

Abstract
Congenital disorders of glycosylation form a rapidly growing group of inherited metabolic diseases. As glycosylation affects proteins all over the organism, a mutation in a single gene leads to a multisystemic disorder. We describe a patient with TMEM165-CDG with facial dysmorphism, nephrotic syndrome, cardiac defects, enlarged cerebral ventricles, feeding problems, and neurological involvement. Having confirmed the diagnosis via prenatal diagnostics, we were able to observe the glycosylation right from birth, finding a pathological pattern already on the first day of life. Within the next few weeks, hypoglycosylation progressed to less sialylated and then also to hypogalactosylated isoforms. On the whole, there has not been much published evidence concerning postnatal glycosylation and its adaptational process. This is the first paper reporting changes in glycosylation patterns over the first postnatal weeks in TMEM165-CDG.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2015_455) contains supplementary material, which is available to authorized users.
Keywords: CDG, Congenital disorders of glycosylation, Glycosylation, Postnatal, TMEM165
Introduction
Glycosylation is a complex and highly conserved way of co- and posttranslational modification of proteins and lipids. More than 50% of eukaryotic proteins are predicted to be glycoproteins, of which more than 90% carry N-linked oligosaccharides (Apweiler et al. 1999). N-Glycosylation is a process taking place in the endoplasmic reticulum (ER) and in the Golgi apparatus. In the ER, an oligosaccharide precursor is synthesized on a lipid anchor, dolichol, and is then transferred to the asparagine residue of a nascent protein. This protein-linked oligosaccharide is further processed in the endoplasmic reticulum and in the Golgi apparatus. The glycans in glycoproteins serve various functions: prevention of protein aggregation during folding, stabilization of the protein, regulation of interaction and recognition processes, regulation of protein degradation, and many more (Helenius and Aebi 2004).
Defects in glycosylation can lead to congenital disorders of glycosylation (CDG), a rapidly growing group of metabolic diseases. In terms of understanding the underlying defects, a distinction can be made between defects in dolichol-linked oligosaccharide synthesis and its transfer to the polypeptide (CDG-I) and in defects in the further processing of the protein-linked oligosaccharide (CDG-II). Since many new variants of glycosylation have been discovered in the last few years – and yet more are expected to be discovered – that do not all fit into this classification, a new nomenclature was proposed (Jaeken et al. 2009). Different forms of CDG should be named after the affected gene with the suffix “-CDG”.
While there are many publications on different kinds of glycosylation disorders, there is only little (and inconsistent) evidence about the results of biochemical examinations in the first days and weeks of life. In this article, we will give evidence for distinct changes in postnatal glycosylation in TMEM165-CDG (OMIM:614726).
Material and Methods
Mutation Analysis
Primers for sequencing of TMEM165 genomic DNA (NG_032881.1), designed with Primer3 software (Rozen and Skaletsky 1998) and purchased from Invitrogen (Carlsbad, CA, USA), are listed in the online supplementary material (see Table 1S). For PCR amplification 1 μL cDNA (fibroblasts 0.2 μg/μL; blood 0.04–0.05 μg/μL) was supplemented with 2 μL PCR buffer 10× (Qiagen), 4 μL Q-solution 5× (Qiagen), 2 mM dNTP mix (GE Healthcare, Buckinghamshire, UK), 9 μL water, 0.1 μL Taq DNA polymerase (Qiagen), 1 μL primer forward, and 1 μL primer reverse (20 pmol/μL). Samples were incubated for 5 min at 94°C. After 35 cycles of 94°C for 1 min, 60°C for 1 min, and 72°C for 1.5 min, a final incubation was performed at 72°C for 10 min. PCR products were purified using the PCR Product Pre-Sequencing Kit (USB Products/Affymetrix, Ohio, USA), and sequencing was performed with the BigDye Terminator Kit 3.1 (Applied Biosystems/Life Technologies, Darmstadt, Germany). 1.5 μL of purified PCR product was supplemented with 1.5 μL sequencing primer (10 pmol/L), 1 μL buffer, 0.5 μL BigDye, and 5.5 μL water. This was heated at 96°C for 2 min and then 25 cycles of 94°C for 10 s, 50°C for 5 s, and 60°C for 2 min followed. For final purification, the Sephadex/Millipore System (GE Healthcare, Buckinghamshire, UK/Merck Millipore, Schwalbach, Germany) was utilized. The sequence was analyzed on the ABI Prism 3700 sequencer (Applied Biosystems, Foster City, CA, USA).
