

Abstract
Understanding how phenotypic differences between males and females arise from the sex-biased expression of nearly identical genomes can reveal important insights into the biology and evolution of a species. Among Anopheles mosquito species, these phenotypic differences include vectorial capacity, as it is only females that blood feed and thus transmit human malaria. Here, we use RNA-seq data from multiple tissues of four vector species spanning the Anopheles phylogeny to explore the genomic and evolutionary properties of sex-biased genes. We find that, in these mosquitoes, in contrast to what has been found in many other organisms, female-biased genes are more rapidly evolving in sequence, expression, and genic turnover than male-biased genes. Our results suggest that this atypical pattern may be due to the combination of sex-specific life history challenges encountered by females, such as blood feeding. Furthermore, female propensity to mate only once in nature in male swarms likely diminishes sexual selection of post-reproductive traits related to sperm competition among males. We also develop a comparative framework to systematically explore tissue- and sex-specific splicing to document its conservation throughout the genus and identify a set of candidate genes for future functional analyses of sex-specific isoform usage. Finally, our data reveal that the deficit of male-biased genes on the X Chromosomes in Anopheles is a conserved feature in this genus and can be directly attributed to chromosome-wide transcriptional regulation that de-masculinizes the X in male reproductive tissues.
One of the most extreme examples of phenotypic variation between individuals of the same species is the difference between the sexes. However, apart from sex-limited Y and W Chromosomes that are typically gene poor and small, males and females share a common genome. Sexual dimorphism is therefore largely achieved through sex-biased expression or splicing of this shared genome throughout development (Connallon and Knowles 2005; Ellegren and Parsch 2007). Genome sharing, however, imposes evolutionary constraints on chromosomal and genic content, because these must respond to different regimes of natural and sexual selection in each sex. Mutations that lead to changes in gene expression or coding sequence can even result in conflicting fitness effects—beneficial in one sex but detrimental in the other—a process called sexual antagonism (Rice 1996, 1998; Chippindale et al. 2001; Chapman et al. 2003).
A common pattern that has emerged from comparative genomic studies of sex-biased genes across metazoans is that male-biased genes are typically more rapidly evolving than female-biased genes: they display higher birth and death rates (turnover), higher rates of expression divergence and sex-bias switching, and a lower conservation of protein sequences (Torgerson et al. 2002; Reinke et al. 2004; Zhang et al. 2004, 2007; Cutter and Ward 2005; Good and Nachman 2005; Khaitovich et al. 2005; Assis et al. 2012; Harrison et al. 2015; Wang et al. 2015). However, among these studied model organisms, the majority of phenotypic variation between the sexes is centered on the act of mating, rather than substantial divergence in other life history traits. In this regard, mosquito species may provide interesting insights for the study of sexual dimorphism within a more diverse context, as adult mosquitoes exhibit a rich repertoire of unique sex-specific biology related to host-seeking, blood-meal ingestion, and encounters with parasites and pathogens, since it is only females that feed on blood to enable egg production. In previous work, we have cataloged sex-biased gene expression of Anopheles gambiae in both adult and immature stages (Magnusson et al. 2011) and in dissected tissues using microarray platforms (Baker et al. 2011). However, the extent and conservation of sex-biased gene expression in other Anopheles species is unknown, thus limiting the comparative analysis to species outside of the Anopheles genus. The recent completion of the genome sequencing of 16 additional Anopheles species (Neafsey et al. 2014) provides an opportunity to study the evolutionary consequences of sex-biased gene expression in much greater detail. Here, we leverage these newly assembled resources and generated complementary transcriptome data based on RNA-seq from four mosquito species spanning the genus, An. gambiae, An. arabiensis, An. minimus, and An. albimanus. These mosquito species span the entire phylogeny and are all principal malaria vectors in the territories where they are endemic. We use these data to explore genome-wide evolutionary rates of sex-biased genes in carcass and reproductive tissues separately and to determine whether their abundance and evolutionary properties reflect phenotypic and functional differences between the sexes.
Results
Cataloging sex-biased gene expression
Total mRNA was obtained from dissected reproductive or carcass tissues of male and female adults from four malaria vectors and differential expression analysis was performed using DESeq2 to identify sex-biased genes (see Supplemental Material for details; Supplemental Figs. S1, S2). Sex-biased genes were subdivided into one of three categories based on the magnitude of sex-biased expression: sex-specific, strong sex-bias (with a fold change [FC] >5) and weak sex-bias (2 > FC < 5) (Supplemental Table S1).
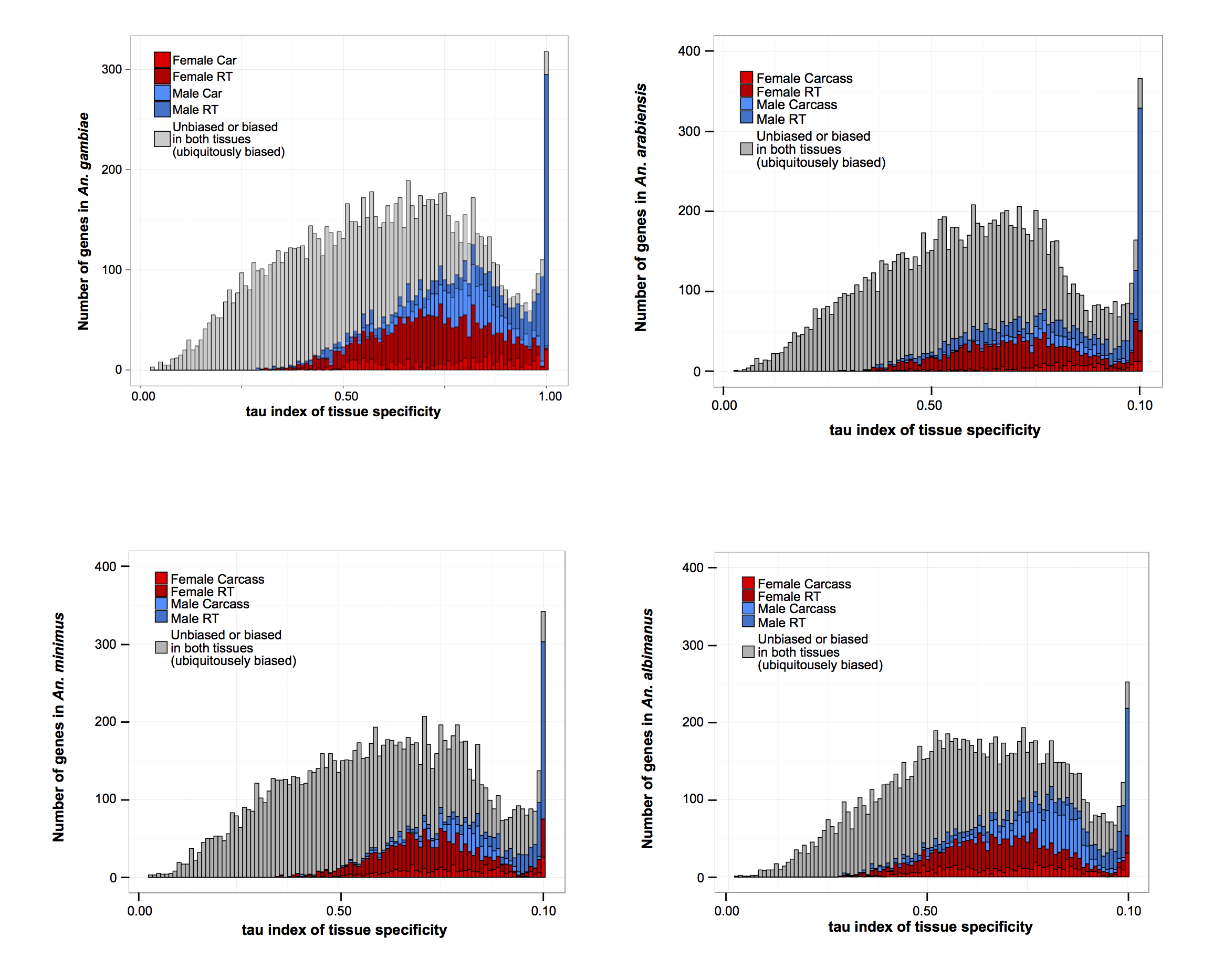
Consistent with findings in other species (Torgerson et al. 2002; Reinke et al. 2004; Zhang et al. 2004, 2007; Cutter and Ward 2005; Good and Nachman 2005; Khaitovich et al. 2005), we found that the majority of the sex-biased gene repertoire is derived from expression in the reproductive tissues, but female-biased genes predominated over male-biased genes (Fig. 1A)—likely as a result of blood-feeding which, as previously described, leads to genome-wide increases in gene expression (Marinotti et al. 2005). There was a higher correlation of levels of gene expression between the carcasses of the sexes (on average, 0.7 Pearson correlation, P-value < 2.2 × 10−16) than between the reproductive tissues of the sexes (average 0.12 Pearson correlation and P-value < 2.2 × 10−16) (Fig. 1B). Similar to other species, genes biased toward the male reproductive tissues were more likely to display sample-specific expression compared to genes biased in the other three tissues (shown for An. gambiae in Fig. 1C and remaining species in Supplemental Fig. S4) and showed a higher magnitude of sex-bias (Fig. 1E). To assess the reliability of our DE calls, we retrieved expression data for An. gambiae from the mosquito atlas (MozAtlas) study (Baker et al. 2011) and found that our classifications were consistent with these results (Fig. 1D).
