


Abstract
It has been previously shown that disruption of RAD26 in yeast strain W303-1B results in a strain that is deficient in transcription-coupled repair (TCR), the preferential repair of the transcribed strand of an expressed gene over the non-transcribed strand and the rest of the genome. RAD26 encodes a protein that is homologous to Cockayne syndrome group B protein (CSB) and is a member of the SWI2/SNF2 family of DNA-dependent ATPases involved in chromatin remodeling. Like the rad26 mutant, cells from Cockayne syndrome patients are defective in TCR. We examined the role of Rad26 in TCR by disrupting RAD26 in two repair-proficient laboratory strains and, remarkably, observed no effect upon TCR. Our results indicate that disruption of RAD26 alone is insufficient to impair TCR. Thus, W303-1B must already possess a mutation that, together with disruption of RAD26, causes a deficiency in TCR. We suggest that other genes are mutated in Cockayne syndrome cells that contribute to the deficiency in TCR. Surprisingly, deletion of RAD26 results in expression of genes that are repressed by flanking transposon δ elements, an Spt– phenotype. The δ elements appear to perturb local chromatin structure. Expression of genes flanked by δ elements in rad26Δ mutants is consistent with a role for Rad26 in chromatin remodeling.
INTRODUCTION
Nucleotide excision repair is a major pathway dealing with the removal of DNA damage found in all organisms examined thus far. The biochemical mechanism of nucleotide excision repair has emerged for prokaryotes and eukaryotes. Identification and cloning of genes involved in nucleotide excision repair, as well as expression of their products, has enabled the reconstitution of repair in vitro (1–4). In a multistep process, eukaryotic nucleotide excision repair proteins interact with the DNA damage, make dual single-strand incisions 3′ and 5′ to the damage in the strand containing the damage, followed by removal of the damage as part of a 24–26 bp oligonucleotide in yeast and a 27–29 bp oligonucleotide in humans (5). Subsequently, DNA polymerases, DNA ligase and additional proteins fill in the resulting gap to regenerate intact duplex DNA.
Nucleotide excision repair does not occur uniformly throughout the genome of prokaryotes and eukaryotes (6,7). The transcribed strands of expressed genes are repaired faster than the non-transcribed strands and the genome overall. This faster repair of the transcribed strand is called transcription-coupled repair (TCR). It has been suggested that repair of the transcribed strand is directly coupled to transcription and that recognition of the DNA lesion by repair proteins may be facilitated by a stalled RNA polymerase complex at the site of DNA damage. While referred to as TCR, it has not been definitively shown that repair is directly coupled to transcription or, rather, the increased repair is a consequence of disrupted chromatin (facilitated by Rad26) preceding or subsequent to transcription through a DNA sequence.
The human disease Cockayne syndrome (CS) is associated with a defect in TCR (8–10). The CSA and CSB genes have been identified as uniquely affecting TCR in mammalian cells. CSB, also known as ERCC6, complements the deficiency in TCR in rodent cells of complementation group 6 and is mutated in CS cells from complementation group B (CS-B) (11,12). A yeast gene with homology to the human CSB gene (RAD26) was isolated, sequenced (13,14) and a rad26 deletion mutant created (14). Consistent with the DNA repair deficiency observed in cultured cells from CS patients, the yeast rad26 mutant lacks TCR (14). When grown in the presence of glucose, a rad26 mutant shows approximately the same rate and extent of removal of cyclobutane pyrimidine dimers (CPDs) from the transcribed strands as from the non-transcribed strands of the repressed GAL7 gene and the constitutive RPB2 gene, i.e. there is no TCR (14–16). However, when grown in the presence of galactose, this same rad26 mutant is proficient for TCR in the induced GAL7 (15) and the constitutive RPB2 genes (16). Therefore, there appear to be Rad26-dependent and Rad26-independent TCR pathways.
To date, repair rates for the rad26 mutant have all been determined in one genetic background, W303-1B. We deleted rad26 in three other strains of different genetic backgrounds and examined their ability to carry out TCR. Surprisingly, we observed no defect in TCR of an expressed gene in these new rad26 mutants. Importantly, deletion of RAD26 enabled expression from genes flanked by transposon δ elements, i.e. an Spt– phenotype. The δ elements alter local chromatin structure and gene expression. Suppression of δ element phenotypes suggests a role for Rad26 in chromatin remodeling or transcription elongation. Our results lead us to suggest that Rad26 (or CSB) is necessary but not sufficient to enable TCR.
