

Abstract
Epstein-Barr virus (EBV) elicits primary CD8+ T cell responses that, by T cell cloning from infectious mononucleosis (IM) patients, appear skewed towards immediate early (IE) and some early (E) lytic cycle proteins, with late (L) proteins rarely targeted. However, L antigen-specific responses have been regularly detected in polyclonal T cell cultures from long-term virus carriers. To resolve this apparent difference between responses to primary and persistent infection, 13 long-term carriers were screened in ex vivo IFN-γ ELISPOT assays using peptides spanning the 2 IE, 6 representative E and 7 representative L proteins. This revealed memory CD8 responses to 44 new lytic cycle epitopes that straddle all three protein classes but, in terms of both frequency and size, maintain the IE > E > L hierarchy of immunodominance. Having identified the HLA restriction of 10 (including 7L) new epitopes using memory CD8+ T cell clones, we looked in HLA-matched IM patients and found such reactivities but typically at low levels, explaining why they had gone undetected in the original IM clonal screens. Wherever tested, all CD8+ T cell clones against these novel lytic cycle epitopes recognised lytically-infected cells naturally expressing their target antigen. Surprisingly, however, clones against the most frequently recognised L antigen, the BNRF1 tegument protein, also recognised latently-infected, growth-transformed cells. We infer that BNRF1 is also a latent antigen that could be targeted in T cell therapy of EBV-driven B-lymphoproliferative disease.
INTRODUCTION
Epstein-Barr virus (EBV), a γ1-herpesvirus with growth-transforming ability for its principal target cell, the B lymphocyte, is widespread in all human populations. Primary infection in infancy, and the life-long carrier state that ensues, is asymptomatic in the vast majority of cases. As a result, many aspects of the virus biology are inferred from individuals in whom late virus acquisition leads to infectious mononucleosis (IM), an immunopathologic disease associated with hyper-expansion of the virus-induced T cell response (1, 2). In IM, orally transmitted virus initiates lytic infection in a permissive cell type in the oropharynx, producing high titres of infectious virions that later subside. Simultaneously, the virus enters the B lymphocyte system and initiates a latent growth-transforming infection, leading to the transient expansion of cells expressing the full range of latent cycle proteins as seen in virus-transformed lymphoblastoid cell lines (LCLs) in vitro. That expansion is eventually contained by the T cell response, but not before some cells down-regulate latent antigen expression and establish a reservoir of true latency in the memory B cell pool (3). Throughout the subsequent virus carrier state, both lytic and growth-transforming latent infections appear subject to T cell surveillance. Thus individuals with profound T cell impairment, for example immuno-suppressed transplant patients or those with late-stage AIDS, show very high levels of virus shedding and are at high risk of EBV-driven B-lymphoproliferative disease (4, 5).
Responses to the 8 growth-transforming latent proteins have attracted most attention because of their therapeutic potential against B-lymphoproliferative lesions and other virus-associated malignancies (2, 4). Yet responses to lytic cycle proteins are larger and account for most of the massive CD8+ T cell expansion seen in acute IM (1, 2). The greater size of lytic cycle responses is perhaps not surprising given that virus replication, characterised by the sequential expression of two immediate early (IE), ~30 early (E) and ~30 late (L) proteins, provides a potentially rich array of foreign antigens for T cell detection. However, screening in vitro-expanded CD8+ T cell clones from IM blood for reactivity to a representative panels of IE, E and L protein targets detected a marked focusing on epitopes derived from the IE and a subset of E antigens, with responses to L antigens being unexpectedly rare (6, 7). That pattern has largely been confirmed in studies screening IM T cell preparations ex vivo for defined epitope-specific responses either by tetramer staining or functional analysis (8-10). Interestingly this IE > E > L hierarchy of immunodominance also directly reflected the efficiency with which these endogenously expressed proteins were presented during lytic cycle, with IE-specific CD8+ T cell clones showing the best recognition of lytically-infected cells and L antigen-specific clones the poorest (6). This implied that antigen processing function was progressively impaired with passage through lytic cycle and prompted the search for virus-coded immune evasion proteins. Several such lytic cycle proteins have now been identified, including BNLF2a that inhibits the TAP-mediated peptide transport onto nascent HLA class I molecules (11, 12), BILF1 that specifically modulates HLA class I trafficking (13, 14), and BGLF5 and the viral IL-10 homologue that have more general effects (15-17).
Given the apparent rarity of L antigen-specific responses in IM patients, it had generally been assumed that they would be at least as rare in memory, particularly given the huge contraction observed in CD8 responses to other lytic antigens following convalescence. Indeed, most surveys of CD8 memory in long-term virus carriers have concentrated primarily on IE/E antigen targets recognised by in vitro-expanded effectors (18-21) and there have been only occasional references to accompanying L antigen reactivities (10, 22-24). However, in 2011 Orlova et al noticed that CD8+ T cells specific for L antigens could be detected at rates comparable to those for E antigens in in vitro-expanded T cell lines from rhesus macaques persistently infected with the EBV-related lymphocryptovirus (LCV) (25). This prompted the authors to use the same approach to screen adult EBV carriers for memory CD8 responses to representative EBV-coded L antigens. Effector T cell lines were generated by repetitive stimulation of peripheral blood mononuclear cells (PBMCs) with the autologous LCL, and then tested in IFN-γ ELISPOT assays for recognition of individual antigens expressed from recombinant vaccinia viruses. All five donors tested showed some L antigen-specific reactivity, with examples of responses against five of the seven L antigens studied. This suggested either that L antigen-specific responses had been missed in the earlier IM-based studies identifying CD8+ T cell targets, or that such responses were indeed absent during primary infection and arose as an accompaniment of persistent virus carriage.
In the present paper, we re-examine the issue of immunodominance among EBV lytic proteins in CD8+ T cell memory. As representative target antigens, we selected the two IE proteins and a panel of 6 E and 7 L proteins; these included some already studied both by ourselves (6) and by Orlova et al (25), and others not hitherto investigated as CD8 targets. To avoid potential artefacts introduced by repeated LCL stimulation and polyclonal T cell expansion in vitro, we began by screening PBMCs from healthy virus carriers for ex vivo reactivity to peptide panels in ELISPOT assays. Our objectives were (i) to determine the relative frequency of IE, E and L peptide-specific responses in CD8+ T cell memory, (ii) to establish the lytic antigen specificity and HLA restriction of selected responses, particularly L antigen-specific responses, by CD8+ T cell cloning, and (iii) to re-examine IM CD8+ T cell preparations ex vivo for these newly-identified reactivities.