Figure 1.

Sex-biased gene expression in the Anopheles genus. (A) Total number of sex-biased genes in the four study species for each of three tissue expression biases: carcass, reproductive tissues, and ubiquitously sex-biased. Female (red hues) and male (blue hues) sex-bias magnitudes are depicted as: dark, sex specific; medium, strongly sex-biased; light, weakly sex-biased. (B) Average Pearson's correlation coefficient of gene expression between sexed tissues in An. gambiae. (C) Distribution of tau (τ) index of tissue specificity in An. gambiae for each sex- and tissue-biased expression classification, where increasing τ values respond to increased sample-specific expression. Genes with very high τ values (∼1) are highly enriched for male reproductive tissue-biased genes. (D) Comparison of sex-bias gene expression categories, produced by this differential expression pipeline using RNA-seq, with results from a previous microarray study for An. gambaie, i.e., MozAtlas (Baker et al. 2011), showing strong congruence of gene expression bias in tissue classifications. (E) Magnitude of gene expression ratios (fold change male/female) for each species. Male-biased genes of the reproductive tissues display the largest magnitude and range of biased expression.
Evolution of sex-biased gene expression
To study the molecular evolution of sex-biased gene expression in the Anopheles genus, we compiled orthology relationships of all annotated genes of the four species and calculated expression and sequence divergence, as well as gene turnover rates separately for the carcass and the reproductive tissues. We cataloged 7400 one-to-one orthologs across the four species, referred to hereafter as the four-species orthologs, which most of the below analysis is focused on (Supplemental Table S2).
Expression divergence
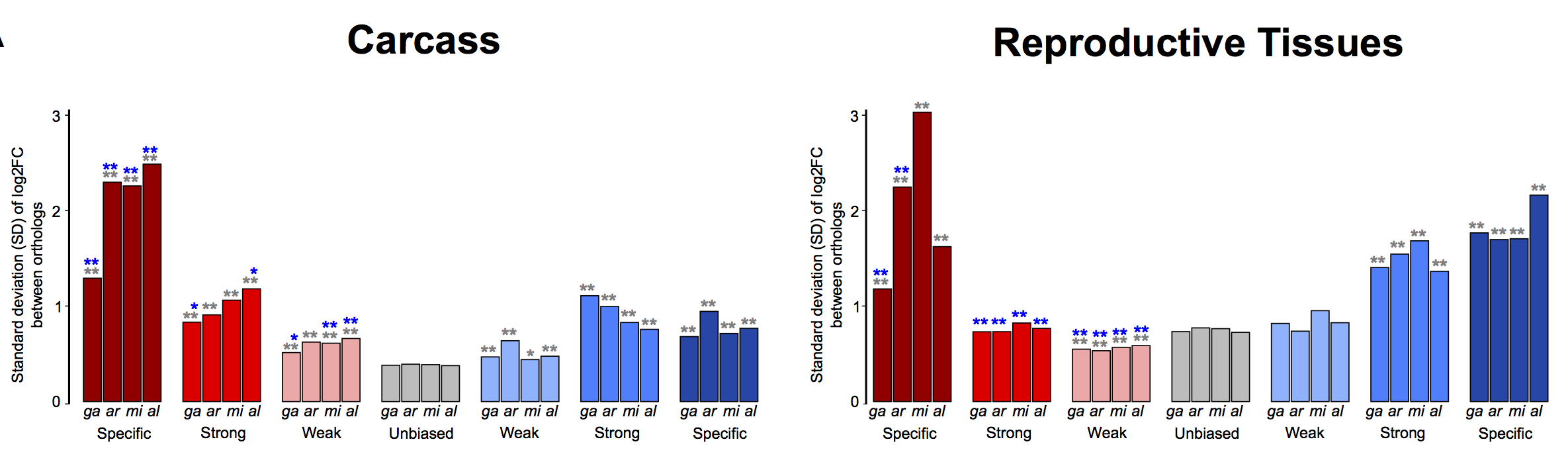
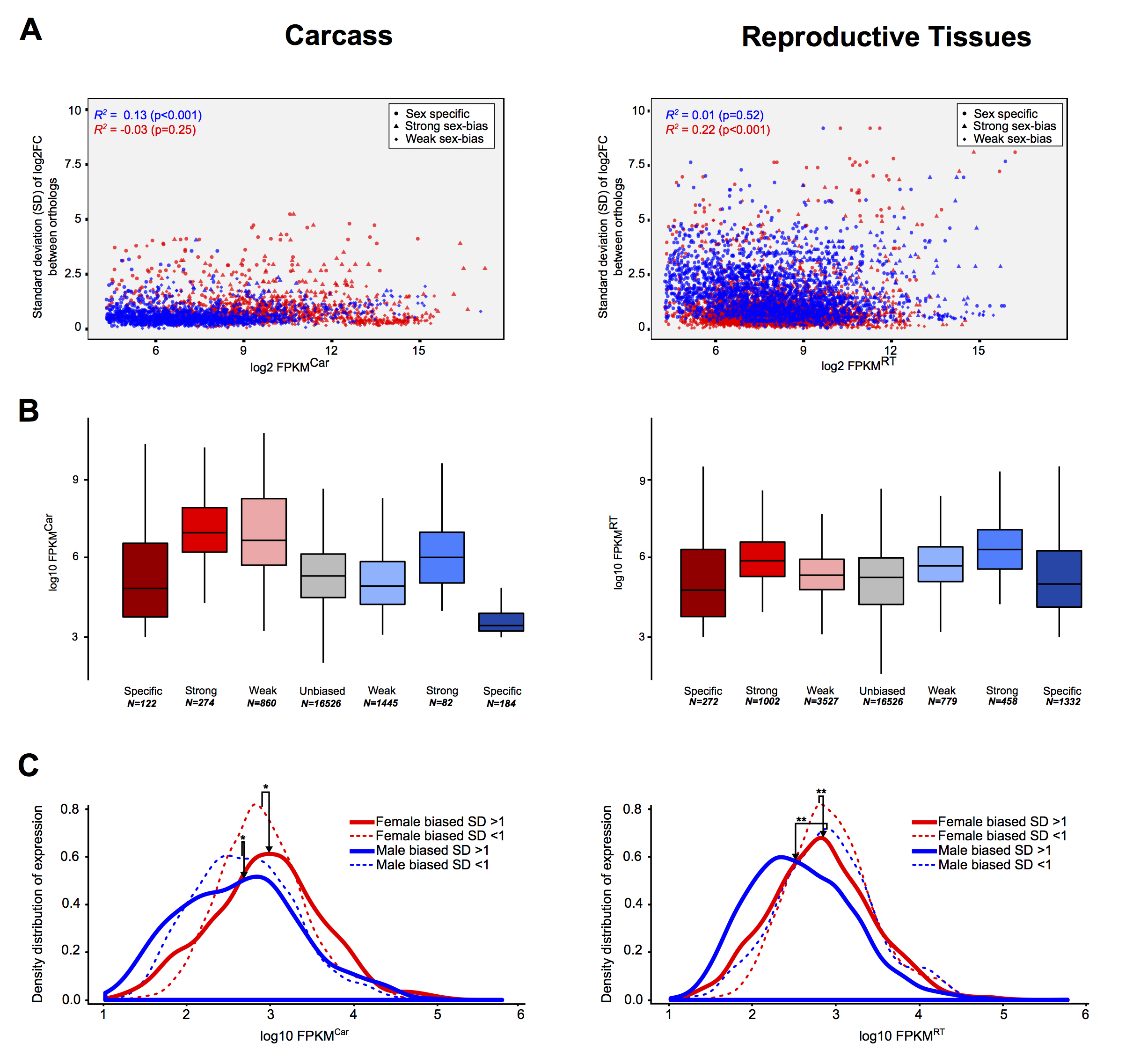
To study how sex-biased gene expression correlated with expression divergence, we calculated the median standard deviation (SD) of the sex expression ratio (log2[male/female]) between the four-species orthologs for each sex-bias classification (including unbiased), similarly to other studies (e.g., Zhang et al. 2007). Overall, we found that female-specific genes expressed in the carcass exhibited the highest levels of expression divergence of all sex-bias categories in all species (Fig. 2A). In the reproductive tissues, female-specific, male-specific, and strongly male-biased genes showed high levels of expression divergence, while all other classes evolve at similar or lower levels compared to unbiased genes (Fig. 2A). These patterns were consistent in each of the four species individually (Supplemental Fig. S5). Using a number of strategies, we assessed the relationship of gene expression levels to expression divergence and found no significant influence (see Supplemental Material; Supplemental Figs. S6, S7). Qualitatively, reversal of sex-biased expression (e.g., male to female) between species occurs infrequently (e.g., seven orthologs between An. gambiae to An. arabiensis show this behavior) and increases with evolutionary distance between species in both the carcass and reproductive tissues (Fig. 2B).
Figure 2.