MATERIALS AND METHODS
Deletion of RAD26, PCR, Southern analyses and RNA probes
Chimeric PCR primers were designed to delete the RAD26 gene. The primers were used to amplify the TRP1 gene and the PCR product was used to transform yeast and delete the RAD26 locus. The rad26Δ::TRP1 deletion mutants were selected on synthetic complete medium (6.7% yeast nitrogen base, 2.0% dextrose and the appropriate bases and amino acids) lacking the amino acid tryptophan (17). Haploid mutant strains were grown in YPD (1.0% yeast extract, 2.0% peptone, 2.0% dextrose; Gibco BRL) and genomic DNA was isolated and purified as described before (18). Genomic DNA was used for PCR reactions to screen mutants for the deletion allele using two oligonucleotides designed to amplify either wild-type or the deleted RAD26 locus (Fig. 2). After PCR, the presence of the deletion allele was confirmed by Southern analysis of the RAD26 locus (data not shown). To make radioactive RNA probes, plasmid pKS212, carrying the 1 kb EcoRI–XhoI fragment of the large subunit of RNA polymerase II (RPB2) in pBluescript (Stratagene), was linearized by digestion with either XhoI for the non-transcribed strand- or EcoRI for the transcribed strand-specific RNA probe of the gene. Transcribed strand and non-transcribed strand RNA probes were synthesized with either T7 RNA polymerase from the T7 promoter or T3 RNA polymerase from the T3 promoter (Boehringer Mannheim), respectively, and incubation with rNTPs and [α-32P]CTP (Amersham Corp.) as recommended by the manufacturer.
Figure 2.
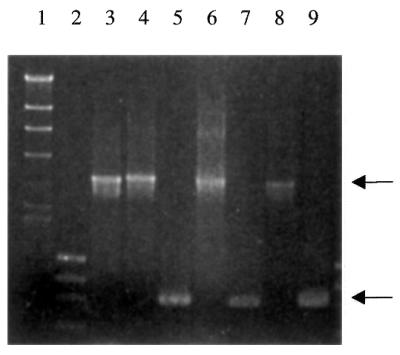
Confirmation of deletion of RAD26 in the SK1 background. PCR primers complementary to sequences immediately upstream and downstream of the RAD26 gene were used to amplify the intervening sequences from the chromosomal locus of haploid wild-type and rad26Δ::TRP1 cells. The wild-type locus yields a product of 3.2 kb (top arrow). The rad26::HIS3 disruption allele yields a product of 3.4 kb and migrates slightly slower than the PCR product from the wild-type locus. The rad26Δ::TRP1 mutant yields a product of 936 bp (bottom arrow), indicative of the deleted allele. Lane 1, λ DNA cut with HindIII; lane 2, φχ DNA cut with HaeIII; lane 3, W303-1B; lane 4, rad26::HIS3 (W303-1B); lane 5, rad26Δ::TRP1 (W303-1B); lane 6, SK1; lane 7, rad26Δ::TRP1 (SK1); lane 8, FY58; lane 9, rad26Δ::TRP1 (FY58).
Strains, meiotic induction and tetrad analysis
Strains used in the current study are described in Table 1. Wild-type strains and heterozygous and homozygous diploid rad26Δ mutants were sporulated in medium containing 1.0% potassium acetate, 0.05% glucose, 0.1% yeast extract and the appropriate amino acids (18). Asci were digested with a β-glucuronidase preparation from Helix pomatia (Sigma Corp.) in suspension in sterile water for 10 min at room temperature. Ten microliters of the suspension was spread across YPD plates and allowed to dry, after which asci were broken apart and spores separated using a Narishige micromanipulator mounted on a CK2 Olympus microscope.
Table 1. Strains used in this study.