Isoelectric Focusing (IEF)
Isoelectric focusing of transferrin was carried out as described in Niehues et al. (1998).
Immunoprecipitation and SDS-PAGE
Immunoprecipitation and sodium dodecyl sulfate–polyacrylamidegel electrophoresis (SDS-PAGE) for investigation of truncated carbohydrate side chains of transferrin was carried out as described in Niehues et al. (1998).
Nano-electrospray Ionization Time-of-Flight Mass Spectrometry (nanoESI-TOF MS)
NanoESI-TOF MS was performed as described previously by Park et al. (2014).
MALDI Time-of-Flight Mass Spectrometry (MALDI-TOF MS)
MALDI-TOF MS was carried out as described by Wada et al. (2012).
2D Gel Electrophoresis
Two plasma samples of the patient were collected at different times of life after parental informed consent was obtained. The sample preparation was performed as follows: 1 μL of plasma was mixed with 1 μL of a sodium dodecyl sulfate/dithioerythritol solution (SDS 10% w/v, DTE 2.3% w/v) and incubated at 95°C for 10 min. Afterward it was mixed with 250 μL of a sample buffer containing 8 M urea, 2% chaps, 50 μM DTT, 0.2% Bio-Lyte 3/10 ampholyte, and 0.001% bromophenol blue (Bio-Rad Laboratories, Hercules, CA, USA) and frozen at −20°C until further use.
The first dimension run (isoelectric focusing) was performed on the Protean i12 IEF System (Bio-Rad). 7 cm linear IPG strips (pH3-10) were rehydrated with 125 μL of the prepared sample solution at 10°C for 12 h at the temperature of 10°C. After rehydration, IEF was performed according to the following protocol.
In a first step, 250 V was applied for 15 min before the voltage was gradually increased to 4,000 V over the course of 1 h. This voltage was maintained until 15,000 volt-hours were reached. Afterward 500 V was applied until the gel strips were removed from the apparatus. The IPG strips were either frozen at −20°C until further use or directly transferred onto Mini-PROTEAN precast gels (Bio-Rad) for the second dimension run. This run was performed on the Mini-PROTEAN system (Bio-Rad). In brief, 50 V was applied until a running front formed and the voltage was consequently increased to 150 V until the front had reached to the bottom of the gels.
Silver staining of the 2D gels was performed as described by Bjellqvist et al. (1993).
Results
Clinical Data
The patient was the second child born to consanguineous parents after their first child had died from TMEM165-CDG at the age of 5 months. At 6 months of gestation, amniotic fluid was drained due to polyhydramnios. In this context, material for genetic analysis was obtained and sequencing of TMEM165 showed homozygosity for the same mutation as previously found in the sibling.
Birth was at term. Directly thereafter, the patient suffered from respiratory distress and CPAP support was necessary. Facial dysmorphic features and a weak abdominal wall were noticed. Echocardiography revealed a small apical VSD, a PFO, and a small PDA with mild signs of right ventricular hypertrophy. Brain ultrasound showed enlarged lateral and third ventricles. In the first days of life, there was suspicion of neonatal seizures, but EEG recording showed no abnormalities. Neurological examination revealed a large, temporarily tensed fontanel, muscular hypertonia with opisthotonic posture, and a sundown position of the eyes.
At the age of 1 week, mild proteinuria was observed, the extent increasing over the next weeks to nephrotic syndrome with slowly progressive renal failure. Edema as well as pericardial and pleural effusions developed, causing an intermittent need for oxygen supply. Infusion of human albumin had only very limited effect. Repeated hospitalizations were necessary due to recurrent edema and feeding problems requiring tube feeding.