MATERIALS and METHODS
EBV target antigens
The following fifteen EBV lytic cycle proteins were selected for the study: the two IE transcription activator proteins, BZLF1 and BRLF1; six representative E proteins, BMLF1 and BMRF1 (both transcriptional transactivators), BHRF1 (bcl2 homologue), BaRF1 (small ribonucleotide reductase), BFRF1 (nuclear envelope protein) and BLLF3 (dUTPase); and seven representative L proteins, gp350/BLLF1, gp85/BXLF2, gp25/BKRF2 and gp42/BZLF2 (all viral glycoproteins), BFRF3 (small capsid protein), BVRF2 (capsid maturational protease) and BNRF1 (major tegument protein). Synthetic peptides based on the B95.8 strain sequence were purchased from either AltaBioscience (University of Birmingham, UK) or Mimotopes (Clayton, Australia), dissolved in DMSO and their concentrations determined by biuret assay. Peptide panels were 15mers overlapping by 10 or 11 aa for most antigens, and 20mers overlapping by 15aa for gp350, gp85 and gp42; both 15mer and 20mer panels were prepared for gp25 for comparison.
Donors
Written, informed consent was given by all donors for the collection and analysis of blood samples and all experiments were approved by the West Midlands (Black Country) Research Ethics Committee (07/Q2702/24). Screening for memory CD8+ T cell responses was carried out on 13 EBV-seropositive donors with no history of infectious mononucleosis (IM) and one EBV-seronegative control donor. Their HLA class I types are recorded in Supplementary Table I and included many alleles commonly found in Caucasian populations. In addition, PBMCs from 8 acute IM patients of known HLA type were used in intracellular cytokine staining experiments.
PBMC preparations
60ml blood samples were collected from all donors and PBMCs separated by Ficoll-Hypaque centrifugation into RPMI 1640 medium + L-glutamine (Invitrogen) supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin and 5% human serum or 10% FCS. PBMCs to be used in ELISPOT screening assays were depleted of CD4+ T cells using CD4 Dynabeads (Invitrogen) in accordance with the manufacturer’s protocol. Efficient depletion was confirmed by flow cytometry and >98% reduction of the CD4+ cells was consistently achieved.
Screening for memory CD8+ T cell responses by IFN-γ ELISPOT assay
CD4+-depleted PBMCs were tested in IFN-γ ELISPOT assays using cytokine capture and detection reagents as previously described (26). Briefly, 96-well nitrocellulose plates (Millipore) were coated with anti-IFN-γ antibodies and CD4+-depleted PBMCs were added to replicate wells in the presence of single or pooled overlapping peptides (8-12 peptides/pool) at a final concentration of 5 μg/ml for each peptide. 10 μg/ml PHA and an equivalent volume of DMSO were used as positive and negative controls, respectively. Generally 150,000 cells were used in each of triplicate wells. After overnight incubation at 37°C in 5% CO2, the cells were discarded and captured IFN-γ was detected with a biotinylated anti-IFN-γ antibody followed by a streptavidin-conjugated alkaline phosphatase and an Alkaline Phosphatase conjugate substrate kit (Bio-Rad). The spots were counted using an automated plate counter (AID). In all experiments, results from ELISPOT assays are expressed as spot-forming cells (SFC) per million CD4+-depleted PBMCs. Positive responses were defined as those where the mean number of spots in replicate wells exceeded the mean number of spots (plus two standard deviations) in the replicate DMSO control wells.
Isolation and culture of T cell clones
Short-term polyclonal cultures of peptide-specific cells were generated by incubating whole PBMCs in RPMI 1640 medium + L-glutamine (Invitrogen) supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin, 5% human serum and 20 ng/ml IL-7 after pre-exposing one fifth of the cells to 5 μg/ml of the appropriate peptide(s) for 1-2 h and washing. IL-2 was added to the cultures on day 2 at a final concentration of 50 U/ml. On day 7, cultures were depleted of CD4+ T cells using anti-CD4 Dynabeads (Invitrogen) and the peptide-specific cells were enriched using a MACS IFN-γ Secretion Assay – Cell Enrichment and Detection Kit (Miltenyi Biotech), all in accordance with the manufacturers’ protocols. T cell clones were isolated from these preparations by limiting dilution seeding in standard culture medium supplemented by IL-2 as previously described (7); irradiated preactivated allogeneic PBMCs (106/ml) were always included as feeders with the addition of an anti-CD3 mAb (OKT3; Unipath, Basingstoke, U.K) to a final concentration of 30 ng/ml. Growing microcultures reactive against the desired peptide(s) in IFN-γ ELISAs were further expanded and cultured by transfer into 2 ml wells using the same stimulation protocol as before.
Characterisation of T cell clones by IFN-γ ELISA
The CD8+ T cell clones (5,000 or 10,000 cells/well) were incubated in V-bottom microtest plate wells with ten times the number of target cells. The supernatant medium was harvested after 18 h and assayed for IFN-γ by ELISA (Endogen), in accordance with the manufacturer’s protocol. For HLA restriction assays, the target cells were autologous, partially HLA-matched or HLA-mismatched LCL cells that had been pre-exposed to either 5 μg/ml epitope peptide or to an equivalent volume of DMSO solvent as a control for 1 h and then washed. All clones were also tested for recognition of lytically-infected target cells using target LCLs of the relevant HLA type transformed with either wild type B95.8 virus or, as a negative control, the replication-deficient BZLF1-knockout (BZ-K/O) derivative strain (27). For some specificities, the minimal epitope recognized by the CD8+ T cells was defined experimentally by testing on HLA-matched LCL cells pre-exposed to a panel of shorter peptides within the 15mer/20mer of interest, each at a range of peptide concentrations.
Identification of primary CD8+ T cell responses to EBV lytic epitopes by flow cytometry
Cryopreserved PBMCs from acute IM patients were thawed, washed and resuspended in RPMI 1640 medium + L-glutamine (Invitrogen) supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin, 10% FCS and 50U/ml IL-2. The cells were transferred to FACS tubes to give 0.5-1×106 cells per tube in 500 μl medium and were incubated at 37°C in 5% CO2 with 5 μg/ml epitope peptide, with the equivalent volume of DMSO as a negative control or with 0.2 g/ml Staphylococcal Enterotoxin B (SEB; Sigma) as a positive control. Brefeldin A (Sigma) was added to each tube after 1 h to a final concentration of 10 g/ml. After a further 5 h incubation period, the cells were washed in PBS and stained with LIVE/DEAD Fixable Far Red Dead Cell Stain (Invitrogen) for 15 min at room temperature. Following a wash with PBS, the cells were further washed with staining buffer (PBS supplemented with 0.5% BSA and 2mM EDTA) before they were stained on ice for 15 min with saturating concentrations of the following anti-human Abs: PerCPCy5.5-conjugated CD8 mAb (clone RPA-T8 eBioscience), PE-conjugated CD4 mAb (clone RPA-T4, BD Pharmingen) and APC-conjugated CD19 mAb (clone HIB19, eBioscience). Cells were then fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.5% saponin for 5 min, and stained with FITC-conjugated IFN-γ mAb (clone 25723.11, BD FastImmune) for 30 min, all at room temperature. After a final wash, the cells were resuspended in 200 μl of staining buffer and analyzed on a BD Accuri C6 flow cytometer (BD Biosciences). All data were processed using FlowJo software (TreeStar).