Expression divergence rates of the four-species orthologs. (A) Median standard deviations of expression ratios (male/female) of carcass and reproductive tissue male-biased (blue) and female-biased (red) among the four-species orthologs. A Wilcoxon rank-sum test is used to test for significant differences when comparing sex-bias gene expression categories between the sexes (blue asterisks) or to unbiased genes (gray asterisks) and indicates: (*) P-value < 0.05, (**) P-value < 0.001. (B) The number of genes that show qualitative sex and/or tissue bias switches compared to their An. gambiae orthologs increases with evolutionary distance.
Sequence divergence
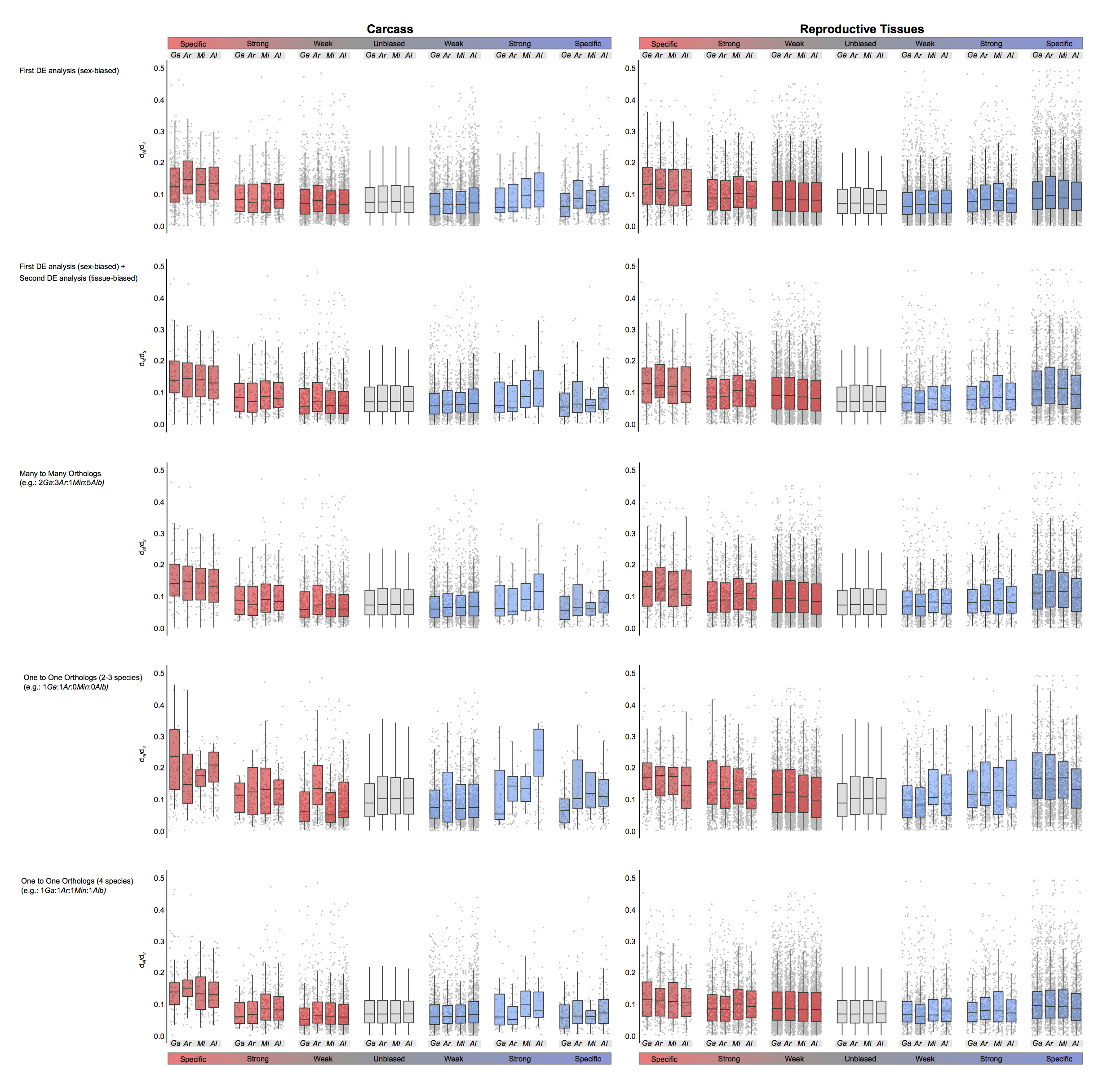
To investigate how rates of sequence divergence relate to sex-biased gene expression, we employed both DNA-level and protein-level measures of divergence calculated across all orthologs from the entire Anopheles genus (Neafsey et al. 2014). Similar to our findings of expression divergence, we found that female-specific genes of the carcass displayed the highest median levels of sequence divergence among all sex-biased genes (Fig. 3A,B), being significantly higher than both male-specific genes and unbiased genes (Wilcoxon rank-sum test P-value < 0.001) in all species. In the reproductive tissues, all three female categories consistently evolved faster than unbiased genes in all species (Wilcoxon rank-sum test P-value < 0.001), and in the males, only the sex-specific genes displayed mean dN/dS ratios and evolutionary rates significantly higher than unbiased genes consistently in all species (Fig. 3A,B). We ruled out that any of our DE analysis and subsequent filtering steps had any substantial effect on the observed patterns of sequence divergence (Supplemental Fig. S8) and also tested for an association between expression and sequence divergence among sex-biased genes of all species, finding that genes displaying rapid expression divergence (SD > 1) also show higher dN/dS ratios compared to those with an SD < 1, as has been observed in mammals (Khaitovich et al. 2005) and Drosophila (Supplemental Fig. S9; Zhang et al. 2007). To distinguish what evolutionary forces drive the rapid rates of evolution we observed, we ran branch site models using the codeml program of the PAML package (Yang 1997) and found that female-biased genes are associated with a higher proportion of positively selected genes compared to male or unbiased genes (Fig. 3C).
Figure 3.

Sequence divergence rates of the four-species orthologs and selection tests. Average DNA sequence (A) and protein sequence (B) divergence levels of the four-species orthologs from each species plotted by the sex-bias expression classification. Wilcoxon rank-sum tests are used to test for significant differences when comparing divergence levels between the sexes (blue asterisks) or between sex-biased categories and unbiased genes (gray asterisks) and indicates: (*) P-value < 0.05, (**) P-value < 0.001. (C) Cross-species average of percentage of genes showing significant positive selection among the four-species orthologs for each sex-biased classification, in the carcass and in the reproductive tissues.
Genic turnover
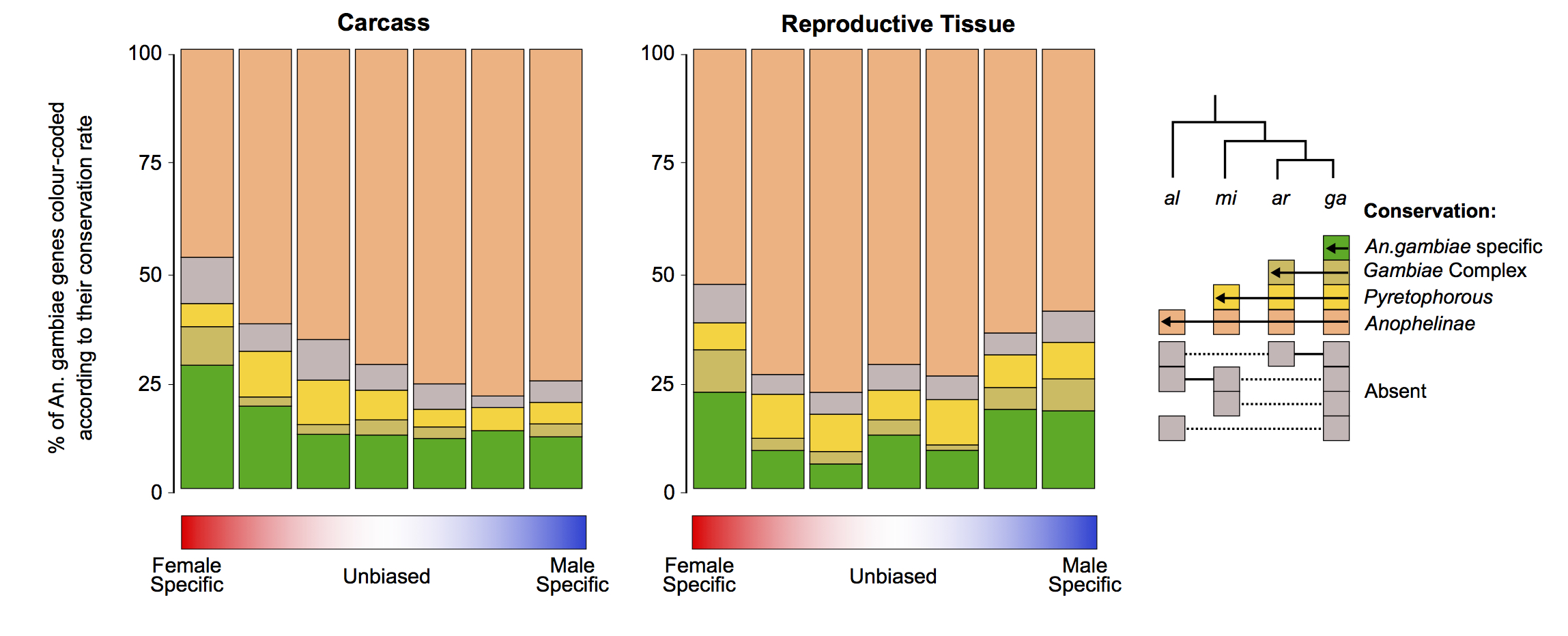
To investigate how patterns of gene gain or loss (turnover) relate to gene expression biases, we assessed the orthology status of all sex-biased genes between the four study species. We first examined species-specific genes, as these likely represent cases of high genic turnover either through species-specific gains or widespread losses, or very high levels of sequence divergence that preclude ortholog identification (Fig. 4A). We also quantified the degrees of conservation of each category of sex-biased gene expression of An. gambiae, ranging from species-specific to present in all four species (Fig. 4B). Comparisons of gene turnover rates for each sex-bias expression category to unbiased genes or between the sexes was consistent with our findings on expression and sequence divergence: in both tissues, the fraction of species-specific genes increased with the magnitude of sex-biased gene expression (except male-specific in the carcass), and female-biased genes displayed higher or similar levels of turnover to male-biased genes in all species (Fig. 4A). Furthermore, the extent of conservation throughout the genus in the carcass was lowest for female-specific genes and in reproductive tissues was similar between males and females, with decreasing conservation as the magnitude of sex-bias increases (Fig. 4B). To test whether this result may have been biased by the inclusion of multigene families, we repeated this analysis removing all orthogroups in which there were any multicopy orthologs and found a similar result (Supplemental Fig. S10).