| Strain |
Genotype |
Source |
| LNY1 | MATa lys2, ura3, leu2::hisG, trp1::hisG, ho::LYS2 | L. Neigeborn |
| LNY2 | MATα lys2, ura3, leu2::hisG, trp1::hisG, ho::LYS2 | L. Neigeborn |
| LNY3 | MATa/α lys2/lys2, ura3/ura3, leu2::hisG/leu2::hisG, trp1::hisG/trp1::hisG, ho::LYS2/ho::LYS2 | L. Neigeborn |
| LNY40 | MATa ade6 mating type testers | L. Neigeborn |
| LNY41 | MATα ade6 mating type testers | L. Neigeborn |
| SGY1 | MATa lys2, ura3, leu2::hisG, trp1::hisG, ho::LYS2 | This study |
| SGY2 | MATa lys2, ura3, leu2::hisG, trp1::hisG, ho::LYS2, rad26Δ::TRP1 | This study |
| SGY5 | MATa/α lys2/lys2, ura3/ura3, leu2::hisG/leu2::hisG, trp1::hisG/trp1::hisG, ho::LYS2/ho::LYS2, rad26Δ::TRP1/RAD26 | This study |
| SGY6 | MATa/α lys2/lys2, ura3/ura3, leu2::hisG/leu2::hisG, trp1::hisG/trp1::hisG, ho::LYS2/ho::LYS2, rad26Δ::TRP1/rad26Δ::TRP1 | This study |
| W303-1B | MATα ho, ade2-1, trp1-1, leu2-3,112, can1-100, his3-11,15, ura3-1 | A. van Gool |
| MGSC102 | MATα ho, ade2-1, trp1-1, leu2-3,112, can1-100, his3-11,15, ura3-1, rad26::HIS3 | A. van Gool |
| SGY7 | MATα ho, ade2-1, trp1-1, leu2-3,112, can1-100, his3-11,15, ura3-1, rad26Δ::TRP1 | This study |
| FY56 | MATα his4-912δ, lys2-128δ, ura3-52 | J. Huibregtse |
| SGY8 | MATα his4-912δ, lys2-128δ, ura3-52, rad26::HIS3 | This study |
| FY58 | MATa his4-912δ, lys2-128δ, ura3-52, trp1Δ63 | M. Hampsey |
| FY111 | MATa his4-912δ, lys2-128δ, ura3-52, trp1Δ63, spt6-140 | M. Hampsey |
| FY114 | MATa his4-912δ, lys2-128δ, ura3-52, trp1Δ63, leu2Δ1, his3Δ200, spt4Δ2::HIS3 | M. Hampsey |
| L735 | MATa his4-912δ, lys2-128δ, ura3-52, trp5, leu1, spt4Δ1::URA3 | M. Hampsey |
| SGY9 | MATa his4-912δ, lys2-128δ, ura3-52, trp1Δ63, rad26Δ::TRP1 | This study |
UV irradiation and isolation and restriction of yeast DNA
Growth and irradiation of strains and purification of DNA were as previously described (19). Cell walls were digested with Zymolyase 100T (ICN Biochemicals) and lysed by addition of sarcosine (20). Chromosomal DNA was purified as described (21). The DNA samples were then restricted to completion with HincII. Restricted DNA was then ethanol precipitated and resuspended in TE (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, pH 8.0).
Repair analysis
The incidence of CPDs in each strand of a specific restriction fragment was determined by previously developed methods (6,19,22). Equal amounts of purified, restricted DNA were either mock treated (10 mM Tris–HCl, pH 7.5, 0.1 M NaCl, 10 mM EDTA, 1 mg/ml BSA) or treated with the enzyme T4 endonuclease V in mock buffer for 30 min at 37°C. T4 endonuclease V is a CPD-specific DNA glycosylase/AP lyase which produces a single-strand break specifically at each CPD. DNA was denatured and electrophoresed through 0.5% alkaline agarose gels to separate full-length restriction fragments from the digestion products of T4 endonuclease V, transferred to Hybond N+ (Amersham) membranes and hybridized with strand-specific 32P-labeled RNA probes generated by in vitro transcription of linearized pKS212. Autoradiograms were generated and signal intensities determined using a Hewlett Packard Scanjet II and the application NIH Image v.1.62. The Poisson expression was applied to calculate the number of CPDs per fragment from the ratio of intensities of bands from T4 endonuclease V-treated and mock-treated DNA samples.
RESULTS
Deletion of RAD26 is insufficient to abolish TCR
To date, repair rates for rad26 mutants have all been determined in one genetic background, W303-1B. A portion of the RAD26 gene was previously deleted and replaced with the HIS3 gene in W303-1B (14). These strains are defective for TCR, as demonstrated previously (14) and shown in Figure 1A and B. The transcribed strand has 40–45% CPDs removed 30 min following UV irradiation. Repair of the non-transcribed strand is slightly slower than the transcribed strand, reaching ∼25% CPDs removed by 30 min post-irradiation. Despite being referred to as TCR-deficient, we observed significantly faster repair of the transcribed strand over the non-transcribed strand with this rad26 mutant (14,15). Consistent with our data, others also noted faster repair of the transcribed strands relative to repair of the non-transcribed strands in the same rad26 mutant (14,15). We disrupted RAD26 in three other strains with different genetic backgrounds and examined their ability to carry out TCR. The entire RAD26 open reading frame was deleted and replaced with the TRP1 gene in W303-1B, FY58 and SK1. The RAD26 gene was replaced with the original rad26::HIS3 allele in strain FY56. Deletions were confirmed using PCR (Fig. 2) and/or hybridization of gene-specific probes to restriction enzyme-digested genomic DNA immobilized on nylon membranes (Southern blots) (data not shown). As has been previously shown for rad26::HIS3 mutants in W303-1B, rad26Δ::TRP1 mutants in W303-1B are also deficient in TCR (Fig. 3A and B). The transcribed strand has 45% CPDs removed 30 min following UV irradiation while repair of the non-transcribed strand is slightly slower, reaching 25–30% CPDs removed by 30 min post-irradiation. Thus, both alleles of RAD26 result in a deficiency in TCR in the W303-1B strain. However, introducing either the original rad26::HIS3 or the new rad26Δ::TRP1 allele into other strain backgrounds gave markedly different results.
