. Lissek et al. (2008), using facial expressions as socially relevant stimuli, but no disorder-unrelated unconditioned stimulus, found that only phobic patients, when compared with healthy controls, develop conditioned fear. In an earlier study (Hermann et al. 2002), aversive conditioning was performed using neutral faces as conditioned stimulus and an unpleasant odor as unconditioned stimulus. Despite the lack of an enhanced conditionability in the phobic group, extinction (i.e., a learning process in which new non-fear memory traces are established, which inhibit the old fear memories) was impaired (Hermann et al. 2002).
Although the above studies do not all demonstrate an enhancement of conditionability in phobia, when this enhancement is present (e.g., Vriends et al. 2012), it is suggested here that it may be caused by metaplasticity in the amygdala. Metaplasticity is defined as a dynamic regulation of synaptic plasticity thresholds in a neuronal population, resulting, for example, in exaggerated or inhibited LTP (Garcia et al. 1997). Metaplasticity tunes the synapses to undergo changes that are necessary prerequisites for memory storage under physiological (Garcia 2001) or pathological (Garcia 2002a,b) conditions. Stress is known as a condition that provokes metaplasticity in several brain regions (e.g., in the hippocampus: Garcia et al. 1997; in the amygdala: Rodríguez Manzanares et al. 2005). In the amygdala, metaplasticity associated with stress results from an elevation in glucocorticoid levels (Karst et al. 2010), which, in turn, increases the norepinephrine release in the amygdala (McReynolds et al. 2010). Of note, norepinephrine is released in the basolateral amygdala from fibers from two structures that are activated by the stress condition: the nucleus of the solitary track and locus coeruleus (de Quervain et al. 2009). In turn, norepinephrine causes a reduction in GABA neurotransmission (Tully et al. 2007). The amygdala, particularly its basolateral complex, is characterized by a high density of GABAA/benzodiazepine receptor binding sites. GABAA receptors operate as gated chloride ion channels, causing influx of negatively charged chloride ions into the neuron, leading to reduce the chance of a successful action potential occurring. In other words, increases of glucocorticoid levels reduce GABAergic inhibition, via norepinephrine action (Liu et al. 2014). Consequently, stress, by altering GABAergic inhibition in amygdala, induces a reduction of the synaptic plasticity threshold that results in exaggerated amygdala LTP (Rodríguez Manzanares et al. 2005). This is also confirmed by studies showing that treatment with the benzodiazepines, which are among the most widely prescribed anxiolytic drugs, enhances GABAergic neurotransmission (Giachero et al. 2015) and suppresses the effect of stress on amygdala synaptic plasticity (Rodríguez Manzanares et al. 2005). The interactions between glucocorticoids and noradrenaline are summarized in Figure 1. Particularly, in the amygdala, glucocorticoids induce metaplasticity, which facilitates fear acquisition (Buchanan and Lovallo 2001; Rodríguez Manzanares et al. 2005) and may explain the enhancement of conditionability found in some phobic patients (Table 2).
Figure 1.

Interactions between glucocorticoids and norepinephrine (NA) in the regulation of GABAergic activity. During stress, adrenal hormones, epinephrine, and glucocorticoids, are released. Epinephrine, which does not cross the blood-brain barrier, induces the release of NA in the basolateral amygdala (BLA) by activating vagal afferents to the nucleus of the solitary tract (NTS). NA neurons in the NTS send direct noradrenergic fibers to the BLA. NA neurons from the NTS project also to the locus coeruleus (LC), which noradrenergic fibers reach directly the BLA. Glucocorticoids freely cross the blood–brain barrier and potentiate NA release in the BLA and facilitate the NA inhibitory effect on GABA interneurons in the BLA (de Quervain et al. 2009). This results in the decrease of excitability threshold of the pyramidal neurons in the BLA, which release the excitatory neurotransmitter, glutamate (Glu), in the central amygdala (CeA).