Figure 4.

Sex-biased gene turnover rates. (A) The fraction of species-specific (orphan) genes is plotted for each sex-biased classification for the carcass and reproductive tissues, either for each species individually (points) or all species (box plot). A Wilcoxon rank-sum test of significance was used comparing sex-biased gene expression to unbiased genes; (*) P-value < 0.05. (B) Conservation of An. gambiae sex-biased genes across the phylogeny.
Rapidly evolving sex-biased genes are confined to a few biological functions
To understand which aspects of mosquito biology drive the rapid evolution of female-biased genes, we investigated the putative functions of the most rapidly evolving and most sex-biased genes in the carcass tissues of An. gambiae. We chose this species because of its more comprehensive functional ontologies and the availability of sets of manual community annotations (Neafsey et al. 2014). We found that a limited subset of functional categories describes the functions of nearly two-thirds of sex-biased genes in the female carcass (Fig. 5). We observed that a similar pattern holds when selecting the most rapid evolving genes using the SD of expression ratio rather than dN/dS (Supplemental Fig. S11). Function or expression in female salivary glands was the most overrepresented annotation, with 34% of rapidly evolving female genes falling in this group (Fisher's exact test for overrepresentation P-value < 0.001) (Fig. 5A). A large fraction of these genes is known to facilitate blood feeding, e.g., apyrase (AGAP011971), gSG6 (AGAP000150), gSG7 (AGAP008216), and the D7 family of genes (AGAP008278, AGAP008279), because they interfere with host homeostasis, inflammation, and immunity (Champagne et al. 1995; Arcà et al. 2005, 2014; Calvo et al. 2006; Isawa et al. 2007). Their rapidly diverging sequences and previous work showing that some of these are under strong positive selection (Arcà et al. 2014) lends support to the notion that host immune pressures are a dominant driving force acting on salivary gland genes essential for blood feeding. At the same time, the presence of saglin (SG1f:AGAP000610) that acts as a binding partner for parasitic TRAP protein (Ghosh et al. 2009; Upton et al. 2015) but also five more members of this SG1 family may indicate the impact of pathogen-driven pressures (Fig. 5B). The second most common functional category was mosquito immunity (15% of female genes; Fisher's exact test P-value < 0.001) (Fig. 5A), and the majority of genes within it have been shown previously to either respond to or influence infection levels (Fig. 5B). Among these are a cluster on Chromosome 2L containing an array of leucine-rich repeat immune proteins (LRIM 8A:AGAP007454, 8B:AGAP007456, 9:AGAP007453, and 10:AGAP007455) (Waterhouse et al. 2010), of which LRIM9 knockdown increases parasite load threefold (Vlachou et al. 2005; Upton et al. 2015). AGAP000151 (IRSP5), whose silencing increases both bacterial and parasite numbers (Dong et al. 2006), and AGAP000570 (AgSGU), which is one of the most abundant peritrophic matrix proteins (Dinglasan et al. 2009) and has important roles in ookinete attachment of Plasmodium falciparum (Mathias et al. 2014), were also identified. Of the remaining genes, four are expressed in the female midgut and are likely involved in blood meal digestion, two are involved in chemosensation and an ABC transporter that is involved in insecticide resistance against permethrin. The putative functions of all other rapidly evolving genes biased toward females are currently unknown or did not fall within these community annotated functional categories.
Figure 5.

Functional analysis of rapidly evolving and highly sex-biased genes from the carcass of An. gambiae. (A) Relative proportions of putative functions among rapidly evolving sex-biased genes (sex-specific and strongly biased) in the carcass of An. gambiae. (B) Sex-biased expression ratios (in log2 fold change male/female) over sequence divergence rates (in dN/dS) for genes displaying strong bias or sex-specific expression in the carcass of An. gambiae. Horizontal box plots indicate dN/dS values for female- (red) and male-biased (blue) genes, highlighting the median values of each sex, which were used as a cutoff for downstream analysis. Genes with dN/dS value greater than the median of each sex are highlighted with putative functions using Gene Ontology (GO), orthology-based predictions, and community annotations (colored circles). Empty circles indicate genes evolving below our threshold of sequence divergence.
The number of rapidly evolving male-biased genes of the carcass without predicted functions was higher compared to females (28%) (Fig. 5A). Functions related to the insect cuticle were overrepresented (11% of male genes; Fisher's exact test P-value < 0.001), including the entire cluster of CPLCG genes on Chromosome 3R, of which CPLCG3 and CPLCG4 mediate through yet unknown mechanisms insecticide resistance in distinct strains and species of Anopheles (Vontas et al. 2007; Awolola et al. 2009). Three additional cuticular proteins, namely CPCFC1 (AGAP007980) and CPR59 (AGAP006829) and CPR140 (AGAP006868), also showed a bias toward males (Fig. 5B). No previous report of male-biased expression for these genes, or any other cuticle protein, exist. We therefore inspected expression profiles of these genes in published expression data and found that all are expressed in females but are down-regulated upon blood feeding, explaining the observed male bias in our data (data not shown). Of the remaining genes, four included candidate immunity genes such as GNBPB4 (AGAP002796) and CLIPB18 (AGAP009215), and two cytochrome P450s (CYP6AF1:AGAP011028 and CYP6AF2:AGAP011029), but this category was not functionally enriched among the rapidly evolving highly male-biased genes.
Sex-specific splicing
An exclusive focus on differential expression of genes between males and females may miss a significant portion of sex-biased gene usage, as many processes could be regulated at the level of transcript usage or splicing, a prime example of which is the sex-determination cascade itself. We therefore expanded the catalog of sex-biased genes in the four species to include those with sex-specific isoform usage and characterized the degree of conservation of splicing patterns between species. Because the annotation of alternative transcript isoforms in these species is incomplete, we first established an isoform-specific gene expression data set, where we predicted novel isoforms of all known genes (Supplemental Fig. S12). We focused our analysis on orthologs that possessed at least two transcripts that were differentially used between the sexes or tissues, so that each isoform was expressed at least at a twofold higher level in the condition in which it was predominant. While sex-specific splicing does occur in the carcass, when considering only genes that show high conservation in alternative isoform usage across the genus, the list is restricted to a few genes, including the well-known doublesex master-switch of the sex determination pathway. We found conserved sex-specific isoform usage between the reproductive tissues of males and females to be at least as common, if not more so, than tissue-specific splicing between the carcass and the reproductive tract in either males or females (Fig. 6A). In An. gambiae, genes that displayed sex-specific isoform usage in both tissues were enriched for functional annotations including behavior, oocyte and embryo development, and the regulation of cellular and organismal anatomy (Fisher's exact test P-value < 0.001) (Fig. 6B). Exemplary transcript structures for genes falling into both classes are shown in Figure 6C, and the complete list of genes with predicted alternative transcript usage is listed in Supplemental Table S3. This analysis confirms that a considerable amount of sex-specific gene usage in the mosquito remains entirely unexplored and we provide candidate loci for future functional analysis.
Figure 6.

Sex- and tissue-specific differential transcript isoform usage. (A) Overview of the number of orthologs displaying conserved alternative isoform usage conserved in four, three, or two species, respectively. (B) Gene Ontology functional enrichment analysis showing enriched GO-terms in An. gambiae among genes displaying sex-specific isoform usage, compared to the entire genome. (C) Two examples of sex-specific isoform usage between males and females are shown for carcass (left) and reproductive tissues (right). In each case, only the predominant isoforms in the two sexes are shown, with the color of the transcript displaying transcript expression levels (in FPKM). The female-specific exon of the well-characterized doublesex (dsx) locus is marked with a black triangle. Although visual inspection of splicing pattern highlights consistent use of alternative sex-specific splicing in all four species, the dsx locus passed the cutoffs only in three species. A similar pattern was also observed for a number of other genes and suggests that this analysis is stringent.