Figure 1.

TCR assay for a rad26::HIS3 mutant and the parent strain W303-1B. (A) Exponentially growing cultures of rad26::HIS3 and the parent strain (W303-1B) at 30°C were irradiated with 65 J/m2 UV radiation and incubated in growth medium for the times indicated. DNA was prepared from the cells as described in Materials and Methods. U, unirradiated. (B) Removal of CPDs from the RPB2 gene in rad26::HIS3 and the parent strain W303-1B. Repair of transcribed and non-transcribed strands was determined from the measured incidences of CPDs in the HincII restriction fragments from RPB2. A graphical representation of repair at the times indicated is shown. Data points represent the average of four independent biological experiments. Open square, W303-1B transcribed strand; filled square, W303-1B non-transcribed strand; open circle, rad26::HIS3 transcribed strand; filled circle, rad26::HIS3 non-transcribed strand.
Figure 3.

TCR assay for a new rad26Δ::TRP1 mutant and the parent strain W303-1B. (A) Exponentially growing cultures of rad26Δ::TRP1 and the parent strain (W303-1B) at 30°C were irradiated with 65 J/m2 UV radiation and incubated in growth medium for the times indicated. DNA was prepared from the cells as described in Materials and Methods. (B) Removal of CPDs from the RPB2 gene in rad26Δ::TRP1 and the parent strain W303-1B. Repair of transcribed and non-transcribed strands was determined from the measured incidences of CPDs in the HincII restriction fragments from RPB2. A graphical representation of repair at the times indicated is shown. Data points represent the average of four independent biological experiments. Open square, W303-1B transcribed strand; filled square, W303-1B non-transcribed strand; open circle, rad26Δ::TRP1 transcribed strand; filled circle, rad26Δ::TRP1 non-transcribed strand.
SK1 strains containing a deletion allele of RAD26 were examined for their ability to perform TCR. Surprisingly, we observed no defect in TCR of an expressed gene in these new rad26Δ mutants. As is apparent in Figure 4A, restoration of hybridization signal in the T4 endonuclease V-treated samples indicates rapid repair of the transcribed strand in both strains. Repair of the non-transcribed strand was slower than repair of the transcribed strand. Data are presented graphically in Figure 4B. The transcribed strand is largely free of CPDs by 60 min following UV irradiation (∼90% removed). Repair of the non-transcribed strand is slower than the transcribed strand, reaching ∼60% CPDs removed by 60 min post-irradiation. Clearly there is no defect in TCR in rad26 mutants in the SK1 background. We note that repair of the non-transcribed strand in rad26 mutants in the SK1 background is significantly elevated at 90 min following UV irradiation relative to the repair observed in the parent strain (>90% versus ∼65% CPDs removed).
Figure 4.

Deletion of RAD26 does not abolish TCR of UV-induced damage in an SK1 strain. (A) TCR assay for a rad26Δ mutant in an SK1 genetic background. Exponentially growing cultures of rad26Δ and the parent strain at 30°C were irradiated with 65 J/m2 UV radiation and incubated in growth medium for the times indicated. DNA was prepared from the cells as described in Materials and Methods. (B) Removal of CPDs from the RPB2 gene in rad26Δ::TRP1 and the parent strain SK1. Repair of transcribed and non-transcribed strands was determined from the measured incidences of CPDs in the HincII restriction fragments from RPB2. A graphical representation of repair at the times indicated is shown. Data points represent the average of four independent biological experiments. Open square, SK1 transcribed strand; filled square, SK1 non-transcribed strand; open circle, rad26Δ::TRP1 transcribed strand; filled circle, rad26Δ::TRP1 non-transcribed strand.