RESULTS
Memory CD8+ T cell responses to EBV lytic cycle antigens
A first set of experiments used IFN-γ ELISPOT assays to screen CD4+-depleted PBMCs from healthy adult donors for ex vivo reactivity to peptide panels representing 15 EBV lytic cycle antigens. These were the two IE proteins (BZLF1 and BRLF1), six E proteins (BMLF1, BMRF1, BHRF1, BaRF1, BFRF1 and BLLF3) and seven L proteins (gp350, gp85, gp25, gp42, BFRF3, BVRF2 and BNRF1). For each protein, peptides were arranged into pools of 8-12 consecutive peptides for the initial screening and, where responses were detected, assays were then repeated on the individual peptides within the pool to identify the relevant epitope region. Figure 1 shows examples of results from two EBV-seropositive individuals. Responses in donor 5 could be detected to peptide pool 5 from the IE protein BZLF1, to pool 4 from the E protein BaRF1, to pool 4 from the L protein BVRF2, and to pools 15 and 20 from a second late protein, BNRF1 (Figure 1A, left panels). Subsequent testing mapped these responses to individual peptides BZLF1 5.2 (aa 209-217), BaRF1 4.1 and 4.2 (sharing aa 149-159); BVRF2 4.2 and 4.3 (sharing aa 153-163), and BNRF1 15.9 and 15.10 (sharing aa 709-719) and 20.5 (aa 929-943) (Figure 1A, right panels). Corresponding data from donor 9, again illustrating responses to peptides within IE (BZLF1), E (BLLF3) and L (BNRF1) antigens, are shown in Figure1B. All the above responses represented novel reactivities. Indeed, of the five target antigens identified in Figure 1, only BZLF1 had previously been reported as a CD8+ T cell target. Such results clearly showed that the peptide screening approach was capable of detecting memory CD8 responses to hitherto unknown epitopes in IE, E and L proteins of the lytic cycle. Note also that, in the examples shown in Figure 1, response size (expressed as the number of SFC per 106 CD4+-depleted PBMCs) fell as one moved from IE through E to L antigen responses.
Figure 1. Identification of memory CD8+ T cell responses to EBV lytic antigens.

CD4+ T cell-depleted PBMCs were screened ex vivo for reactivity against overlapping peptides spanning the B95.8 strain sequences of 15 EBV lytic proteins by IFN-γ ELISPOT. Representative results from donor 5 (A) and donor 9 (B) are shown here. Responses to pools of overlapping peptides were identified first (left panels) and then mapped to individual peptides within those pools (right panels). Results are shown as the mean number of spot-forming cells (SFC) per million CD4+-depleted PMBCs from replicate wells after subtraction of baseline reactivity to DMSO (mean of replicate DMSO wells + 2 SD).
In total, 13 EBV-seropositive donors were screened in this way, alongside one EBV-seronegative donor as a control. Throughout this work, all assays on peptide pools and/or individual peptides were repeated using further bleeds of the donors in question to confirm that the reported responses were reproducible. Table I shows the combination of individual antigen responses seen in each donor. All 13 seropositive donors showed reactivity to at least 1 of the 15 lytic cycle antigens screened, with responses to a median of 6 antigens per donor, while the seronegative donor gave uniformly negative results. In line with their previously reported immunodominance, the IE proteins BZLF1 and BRLF1 were the most frequently recognised, with reactivities detectable in 12/13 and 10/13 seropositive subjects respectively. Among the E proteins, the incidence of recognition among seropositive donors ranged from frequent (10/13) for BMLF1 to rare (1/12) to BHRF1, again reminiscent of the breadth of results obtained in earlier work examining E proteins as CD8 targets. Most interesting, however, were the data obtained with the L antigen peptide panel. While peptides from some antigens such as the viral envelope glycoproteins were rarely recognised, we found examples of 4 donors responding to peptides within the virus assembly protein BVRF2, 5 donors to peptides within the capsid protein BFRF3, and a remarkable total of 9 donors to peptides within the tegument protein BNRF1. Overall, positive responses were obtained from 85% of individual donor/antigen combinations tested involving IE proteins, 38% of those involving E proteins, and 25% of those involving L proteins.
Table I. Summary of all memory CD8+ T cell responses to EBV lytic antigens identified by ex vivo IFN-γ ELISPOT.
| Sero-positive donors | Sero-neg | Number of sero-positive responders |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | ||||
| IE | BZLF1 | * | * | * | * | * | * | * | * | * | * | 12/13 | ||||
| BRLF1 | * | * | * | * | * | * | * | * | * | * | 10/13 | |||||
| E | BMLF1 | * | * | * | * | * | * | * | * | * | * | 10/13 | ||||
| BHRF1 | nt | * | 1/12 | |||||||||||||
| BMRF1 | * | * | * | 5/13 | ||||||||||||
| BaRF1 | * | * | * | * | * | * | * | 8/13 | ||||||||
| BFRF1 | * | 3/13 | ||||||||||||||
| BLLF3 | nt | * | * | 2/12 | ||||||||||||
| L | gp350 | 0/13 | ||||||||||||||
| gp85 | nt | * | 1/12 | |||||||||||||
| gp25 | * | * | * | 3/13 | ||||||||||||
| gp42 | nt | 0/12 | ||||||||||||||
| BFRF3 | * | * | * | * | 5/13 | |||||||||||
| BVRF2 | * | * | * | 4/13 | ||||||||||||
| BNRF1 | * | * | * | * | * | * | * | * | 9/13 | |||||||
Thirteen healthy EBV-seropositve donors and one healthy EBV-seronegative donor were analyzed for memory CD8+ T cell reactivity to two IE proteins (BZLF1 and BRLF1), six E proteins (BMLF1, BHRF1, BMRF1, BaRF1, BFRF1, BLLF3) and seven L proteins (gp350, gp85, gp25, gp42, BNRF1, BFRF3 and BVRF2). For each donor, reactivity to the different proteins is represented as a shaded box. Where these reactivities have been mapped to individual peptide(s) within the pool(s) an asterisk is shown within the shaded box. Donor 1 has not been screened against the complete panel of antigens; those antigens that have not been tested are denoted by ‘nt’.