The patient died at the age of 5 months due to complications of nephrotic syndrome and renal failure.
Mutation Analysis of TMEM165
Sequence analysis of TMEM165 showed homozygosity for the missense mutation c.323 A>G (p.E108G) in exon 2. The same mutation had been found in a homozygous state in the older sister, both parents have been shown to be heterozygous carriers of this mutation. In exome variation databases (Exome Variant Server), this mutation was not found in more than 5,000 sequenced probands.
IEF and SDS-PAGE
Isoelectric focusing (IEF) of transferrin showed a pathological pattern with increased amounts of tri-, di-, and monosialotransferrin already in the first blood sample taken 3 min after birth. In the follow-up controls, the pattern changed with increasing amounts of di-, mono-, and asialotransferrin and decreasing tetra- and trisialotransferrin until there was hardly any tetrasialotransferrin left (Fig. 1).
Fig. 1.
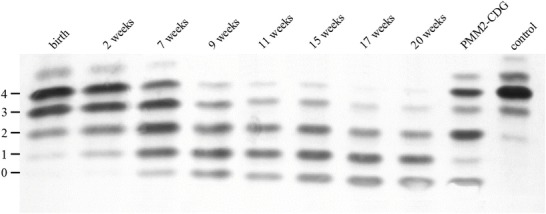
IEF of transferrin (Coomassie staining). Numbers at the left represent the number of negatively charged sialic acid residues. A distinct change to less sialylated specimens is visible over time
In SDS-PAGE, changes in apparent molecular mass led to a slight shift of the patient’s transferrin toward faster migrating isoforms. In comparison to a patient with PMM2-CDG where the bands represent normally glycosylated transferrin and transferrin missing one or two carbohydrate side chains, respectively, the loss in our patient was less than a whole carbohydrate side chain (2 kDa) (Fig. 2).
Fig. 2.
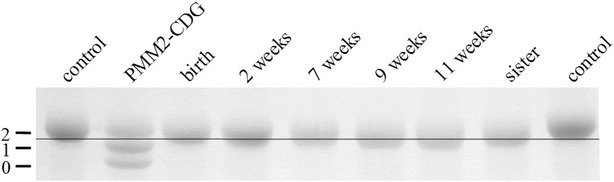
SDS-PAGE of transferrin (Coomassie staining). Numbers at the left indicate the amount of glycans. The band shifts toward the anode, indicating a loss of molecular mass of less than 2 kDa
Mass Spectrometry
Mass spectrometry (nanoESI-TOF MS) of transferrin from the day of birth detected normally glycosylated transferrin and another isoform missing one sialic acid. In the follow-up controls, the proportion of truncated forms steadily increased with different forms missing one or more sialic acids and then also galactose residues (Fig. 3). This was also observed in samples of the affected sister (not shown).
Fig. 3.
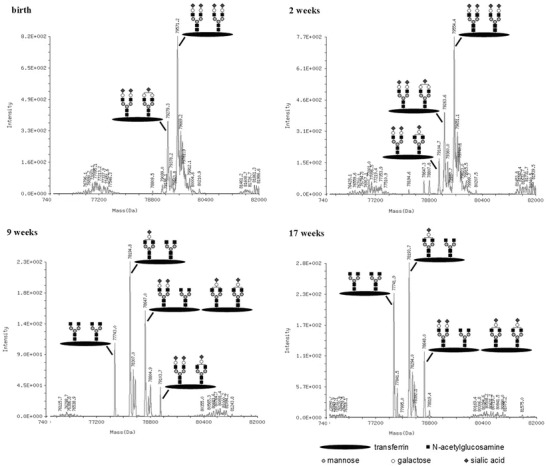
nanoESI-TOF MS of transferrin: a shift to less sialylated (and galactosylated) specimen is obvious in this analysis
Mass spectrometry of apolipoprotein C3 (MALDI-TOF MS) showed an increasing proportion of truncated and unglycosylated apoprotein C3 as a marker for a defect in O-glycosylation. These findings also increased over time (not shown).
2D Gel Electrophoresis
In general, the second sample showed a fainter pattern compared to sample 1 while being incubated for the same time in silver staining solution.