We examined the ability of the rad26Δ mutants in the FY56 and FY58 backgrounds to perform TCR and global genomic repair. As shown in Figure 5A, repair of the transcribed strand in the rad26Δ::TRP1 mutant was faster than repair of the non-transcribed strand in the rad26Δ::TRP1 mutant and the parent strain FY58. Similar repair was observed in a rad26::HIS3 mutant and its parent strain FY56 (data not shown). Repair in both rad26Δ::TRP1 and rad26::HIS3 mutants and the parent strains is shown graphically in Figure 5B. Greater than 70% of the CPDs were removed from the transcribed strand by 30 min following UV irradiation. The non-transcribed strand showed ∼35% CPDs removed by 30 min following UV irradiation (Fig. 5B). The repair observed is indicative of proficient TCR in these rad26Δ strains.
Figure 5.

Deletion of RAD26 does not abolish TCR of UV-induced damage in FY58 and FY56 strains. (A) TCR assay for rad26Δ mutants in an FY58 strain. Exponentially growing cultures of a rad26Δ::TRP1 mutant and the parent strain FY58 at 30°C were irradiated with 65 J/m2 UV radiation and incubated in growth medium for the times indicated. DNA was prepared from the cells as described in Materials and Methods. (B) Removal of CPDs from the RPB2 gene in rad26 mutants and the parent strains. Repair of transcribed and non-transcribed strands was determined from the measured incidences of CPDs in the HincII restriction fragments from RPB2. A graphical representation of repair at the times indicated is shown. Data points represent the average of four independent biological experiments, except for rad26Δ::TRP1, which was performed three times. Open square, FY58 transcribed strand; filled square, FY58 non-transcribed strand; open circle, rad26Δ::TRP1 transcribed strand; filled circle, rad26Δ::TRP1 non-transcribed strand; open triangle, FY56 transcribed strand; filled triangle, FY56 non-transcribed strand; open diamond, rad26::HIS3 transcribed strand; filled diamond, rad26::HIS3 non-transcribed strand.
Deletion of RAD26 yields an Spt– phenotype
The original rad26::HIS3 deletion allele (14) was amplified by PCR and the linear PCR product was used to transform yeast strain FY56. Confirmation of the rad26::HIS3 allele in FY56 was by PCR (data not shown). The HIS3 gene should not complement the histidine auxotrophy of cells carrying the his4-912δ allele in FY56 unless deletion of RAD26 yields the Spt– phenotype (23–25). Similarly, expression of Lys2 from lys2-128δ enables growth on synthetic complete medium lacking lysine only if deletion of RAD26 gives the Spt– phenotype. The Spt– phenotype appears to be dependent upon changes in the cellular capacity to remodel chromatin. Rad26 is a member of the Swi2/Snf2 family of DNA-dependent ATPases that are thought to function in chromatin remodeling. We examined both FY56 rad26::HIS3 and FY58 rad26Δ::TRP1 mutants for ability to grow on synthetic complete medium lacking lysine and histidine, i.e. exhibit a Spt– phenotype. Both the FY56 rad26::HIS3 and FY58 rad26Δ::TRP1 mutants were able to grow on medium lacking histidine and lysine. The parent strains were unable to grow on medium lacking either histidine or lysine (Fig. 6). Thus, deletion of RAD26 results in a Spt– phenotype.
Figure 6.
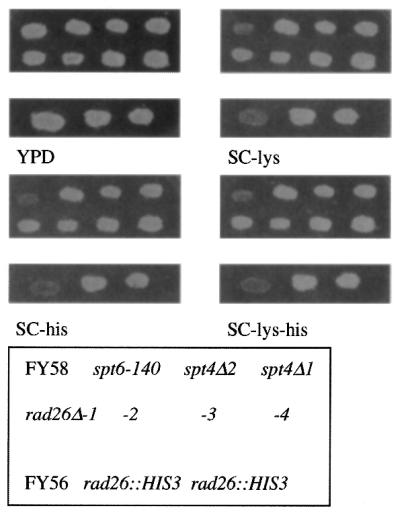
Deletion of RAD26 results in a Spt– phenotype, indicative of chromatin alterations. Two different rad26Δ mutants and their respective wild-type parent strains were grown on YPD plates and then replica-plated onto YPD, SC-his, SC-lys and SC-his,lys plates. Plates were incubated at 30°C for 3–5 days.
Strains carrying mutations in either SPT4 or SPT6 are repair proficient
Given the Spt– phenotype we observed for rad26 mutants, we wished to see if spt mutants, thought to be involved in chromatin remodeling and/or transcription elongation, might be defective in TCR or global genome repair. Repair experiments with spt mutants derived from strain FY58 were performed as above. Repair of the transcribed strand of RPB2 in spt4Δ1::URA3 and spt4Δ2::HIS3 was as rapid as in the parent strains (Fig. 7). Likewise, repair of the non-transcribed strand of RPB2 was at the same rate as repair in the parent strains. Therefore, mutations in the SPT4 or SPT6 gene do not singly affect the ability of these strains to efficiently repair the transcribed and non-transcribed strands of an expressed gene (Fig. 7).