Most of the above responses were mapped to single epitope regions in assays on individual peptides within the original positive pool, and the results are summarised in Figure 2. Each protein is drawn to show its relative size and the locations of CD8 epitopes within its sequence are identified as vertical bars. The two IE proteins, and in particular BZLF1 (the smaller of the two) contained multiple epitopes, while epitope numbers in the E proteins ranged between 1 and 5. Patterns of epitope density were even more varied among the L proteins. Of the glycoproteins, gp350 and gp42 were completely devoid of detectable epitopes while the occasional responses to gp85 and gp25 in each case mapped to a single peptide in the sequence. By contrast the more frequently recognised BVRF2 and BFRF3 proteins each contained 3 epitopes, while the many responses detected against the large BNRF1 protein mapped to a total of 12 separate epitope regions. Overall, these screening assays had detected memory CD8 responses to a total of 53 epitopes in the lytic cycle antigens chosen for study. The detailed locations and sequences of these epitopes, and the individual donors that responded to them, are recorded in Supplementary Table II. Remarkably, only 9 of these responses targeted previously identified epitopes, all within the IE (BZLF1, BRLF1) or dominant E (BMLF1, BMRF1) proteins (2, 28). The remaining 44, marked with asterisks both in Figure 2 and in Supplementary Table II, represent novel reactivities. Clearly the CD8 T cell pool of EBV carriers contained memory to multiple lytic cycle epitopes, many of which had not been identified before.
Figure 2. CD8 epitope maps of the lytic antigens screened, showing all the CD8 epitopes identified in healthy EBV-seropositive donors by ex vivo IFN-γ ELISPOT assay.

Each protein is represented according to its relative size and epitopes are illustrated as vertical bars. The 44 newly identified epitopes are denoted by an asterisk. Of these 44, the 10 epitopes against which we subsequently isolated CD8+ T cell clones are identified by a second asterisk.
For each epitope region, the size of the IFN-γ ELISPOT response to the relevant screening peptide (15- or 20-mer) was expressed as mean SFC per 106 CD4+-depleted PBMCs (minus the average DMSO control + 2SD). Combining results from all positive responses observed in the above assays, Supplementary Table II shows the range and median size of responses against each epitope, and the data are summarized in Figure 3 in terms of response size to antigens either grouped by kinetic class (Figure 3A) or individually (Figure 3B). Overall IE-specific responses were slightly larger in size than responses to E proteins, and both were significantly larger than L-specific responses. Interestingly, when examined at the level of individual antigens, both IE proteins and the three most frequently recognised E proteins (BMLF1, BMRF1, BaRF1) could elicit numerically strong responses (>1000 SFC per 106 CD4+-depleted PBMCs). However responses to the L proteins, including the frequently recognised BNRF1, are typically very low, the one exception being responses to the single epitope in gp25.
Figure 3. Magnitude of memory CD8+ T cell responses to EBV lytic antigens identified in the 13 healthy EBV-seropositive donors screened by ex vivo IFN-γ ELISPOT assay.
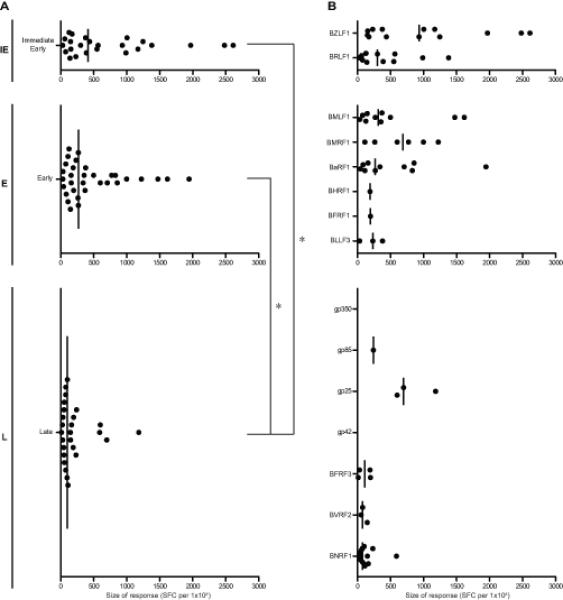
(A) Scatter dot plots (with line at median) show the size of responses to individual epitopes from antigens expressed during IE, E and L phases of the lytic cycle, as determined in assays using the original screening peptide (15- or 20-mer). Each dot shows the mean number of spot-forming cells per million CD4+-depleted PMBCs from replicate wells after subtraction of baseline reactivity to DMSO (mean of replicate DMSO wells + 2 SD). When analysed statistically by Kruskal-Wallis test with post-hoc Dunn’s test, both the IE- and E-antigen-specific responses were significantly larger than the L antigen-specific responses, as indicated with asterisks on brackets (p<0.05). Where a response to two adjacent screening peptides has been identified, the larger response is shown. Reactivities that have only been identified against pool(s) of overlapping peptides and have not yet been mapped to individual peptides within the pool(s) are not included. (B) The same results are further subdivided to show responses to the individual antigens expressed within each phase.
CD8+ T cell clones against lytic antigen-derived epitopes
To further characterise some of these responses, we selected 10 of the 44 novel reactivities identified in the initial screening assays for detailed analysis by CD8+ T cell cloning. These 10 epitopes are marked by double asterisks in Figure 2 and are named in subsequent experiments using the first three amino acids of the sequence; two are IE epitopes (one in BZLF1 and one in BRLF1), one is an E epitope (in BaRF1) and seven are L epitopes (one each in gp25 and BFRF3, two in BVRF2 and three in BNRF1). In each case, CD8+ T cells reactive to peptide stimulation in short-term PBMC cultures were selected using IFN-γ-capture assay, seeded by limiting dilution into cloning medium; thereafter, growing cultures were screened for peptide-specificity by IFN-γ ELISA and selected clones further expanded as previously described (7). All selected clones were confirmed as uniformly CD8+ by mAb staining and, in many but not all cases, the optimal epitope was identified in titration assays using shorter peptides within the originally recognised sequence (data not shown). For all 10 cloned responses, Table II gives the currently defined epitope sequence and its coordinates within the source antigen. Access to cloned populations made two objectives possible: firstly to identify the epitope’s HLA restricting allele and secondly, using an appropriate panel of target LCLs, to check whether these lytic epitope-specific T cells could indeed recognise lytically-infected cells within LCL populations.