As seen above, the other phenomenon that characterizes phobia is poor fear extinction learning (Hermann et al. 2002). It has been hypothesized that poor extinction contributes to the maintenance of anxiety disorders (Mineka and Zinbarg 1996). Phobias are chronic disorders with a high rate of resistance to pharmacotherapy (Slaap et al. 1996; Van Ameringen et al. 2004; Stein et al. 2005) and psychotherapy (Van Ameringen et al. 2004), including exposure therapy (Lass-Hennemann and Michael 2014). Exposure therapy is thought to rely on the extinction of the fear memory. Even though this extinction-based therapy is a highly successful treatment option, not all patients profit from it (Lass-Hennemann and Michael 2014), showing therefore poor extinction mechanisms. Here, it is suggested that poor extinction mechanisms may include dysfunction in the serotonergic system for at least two main reasons. First, as previously mentioned, SSRI, by increasing concentrations of 5-HT in the synapse cleft, have been found effective for phobias (Cassano et al. 2002). Moreover, as demonstrated with knockout mice, the ionotropic 5-HT receptor, which is expressed in limbic regions including the amygdala, is not required for the acquisition or retention of fear memory, but is essential for the extinction of conditioned fear (Kondo et al. 2014). Other studies have shown that chronic treatment with fluoxetine, a selective serotonin reuptake inhibitor, facilitates fear extinction (Karpova et al. 2011; Popova et al. 2014; Gunduz-Cinar et al. 2016). Second, the serotonin system interacts within the amygdala with the GABAergic and endocannabinoid systems. Indeed, activation of the serotonergic system has been reported to stimulate the release of GABA in this structure (Koyama et al. 2002). Similarly, activation of the serotonergic system has been found to produce a significant and selective increase in levels of anandamide, an endogenous ligand of cannabinoid receptors, in the basolateral amygdala (Gunduz-Cinar et al. 2016). As for the serotonin system, blockade of each system (GABAergic or endocannabinoid) is known to impair fear extinction, whereas their activation facilitates fear extinction. For example, in an animal study, it has been reported that infusion of a low dose of GABAergic agonist muscimol into the basolateral amygdala complex, prior to fear extinction training, facilitates extinction learning (Akirav et al. 2006). Regarding the endocannabinoid system, poor fear extinction has also been reported in knockout mice with reduced anandamide levels in the amygdala (Marsicano et al. 2002; Jenniches et al. 2016). In other words, extinction may require activation of the serotonin system that may induce, in the basolateral amygdala, increased endocannabinoid levels and increased GABAergic activity (Fig. 2).
Figure 2.
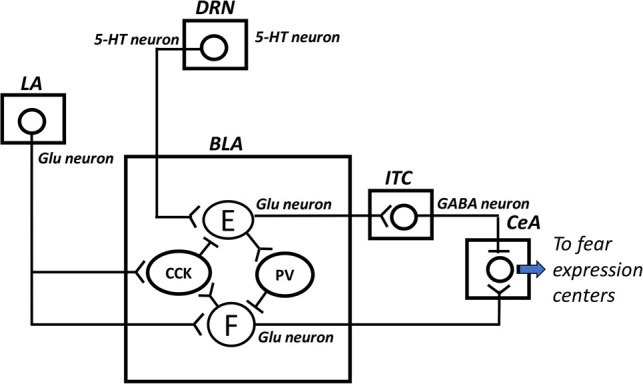
Interactions between serotonin (5-HT) and GABAergic systems in the basolateral amygdala (BLA) during fear expression and extinction. Following fear conditioning, presentation of the conditioned cue activates glutamatergic (Glu) neurons in the lateral amygdala (LA) that project to fear neurons (F) in the basolateral amygdala (BLA). The fear neurons activate, in turn, neurons in the central amygdala (CeA) for fear expression. The LA neurons also activate a BLA cell population that expresses cholecystokinin (CCK) and establishes GABAergic synapses with extinction neurons (E) within the BLA, inhibiting therefore these extinction neurons. Consequently, extinction neurons cannot inhibit fear expression through their Glu projections to BLA interneurons containing parvalbumin (PV), which establish a GABAergic connection with the fear neurons, or through their Glu projections to intercalated cells (ITC), which send GABAergic fibers to the CeA. It is well known that endocannabinoid signaling results in retrograde inhibition of afferent neurotransmission. Interestingly, the cannabinoid type 1 receptors are located presynaptically on CCK-containing cells. However, endocannabinoid synthesis is initiated via voltage-dependent mechanisms (Kano et al. 2009) and because of the inhibitory effect of CCK-containing cells on extinction neurons, endocannabinoids are not released. Here, it is hypothesized that, during extinction training, repeated presentations of the conditioned stimulus alone, indicating safety, may induce progressive enhancement of serotonin (5-HT) release in the BLA, which may partially activate the extinction neurons, hence provoking the release of endocannabinoids. The activation of the extinction neurons may be achieved with the inhibitory effect of endocannabinoids on CCK-containing cells via their cannabinoid type 1 receptors, resulting in inhibition of fear expression.
Using single-unit recording in mice, Herry et al. (2008) demonstrated that neurons in the basolateral amygdala can be divided into distinct functional classes: fear neurons and extinction neurons. Fear neurons display a selective enhancement of activity in response to the conditioned stimulus. During extinction training, there is a progressive decrease of this activity. At synaptic plasticity level, this can be interpreted as a suppression of LTP-like changes or depotentiation. Extinction neurons exhibit, on the contrary, a progressive increase of their activity during extinction; this being interpreted here as a progressive development of LTP-like changes.
Regarding extinction training-associated depotentiation, it has already been shown that conditioning-induced LTP-like changes in the thalamo–amygdala pathway are reversed with extinction (Kim et al. 2007). At the basolateral amygdala, conditioning-associated reduction of GABA levels is also reversed with extinction (Stork et al. 2002). Fear extinction may require this change (increased GABA levels) to initiate depotentiation mechanisms, among which activation of NMDARs, increased calcium entry in postsynaptic cells and calcineurin Aα activation (Zhuo et al. 1999), leading to internalization of AMPARs (Kim et al. 2007). Therefore, another way to improve extinction, or exposure therapy in phobics, in addition to GABAergic interventions, is the use of NMDAR agonists. D-cycloserine, a partial agonist of the glycine site, is one example of NMDAR agonists that has been often tested in studies on fear extinction in rodents (e.g., Ledgerwood et al. 2003; Lehner et al. 2010) and in humans (e.g., Ressler et al. 2004; Smits et al. 2013) studies. For example, in human studies, D-cycloserine has been reported to facilitate exposure therapy in height-phobic subjects, when compared with the placebo condition (Ressler et al. 2004; Smits et al. 2013).