Figure 7.

Mutation of SPT4 or SPT6 does not affect TCR of UV-induced damage in the FY58 background. (A) TCR assay for spt4 and spt6 mutants in an FY58 strain. Exponentially growing cultures of spt4 or spt6 mutants and the parent strain FY58 at 30°C were irradiated with 65 J/m2 UV radiation and incubated in growth medium for the times indicated. DNA was prepared from the cells as described in Materials and Methods. (B) Removal of CPDs from the RPB2 gene in spt mutants and the parent strain. Repair of transcribed and non-transcribed strands was determined from the measured incidences of CPDs in the HincII restriction fragments from RPB2. A graphical representation of repair at the times indicated is shown. Data points represent the average of four independent biological experiments. Open diamond, FY58 transcribed strand; filled diamond, FY58 non-transcribed strand; open circle, spt6-140 transcribed strand; filled circle, spt6-140 non-transcribed strand; open square, spt4Δ2 transcribed strand; filled square, spt4Δ2 non-transcribed strand; open triangle, spt4Δ1 transcribed strand; filled triangle, spt4Δ1 non-transcribed strand.
Deletion of RAD26 causes no defects in mating or conditional growth phenotypes
Haploid rad26Δ::TRP1 mutants were mated with wild-type and rad26Δ::TRP1 strains. Mating efficiencies were the same for wild-type and rad26Δ::TRP1 strains as compared to mating of wild-type to wild-type strains. Thus, there does not appear to be significant derepression of the silent mating loci in the rad26Δ::TRP1 mutants relative to the wild-type strains.
We also observed no cold-sensitive or heat-sensitive phenotypes at 15 or 37°C, respectively, for the rad26Δ strains, as has been reported previously for spt mutations. We conclude that Rad26 may not form an integral part of protein complexes involved in chromatin remodeling and/or transcription, as reported for the Spt4, Spt5 and Spt6 proteins (26).
DISCUSSION
The RAD26 gene, the homolog of the human CS group B gene, was reported to enable TCR in yeast. Like cells from CS patients, yeast rad26::HIS3 mutants lack TCR. To date, all repair studies with yeast carrying the rad26::HIS3 deletion were in the W303 background. We have examined TCR in rad26 deletion alleles in different genetic backgrounds and found that the defect in TCR was strain dependent. We found that the rad26::HIS3 mutant and a new deletion mutant, rad26Δ::TRP1, in the W303-1B background exhibit a deficiency in TCR relative to other repair-proficient laboratory strains. No defect was observed in these strains for repair of the non-transcribed strands of expressed genes. Thus, repair of the genome overall is proficient based on the strong correlation between repair of the non-transcribed strand and overall genomic repair (27). Deletion of RAD26 in the three other strains had no effect upon TCR. Based on our results, we suggest that another protein is required to enable TCR, along with Rad26. Deficiency in either activity is insufficient to abolish TCR. Deficiency in both activities leads to a loss of TCR. It may be that W303 is already defective for this second activity. Our results suggest that suppressors of a TCR defect in the W303 background might not suppress rad26 (28), but may be suppressing another mutation already present in W303-1B. Such a possibility remains to be demonstrated.
A role for Rad26 and CSB in TCR was suggested based on their homology to members of the Swi/Snf family of DNA-dependent ATPases (29). That model stated that Rad26 and CSB might be responsible for displacing the RNA polymerase II stalled at the site of DNA damage, enabling repair of the newly uncovered damage. Several members of the Swi/Snf family are proteins with demonstrated roles in chromatin remodeling and removal of proteins bound to chromatin (30–33). Remodeling of chromatin enhances the binding of transcription factors and presumably DNA repair proteins to nucleosomal DNA. Such properties suggest that Rad26 also functions in chromatin remodeling and/or protein displacement. Does Rad26 act as a DNA-dependent ATPase which is activated by binding to damaged DNA in the relatively nucleosome-free template strand that is undergoing transcription? Recently, Vermeulen and colleagues demonstrated that CSB protein could remodel nucleosomes in vitro (34). Furthermore, can other members of the Swi/Snf family fulfill the role of Rad26 in TCR?