Table II. Summary of epitope-specific CD8+ T cell clones.
| Phase | Protein | Function | Epitope Coordinates |
Sequence | HLA Restriction | Responders |
|---|---|---|---|---|---|---|
| IE | BZLF1 | Transcription activator | 66-75† | VSTAPTGSWF† | B58.01 | 1/2 |
| BRLF1 | Transactivator | 101-115 | IACPIVMRYVLDHLI | B58.01 | 1/2 | |
|
| ||||||
| E | BaRF1 | Ribonucleotide reductase | 151-159 | LLIEGIFFI | A2.01 | 4/10 |
|
| ||||||
| L | gp25 | Membrane fusion (gL) | 120-128 | VEDLFGANL | B60 | 3/3 |
| BNRF1 | Tegument | 709-719† 1247-1257 1281-1289 |
GEQGYKVSLDL† YPRNPTEQGNI WQWEHIPPA |
B41 B7.02 A2.01 |
1/1 3/4 2/10 |
|
| BFRF3 | Capsid | 127-137 | TPSVSSSISSL | B7.02 | 2/4 | |
| BVRF2 | Assembly | 153-163† 341-351† |
AVYGTDLAWVL† GYFTSPGGYYA† |
C3 A29 |
1/5 1/1 |
|
Epitopes are hereby referred to by the first three amino acids of their sequence, as underlined,
Minimal epitopes have not been defined experimentally; coordinates and sequences given are the overlapping amino acids from the peptides the clones respond to. ‘Number of Responders’ column gives the number of HDs that were shown to have a CD8+ cell response to the specified peptide by IFN-γ ELISPOT / number of HDs screened with the correct HLA class I-restriction (N.B. The subtype of the HLA-C3-positive donors has not been determined).
Representative results from the analyses of HLA restriction are shown in Figure 4A, with data from clones against four of the selected epitopes. Using IFN-γ ELISA as the readout, clones were tested for recognition of epitope peptide-loaded target cells either from the autologous donor or from allogeneic donors partially matched through specific HLA-A, -B, or -C alleles. For example, the epitope recognised by the BZLF1 (VST)-specific clone (top panel) was only presented by target cells sharing HLA-B58 with the T cell donor, showing this to be the restricting allele. Likewise, the results shown for other clones identified HLA-A*0201 as the restricting allele for the BaRF1 (LLI) epitope, HLA-B*0702 for the BFRF3 (TPS) epitope, and HLA- B60 for the gp25 (VED) epitope. All 10 cloned reactivities were studied in this way and their restricting alleles are shown alongside the epitopes in Table II. Overall, two of these novel epitopes were restricted through HLA-A*0201, two through HLA-B*0702, and two through HLA-B58 (subtype not determined), while the other four were restricted through individual HLA-A, B or C alleles. Knowing the restriction, it was now possible to go back to the original panel of 13 seropositive donors and ask how many of those with the relevant HLA restricting allele actually made an epitope response. As shown in Table II, several of these newly defined epitopes were inducing detectable responses in only a subset of individuals with the relevant HLA allele, a pattern often associated with sub-dominant epitopes (10). For example, typically weak responses to the TPS epitope in L antigen BFRF3 were detected in just 2/4 B*0702-positive donors. Even the intermediate size response to the LLI epitope in the E protein BaRF1was seen in only 4/8 A*0201-positive donors, clearly showing that this epitope is sub-ordinate to the well known YVL (BRLF1) and GLC (BMLF1) epitopes against which most A*0201-positive donors respond (see Supplementary Table II). However the gp25 epitope VED (B60-restricted) was again an interesting exception; all three B60-positive donors tested had a detectable memory response to this L epitope.
Figure 4. Characterization of representative IE, E and L antigen-specific CD8+ T cell clones.
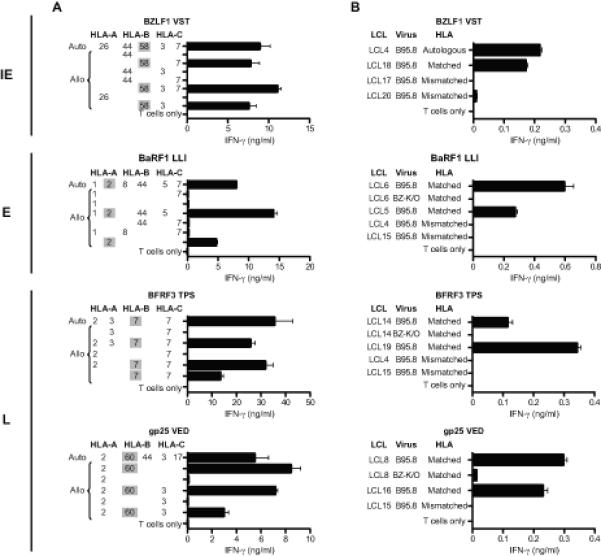
Recognition of target LCLs was measured by IFN-γ ELISA and results are mean ± 1 SD of duplicate wells. (A) HLA restriction of CD8+ T cell clones. T cells were incubated overnight with peptide-loaded cells of the autologous B95.8 LCL and of partially HLA class I-matched allogeneic LCLs (for which the matching alleles are given). (B) CD8+ T cell recognition of unmanipulated LCL targets. T cells were incubated overnight with LCL cells from HLA-matched (or autologous) and HLA-mismatched donors. Where possible, a pair of B95.8 and BZ-K/O LCLs from an HLA-matched donor was included. All HLA-matched B95.8 and BZ-K/O LCLs were strongly recognised by the relevant T cells if they were pre-exposed to target peptide, with > 8ng/ml IFN-γ released in all cases (data not shown). N.B. Levels of B95.8 LCL recognition by IE versus E versus L antigen-specific clones cannot be directly compared since the percentage of lytically-infected cells in these LCLs differs between cell lines.
All clones were then tested for their ability to recognize lytically-infected cells. Assays were conducted on LCL target lines with and without the relevant HLA restricting allele and transformed with the wild-type B95.8 EBV strain; note that such lines are semi-permissive for virus replication, with typically 1-5% cells in lytic cycle. As a control, the assays included matched LCLs established using BZ-K/O-deleted B95.8 virus (27) and therefore devoid of lytically-infected cells. Examples of results from such assays are presented in Figure 4B, using clones directed against the same lytic epitopes as in Figure 4A. In each case, there was clear recognition of wild-type B95.8 LCLs with the relevant restricting allele, but not of matching BZ-K/O LCLs. This pattern of results was seen in all assays using clones against the IE proteins BZLF1 and BRLF1, the E protein BaRF1 and the L proteins gp25, BFRF3 and BVRF2. In each case, such results were consistent with the clone’s presumed lytic antigen specificity and with its ability to recognize that antigen when expressed in lytically-infected cells.