Chromosomal distribution of sex-biased genes
Sex-biased genes have been shown in a number of species to be nonrandomly distributed across chromosomes, and typically, in species where males are heterogametic (XY), male-biased genes are underrepresented on the X Chromosome (Parisi et al. 2003; Wu and Xu 2003; Ellegren and Parsch 2007; Sturgill et al. 2007). We observed a significant paucity for male RT-biased genes on the X Chromosome in all species (χ2 test P < 0.001) but found no similar deficit among male-biased genes of the carcass (Fig. 7A). These results extend our previous findings in An. gambiae (Magnusson et al. 2012) in two ways. First, they demonstrate that the underrepresentation of male-biased genes on X Chromosomes is a conserved feature in the genus. Second, they show that the underrepresentation detected previously using whole-body data is derived from differences in the abundance of sex-biased genes in the reproductive tissues, confirming previous data from the Baker and Russell (2011) study. While the X Chromosomes did show a higher than expected number of female-biased genes in some cases (Fig. 7A), this apparent “feminization” was not as significant and not consistently found in all four species (χ2 test P-value < 0.05). In the Drosophila male germline, the deficit of male-biased genes on the X Chromosome has been attributed to chromosome-wide transcriptional regulation(s) in the male germline, namely lack of dosage compensation and transcriptional suppression of the X—in a process similar to meiotic sex chromosome inactivation (MSCI), except that the suppression appears to be initiated in premeiotic cells (Meiklejohn et al. 2011; Meiklejohn and Presgraves 2012). In combination, these processes result in a significant reduction in gene expression along the entire X Chromosome in the male germline, observed as a paucity of male-biased genes on the X Chromosome, when expression is compared to females or other tissues. It has been argued that the use of the term “evolutionary X de-masculinization” may thus be misleading to describe the forces acting on gene expression in the male testis, because it implies that selective forces, such as sexual antagonism, drive genes beneficial for males off the X (Meiklejohn et al. 2011; Meiklejohn and Presgraves 2012). This argument is supported by findings that testis-specific genes are, in fact, not underrepresented on the X of Drosophila (Meiklejohn and Presgraves 2012). Instead, testis-specific genes have recruited strong cis-regulatory elements to overcome the transcriptional silencing (Landeen et al. 2016).
Figure 7.

Chromosomal distribution of sex-biased genes. (A) Proportions of observed/expected numbers of male-biased (blue) and female-biased (red) genes in the carcass and reproductive tissues located on autosomes or the X Chromosomes of each species. The horizontal dashed line indicates an equal number of observed and expected genes, and ratios greater or lower than this indicate an enrichment or depletion, respectively. Asterisks indicate: (*) P-value < 0.05, (**) P-value < 0.001, χ2 test comparing ratios between the X Chromosome and the autosomes. (B) Violin plots of expression levels (in log2FPKM) of all genes in the female (red) or male (blue) carcass and reproductive tissues for autosomes and X Chromosomes. (C) Pseudonormalization of X Chromosome male expression in the reproductive tissues using the A:X ratio eliminates the de-masculinization effect.
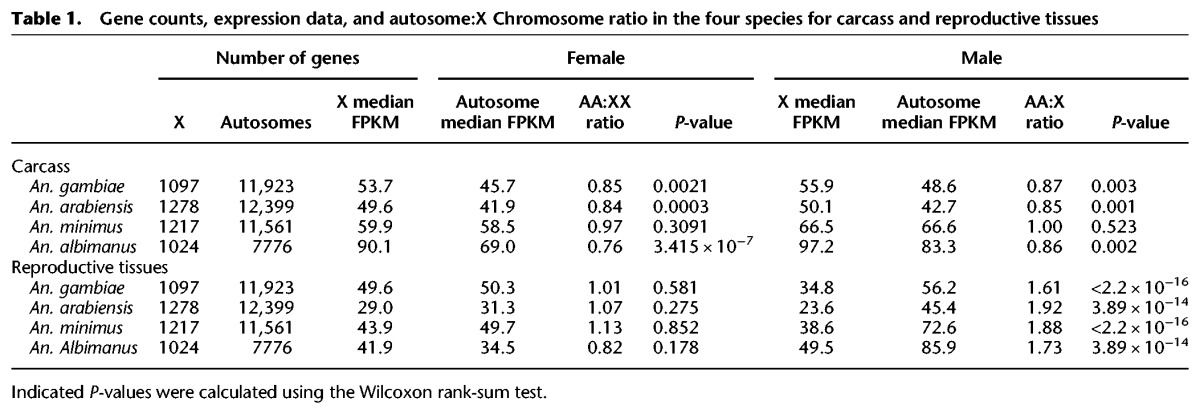
To confirm previous findings in An. gambiae of the lack of dosage compensation in the male germline and to test this at the genus level, we explored the evidence for chromosome-wide transcriptional regulation. Similarly to previous data from Baker and Russell (2011), we found that the median expression of all genes on the X Chromosomes is, on average, 0.3-fold lower in males compared to females in the reproductive tissues in all four species (Fig. 7B). We calculated the autosome to X Chromosome (A:X) ratio of median gene expression and found that in all species the A:X ratio ranged between 1.61 and 1.92 in the male reproductive tissues (Table 1). In comparison, median expression of X genes in the carcass tissues and autosomal genes in both tissues were not significantly different between the two sexes (Fig. 7B). Together, these results confirmed the signal previously detected in An. gambiae and suggest that within the male reproductive tissues of Anopheles, likely the testis, X Chromosomes undergo chromosome-wide transcriptional regulation, similarly to Drosophila (Landeen et al. 2016). We tested whether such X-specific regulation can explain the underrepresentation of male-biased genes on the X Chromosomes of Anopheles species by simulating the absence of X-regulation in the male reproductive tissues, following the rationale and methods previously used to explore this question using the MozAtlas data in the Meiklejohn and Presgraves (2012) study. To do this, we first normalized the raw read counts of male X Chromosome genes with the respective A:X expression ratio to pseudocorrect for X transcriptional regulation. Using our data, we found that the ratios of observed/expected genes no longer support the underrepresentation of male-biased genes on the X Chromosomes in the male reproductive tissues of these species (Fig. 7C). We also assessed the chromosomal distribution of genes specifically expressed in the male reproductive tissues: We calculated the index of tissue specificity (τ) for each gene and selected genes with a τ index > 0.95 as being specific to a tissue. Unlike previous work (Meiklejohn and Presgraves 2012), when we tested for evidence of X de-masculinization for RT-specific genes using the restricted range of τ > 0.95, we found no significant paucity on the X compared to autosomes (χ2 test P = 0.26). By comparing our DE and tissue specificity calls with those from the MozAtlas study (Supplemental Material; Supplemental Table S4; Supplemental Fig. S14), we concluded that our signal of no significant paucity of X-linked male RT-specific genes is in part due to a higher dynamic range afforded by RNA-seq resulting in a lowering of false positive male-RT calls and improved genome assemblies. We conclude therefore that the paucity of male-biased genes on the X Chromosome in reproductive tissues results directly from X Chromosome transcriptional regulation and its effect on DE analyses, and is not likely a result of selective forces such as sexual antagonism.
Table 1.
Gene counts, expression data, and autosome:X Chromosome ratio in the four species for carcass and reproductive tissues
Discussion
Studies in many different organisms investigating the relationship between expression patterns and rates of molecular evolution have consistently found evidence for complex selective pressures acting differentially on male- and female-biased genes. Typically, genes biased toward males evolve more rapidly than female-biased genes, with those expressed during spermatogenesis showing the fastest rates of evolution in the genome. Amazingly, this pattern is observed in different taxa ranging from primates (Khaitovich et al. 2005) to worms (Cutter and Ward 2005), i.e., even in species in which the two sexes coexist within a single individual.
Our findings suggest that Anopheles mosquitoes do not follow this common pattern. In all four sequenced species, genes belonging to the category of female-CA specific consistently display the highest overall levels of DNA- and protein sequence divergence, positive selection, expression divergence, and genic turnover. By exploring the putative functions of these genes using manual annotations and expression data, we propose that this pattern is driven by the strong selective forces acting on female-specific phenotypic characters that facilitate blood-feeding and thus underlie vectorial capacity, as nearly two-thirds of female genes that are rapidly evolving in An. gambiae have predicted functions involving host seeking, blood feeding, digestion, and mounting an immune response to control pathogens. Genes with functions in the female salivary glands, whose products are either injected into the host or are located at the interface with the ingested blood meal within salivary gland tissues, are significantly overrepresented among rapidly evolving female genes (Fig. 5). This finding reinforces the notion that both host pressures and mosquito-pathogen interactions are principal forces acting on the evolution of mosquito genomes (Arcà et al. 2014; Neafsey et al. 2014). Our data also highlight that male biology is not under similar selection pressures as those of females, but the less-well-studied biology of the male somatic tissues makes it harder to establish a link between rapidly evolving male genes in the carcass and their role in male-specific processes or behaviors—for example, their aggregation and mating in swarms.