Regarding extinction training-association LTP-like changes, it has also been shown that extinction is associated with a potentiation of extinction neurons projecting to GABAergic intercalated amygdala neurons that project, in turn, to the central amygdala (Amano et al. 2010). Of note, the so-called extinction neurons in the basolateral amygdala correspond to pyramidal neurons that are powerfully inhibited by cholecystokinin-containing interneurons (Marsicano et al. 2002). Extinction can take place only if GABA release by these interneurons is reduced (see legend of Fig. 2). Marsicano et al. (2002) found that low-frequency stimulation of the cholecystokinin-containing interneurons leads to a long-term reduction in the release of GABA, which, in turn, leads to less inhibition of the extinction neurons. Interestingly, the authors reported that this long-term depression of GABAergic transmission was completely blocked by the cannabinoid type 1 receptor agonist. In addition, this synaptic plasticity was observed in cannabinoid type 1 receptor-deficient mice (Marsicano et al. 2002). One can suggest that during extinction training, serotonin may induce strong release of endocannabinoids that may generate long-term depression of GABAergic release by cholecystokinin-containing interneurons. This may thereby help development of LTP-like of synapses between extinction neurons and their GABAergic target cells (Fig. 2).
Collectively, these data support the view that serotonergic dysfunction, which characterized phobic patients, may indirectly contribute, via the lack of activation of endocannabinoid system in the basolateral amygdala, in poor extinction that is reported in phobia.
Concluding remarks
While fear is an adaptive component of response to potentially threatening stimuli, too much or inappropriate fear accounts for chronic psychiatric disorders, including PTSD and phobias. Understanding the neurobiological basis of fear is therefore critical at elucidating the mechanisms improving treatments of these fear-related pathologies. Concerning phobias, nonexperiential, engaging innate fear, and experiential, engaging conditioned fear, disorders can be distinguished. However, so far, we know a lot about how the brain processes fear that is conditioned, while much less is known about innate fear. An increase of research on innate fear is therefore necessary.
Based on the research reviewed in this article, it appears that the amygdala is central to two phenomena that may support pathological innate fear: fear sensitization (with decreased amygdala threshold activity and potentiation of amygdala activity) and failure of fear habituation (with decreased amygdala habituation). Mechanisms of fear sensitization may contribute to decreased amygdala habituation. As current research is limited, this hypothesis is not fully supported here.
The cause of experiential phobia is attributed to some external learning experience. However, although phobia-like fears can be produced in laboratory with fear conditioning paradigms, experimental findings do not prove that in everyday life, classical fear conditioning is the cause of phobias (Merckelbach et al. 1996). For example, not all people who have had fearful events go on to develop phobias (Aitken et al. 1981), why? This may result from exaggerated fear conditionability, due to amygdala metaplastic changes (i.e., exaggerated LTP-like changes) in vulnerable individuals. However, not all phobic patients show exaggerated fear conditionability in laboratory experiences (Hermann et al. 2002). Poor extinction, due to impairment in mechanisms of depotentiation in the amygdala of vulnerable individuals, is also hypothesized as a potential factor maintaining pathological phobia. In experiments done by Öhman (1986), it was observed that people conditioned to angry faces showed significant resistance to extinction compared with those exposed to happy and neutral faces. So, nonphobic individuals can also present resistance to extinction.
As mentioned in the introduction, the development of experiential phobia seems to involve two stages. During the first stage, a pairing of a neutral stimulus and an aversive event results in a conditioned fear response to the neutral stimulus, which becomes a conditioned stimulus. During the second stage, the person learns that fear responses to the conditioned stimulus can be reduced by avoiding this stimulus. However, as shown in rats, strong avoidance causes resistance to extinction of classical fear conditioning (Nachon et al. 2014). Mechanisms that are involved need also to be examined in future studies. It would also be good to consider, in this context, other pathologies that contribute to pathological fear, such as relapse of fear after extinction (e.g., renewal, recovery, and reinstatement).
Despite these critical remarks, poor habituation and poor extinction, support, at least in part, fear dysfunction in phobia.
Acknowledgments
I thank Professor Stephen Maren for critically reading an earlier version of this paper.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.044115.116.