Consistent with a role in chromatin remodeling is our observation that deletion of RAD26 results in suppression of δ insertion mutations in the 5′-non-coding region of the HIS4 and LYS2 genes (Spt– phenotype) (23–25). The δ element is a copy of the Ty transposon long terminal repeat. In wild-type cells insertion of Ty or solo δ elements upstream of genes leads to altered transcription initiation start sites and non-functional transcripts. Transcription can begin at the normal transcription start site and terminate within the δ element or transcription can begin within the δ element and terminate at the normal termination site. The result is either a truncated mRNA or an mRNA that is not read in the normal reading frame, respectively. Thus, wild-type cells containing his4-912δ or lys2-128δ alleles cannot grow in the absence of exogenous histidine or lysine, i.e. they have a His– or Lys– phenotype. Mutations in a number of genes encoding proteins involved in chromatin structure or remodeling alter the transcription start site such that mRNAs encoding functional His4 and Lys2 proteins are produced. For example, mutations in the SWI2/SNF2 or SPT6 genes result in his4-912δ or lys2-128δ strains that are able to grow on selective media lacking histidine or lysine, respectively. They have His+ and Lys+ phenotypes. We found that partial deletion of RAD26 with HIS3 or complete deletion of RAD26 with TRP1 in an FY56 or FY58 strain, respectively, enables growth on selective medium lacking histidine or lysine, or both. Thus, rad26 mutants have an Spt– phenotype. However, the fact that deletion of RAD26 does not result in a TCR defect suggests that another protein, perhaps another member of the Swi/Snf family, is able to substitute for Rad26 and enable TCR.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Lori Lommel, Miriam Bucheli, Melvin Limson, Kiran Madura and Lenore Neigeborn for helpful suggestions and critical reading of this manuscript. This work was supported in part by Public Health Service grant R29 GM53717 from the National Institutes of Health.
References
- 1.Lin J.J. and Sancar,A. (1992) Active site of (A)BC excinuclease. 1. Evidence for 5′ incision by UvrC through a catalytic site involving Asp399, Asp438, Asp466, His538 residues. J. Biol. Chem., 267, 17688–17692. [PubMed] [Google Scholar]
- 2.Lin J.J., Phillips,A.M., Hearst,J.E. and Sancar,A. (1992) Active site of (A)BC excinuclease. 2. Binding, bending and catalysis mutants of UvrB reveal a direct role in 3′ and an indirect role in 5′ incision. J. Biol. Chem., 267, 17693–17700. [PubMed] [Google Scholar]
- 3.Guzder S.N., Habraken,Y., Sung,P., Prakash,L. and Prakash,S. (1995) Reconstitution of yeast nucleotide excision repair with purified Rad proteins, replication protein A and transcription factor TFIIH. J. Biol. Chem., 270, 12973–12976. [DOI] [PubMed] [Google Scholar]
- 4.Aboussekhra A., Biggerstaff,M., Shivji,M.K., Vilpo,J.A., Moncollin,V., Podust,V.N., Protic,M., Hubscher,U., Egly,J.M. and Wood,R.D. (1995) Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell, 80, 859–868. [DOI] [PubMed] [Google Scholar]
- 5.Huang J.-C., Svoboda,D.L., Reardon,J.T. and Sancar,A. (1992) Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc. Natl Acad. Sci. USA, 89, 3664–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellon I., Spivak,G. and Hanawalt,P.C. (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell, 51, 241–249. [DOI] [PubMed] [Google Scholar]
- 7.Mellon I. and Hanawalt,P.C. (1989) Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature, 342, 95–98. [DOI] [PubMed] [Google Scholar]
- 8.Venema J., Mullenders,L.H.F., Natarajan,A.T., van Zeeland,A.A. and Mayne,L.V. (1990) The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl Acad. Sci. USA, 87, 4707–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Hoffen A., Natarajan,A.T., Mayne,L.V., van Zeeland,A.A., Mullenders,L.H.F. and Venema,J. (1993) Deficient repair of the transcribed strand of active genes in Cockayne’s syndrome cells. Nucleic Acids Res., 21, 5890–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tu Y., Bates,S. and Pfeifer,G.P. (1997) Sequence-specific and domain-specific DNA repair in xeroderma pigmentosum and Cockayne syndrome cells. J. Biol. Chem., 272, 20747–20755. [DOI] [PubMed] [Google Scholar]
- 11.Troelstra C., Odijk,H., de Wit,J., Westerveld,A., Thompson,L.H., Bootsma,D. and Hoeijmakers,J.H.J. (1990) Molecular cloning of the human DNA excision repair gene ERCC-6. Mol. Cell. Biol., 10, 5806–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Troelstra C., van Gool,A., de Wit,J., Vermeulen,W., Bootsma,D. and Hoeijmakers,J.H.J. (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell, 71, 939–953. [DOI] [PubMed] [Google Scholar]