Changes in charge and molecular mass were present in various proteins. Most notably, α1-antitrypsin was shown to present with three more negatively charged subspecies in the later sample (Fig. 4).
Fig. 4.
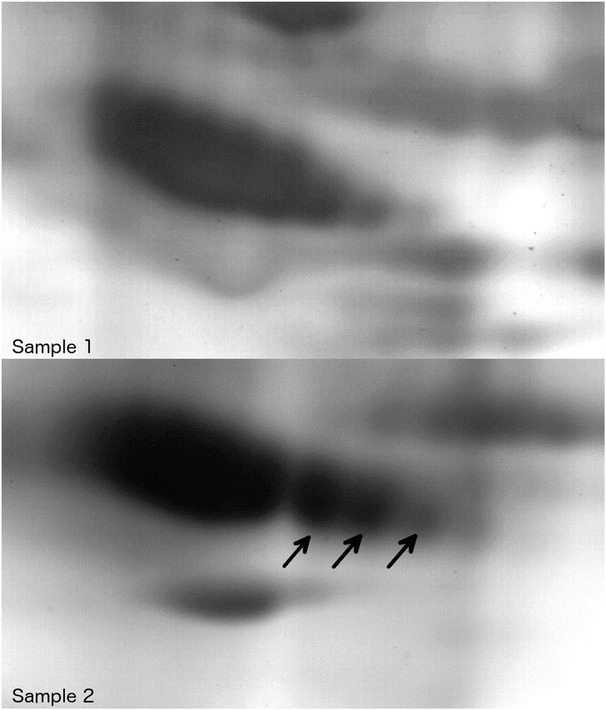
2D gel electrophoresis: comparison of α1-antitrypsin in plasma samples 1 (above) and 2 (below) of the patient. Note the different migration patterns in the sample collected later during the course of the disease
Discussion
Up to date, there is no CDG screening test for newborns, due to the fact that (1) there is no test available for a reliable screening on the third day of life and (2) no treatment for most types of CDG syndromes is available. Only children with conspicuous clinical features are selectively screened for CDG. Thus, CDG syndromes are often diagnosed in the first year of life, but normally not within the neonatal period or the first 3 months of life (Funke et al. 2013). In the present case, a sibling already diagnosed with TMEM165-CDG allowed prenatal diagnosis. This gave us the opportunity to monitor the changes in glycosylation patterns directly from birth.
The TMEM165 gene (HGNC:30760) is located on chromosome 4q12. The gene consists of six exons, the corresponding protein has 324 amino acid residues. It belongs to the highly conserved UPF0016 family of membrane proteins. Members of this family are found among many organisms, bacteria as well as eukaryotes (Fig. 5). TMEM165 is supposed to be a transporter for calcium ions, involved in Golgi pH homeostasis by a Ca2+/H+ antiport (Demaegd et al. 2013). It is located within the Golgi apparatus, plasma membrane, and late endosomes/lysosomes (Rosnoblet et al. 2013). Up to now, five patients affected with TMEM165-CDG have been described with skeletal dysplasia, growth retardation, and psychomotor retardation as key features (Foulquier et al. 2012). Early fatal outcome due to nephrotic syndrome has not been described before.
Fig. 5.