Intriguingly, we found that genes biased toward male RT relative to female RT did not display overall faster rates of evolution, as has been observed so consistently in other species. However, because the total number of male RT-specific genes is much higher, median levels of evolutionary divergence may underestimate the signatures of selection. Generally, increased evolutionary rates among male-biased reproductive genes are explained by selection arising from male-male competition (Zhang and Parsch 2005; Pröschel et al. 2006; Butler et al. 2007) and/or reduced pleiotropy of genes expressed in male reproductive tissues (Tripet et al. 2003; Zhang et al. 2007; Pondeville et al. 2008; Meiklejohn and Presgraves 2012). Our analysis on tissue specificity in Anopheles shows that male RT-biased genes display the highest overall tissue specificity of all sex- and tissue-bias categories (Fig. 1C; Supplemental Fig. S4), so increased pleiotropy of male RT genes in these mosquitoes is unlikely to explain this result. More likely, the fact that Anopheles females typically mate once during their lifetime under natural conditions (Tripet et al. 2003) results in the absence of selection on sperm competition that typically drives increased rates of evolution in male RT genes in polygamous species. Males of several Anopheles species, including An. gambiae and An. arabiensis, produce a mating plug that is deposited in the female reproductive tract during mating, enhancing sperm storage (Rogers et al. 2009) and modifying female physiology (Pondeville et al. 2008; Baldini et al. 2013) such that females are no longer receptive to further matings. It will be interesting to compare rates of evolution of male- and female-biased genes in other mosquito species where females are polyandrous (Boyer et al. 2012) or other insect taxa where both sexes blood-feed (Yuval 2006). Our results do not necessarily imply a relaxation of purifying selection acting on male RT-specific genes, as these still have to perform important functions and we cannot preclude the possibility that future improvements in genome annotations of the Anopheles genus may lead to the discovery of currently unknown male RT-specific genes not included in this work that may balance the evidence of selection back toward male reproduction.
In addition to sex-biased gene expression, differential isoform usage between sexes, resulting from alternative splicing or alternative transcription start sites, can also account for sexual dimorphism. This area remains understudied due to the difficulty of extracting isoform-level data from whole-genome expression data. In mosquitoes, only two genes, doublesex and fruitless, initially characterized in Drosophila, are known to behave in this manner (Scali et al. 2005), but no systematic analysis has been performed to date. Making use of novel tools for isoform-level differential expression analysis and combining it within a comparative analysis across four malaria mosquito species, we have cataloged conserved genes that display sex-specific isoform usage in the carcass and the reproductive tissues. These candidate genes form the basis for future functional investigations into processes such as sex determination or behavior, for example, by the depletion of sex-specific transcripts or exons using CRISPR/Cas9.
Finally, we have also used our expression data to re-examine evidence of the nonrandom chromosomal distribution of sex-biased genes in Anopheles mosquitoes and to begin to characterize its underlying causes. We and others have previously shown, using whole-body expression data from adults and larvae, that the X Chromosome of An. gambiae has a significant paucity of male-biased genes and that transgenes that are normally expressed strongly in the male testis are transcriptionally suppressed in sperm when the transgenic construct is located on the X Chromosome, in a process resembling MSCI (Baker and Russell 2011; Baker et al. 2011; Magnusson et al. 2012). The results from the present study reveal that the paucity of male RT-biased genes on the X Chromosome is a conserved feature in the genus and that this deficit is based on differences in chromosome-wide transcriptional regulation of the X. In the carcass, the difference in ploidy of the X between males and females, or the X and autosomes in males, does not translate into chromosome-wide gene expression differences, suggesting that these tissues are actively dosage-compensated—probably through X Chromosome hypertranscription in males as in Drosophila. Conversely, in the male reproductive tissues, we find that gene expression from the X Chromosome is, on average, 0.3-fold lower in males compared to females and, on average, 1.8-fold lower compared to gene expression from the autosomes for all four species (Fig. 7B; Table 1). We have shown here that the X Chromosome transcriptional regulation in the male reproductive tissues is directly responsible for the deficit of male-biased genes in the male reproductive tissues and is not merely a contributing factor favoring X-to-autosome retro-movement of male genes leading to the evolutionarily de-masculinization of the X. We show this by computationally simulating the absence of the observed X Chromosome transcriptional regulation, which results in eliminating the underrepresentation of male-biased genes on the X. Furthermore, when analyzing the chromosomal distribution of tissue-specific genes, which are not similarly sensitive to DE-biases that assume equal ploidy or chromosomal transcriptional regulation between conditions, we find no support that male reproductive tissue specific-genes are underrepresented on the X Chromosome, again similarly to Drosophila (Meiklejohn et al. 2011) but distinct from the MozAtlas-based metaanalysis of Meiklejohn and Presgraves (2012). Taken together, these results indicate that the X Chromosomes of Anopheles mosquitoes are, in effect, “transcriptionally de-masculinized” in the male germline by chromosome-wide transcriptional regulation that leads to reduction of X Chromosome gene expression. In the absence of mutant strains or markers for the chromatin-modifications that govern the levels of X transcription in the male germline, it is currently difficult to quantify the individual contributions of the lack of X-dosage compensation or X-suppression (via processes like MSCI), especially since these have similar outcomes. Dissecting further the mechanisms and functions of X-specific transcriptional regulation will likely reveal important insights into the evolution of sex chromosomes, including addressing whether similar transcriptional regulation occurs on the Y Chromosomes, whose content and evolutionary history we have recently described (Hall et al. 2016).
Methods
RNA-seq and differential expression analysis
Total mRNA was extracted from sexed samples of An. gambiae, An. arabiensis, An. minimus, and An. albimanus. Individuals were dissected to obtain the reproductive tissues, which consisted of ovaries and common oviduct for females and testis and accessory glands for males (see Supplementary Material for further details; Supplemental Fig. S1C). Sex-biased differential expression analysis was performed using the DESeq2 (Love et al. 2014) with a FDR < 0.05 after mapping reads to the genomes using TopHat2 (Kim et al. 2013) and quantifying the expression using HTSeq (Anders et al. 2014). Results were then filtered using a custom pipeline (see Supplemental Material; Supplemental Fig. S2). Genes that did not pass our selection criteria because of discordancy between our filtering were individually analyzed (see Supplemental Material; Supplemental Fig. S3) and shown to evolve similarly to unbiased genes and were thus excluded from the analysis.
Evolution of sex-biased genes
We retrieved OrthoDB-delineated (Kriventseva et al. 2015) orthology relationships of all genes between the four species (Neafsey et al. 2014) and calculated expression, sequence, and gene turnover rates of sex-biased and unbiased genes of all species. Expression divergence was estimated by computing the median standard deviation of the log2 FC between the four-species orthologs for each sex-bias category. Sequence divergence levels were calculated both at the DNA level (dN/dS) and the protein level (evolutionary rate) (Waterhouse et al. 2011; Kriventseva et al. 2015) for each species and each sex-bias category. We used the Wilcoxon test to compare divergence estimates between unbiased and sex-bias categories or between identical sex-biased categories in the two sexes. Average turnover rates were estimated for each species and for each sex-bias expression category by calculating the fraction of species-specific genes from the total number of genes per category. Genic turnover rates were also calculated by dividing the number of genes in each sex-bias category displaying distinct conservation levels in the genus (e.g., species-specific or present in all species) by the total number genes in that category. To infer events of positive selection and distinguish it from relaxed selective constraint, we used the codeml program of the PAML package (Yang 1997) and ran a branch site models test (see Supplemental Material for additional information).
Functional analysis
Gene Ontology (GO) (Ashburner et al. 2000; Blake et al. 2015) and InterPro (Mitchell et al. 2015) terms were retrieved from Vectorbase (Giraldo-Calderón et al. 2015) for each species. We expanded these ontology data sets using Blast2GO (Conesa et al. 2005) using the nonredundant NCBI protein database. For the functional analysis of An. gambiae sex-biased genes, we also incorporated manual functional annotations provided generously from the community as well as gene expression data sets in the female salivary gland and male accessory gland.