However, a different pattern of results to the above was observed using clones specific for epitopes in BNRF1, the L antigen against which memory CD8+ T cell responses had been most frequently detected. Figure 5A shows representative data from multiple experiments with clones specific for the HLA-A*0201-restricted WQW epitope in BNRF1. These again show the expected recognition of wild-type B95.8 LCLs from HLA-A*0201-positive (but not negative) donors. However, this is always accompanied by significant, albeit slightly lower, recognition of the corresponding A*0201-matched BZ-K/O LCL. Likewise in Figure 5B, the same result was reproducibly obtained using clones to the HLA-B*0702-restricted YPR epitope in BNRF1, with recognition of B*0702-matched targets whether transformed with wild-type or BZ-K/O virus strains. Both sets of assays included, as an additional control, CD8 clones with the same restricting allele as the BNRF1 effectors but specific for other lytic cycle antigens. Figure 5C and 5D show the control data, from an A*0201-restricted clone against a previously identified epitope FLD in the late lytic glycoprotein gp110 (6) and from a B*0702-restricted clone against a recently identified epitope RPG (RPGRPLAGFYA) in the early lytic protein BNLF2b (L. Quinn, unpublished observations), respectively. Just as noted with the other lytic cycle effectors tested in Figure 4, CD8+ T cells specific for gp110 and BNLF2b again showed no recognition of BZ-K/O LCLs, confirming that these target lines are indeed devoid of lytically-infected cells. As a further check, we searched among a panel of A*0201- and/or B*0702-positive wild-type B95.8 LCLs to select lines lacking spontaneous lytic cycle entry and found that these were also recognised by the BNRF1 effectors but not by effectors against other lytic cycle antigens (data not shown). This showed that the unexpected detection of BNRF1 in a latent growth-transforming infection was not simply an artifact of the recombinant BZ-K/O virus construct.
Figure 5. Recognition of lytic cycle-deficient BZ-K/O LCLs by BNRF1-specific CD8+ T cell clones.

T cells were incubated overnight with pairs of B95.8 and BZ-K/O LCL cells from HLA-matched donors and, as a negative control, B95.8 LCL cells from an HLA-mismatched donor. Results are mean ± 1 SD of duplicate or triplicate wells and are expressed as IFN-γ release, as determined by ELISA. All HLA-matched B95.8 and BZ-K/O LCLs were strongly recognised by the relevant T cells if they were pre-exposed to target peptide, with > 8ng/ml IFN-γ released in all cases (data not shown). The BNRF1-specific CD8+ T cell clones used recognise: (A) the HLA-A*0201-restricted WQW epitope, and (B) the HLA-B*0702-restricted YPR epitope. As further controls to check the lytic cycle-deficient status of the BZ-K/O LCLs, the experiments included lytic antigen-specific CD8+ T cell clones recognising (C) the HLA-A*0201-restricted FLD epitope in BALF4, and (D) a newly-identified HLAB*0702-restricted RPG epitope in BNLF2b.
Primary CD8+ T cell responses to EBV lytic cycle antigens in IM patients
The work to this point had revealed a surprisingly large number of CD8 responses to IE, E and also L antigens in healthy virus carriers, many more than had been identified in the original T cell cloning work with IM patients. We therefore turned our attention back to IM, asking whether responses to these novel epitopes were indeed detectable during primary infection or had arisen as accompaniments of long-term virus carriage. We again used peptide-induced IFN-γ production as the marker of a response but in this case stained for intracellular IFN-γ after a 6 hr peptide exposure because activated, apotosis-prone, CD8+ T cells in IM blood are inefficiently detected in an 18 hr ELISPOT assay. First we focused on HLA-A*0201-positive IM patients, where earlier ex vivo cloning assays have already identified abundant responses to two A*0201-restricted epitopes, YVL from the IE protein BRLF1 and TLD from the E protein BMRF1 (9). These peptides were used to stimulate IM PBMC preparations in parallel with two A*0201-restricted epitopes identified in the present work, LLI in the E protein BaRF1 and WQW in the L protein BNRF1, and with a previously identified weak A*0201 epitope from another L protein, gp110 (6). Figure 6A shows examples of staining profiles from one such patient IM223, and Figure 6B summarises the overall data from six A*0201-positive patients. Responses to the already-known YVL and TLD epitopes ranged up to almost 1% of CD8+ T cells with median values of 0.57 and 0.17%. Interestingly, responses to the new E epitope LLI were also easily detectable, with a median value of 0.2% CD8+ T cells, while responses to the L epitopes WQW and FLD were detectable in at least some patients at low but significant levels. Figure 6C shows the corresponding results obtained from five IM patients screened for B*0702-restricted reactivity to the known RPQ epitope in E protein BMRF1 and to the new L protein epitopes YPR and TPS. Again a reasonably strong RPQ response was seen in most patients, but smaller responses to the L epitopes were also detectable. These novel A*0201- and B*0702-resticted responses had been identified as typically sub-dominant components of long-term memory and so their detection in IM, even at low levels, is significant. These studies were then extended to include the one L antigen-specific response, against the B60-restricted VED epitope in gp25, which we had found was reasonably abundant in long-term memory. We sought to detect this response in acute IM, comparing it with that seen against another B60-restricted IE epitope, SEN in BZLF1 (29); note that SEN is a weak epitope against which responses are detectable in some but not all B60-positive carriers (Supplementary Table II). Figure 6D shows the data examining these two epitope responses in three B60-positive IM patients. The VED response is detectable in all three cases, albeit at quite low levels but always larger than that seen against SEN. We conclude that the novel L antigen responses, which the present work has identified by memory screening, are not products of long-term virus carriage but are already present as components of the primary response.
Figure 6. Identification of primary CD8+ T cell responses to EBV lytic epitopes restricted through HLA-A*0201, HLA-B*0702 and HLA-B60 in acute IM patients.
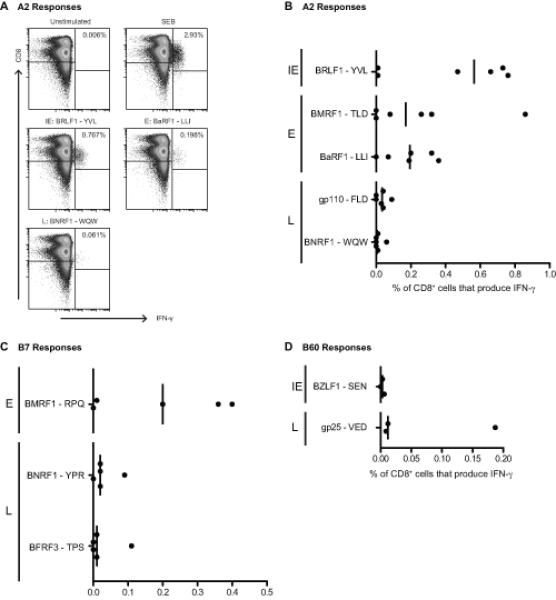
Ex vivo PBMCs from A*0201+, B*0702+ and/or B60+ IM donors were incubated for six hours with 5 μg/ml epitope peptide, or with Staphylococcal Enterotoxin B (SEB) as a positive control, and then analysed for reactivity by intracellular IFN-γ staining. A: Representative dot plots from an A*0201+ donor, showing CD8 and intracellular IFN-γ staining of lymphocytes remaining after exclusion of CD4+, CD19+ and dead cells. The numbers indicate percentages of IFN-γ+ cells. B-D: Scatter dot plots showing the percentage of CD8+ T cells that produce IFN-γ in response to five A*0201 (B), three B*0702 (C) and two B60 (D) epitopes in acute IM patients with the relevant HLA allele; results compiled from a total of eight acute IM donors (six A*0201+, five B*0702+ and three B60+).