Cross-species alignment of TMEM165 and other members of the UPF0016 family. Alignment was prepared using the alignment tool from UniProt homepage (The UniProt Consortium 2014). Reference sequences for human (NP_060945.2), mouse (NP_035756.2), zebra fish (NP_997848.1), fruit fly (NP_650426.1), baker’s yeast (Saccharomyces cerevisiae, NP_009746.1), cyanobacteria (Synechocystis sp., NP_442278.1), and rice (NP_001068053.1). Columns below indicate the degree of conversation. The motif ELGDK (light gray box) is highly conserved and characteristic for proteins of the cation transporter UPF0016 family, indicating its functional importance. Most proteins of this family have two regions containing this motif, each with three predicted transmembrane regions and a central hydrophilic loop (Demaegd et al. 2013). Rosnoblet et al. (2013) suggested that the E108LGDK motif might be crucial for putative cation recognition signals. Since the patient’s mutation affects the glutamic acid on position 108 (“E”, dark gray box) in human TMEM165, one of the two negatively charged amino acids in this motif, this mutation might critically interact with the normal protein function as a calcium transporter. This could explain the severe phenotype in the patient
Directly after birth, glycosylation was abnormal in this case with increased amounts of tri-, di-, and monosialotransferrin, with tetra- and trisialotransferrin still forming the largest fractions. Over the next few weeks, the spectrum of transferrin changed to less sialylated and then also to hypogalactosylated forms, resulting in the nearly complete loss of tetrasialotransferrin. From week 9 forward, galactose and sialic acid residues are missing in equal proportions, indicating that the critical point was galactosylation rather than sialylation. We do not know if this pattern would have further changed since the patient died a few days after the last blood sample was collected. O-Glycosylation was also impaired right from birth with patterns deteriorating over time.
It is remarkable that an abnormal glycosylation pattern was already observed only 3 min after birth. It has previously been reported that abnormal glycosylation in PMM2-CDG develops not until the second or third week of life (Clayton et al. 1993). Fetal blood obtained in the 19th week of gestation showed normal transferrin glycosylation. Only in the second to third postnatal week the pattern changed in the pathological way normally observed in PMM2-CDG. Denecke et al. (2005) confirmed that IEF of transferrin in the 19th gestational week was normal in a fetus affected with ALG3-CDG. Other proteins like α1-antitrypsin were also shown to be normally glycosylated in an affected individual in utero. Other examples for negative IEF screening for different forms of CDG syndrome (PMM2-CDG, ATP6V0A2-CDG, SRDA3-CDG) in the first weeks of life have been reported (Funke et al. 2013). Why transferrin IEF is a poor marker neonatally for various CDGs is not entirely clear. Possible explanations are maternal proteins crossing of the placental barrier or a maternal factor influencing fetal glycosylation.
Synthesis of transferrin in the fetus has been observed as early as the ninth week of gestation but is quantitatively different compared to adults (Melartin et al. 1966). Relatively stable concentrations of transferrin postnatally without a significant decrease indicate sufficient synthesis by the fetus and neonate (Hitzig 1961). Transferrin has been shown to be able to cross the placenta, but only to a very limited extent (Gitlin et al. 1964). The concentration of radioiodinated transferrin in cord plasma was below 20% of the concentration in the mother at delivery. In some cases, little or no labeled transferrin was found in fetal plasma at all. Due to missing data concerning rates of degradation in the fetus and rates of transfer from fetus to mother, the plasma concentrations could not be exactly translated into placental transfer rates.
Placental transfer of transferrin alone cannot explain why the IEF of transferrin is entirely normal in the reported cases, as the predominant amount of transferrin is from fetal origin. With a half-life of 8–10 days (Chung 1984), even if transferrin in the newborn is in part of maternal origin, the proportion of maternal transferrin is negligible after 3–4 weeks. This total loss of maternal transferrin can explain a postnatal change to a more pathological glycosylation pattern, but not the initially normal results observed in different types of CDG.
de Jong and van Eijk (1988) demonstrated that during pregnancy, highly sialylated transferrin is increased in relative as well as in absolute amounts. The increase was also detectable in women using oral contraceptives, indicating that it may be caused by hormonal changes. It was also shown that this change could be observed in guinea pig fetuses as well, with the transferrin spectrum normalizing within 20 days after birth. These results point to a hormonal factor favoring higher degrees of sialylation in the maternal as well as in the fetal organism. Yet there was a difference between women in pregnancy and those taking oral contraceptives – while the absolute concentration of transferrin grew in the same extent, the relative gain of highly sialylated transferrin was less pronounced than in pregnancy, maybe due to a different estrogen/progesteron ratio or other hormones (de Jong et al. 1992). The existence of hormone-responsive elements within the promotor regions that might be the mediators for this hormonal influence on glycosylation has been proven for some glycosyltransferases (Medvedova et al. 2003), and that applies also to different expression patterns of glycosyltransferases in embryonic tissues (Uehara and Thelu 2001; Zhou et al. 1998; Granovsky et al. 1995) that might arise from this hormonal influence.