- 13.Huang M.E., Chuat,J.C. and Galibert,F. (1994) A possible yeast homolog of human active-gene-repairing helicase ERCC6+. Biochem. Biophys. Res. Commun., 201, 310–317. [DOI] [PubMed] [Google Scholar]
- 14.van Gool A.J., Verhage,R., Swagemakers,S.M., van de Putte,P., Brouwer,J., Troelstra,C., Bootsma,D. and Hoeijmakers,J.H. (1994) RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J., 13, 5361–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verhage R.A., van Gool,A.J., de Groot,N., Hoeijmakers,J.H., van de Putte,P. and Brouwer,J. (1996) Double mutants of Saccharomyces cerevisiae with alterations in global genome and transcription-coupled repair. Mol. Cell. Biol., 16, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bucheli M., Lommel,L. and Sweder,K. (2001) The defect in transcription-coupled repair displayed by a rad26 mutant is dependent on carbon source and is not associated with a lack of transcription. Genetics, 158, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothstein R.J. (1983) One-step gene disruption in yeast. Methods Enzymol., 101, 202–211. [DOI] [PubMed] [Google Scholar]
- 18.Sherman F., Fink,G. and Hicks,J. (1986) Laboratory Course Manual for Methods In Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 19.Sweder K.S. and Hanawalt,P.C. (1992) Preferential repair of cyclobutane pyrimidine dimers in the transcribed strand of a gene in yeast chromosomes and plasmids is dependent on transcription. Proc. Natl Acad. Sci. USA, 89, 10696–10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nasmyth K.A. and Reed,S.I. (1980) Isolation of genes by complementation in yeast: molecular cloning of a cell-cycle gene. Proc. Natl Acad. Sci. USA, 77, 2119–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 22.Bohr V.A., Smith,C.A., Okumoto,D.S. and Hanawalt,P.C. (1985) DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell, 40, 359–369. [DOI] [PubMed] [Google Scholar]
- 23.Simchen G., Winston,F., Styles,C.A. and Fink,G.R. (1984) Ty-mediated gene expression of the LYS2 and HIS4 genes of Saccharomyces cerevisiae is controlled by the same SPT genes. Proc. Natl Acad. Sci. USA, 81, 2431–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winston F., Chaleff,D.T., Valent,B. and Fink,G.R. (1984) Mutations affecting Ty-mediated expression of the HIS4 gene of Saccharomyces cerevisiae. Genetics, 107, 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winston F., Durbin,K.J. and Fink,G.R. (1984) The SPT3 gene is required for normal transcription of Ty elements in S. cerevisiae. Cell, 39, 675–682. [DOI] [PubMed] [Google Scholar]
- 26.Bortvin A. and Winston,F. (1996) Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science, 272, 1473–1476. [DOI] [PubMed] [Google Scholar]
- 27.Sweder K.S. and Hanawalt,P.C. (1994) The COOH terminus of suppressor of stem loop (SSL2/Rad25) in yeast is essential for overall genomic excision repair and transcription-coupled repair. J. Biol. Chem., 269, 1852–1857. [PubMed] [Google Scholar]
- 28.Jansen L.E., den Dulk,H., Brouns,R.M., de Ruijter,M., Brandsma,J.A. and Brouwer,J. (2000) Spt4 modulates Rad26 requirement in transcription-coupled nucleotide excision repair. EMBO J., 19, 6498–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisen J.A., Sweder,K.S. and Hanawalt,P.C. (1995) Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res., 23, 2715–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winston F. and Carlson,M. (1992) Yeast SNF/SWI transcriptional activators and the SPT/SIN chromatin connection. Trends Genet., 8, 387–391. [DOI] [PubMed] [Google Scholar]
- 31.Struhl K. (1996) Chromatin structure and RNA polymerase II connection: implications for transcription. Cell, 84, 179–182. [DOI] [PubMed] [Google Scholar]
- 32.Auble D.T., Hansen,K.E., Mueller,C.G., Lane,W.S., Thorner,J. and Hahn,S. (1994) Mot1, a global repressor of RNA polymerase II transcription, inhibits TBP binding to DNA by an ATP-dependent mechanism. Genes Dev., 8, 1920–1934. [DOI] [PubMed] [Google Scholar]
- 33.Owen-Hughes T., Utley,R.T., Côté,J., Peterson,C.L. and Workman,J.L. (1996) Persistent site-specific remodelling of a nucleosome array by transient action of the SWI/SNF complex. Science, 273, 513–516. [DOI] [PubMed] [Google Scholar]
- 34.Citterio E., Van den Boom,V., Schnitzler,G., Kanaar,R., Bonte,E., Kingston,R.E., Hoeijmakers,J.H.J. and Vermeulen,W. (2000) ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol., 20, 7643–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]