References
- Adamec R, Bartoszyk GD, Burton P. 2004. Effects of systemic injections of Vilazodone, a selective serotonin reuptake inhibitor and serotonin 1A receptor agonist, on anxiety induced by predator stress in rats. Eur J Pharmacol 504: 65–77. [DOI] [PubMed] [Google Scholar]
- Adamec R, Blundell J, Burton P. 2005. Role of NMDA receptors in the lateralized potentiation of amygdala afferent and efferent neural transmission produced by predator stress. Physiol Behav 86: 75–91. [DOI] [PubMed] [Google Scholar]
- Adamec R, Holmes A, Blundell J. 2008. Vulnerability to lasting anxiogenic effects of brief exposure to predator stimuli: sex, serotonin and other factors—relevance to PTSD. Neurosci Biobehav Rev 32: 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolphs R, Tranel D, Damasio H, Damasio AR. 1995. Fear and the human amygdala. J Neurosci 15: 5879–5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken RC, Lister JA, Main CJ. 1981. Identification of features associated with flying phobia in aircrew. Br J Psychiatry 139: 38–42. [DOI] [PubMed] [Google Scholar]
- Akirav I, Raizel H, Maroun M. 2006. Enhancement of conditioned fear extinction by infusion of the GABAA agonist muscimol into the rat prefrontal cortex and amygdala. Eur J Neurosci 23: 758–764. [DOI] [PubMed] [Google Scholar]
- Amano T, Unal CT, Paré D. 2010. Synaptic correlates of fear extinction in the amygdala. Nat Neurosci 13: 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE. 2002. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 22: 5239–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu TC, Busti D, Micklem BR, Mansouri M, Magill PJ, Ferraguti F, Capogna M. 2015. Large intercalated neurons of amygdala relay noxious sensory information. J Neurosci 35: 2044–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracha HS. 2004. Freeze, flight, fight, fright, faint: adaptationist perspectives on the acute stress response spectrum. CNS Spectr 9: 679–685. [DOI] [PubMed] [Google Scholar]
- Buchanan TW, Lovallo WR. 2001. Enhanced memory for emotional material following stress-level cortisol treatment in humans. Psychoneuroendocrinology 26: 307–317. [DOI] [PubMed] [Google Scholar]
- Cassano GB, Baldini Rossi N, Pini S. 2002. Psychopharmacology of anxiety disorders. Dialogues Clin Neurosci 4: 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan T, Kyere K, Davis BR, Shemyakin A, Kabitzke PA, Shair HN, Barr GA, Wiedenmayer CP. 2011. The role of the medial prefrontal cortex in innate fear regulation in infants, juveniles, and adolescents. J Neurosci 31: 4991–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Panksepp JB, Lahvis GP. 2009. Empathy is moderated by genetic background in mice. PLoS One 4: e4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook M, Mineka S, Wolkenstein B, Laitsch K. 1985. Observational conditioning of snake fear in unrelated rhesus monkeys. J Abnorm Psychol 94: 591–610. [DOI] [PubMed] [Google Scholar]
- Crestani CC, Alves FH, Gomes FV, Resstel LB, Correa FM, Herman JP. 2013. Mechanisms in the bed nucleus of the stria terminalis involved in control of autonomic and neuroendocrine functions: a review. Curr Neuropharmacol 11: 141–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny BT, Fan J, Liu X, Guerreri S, Mayson SJ, Rimsky L, New AS, Siever LJ, Koenigsberg HW. 2014. Insula-amygdala functional connectivity is correlated with habituation to repeated negative images. Soc Cogn Affect Neurosci 9: 1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Quervain DJ, Aerni A, Schelling G, Roozendaal B. 2009. Glucocorticoids and the regulation of memory in health and disease. Front Neuroendocrinol 30: 358–370. [DOI] [PubMed] [Google Scholar]
- Dielenberg RA, McGregor IS. 1999. Habituation of the hiding response to cat odor in rats (Rattus norvegicus). J Comp Psychol 113: 376–387. [DOI] [PubMed] [Google Scholar]
- Dielenberg RA, Hunt GE, McGregor IS. 2001. “When a rat smells a cat”: the distribution of Fos immunoreactivity in rat brain following exposure to a predatory odor. Neuroscience 104: 1085–1097. [DOI] [PubMed] [Google Scholar]
- Do Monte FH, Canteras NS, Fernandes D, Assreuy J, Carobrez AP. 2008. New perspectives on β-adrenergic mediation of innate and learned fear responses to predator odor. J Neurosci 28: 13296–13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop BW, Papp L, Garlow SJ, Weiss PS, Knight BT, Ninan PT. 2007. Tiagabine for social anxiety disorder. Hum Psychopharmacol 22: 241–244. [DOI] [PubMed] [Google Scholar]
- Fenchel D, Levkovitz Y, Vainer E, Kaplan Z, Zohar J, Cohen H. 2015. Beyond the HPA-axis: the role of the gonadal steroid hormone receptors in modulating stress-related responses in an animal model of PTSD. Eur Neuropsychopharmacol 25: 944–957. [DOI] [PubMed] [Google Scholar]
- Fischer H, Wright CI, Whalen PJ, McInerney SC, Shin LM, Rauch SL. 2003. Brain habituation during repeated exposure to fearful and neutral faces: a functional MRI study. Brain Res Bull 59: 387–392. [DOI] [PubMed] [Google Scholar]