Stibler and Skovby (1994) reported another case of prenatal diagnostics in whom chorionic villus biopsy and amniotic fluid at gestational weeks 11 and 17 in twins with PMM2-CDG showed normal results for transferrin. In contrast to what was mentioned above, already on the first day of life, a CDG type I pattern was observed in transferrin IEF of blood samples. During the following 2 months, an increase in the concentration of carbohydrate-deficient transferrin was observed from 3.5/4.9 mg/L to 189/197 mg/L (data for twin 1/twin 2).
A pathological pattern in IEF of transferrin directly after birth, followed by an aggravation in the following weeks, was also observed by van de Kamp et al. (2007) with regard to two siblings affected with PMM2-CDG. In both cases, a pathological transferrin IEF pattern was observed on the second day of life. In the second child, there were follow-ups on days 22 and 39, respectively, when an increase in di- and asialotransferrin was noticed.
Other authors found abnormally glycosylated transferrin already in utero in the 27th and 30th gestational week, respectively (Edwards et al. 2006; Léticée et al. 2010), in patients affected with PMM2-CDG.
2D electrophoresis revealed a change in glycosylation not only for the common CDG biomarker transferrin but also for a variety of other glycoproteins such as the example shown in Fig. 4 (α1-antitrypsin). This indicates that TMEM165 deficiency affects the glycosylation of various proteins and that the changes are not restricted to transferrin. The faint pattern of the second sample can be explained by the patient’s nephrotic syndrome since it has been observed that the consequent protein loss can account for faint patterns in IEF (Kranz et al. 2004).
Results in our case show that glycosylation defects can be diagnosed by IEF already directly after birth (and probably already in the late intrauterine period) at least in some cases. The current opinion is that prenatal diagnosis for CDGs should only be made by molecular analysis (Matthijs et al. 2004). Of course this is the best way, giving a definite and reliable answer. In cases, however, in whom the underlying molecular defect is not known, IEF of transferrin might also give some information in the late prenatal and in the early postnatal period. With regard to the false-negative results described earlier, a negative result cannot definitely exclude a CDG syndrome. In case of ongoing suspicion, diagnostics should be repeated up to the age of at least 1 month for a definite exclusion of CDG. But in case of a positive result, transferrin IEF might shorten the time of uncertainty for parents.
It can only be speculated why there are CDG patients with normal IEF in the first days and weeks of life and others with a pathological pattern already in utero. This phenomenon is not restricted to specific types of CDG, but witnessed over a broad spectrum of different forms. A possible answer is that the more severe the impact of the underlying molecular defect is, the earlier a pathological pattern can be detected in transferrin analysis. Yet it seems that there is indeed a protecting factor during intrauterine life. Since it is hard to imagine that a fundamental pathway like glycosylation has major differences in the fetus compared to the neonate circumventing different metabolic blocks, it is likely that there is a protecting factor coming from the mother.
It would be very interesting to find out the nature of this protecting factor of neonatal glycosylation. From what we know, it should be a factor coming from the mother that is withdrawn by the time of birth, maybe a hormonal change. Cell culture work with CDG fibroblasts and maternal serum might be a way to identification of this factor which could render new therapeutic approaches to different types of CDG.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgement
We dedicate this paper to our friend and colleague Christian Körner. He was one of the founders of CDG research and will be deeply missed by colleagues and patients.
Take-Home Message
Glycosylation patterns undergo an adaptational process over the first postnatal weeks that can be shown by transferrin analysis. Postnatal screening for CDG has pitfalls due to a correcting factor likely derived from the mother.
Compliance with Ethic Guidelines
Conflict of Interest
All authors declare no conflict of interests.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from the parents.