DISCUSSION
This paper addresses the issue of immunodominance among EBV lytic cycle proteins as targets for CD8+ T cells. Our earlier studies, cloning T cells from IM blood and screening on a limited range of IE, E and L antigens, had suggested that the primary CD8 response was markedly skewed towards the two IE proteins and a subset of E proteins, while L proteins constituted only rare sub-dominant targets (6, 7, 9). However, studying memory responses in the rhesus model of LCV infection and subsequently in healthy EBV carriers, Orlova et al found that polyclonal T cell lines made by repeatedly stimulating PBMCs with the autologous semi-permissive LCL regularly contained CD8+ T cell reactivities against one or more L proteins (25). Interestingly, in the rhesus LCV model, such reactivities were detected more frequently in older than in younger animals, prompting the authors to suggest that L antigen-specific responses may develop slowly over time, perhaps as a result of ongoing virus replication during the virus carrier state (25). This certainly seemed plausible in the context of EBV infection since prospective studies on IM patients had already provided examples from where the balance between CD8 responses to individual antigens or epitopes appeared to change over time (9, 30); furthermore with another human herpesvirus, cytomegalovirus, some but not all components of the CD8 response expand considerably with life-long viral carriage (31, 32). The present work therefore sought to re-examine the CD8+ T cell memory of healthy EBV carriers to selected IE, E and L antigens, to identify a representative range of new IE, E and L target epitopes and their restricting alleles, and then to compare the relative abundance of these reactivities in primary versus persistent infection. The results provide a much more detailed picture of CD8 responses to lytic cycle antigens, with multiple subdominant IE, E and L epitope reactivities present alongside those against dominant IE/E epitopes. Importantly, however, the same hierarchy of immunodominance (IE > E>L) is apparent in both primary and memory phases of the response.
To avoid the possibility that in vitro-expansion might distort the composition of T cell memory, we screened CD4+-depleted PBMCs from long-term EBV carriers ex vivo using IFN-γ ELISPOT assays and peptides covering the primary sequences of 15 selected lytic cycle antigens. In addition to EBV’s two IE transactivator proteins, the antigen panel included six E proteins with a range of different lytic cycle functions and seven L proteins ranging from virion capsid (BFRF3), tegument (BNRF1) and envelope (gp25, gp42, gp85 and gp350) components to an assembly protein (BVRF2) that is not detected in mature virus particles (33). While there was some overlap with previous panels used by ourselves and others (6, 25), three E (BaRF1, BFRF1, BLLF3) and three L proteins (gp25, gp42, BFRF3) were examined as CD8+ T cell targets for the first time. The ELISPOT assays quickly showed that EBV carriers had detectable CD8 memory cells against a broad array of lytic cycle antigens; overall, individuals recognised a mean of 6 antigens on the panel and every individual tested had at least one response. The relative importance of IE, E and L target antigens can be judged in terms of the frequency of positive responses among donors tested (Table I), the number of epitopes identified in these proteins (Figure 2) and the size of response to those epitopes (Figure 3, Supplementary table II). The IE antigens, BZLF1 and BRLF1, remain the strongest targets by all three criteria, with responses detectable to one or both proteins in all 13 individuals, with 8 new epitopes added to the 6 already known, and with the largest median size of response; interestingly, BZLF1 was stronger than BRLF1 in all three respects. As observed in the original IM clonal analysis, E antigens were more variable as CD8 targets. Thus the two transactivator proteins, BMLF1 and BMRF1, and a newly studied protein BaRF1 were more frequently recognised than the other three E antigens tested, contained more epitopes than they did (including 11 of the 16 new E epitopes identified) and elicited numerically stronger responses. Leaving aside the special case of BNRF1 (discussed below), the other five L proteins tested were also variable as CD8 targets. Overall, however, they showed lower frequencies of recognition than the six E proteins, contained just 8 epitopes (all newly identified) and, for 7 of these 8 epitopes, elicited weak responses.
The above findings are therefore broadly consistent with an IE > E > L hierarchy of immunodominance among lytic cycle antigens as targets for the memory CD8 response, in line with the hierarchy first suggested from studies of the primary response in acute IM (6, 7). That said, there are important caveats to the conclusions drawn from a study of this kind. Firstly, we have detected memory responses using IFN-γ secretion which, though the most sensitive cytokine in this viral system, nevertheless under-estimates the true number of epitope-specific cells (26). Secondly, given the limited size of the donor panel (13 individuals), our study covers most but certainly not all HLA types that are relatively common in Caucasian populations and so the analysis is incomplete. Indeed, just as seen with responses to latent cycle antigens (2, 34), individual HLA alleles can have idiosyncratic preferences among lytic cycle targets that appear to contradict the general trend; for example the three L antigen-specific memory responses that are clearly larger than the rest all came from B60-positive donors responding to a single B60-restricted epitope in gp25. Thirdly, our target antigen panel represents just a subset (20-25%) of all E and L proteins, and there is a clear need to extend the panel before drawing too firm conclusions. In that regard, among the L proteins tested, the apparent difference in antigenicity between the four viral envelope glycoproteins (gp25, gp42, gp85, gp350) and the non-envelope BFRF3 (capsid) and BVRF2 (assembly) proteins raises the possibility that a glycoprotein bias has lowered the overall frequency of L antigen responses. Here it should be noted that three of these glycoproteins were represented by 20-mer rather than 15-mer peptide libraries; using the longer peptides is reported in some studies to slightly reduce the efficiency of detection in CD8 memory screens (35) whereas in others no such difference was found (36). We conducted several preliminary experiments (including screening gp25 with both 15- and 20-mers) to show that, under our assay conditions, 20-mer screening detected the same epitope responses as did 15-mers but tended to give slightly lower spot-forming cell counts. On these grounds, we are confident that the infrequency of detectable ELISPOT responses seen against these glycoproteins genuinely reflects their poor immunogenicity for CD8+ T cells. In that regard, others have reported the existence of sub-dominant A2-restricted CD8 epitopes in gp350 and gp85 (23), but even re-screening the A2-positive donors in our study with the published minimal epitope peptides did not detect such gp350 and gp85 responses (data not shown).