- Fredrikson M, Furmark T. 2003. Amygdaloid regional cerebral blood flow and subjective fear during symptom provocation in anxiety disorders. Ann N Y Acad Sci 985: 341–347. [DOI] [PubMed] [Google Scholar]
- Garcia R. 2001. Stress, hippocampal plasticity and spatial learning. Synapse 40: 180–183. [DOI] [PubMed] [Google Scholar]
- Garcia R. 2002a. Stress, metaplasticity, and antidepressants. Curr Mol Med 2: 629–638. [DOI] [PubMed] [Google Scholar]
- Garcia R. 2002b. Stress, synaptic plasticity, and psychopathology. Rev Neurosci 13: 195–208. [DOI] [PubMed] [Google Scholar]
- Garcia R, Musleh W, Tocco G, Thompson RF, Baudry M. 1997. Time-dependent blockade of STP and LTP in hippocampal slices following acute stress in mice. Neurosci Lett 233: 41–44. [DOI] [PubMed] [Google Scholar]
- Giachero M, Calfa GD, Molina VA. 2015. Hippocampal dendritic spines remodeling and fear memory are modulated by GABAergic signaling within the basolateral amygdala complex. Hippocampus 25: 545–555. [DOI] [PubMed] [Google Scholar]
- Gray TS, Carney ME, Magnuson DJ. 1989. Direct projections from the central amygdaloid nucleus to the hypothalamic paraventricular nucleus: possible role in stress-induced adrenocorticotropin release. Neuroendocrinology 50: 433–446. [DOI] [PubMed] [Google Scholar]
- Gunduz-Cinar O, Flynn S, Brockway E, Kaugars K, Baldi R, Ramikie TS, Cinar R, Kunos G, Patel S, Holmes A. 2016. Fluoxetine facilitates fear extinction through amygdala endocannabinoids. Neuropsychopharmacology 41: 1598–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann C, Ziegler S, Birbaumer N, Flor H. 2002. Psychophysiological and subjective indicators of aversive pavlovian conditioning in generalized social phobia. Biol Psychiatry 52: 328–337. [DOI] [PubMed] [Google Scholar]
- Hermann A, Schäfer A, Walter B, Stark R, Vaitl D, Schienle A. 2007. Diminished medial prefrontal cortex activity in blood-injection-injury phobia. Biol Psychol 75: 124–130. [DOI] [PubMed] [Google Scholar]
- Hermann A, Schäfer A, Walter B, Stark R, Vaitl D, Schienle A. 2009. Emotion regulation in spider phobia: role of the medial prefrontal cortex. Soc Cogn Affect Neurosci 4: 257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Bach DR, Esposito F, Di Salle F, Perrig WJ, Scheffler K, Lüthi A, Seifritz E. 2007. Processing of temporal unpredictability in human and animal amygdala. J Neurosci 27: 5958–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. 2008. Switching on and off fear by distinct neuronal circuits. Nature 454: 600–606. [DOI] [PubMed] [Google Scholar]
- Huang YY, Martin KC, Kandel ER. 2000. Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci 20: 6317–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Reisel D, Johnson AW, Borchardt T, Jensen V, Gebhardt C, Bosch V, Gass P, Bannerman DM, Good MA, et al. 2007. A pathway-specific function for different AMPA receptor subunits in amygdala long-term potentiation and fear conditioning. J Neurosci 27: 10947–10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlemann R, Walter H, Rehme AK, Kukolja J, Santoro SC, Schmidt C, Schnell K, Musshoff F, Keysers C, Maier W, et al. 2010. Human amygdala reactivity is diminished by the β-noradrenergic antagonist propranolol. Psychol Med 40: 1839–1848. [DOI] [PubMed] [Google Scholar]
- Ipser JC, Kariuki CM, Stein DJ. 2008. Pharmacotherapy for social anxiety disorder: a systematic review. Expert Rev Neurother 8: 235–257. [DOI] [PubMed] [Google Scholar]
- Janitzky K, D'Hanis W, Kröber A, Schwegler H. 2015. TMT predator odor activated neural circuit in C57BL/6J mice indicates TMT-stress as a suitable model for uncontrollable intense stress. Brain Res 1599: 1–8. [DOI] [PubMed] [Google Scholar]
- Jenniches I, Ternes S, Albayram O, Otte DM, Bach K, Bindila L, Michel K, Lutz B, Bilkei-Gorzo A, Zimmer A. 2016. Anxiety, stress, and fear response in mice with reduced endocannabinoid levels. Biol Psychiatry 79: 858–868. [DOI] [PubMed] [Google Scholar]
- Jeon D, Kim S, Chetana M, Jo D, Ruley HE, Lin SY, Rabah D, Kinet JP, Shin HS. 2010. Observational fear learning involves affective pain system and Cav1.2 Ca2+ channels in ACC. Nat Neurosci 13: 482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Tarpley JW, LeDoux JE, Blair HT. 2010. Neural substrates for expectation-modulated fear learning in the amygdala and periaqueductal gray. Nat Neurosci 13: 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin NH, Shelton SE, Davidson RJ. 2004. The role of the central nucleus of the amygdala in mediating fear and anxiety in the primate. J Neurosci 24: 5506–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. 2009. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89: 309–380. [DOI] [PubMed] [Google Scholar]
- Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, Agústsdóttir A, Antila H, Popova D, Akamine Y, Sullivan R, et al. 2011. Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science 334: 1731–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst H, Berger S, Erdmann G, Schütz G, Joëls M. 2010. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci 107: 14449–14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DL, McLaren DG, Mathy RM, Nitschke JB. 2012. Controllability modulates the anticipatory response in the human ventromedial prefrontal cortex. Front Psychol 3: 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, et al. 2007. Amygdala depotentiation and fear extinction. Proc Natl Acad Sci 104: 20955–20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klahn AL, Klinkenberg IA, Lueken U, Notzon S, Arolt V, Pantev C, Zwanzger P, Junghoefer M. 2017. Commonalities and differences in the neural substrates of threat predictability in panic disorder and specific phobia. Neuroimage Clin 14: 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Nakamura Y, Ishida Y, Yamada T, Shimada S. 2014. The 5-HT3A receptor is essential for fear extinction. Learn Mem 21: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Matsumoto N, Murakami N, Kubo C, Nabekura J, Akaike N. 2002. Role of presynaptic 5-HT1A and 5-HT3 receptors in modulation of synaptic GABA transmission in dissociated rat basolateral amygdala neurons. Life Sci 72: 375–387. [DOI] [PubMed] [Google Scholar]
- Lanzenberger RR, Mitterhauser M, Spindelegger C, Wadsak W, Klein N, Mien LK, Holik A, Attarbaschi T, Mossaheb N, Sacher J, et al. 2007. Reduced serotonin-1A receptor binding in social anxiety disorder. Biol Psychiatry 61: 1081–1089. [DOI] [PubMed] [Google Scholar]
- Lass-Hennemann J, Michael T. 2014. Endogenous cortisol levels influence exposure therapy in spider phobia. Behav Res Ther 60: 39–45. [DOI] [PubMed] [Google Scholar]
- Ledgerwood L, Richardson R, Cranney J. 2003. Effects of D-cycloserine on extinction of conditioned freezing. Behav Neurosci 117: 341–349. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. 2014. Coming to terms with fear. Proc Natl Acad Sci 111: 2871–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Cicchetti P, Xagoraris A, Romanski LM. 1990. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci 10: 1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim SJ, Kwon OB, Lee JH, Kim JH. 2013. Inhibitory networks of the amygdala for emotional memory. Front Neural Circuits 7: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner M, Wisłowska-Stanek A, Taracha E, Maciejak P, Szyndler J, Skórzewska A, Turzyńska D, Sobolewska A, Hamed A, Bidziński A, et al. 2010. The effects of midazolam and D-cycloserine on the release of glutamate and GABA in the basolateral amygdala of low and high anxiety rats during extinction trial of a conditioned fear test. Neurobiol Learn Mem 94: 468–480. [DOI] [PubMed] [Google Scholar]
- Li Q, Wichems C, Heils A, Lesch KP, Murphy DL. 2000. Reduction in the density and expression, but not G-protein coupling, of serotonin receptors (5-HT1A) in 5-HT transporter knock-out mice: gender and brain region differences. J Neurosci 20: 7888–7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissek S, Levenson J, Biggs AL, Johnson LL, Ameli R, Pine DS, Grillon C. 2008. Elevated fear conditioning to socially relevant unconditioned stimuli in social anxiety disorder. Am J Psychiatry 165: 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZP, Song C, Wang M, He Y, Xu XB, Pan HQ, Chen WB, Peng WJ, Pan BX. 2014. Chronic stress impairs GABAergic control of amygdala through suppressing the tonic GABAA receptor currents. Mol Brain 7: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. 2004. Long-term potentiation and memory. Physiol Rev 84: 87–136. [DOI] [PubMed] [Google Scholar]
- Maren S. 2015. Out with the old and in with the new: synaptic mechanisms of extinction in the amygdala. Brain Res 1621: 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marowsky A, Yanagawa Y, Obata K, Vogt KE. 2005. A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron 48: 1025–1037. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, et al. 2002. The endogenous cannabinoid system controls extinction of aversive memories. Nature 418: 530–534. [DOI] [PubMed] [Google Scholar]
- McGregor IS, Hargreaves GA, Apfelbach R, Hunt GE. 2004. Neural correlates of cat odor-induced anxiety in rats: region-specific effects of the benzodiazepine midazolam. J Neurosci 24: 4134–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McReynolds JR, Donowho K, Abdi A, McGaugh JL, Roozendaal B, McIntyre CK. 2010. Memory-enhancing corticosterone treatment increases amygdala norepinephrine and Arc protein expression in hippocampal synaptic fractions. Neurobiol Learn Mem 93: 312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merckelbach H, Muris P, Schouten E. 1996. Pathways to fear in spider phobic children. Behav Res Ther 34: 935–938. [DOI] [PubMed] [Google Scholar]
- Mineka S, Cook M. 1993. Mechanisms involved in the observational conditioning of fear. J Exp Psychol Gen 122: 23–38. [DOI] [PubMed] [Google Scholar]