Details of the Contributions of Individual Authors
S. Schulte Althoff: acquisition and analysis of data, drafting and revision of the manuscript
M. Grüneberg, J. Reunert, J. H. Park, S. Rust, Y. Wada: acquisition and analysis of data, revision of the manuscript
C. Mühlhausen, R. Santer: medical treatment of the patient, acquisition and analysis of data, revision of the manuscript
T. Marquardt: supervision, data acquisition and interpretation, revision of the manuscript
Footnotes
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2015_455) contains supplementary material, which is available to authorized users.
Competing interests: None declared
Contributor Information
T. Marquardt, Email: marquat@uni-muenster.de
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473(1):4–8. doi: 10.1016/S0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- Bjellqvist B, Pasquali C, Ravier F, Sanchez JC, Hochstrasser D. A nonlinear wide-range immobilized pH gradient for two-dimensional electrophoresis and its definition in a relevant pH scale. Electrophoresis. 1993;14(12):1357–1365. doi: 10.1002/elps.11501401209. [DOI] [PubMed] [Google Scholar]
- Chung MC-M. Structure and function of transferrin. Biochem Educ. 1984;12:146–154. doi: 10.1016/0307-4412(84)90118-3. [DOI] [Google Scholar]
- Clayton P, Winchester B, Di Tomaso E, Young E, Keir G, Rodeck C. Carbohydrate-deficient glycoprotein syndrome: normal glycosylation in the fetus. Lancet. 1993;341(8850):956. doi: 10.1016/0140-6736(93)91244-G. [DOI] [PubMed] [Google Scholar]
- de Jong G, van Eijk HG. Microheterogeneity of human serum transferrin: a biological phenomenon studied by isoelectric focusing in immobilized pH gradients. Electrophoresis. 1988;9(9):589–598. doi: 10.1002/elps.1150090921. [DOI] [PubMed] [Google Scholar]
- de Jong G, van Noort WL, Feelders RA, de Jeu-Jaspars CM, van Eijk HG. Adaptation of transferrin protein and glycan synthesis. Clin Chim Acta. 1992;212(1–2):27–45. doi: 10.1016/0009-8981(92)90135-D. [DOI] [PubMed] [Google Scholar]
- Demaegd D, Foulquier F, Colinet A-S, Gremillon L, Legrand D, Mariot P, et al. Newly characterized Golgi-localized family of proteins is involved in calcium and pH homeostasis in yeast and human cells. Proc Natl Acad Sci U S A. 2013;110(17):6859–6864. doi: 10.1073/pnas.1219871110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denecke J, Kranz C, Von Kleist-Retzow JC, et al. Congenital disorder of glycosylation type Id: clinical phenotype, molecular analysis, prenatal diagnosis, and glycosylation of fetal proteins. Pediatr Res. 2005;58:248–253. doi: 10.1203/01.PDR.0000169963.94378.B6. [DOI] [PubMed] [Google Scholar]
- Edwards M, McKenzie F, O’Callaghan S, et al. Prenatal diagnosis of congenital disorder of glycosylation type Ia (CDG-Ia) by cordocentesis and transferrin isoelectric focussing of serum of a 27-week fetus with non-immune hydrops. Prenat Diagn. 2006;26:985–988. doi: 10.1002/pd.1543. [DOI] [PubMed] [Google Scholar]
- Exome Variant Server, NHLBI GO Exome sequencing project (ESP), Seattle, WA. http://evs.gs.washington.edu/EVS/. Accessed Jan 2015
- Foulquier F, Amyere M, Jaeken J, et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am J Hum Genet. 2012;91(1):15–26. doi: 10.1016/j.ajhg.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke S, Gardeitchik T, Kouwenberg D, et al. Perinatal and early infantile symptoms in congenital disorders of glycosylation. Am J Med Genet A. 2013;161A:578–584. doi: 10.1002/ajmg.a.35702. [DOI] [PubMed] [Google Scholar]
- Gitlin D, Kumate J, Urrusti J, Morales C. The selectivity of the human placenta in the transfer of plasma proteins from mother to fetus. J Clin Invest. 1964;43(10):1938–1951. doi: 10.1172/JCI105068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granovsky M, Fode C, Warren CE, et al. GlcNAc-transferase V and core 2 GlcNAc-transferase expression in the developing mouse embryo. Glycobiology. 1995;5:797–806. doi: 10.1093/glycob/5.8.797. [DOI] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- Hitzig WH. Das Bluteiweissbild beim gesunden Säugling. Spezifische Proteinbestimmungen mit besonderer Berücksichtigung immunochemischer Methoden. Helv Paediatr Acta. 1961;16:46–81. [Google Scholar]