Chromosomal distribution and expression of sex-biased genes
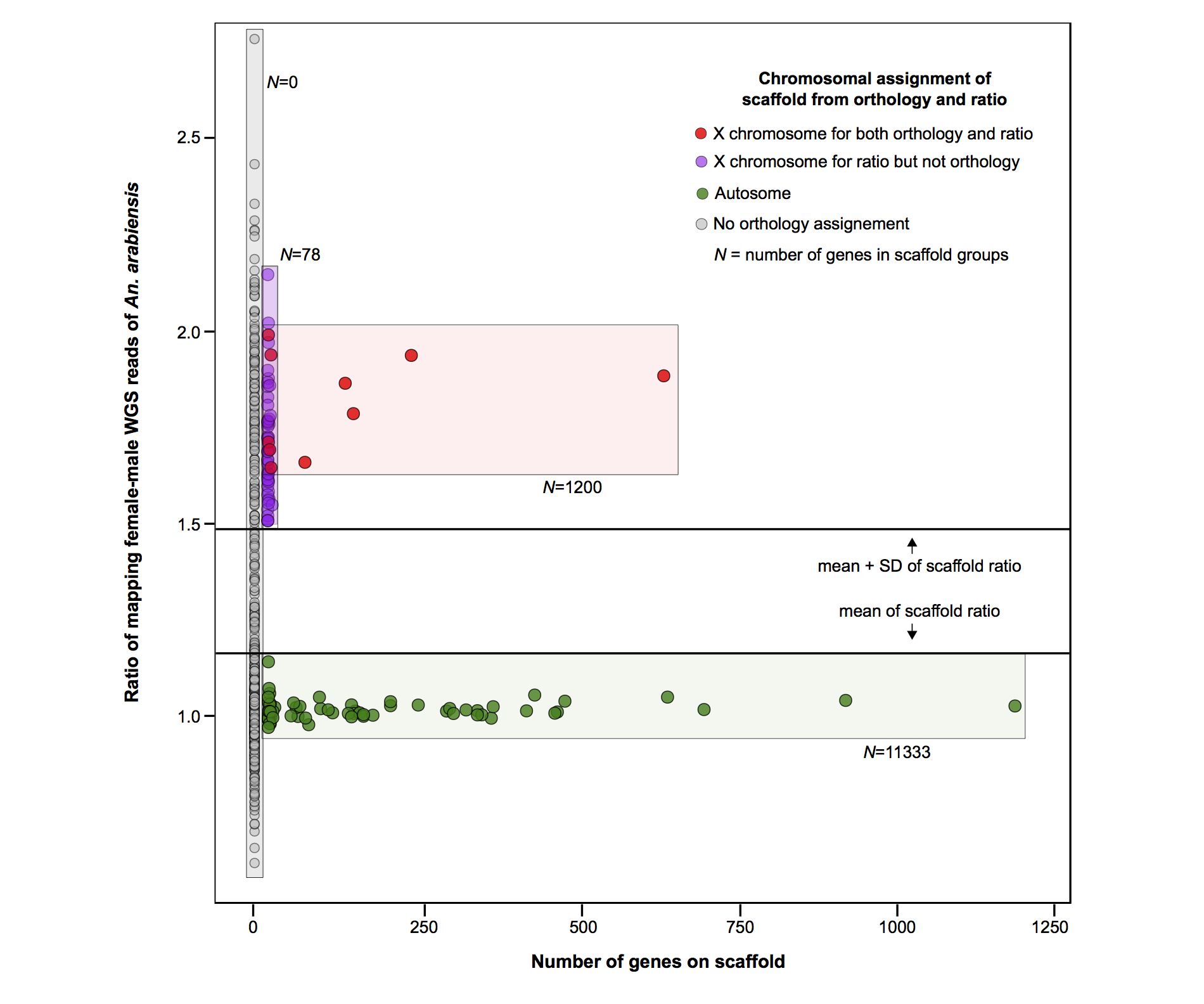
Gene-to-chromosome assignments are currently only available for An. gambiae. To identify the chromosomal origin of scaffolds in the other species, scaffolds were assigned to a chromosomal arm if more than half of the genes on a scaffold had orthologs in An. gambiae, and of these, if two thirds had hits to the same chromosomal arm of An. gambiae. We tested this approach using whole-genome sequencing (WGS) from male and female pools of An. arabiensis NCBI SRP044010 to predict X-linked scaffolds as the ones showing the ratio of females mapping reads over males for each higher than 1.48 (see Supplemental Material for more details; Supplemental Fig. S13). We divided the number of observed sex-biased genes for each chromosome by the number of expected sex-biased genes by random distribution in the genome. A χ2 test was performed to determine whether the ratio of observed over expected numbers of male- and female-biased genes was significantly different between the autosomes and the X Chromosome for the carcass and reproductive tissues for each sex. To understand if the apparent de-masculinization of the X Chromosome was due to a lack of dosage compensation in the male reproductive tissues, we “pseudonormalized” the numbers of mapping raw read counts for X-linked genes in the male reproductive tissues of each species by multiplying them by the A:X ratio from each species. This modeled dosage compensation, showed no evidence of an underrepresentation of observed male-biased genes/expected on the X Chromosomes of each species.
Analysis of sex-specific isoform usage
RNA-seq reads were aligned separately to the aforementioned downloaded genomes using HISAT (Kim et al. 2015). We then used Stringtie (Pertea et al. 2015) to predict novel isoforms of known genes separately for each of the four species across the samples. For each species, predicted isoforms for all genes were merged into a combined gene set using Cuffmerge (Trapnell et al. 2010). Expression of all isoforms was then quantified using Stringtie. We used the Ballgown (Frazee et al. 2015) suite to determine significantly different isoform expression using a cutoff P-value < 0.05 and a mean expression > 1 FPKM across the samples in a way that each gene was required to have, at a minimum, one male- and one female-biased isoform with a log2 fold change > 1 between the two sexes.
Data access
All newly generated sequence data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP083856.
Supplementary Material
Acknowledgments
We thank Scott Cornman, Sara Mitchell, Adam Jenkins, Michael Riehle, Craig Wilding, Xiofan Zhou, and Jose Ribeiro for providing us with manual community annotations of An. gambiae gene functions, and Paul Howell at the MR4/BEI for support on rearing of the mosquito species. This study has received funding from the European Union's Seventh Framework Programme (FP7 2007–2013 Infravec) under the GA no. 228421. This study was also funded by the European Union's Seventh Framework Programme (FP7 2007–2013) Marie Curie Actions cofund (Project I-Move) under GA no. 267232. This study was funded in part by a grant from the Foundation for the National Institutes of Health through the Vector-Based Control of Transmission: Discovery Research (VCTR) program of the Grand Challenges in Global Health initiative of the Bill & Melinda Gates Foundation. P.A.P. was supported by a Rita Levi Montalcini award from the Ministry of Education, University and Research (MIUR – D.M. no. 79 04.02.2014). This study was also funded by the European Research Council under the European Union's Seventh Framework Programme ERC grant no. 335724 awarded to N.W. R.M.W. was supported by Marie Curie International Outgoing Fellowship (European Commission) PIOF-GA-2011-303312. M.K.N.L. was supported by a Medical Research Council (MRC) Career Development Award (G1100339) and by the Wellcome Trust (098051). R.M.W. was supported by Swiss National Science Foundation grant PP00P3_170664.
Author contributions: F.P., N.W., R.M.W., and P.A.P. performed all analysis and wrote the paper with help from T.N. and M.K.N.L. T.N. and M.K.N.L. contributed to the experimental design of the study. A.C., R.D., and T.P. performed tissue dissections and RNA-seq library preparations.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.217216.116.
Freely available online through the Genome Research Open Access option.
References
- Anders S, Pyl PT, Huber W. 2014. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcà B, Lombardo F, Valenzuela JG, Francischetti IMB, Marinotti O, Coluzzi M, Ribeiro JMC. 2005. An updated catalogue of salivary gland transcripts in the adult female mosquito, Anopheles gambiae. J Exp Biol 208: 3971–3986. [DOI] [PubMed] [Google Scholar]
- Arcà B, Struchiner CJ, Pham VM, Sferra G, Lombardo F, Pombi M, Ribeiro JMC. 2014. Positive selection drives accelerated evolution of mosquito salivary genes associated with blood-feeding. Insect Mol Biol 23: 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene Ontology: tool for the unification of biology. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assis R, Zhou Q, Bachtrog D. 2012. Sex-biased transcriptome evolution in Drosophila. Genome Biol Evol 4: 1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awolola TS, Oduola OA, Strode C, Koekemoer LL, Brooke B, Ranson H. 2009. Evidence of multiple pyrethroid resistance mechanisms in the malaria vector Anopheles gambiae sensu stricto from Nigeria. Trans R Soc Trop Med Hyg 103: 1139–1145. [DOI] [PubMed] [Google Scholar]
- Baker DA, Russell S. 2011. Role of testis-specific gene expression in sex-chromosome evolution of Anopheles gambiae. Genetics 189: 1117–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Nolan T, Fischer B, Pinder A, Crisanti A, Russell S. 2011. A comprehensive gene expression atlas of sex- and tissue-specificity in the malaria vector, Anopheles gambiae. BMC Genomics 12: 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldini F, Gabrieli P, South A, Valim C, Mancini F, Catteruccia F. 2013. The interaction between a sexually transferred steroid hormone and a female protein regulates oogenesis in the malaria mosquito Anopheles gambiae. PLoS Biol 11: e1001695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Christie KR, Dolan ME, Drabkin HJ, Hill DP, Ni L, Sitnikov D, Burgess S, Buza T, Gresham C, et al. 2015. Gene Ontology Consortium: going forward. Nucleic Acids Res 43: D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer S, Toty C, Jacquet M, Lempérière G, Fontenille D. 2012. Evidence of multiple inseminations in the field in Aedes albopictus. PLoS One 7: e42040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MA, Sawyer SA, Losos JB. 2007. Sexual dimorphism and adaptive radiation in Anolis lizards. Nature 447: 202–205. [DOI] [PubMed] [Google Scholar]
- Calvo E, Pham VM, Lombardo F, Arcà B, Ribeiro JMC. 2006. The sialotranscriptome of adult male Anopheles gambiae mosquitoes. Insect Biochem Mol Biol 36: 570–575. [DOI] [PubMed] [Google Scholar]
- Champagne DE, Smartt CT, Ribeiro JM, James AA. 1995. The salivary gland-specific apyrase of the mosquito Aedes aegypti is a member of the 5′-nucleotidase family. Proc Natl Acad Sci 92: 694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T, Arnqvist G, Bangham J, Rowe L. 2003. Sexual conflict. Trends Ecol Evol 18: 41–47. [Google Scholar]
- Chippindale AK, Gibson JR, Rice WR. 2001. Negative genetic correlation for adult fitness between sexes reveals ontogenetic conflict in Drosophila. Proc Natl Acad Sci 98: 1671–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- Connallon T, Knowles LL. 2005. Intergenomic conflict revealed by patterns of sex-biased gene expression. Trends Genet 21: 492–495. [DOI] [PubMed] [Google Scholar]
- Cutter AD, Ward S. 2005. Sexual and temporal dynamics of molecular evolution in C. elegans development. Mol Biol Evol 22: 178–188. [DOI] [PubMed] [Google Scholar]
- Dinglasan RR, Devenport M, Florens L, Johnson JR, McHugh CA, Donnelly-Doman M, Carucci DJ, Yates JR, Jacobs-Lorena M. 2009. The Anopheles gambiae adult midgut peritrophic matrix proteome. Insect Biochem Mol Biol 39: 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Aguilar R, Xi Z, Warr E, Mongin E, Dimopoulos G. 2006. Anopheles gambiae immune responses to human and rodent Plasmodium parasite species. PLoS Pathog 2: e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. 2007. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet 8: 689–698. [DOI] [PubMed] [Google Scholar]
- Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT. 2015. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol 33: 243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Devenport M, Jethwaney D, Kalume DE, Pandey A, Anderson VE, Sultan AA, Kumar N, Jacobs-Lorena M. 2009. Malaria parasite invasion of the mosquito salivary gland requires interaction between the Plasmodium TRAP and the Anopheles saglin proteins. PLoS Pathog 5: e1000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldo-Calderón GI, Emrich SJ, MacCallum RM, Maslen G, Emrich S, Collins F, Dialynas E, Topalis P, Ho N, Gesing S, et al. 2015. VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res 43: D707–D713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM, Nachman MW. 2005. Rates of protein evolution are positively correlated with developmental timing of expression during mouse spermatogenesis. Mol Biol Evol 22: 1044–1052. [DOI] [PubMed] [Google Scholar]
- Hall AB, Papathanos P-A, Sharma A, Cheng C, Akbari OS, Assour L, Bergman NH, Cagnetti A, Crisanti A, Dottorini T, et al. 2016. Radical remodeling of the Y chromosome in a recent radiation of malaria mosquitoes. Proc Natl Acad Sci 113: E2114–E2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PW, Wright AE, Zimmer F, Dean R, Montgomery SH, Pointer MA, Mank JE. 2015. Sexual selection drives evolution and rapid turnover of male gene expression. Proc Natl Acad Sci 112: 4393–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isawa H, Orito Y, Iwanaga S, Jingushi N, Morita A, Chinzei Y, Yuda M. 2007. Identification and characterization of a new kallikrein-kinin system inhibitor from the salivary glands of the malaria vector mosquito Anopheles stephensi. Insect Biochem Mol Biol 37: 466–477. [DOI] [PubMed] [Google Scholar]
- Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, Weiss G, Lachmann M, Pääbo S. 2005. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 309: 1850–1854. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva EV, Tegenfeldt F, Petty TJ, Waterhouse RM, Simão FA, Pozdnyakov IA, Ioannidis P, Zdobnov EM. 2015. OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res 43: D250–D256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeen EL, Muirhead CA, Wright L, Meiklejohn CD, Presgraves DC. 2016. Sex chromosome-wide transcriptional suppression and compensatory cis-regulatory evolution mediate gene expression in the Drosophila male germline. PLoS Biol 14: e1002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson K, Mendes AM, Windbichler N, Papathanos PA, Nolan T, Dottorini T, Rizzi E, Christophides GK, Crisanti A. 2011. Transcription regulation of sex-biased genes during ontogeny in the Malaria vector Anopheles gambiae. PLoS One 6: e21572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson K, Lycett GJ, Mendes AM, Lynd A, Papathanos P-A, Crisanti A, Windbichler N. 2012. Demasculinization of the Anopheles gambiae X chromosome. BMC Evol Biol 12: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinotti O, Nguyen QK, Calvo E, James AA, Ribeiro JM. 2005. Microarray analysis of genes showing variable expression following a blood meal in Anopheles gambiae. Insect Mol Biol 4: 365–373. [DOI] [PubMed] [Google Scholar]
- Mathias DK, Jardim JG, Parish LA, Armistead JS, Trinh HV, Kumpitak C, Sattabongkot J, Dinglasan RR. 2014. Differential roles of an Anopheline midgut GPI-anchored protein in mediating Plasmodium falciparum and Plasmodium vivax ookinete invasion. Infect Genet Evol 28: 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Presgraves DC. 2012. Little evidence for demasculinization of the Drosophila X chromosome among genes expressed in the male germline. Genome Biol Evol 4: 1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Landeen EL, Cook JM, Kingan SB, Presgraves DC. 2011. Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol 9: e1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, et al. 2015. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res 43: D213–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Waterhouse RM, Abai MR, Aganezov SS, Alekseyev MA, Allen JE, Amon J, Arca B, Arensburger P, Artemov G, et al. 2014. Highly evolvable malaria vectors: the genomes of 16 Anopheles mosquitoes. Science 347: 1258522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, Eastman S, Oliver B. 2003. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299: 697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pondeville E, Maria A, Jacques J-C, Bourgouin C, Dauphin-Villemant C. 2008. Anopheles gambiae males produce and transfer the vitellogenic steroid hormone 20-hydroxyecdysone to females during mating. Proc Natl Acad Sci 105: 19631–19636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pröschel M, Zhang Z, Parsch J. 2006. Widespread adaptive evolution of Drosophila genes with sex-biased expression. Genetics 174: 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke V, Gil IS, Ward S, Kazmer K. 2004. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development 131: 311–323. [DOI] [PubMed] [Google Scholar]
- Rice WR. 1996. Sexually antagonistic male adaptation triggered by experimental arrest of female evolution. Nature 381: 232–234. [DOI] [PubMed] [Google Scholar]
- Rice WR. 1998. Intergenomic conflict, interlocus antagonistic coevolution, and the evolution of reproductive isolation. Endless Forms Species Speciat 261–270. [Google Scholar]
- Rogers DW, Baldini F, Battaglia F, Panico M, Dell A, Morris HR, Catteruccia F. 2009. Transglutaminase-mediated semen coagulation controls sperm storage in the malaria mosquito. PLoS Biol 7: e1000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scali C, Catteruccia F, Li Q, Crisanti A. 2005. Identification of sex-specific transcripts of the Anopheles gambiae doublesex gene. J Exp Biol 208: 3701–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill D, Zhang Y, Parisi M, Oliver B. 2007. Demasculinization of X chromosomes in the Drosophila genus. Nature 450: 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgerson DG, Kulathinal RJ, Singh RS. 2002. Mammalian sperm proteins are rapidly evolving: evidence of positive selection in functionally diverse genes. Mol Biol Evol 19: 1973–1980. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripet F, Touré YT, Dolo G, Lanzaro GC. 2003. Frequency of multiple inseminations in field-collected Anopheles gambiae females revealed by DNA analysis of transferred sperm. Am J Trop Med Hyg 68: 1–5. [PubMed] [Google Scholar]
- Upton LM, Povelones M, Christophides GK. 2015. Anopheles gambiae blood feeding initiates an anticipatory defense response to Plasmodium berghei. J Innate Immun 7: 74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachou D, Schlegelmilch T, Christophides GK, Kafatos FC. 2005. Functional genomic analysis of midgut epithelial responses in Anopheles during Plasmodium invasion. Curr Biol 15: 1185–1195. [DOI] [PubMed] [Google Scholar]
- Vontas J, David JP, Nikou D, Hemingway J, Christophides GK, Louis C, Ranson H. 2007. Transcriptional analysis of insecticide resistance in Anopheles stephensi using cross-species microarray hybridization. Insect Mol Biol 16: 315–324. [DOI] [PubMed] [Google Scholar]
- Wang X, Werren JH, Clark AG. 2015. Genetic and epigenetic architecture of sex-biased expression in the jewel wasps Nasonia vitripennis and giraulti. Proc Natl Acad Sci 112: E3545–E3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Povelones M, Christophides GK. 2010. Sequence-structure-function relations of the mosquito leucine-rich repeat immune proteins. BMC Genomics 11: 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV. 2011. OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res 39: D283–D288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CI, Xu EY. 2003. Sexual antagonism and X inactivation—the SAXI hypothesis. Trends Genet 19: 243–247. [DOI] [PubMed] [Google Scholar]
- Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 5: 555–556. [DOI] [PubMed] [Google Scholar]
- Yuval B. 2006. Mating systems of blood-feeding flies. Annu Rev Entomol 51: 413–440. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Parsch J. 2005. Positive correlation between evolutionary rate and recombination rate in Drosophila genes with male-biased expression. Mol Biol Evol 22: 1945–1947. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hambuch TM, Parsch J. 2004. Molecular evolution of sex-biased genes in Drosophila. Mol Biol Evol 21: 2130–2139. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. 2007. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature 450: 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}