- Mineka S, Zinbarg R. 1996. Conditioning and ethological models of anxiety disorders: stress-in-dynamic-context anxiety models. Nebr Symp Motiv 43: 135–210. [PubMed] [Google Scholar]
- Mobbs D, Yu R, Rowe JB, Eich H, FeldmanHall O, Dalgleish T. 2010. Neural activity associated with monitoring the oscillating threat value of a tarantula. Proc Natl Acad Sci 107: 20582–20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow BA, Redmond AJ, Roth RH, Elsworth JD. 2000. The predator odor, TMT, displays a unique, stress-like pattern of dopaminergic and endocrinological activation in the rat. Brain Res 864: 146–151. [DOI] [PubMed] [Google Scholar]
- Müller M, Fendt M. 2006. Temporary inactivation of the medial and basolateral amygdala differentially affects TMT-induced fear behavior in rats. Behav Brain Res 167: 57–62. [DOI] [PubMed] [Google Scholar]
- Muñoz-Abellán C, Rabasa C, Daviu N, Nadal R, Armario A. 2011. Behavioral and endocrine consequences of simultaneous exposure to two different stressors in rats: interaction or independence? PLoS One 6: e21426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münsterkötter AL, Notzon S, Redlich R, Grotegerd D, Dohm K, Arolt V, Kugel H, Zwanzger P, Dannlowski U. 2015. Spider or no spider? Neural correlates of sustained and phasic fear in spider phobia. Depress Anxiety 32: 656–663. [DOI] [PubMed] [Google Scholar]
- Nachon O, Cleren C, Husson S, Huguet C, Auclair J, Faure S, Akirav I, Moreau JL, Garcia R. 2014. Prefrontal tetanic stimulation, following fear reconditioning, facilitates expression of previously acquired extinction. Neurobiol Learn Mem 113: 62–68. [DOI] [PubMed] [Google Scholar]
- Öhman A. 1986. Face the beast and fear the face: animal and social fears as prototypes for evolutionary analyses of emotion. Psychophysiology 23: 123–145. [DOI] [PubMed] [Google Scholar]
- Onur OA, Walter H, Schlaepfer TE, Rehme AK, Schmidt C, Keysers C, Maier W, Hurlemann R. 2009. Noradrenergic enhancement of amygdala responses to fear. Soc Cogn Affect Neurosci 4: 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré D, Quirk GJ, Ledoux JE. 2004. New vistas on amygdala networks in conditioned fear. J Neurophysiol 92: 1–9. [DOI] [PubMed] [Google Scholar]
- Paul C, Schöberl F, Weinmeister P, Micale V, Wotjak CT, Hofmann F, Kleppisch T. 2008. Signaling through cGMP-dependent protein kinase I in the amygdala is critical for auditory-cued fear memory and long-term potentiation. J Neurosci 28: 14202–14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova D, Ágústsdóttir A, Lindholm J, Mazulis U, Akamine Y, Castrén E, Karpova NN. 2014. Combination of fluoxetine and extinction treatments forms a unique synaptic protein profile that correlates with long-term fear reduction in adult mice. Eur Neuropsychopharmacol 24: 1162–1174. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Gehlert DR. 2003. Inhibition of the amygdala: key to pathological states? Ann N Y Acad Sci 985: 263–272. [DOI] [PubMed] [Google Scholar]
- Rajbhandari AK, Zhu R, Adling C, Fanselow MS, Waschek JA. 2016. Graded fear generalization enhances the level of cfos-positive neurons specifically in the basolateral amygdala. J Neurosci Res 94: 1393–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. 2004. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry 61: 1136–1144. [DOI] [PubMed] [Google Scholar]
- Richard IH. 2005. Anxiety disorders in Parkinson's disease. Adv Neurol 96: 42–55. [PubMed] [Google Scholar]
- Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE. 2004. Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci 24: 3281–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez Manzanares PA, Isoardi NA, Carrer HF, Molina VA. 2005. Previous stress facilitates fear memory, attenuates GABAergic inhibition, and increases synaptic plasticity in the rat basolateral amygdala. J Neurosci 25: 8725–8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Stäubli UV, LeDoux JE. 1997. Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390: 604–607. [DOI] [PubMed] [Google Scholar]
- Rosen JB, Asok A, Chakraborty T. 2015. The smell of fear: innate threat of 2,5-dihydro-2,4,5-trimethylthiazoline, a single molecule component of a predator odor. Front Neurosci 9: 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadikot AF, Parent A. 1990. The monoaminergic innervation of the amygdala in the squirrel monkey: an immunohistochemical study. Neuroscience 36: 431–447. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. 2000. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev 21: 55–89. [DOI] [PubMed] [Google Scholar]
- Sargin D, Mercaldo V, Yiu AP, Higgs G, Han JH, Frankland PW, Josselyn SA. 2013. CREB regulates spine density of lateral amygdala neurons: implications for memory allocation. Front Behav Neurosci 7: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Bauer EP, Rosis S, Farb CR, Rodrigues SM, LeDoux JE. 2005. Memory consolidation of Pavlovian fear conditioning requires nitric oxide signaling in the lateral amygdala. Eur J Neurosci 22: 201–211. [DOI] [PubMed] [Google Scholar]