- Jaeken J, Hennet T, Matthijs G, Freeze HH. CDG nomenclature: time for a change. Biochim Biophys Acta. 2009;1792(9):825–826. doi: 10.1016/j.bbadis.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz C, Denecke J, Lehle L, et al. Congenital disorder of glycosylation type Ik (CDG-Ik): a defect of mannosyltransferase I. Am J Hum Genet. 2004;74(3):545–551. doi: 10.1086/382493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léticée N, Bessières-Grattagliano B, Dupré T, et al. Should PMM2-deficiency (CDG Ia) be searched in every case of unexplained hydrops fetalis? Mol Genet Metab. 2010;101(2–3):253–257. doi: 10.1016/j.ymgme.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Matthijs G, Schollen E, Van Schaftingen E. The prenatal diagnosis of congenital disorders of glycosylation (CDG) Prenat Diagn. 2004;24:114–116. doi: 10.1002/pd.815. [DOI] [PubMed] [Google Scholar]
- Medvedova L, Knopp J, Farkas R. Steroid regulation of terminal protein glycosyltransferase genes: molecular and functional homologies within sialyltransferase and fucosyltransferase families. Endocr Regul. 2003;37:203–210. [PubMed] [Google Scholar]
- Melartin L, Hirvonen T, Kaarsalo E, Toivanen P. Group-specific components and transferrins in human fetal sera. Scand J Haematol. 1966;3:8. doi: 10.1111/j.1600-0609.1966.tb01432.x. [DOI] [PubMed] [Google Scholar]
- Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest. 1998;101(7):1414–1420. doi: 10.1172/JCI2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Zühlsdorf A, Wada Y, et al. The novel transferrin E592A variant impairs the diagnostics of congenital disorders of glycosylation. Clin Chim Acta. 2014;436:135–139. doi: 10.1016/j.cca.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Rosnoblet C, Legrand D, Demaegd D, et al. Impact of disease-causing mutations on TMEM165 subcellular localization, a recently identified protein involved in CDG-II. Hum Mol Genet. 2013;22(14):2914–2928. doi: 10.1093/hmg/ddt146. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky HJ (1998) Primer3. Code available at http://www-genome.wi.mit.edu/genome_software/other/primer3.html
- Stibler H, Skovby F. Failure to diagnose carbohydrate-deficient glycoprotein syndrome prenatally. Pediatr Neurol. 1994;11(1):71. doi: 10.1016/0887-8994(94)90097-3. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium Activities at the universal protein resource (UniProt) Nucleic Acids Res. 2014;42:D191–D198. doi: 10.1093/nar/gku469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara K, Thelu J. Stage- and tissue-specific expression of a beta-1,4-galactosyltransferase in the embryonic epidermis. In Vitro Cell Dev Biol Anim. 2001;37:613–617. doi: 10.1290/1071-2690(2001)037<0613:SATSEO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Van de Kamp JM, Lefeber DJ, Ruijter GJG, et al. Congenital disorder of glycosylation type Ia presenting with hydrops fetalis. J Med Genet. 2007;44(4):277–280. doi: 10.1136/jmg.2006.044735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Kadoya M, Okamoto N. Mass spectrometry of apolipoprotein C-III, a simple analytical method for mucin-type O-glycosylation and its application to an autosomal recessive cutis laxa type-2 (ARCL2) patient. Glycobiology. 2012;22(8):1140–1144. doi: 10.1093/glycob/cws086. [DOI] [PubMed] [Google Scholar]
- Zhou D, Chen C, Jiang S, Shen Z, Chi Z, Gu J. Expression of 1,4-galactosyltransferase in the development of mouse brain. Biochim Biophys Acta. 1998;1425:204–208. doi: 10.1016/S0304-4165(98)00070-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.