- Schweckendiek J, Klucken T, Merz CJ, Tabbert K, Walter B, Ambach W, Vaitl D, Stark R. 2011. Weaving the (neuronal) web: fear learning in spider phobia. Neuroimage 54: 681–688. [DOI] [PubMed] [Google Scholar]
- Sheng T, Zhang X, Wang S, Zhang J, Lu W, Dai Y. 2015. Endothelin-1-induced mini-stroke in the dorsal hippocampus or lateral amygdala results in deficits in learning and memory. J Biomed Res 29: 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson T, Doyère V, Cain CK, LeDoux JE. 2007. Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology 52: 215–227. [DOI] [PubMed] [Google Scholar]
- Silva BA, Gross CT, Gräff J. 2016. The neural circuits of innate fear: detection, integration, action, and memorization. Learn Mem 23: 544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singewald N, Schmuckermair C, Whittle N, Holmes A, Ressler KJ. 2015. Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther 149: 150–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly MJ, Ariwodola OJ, Weiner JL. 2017. Fear conditioning selectively disrupts noradrenergic facilitation of GABAergic inhibition in the basolateral amygdala. Neuropharmacology 113: 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaap BR, van Vliet IM, Westenberg HG, den Boer JA. 1996. The prediction of response in the anxiety disorders. Acta Neuropsychiatr 8: 102–104. [DOI] [PubMed] [Google Scholar]
- Smits JA, Rosenfield D, Otto MW, Powers MB, Hofmann SG, Telch MJ, Pollack MH, Tart CD. 2013. D-cycloserine enhancement of fear extinction is specific to successful exposure sessions: evidence from the treatment of height phobia. Biol Psychiatry 73: 1054–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein MB, Pollack MH, Bystritsky A, Kelsey JE, Mangano RM. 2005. Efficacy of low and higher dose extended-release venlafaxine in generalized social anxiety disorder: a 6-month randomized controlled trial. Psychopharmacology (Berl) 177: 280–288. [DOI] [PubMed] [Google Scholar]
- Stork O, Ji FY, Obata K. 2002. Reduction of extracellular GABA in the mouse amygdala during and following confrontation with a conditioned fear stimulus. Neurosci Lett 327: 138–142. [DOI] [PubMed] [Google Scholar]
- Suarez-Jimenez B, Gore HE, Hachey J, King HM, Lacreuse A. 2013. Testosterone modulation of anxiety in gonadally-suppressed male rhesus monkeys: a role for gonadotropins? Pharmacol Biochem Behav 104: 97–104. [DOI] [PubMed] [Google Scholar]
- Taylor SE, Klein LC, Lewis BP, Gruenewald TL, Gurung RA, Updegraff JA. 2000. Biobehavioral responses to stress in females: tend-and-befriend, not fight-or-flight. Psychol Rev 107: 411–429. [DOI] [PubMed] [Google Scholar]
- Tillfors M. 2004. Why do some individuals develop social phobia? A review with emphasis on the neurobiological influences. Nord J Psychiatry 58: 267–276. [DOI] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E, Bolshakov VY. 2007. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc Natl Acad Sci 104: 14146–14150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ameringen M, Mancini C, Pipe B, Bennett M. 2004. Optimizing treatment in social phobia: a review of treatment resistance. CNS Spectr 9: 753–762. [DOI] [PubMed] [Google Scholar]
- Venton BJ, Robinson TE, Kennedy RT, Maren S. 2006. Dynamic amino acid increases in the basolateral amygdala during acquisition and expression of conditioned fear. Eur J Neurosci 23: 3391–3398. [DOI] [PubMed] [Google Scholar]
- Vianna DM, Brandão ML. 2003. Anatomical connections of the periaqueductal gray: specific neural substrates for different kinds of fear. Braz J Med Biol Res 36: 557–566. [DOI] [PubMed] [Google Scholar]
- Viau V. 2002. Functional cross-talk between the hypothalamic-pituitary-gonadal and -adrenal axes. J Neuroendocrinol 14: 506–513. [DOI] [PubMed] [Google Scholar]
- Vriends N, Michael T, Schindler B, Margraf J. 2012. Associative learning in flying phobia. J Behav Ther Exp Psychiatry 43: 838–843. [DOI] [PubMed] [Google Scholar]
- Wallace KJ, Rosen JB. 2000. Predator odor as an unconditioned fear stimulus in rats: elicitation of freezing by trimethylthiazoline, a component of fox feces. Behav Neurosci 114: 912–922. [DOI] [PubMed] [Google Scholar]
- Wright CI, Wedig MM, Williams D, Rauch SL, Albert MS. 2006. Novel fearful faces activate the amygdala in healthy young and elderly adults. Neurobiol Aging 27: 361–374. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Zhang W, Son H, Mansuy I, Sobel RA, Seidman J, Kandel ER. 1999. A selective role of calcineurin Aα in synaptic depotentiation in hippocampus. Proc Natl Acad Sci 96: 4650–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilverstand A, Sorger B, Kaemingk A, Goebel R. 2017. Quantitative representations of an exaggerated anxiety response in the brain of female spider phobics—a parametric fMRI study. Hum Brain Mapp 38: 3025–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]