We then went on to establish CD8+ T cell clones against 10 of the 44 new epitopes identified by ELISPOT screening, thereby opening the way to functional studies. One particular issue, bearing upon the biological relevance of these newly identified responses, was to determine whether they were capable of recognising lytically-infected cells. The results (Figure 4B) show that this was indeed the case, not just for IE- and E-epitope-specific effectors but even for clones directed against L epitopes. Semi-permissive B95.8 LCL targets with the correct HLA restricting allele were recognised whereas (for all but the BNRF1 effectors) the matching BZ-K/O LCLs were not. That recognition of semi-permissive lines must have been directed towards the lytically-infected cells themselves since, within LCL cultures, we have never observed any cross-presentation of released lytic cycle antigens to CD8+ T cells ((37) and data not shown). Therefore, despite the virus’ immune evasion strategies becoming ever more effective with progress through lytic cycle (38), there is still enough epitope display, even of L antigen-derived epitopes, to allow some level of T cell recognition. This no doubt attests to the potent antigen presenting function of LCL cells and the difficulty of completely eliminating epitope display. We conclude that even L antigen-specific responses, though a minor component of the total lytic antigen response, have the capacity to recognise lytically-infected cells of the kind found within EBV-driven lymphoproliferative lesions in vivo (39-41); this could be important in preventing virus release, secondary infection and the recruitment of new B cell transformants into these lesions.
More interesting in that regard, however, were the unexpected findings with clones specific for BNRF1, a virion tegument protein (42, 43) hitherto thought to be expressed exclusively in late lytic cycle but encoded by a gene lying immediately downstream of the latent membrane protein LMP2 gene in the EBV genome. Clones against two independent BNRF1 epitopes recognised not just semi-permissive B95.8 LCL targets of the appropriate HLA type but also the matched BZ-K/O LCLs included as lytic cycle-deficient controls (Figure 5), as well as matched LCLs carrying wild type virus but identified as tightly latent through their absence of BZLF1 expression (data not shown). We infer that, besides its established status as a late lytic cycle protein, BNRF1 is also expressed as a latent protein in growth-transforming infection. Indeed recognition of the BZ-K/O LCL in the above experiments was in many cases only slightly lower than that of the semi-permissive line, indicating that most recognition reflects epitope display on latently-infected cells with only a small contribution from cells in lytic cycle. Note that these findings are not indicative of a general promiscuity of lytic gene expression in BZ-K/O LCLs or indeed in the BZLF1-negative B95.8 LCLs tested because, both in this study (Figures 4B and 5) and elsewhere (11), T cell assays with other lytic antigen-specific effectors make it clear that genuine lytic cycle proteins are not detectably expressed in such lines. Fortuitously therefore, the present work has identified BNRF1 as a protein expressed in both latent and lytic infections, which contains multiple CD8 epitopes and elicits detectable, though usually weak, responses in the majority of EBV carriers. Such BNRF1 epitope responses can be amplified in vitro and thereby add to the arsenal of latent antigen-specific T cells with therapeutic potential against EBV-driven B lymphoproliferative disease (4).
Access to T cell clones also allowed us to identify the HLA restricting alleles through which 10 of the new lytic epitope responses were mediated (Figure 4A). It was then possible to return to one of our original objectives and ask whether epitope specificities that we had identified in memory, particularly those directed against L antigens, were also present in the primary response. We addressed this point comparing, wherever possible, the abundance of responses to IE, E and L epitopes restricted through the same HLA allele. In the absence of peptide-HLA I tetramer reagents for all of the epitopes, we again had to rely on peptide-induced IFN-γ production to detect responses, accepting that such functional assays only detect a proportion of the activated epitope-specific CD8+ T cells in IM blood (44). With this caveat, the results (Figure 6) support the view that IE, E and L epitope-specific reactivities are all present in the primary response in proportions that broadly reflect their order of abundance in memory. These findings suggest that L antigen-specific responses are not selectively expanded with long-term virus carriage (25) but, like most of the better studied IE and E antigen responses (9), undergo a phase of primary expansion followed by contraction into a smaller memory population that is then stably maintained. Observations on rare responses to A2-restricted gp350 and gp85 epitopes (22, 23) would also support this conclusion. However, the way is now open to address the issue more systematically using tetramers to track L epitope-specific responses in individuals following IM.
In summary, our work makes it clear that CD8+ T cell responses to lytic EBV infection contain considerably more components than were apparent from earlier studies based on T cell cloning from IM patients. This attests to the greater ability of the ELISPOT assay to detect low abundance responses, compared to CD8+ T cell cloning which typically will pick up numerically dominant components of the response, but, given the finite number of clones that can be expanded and screened, will by chance miss rare reactivities. Nevertheless the hierarchy of immunodominance among lytic cycle proteins first proposed on the basis of the IM cloning studies still holds, and does so in both primary and persistent infection. Thus the numerically dominant responses tend to be directed towards epitopes drawn from the two IE antigens and a subset of E proteins. However, these are accompanied by many low abundance responses directed towards sub-dominant epitopes, some drawn from IE, some from E and some from L proteins. The relative paucity of L antigen-specific CD8+ T cell responses is particularly striking given the abundant expression of these proteins in lytically-infected cells and their capacity to elicit strong CD4+ T cell (37, 45, 46) and humoral (5) responses. Therefore, rather than reflecting the amount of exogenous antigen available for re-processing by the dendritic cell system, the CD8 response to lytic cycle antigens more closely reflects the efficiency with which the endogenously expressed proteins are presented on lytically-infected cells (6). This reinforces the notion that the main driver of CD8 responses in the EBV system is not antigen displayed via the cross-priming pathway (47) but direct contact with infected cells.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Graham S. Taylor for statistical analysis and Andrew D. Hislop for general help and advice.
This work was supported by a program grant (G0901755) from the Medical Research Council, U.K.
Abbreviations used in this article
- BZ-K/O
BZLF1-knockout
- E
early
- IE
immediate early
- IM
infectious mononucleosis
- L
late
- LCL
lymphoproliferative cell line
- LCV
lymphocryptovirus
- SEB
Staphylococcal Enterotoxin B
- SFC
spot-forming cells
REFERENCES
- 1.Callan MF, Tan L, Annels N, Ogg GS, Wilson JD, O’Callaghan CA, Steven N, McMichael AJ, Rickinson AB. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J. Exp. Med. 1998;187:1395–1402. doi: 10.1084/jem.187.9.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007;25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553. [DOI] [PubMed] [Google Scholar]
- 3.Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. EBV persistence in memory B cells in vivo. Immunity. 1998;9:395–404. doi: 10.1016/s1074-7613(00)80622-6. [DOI] [PubMed] [Google Scholar]
- 4.Gottschalk S, Rooney CM, Heslop HE. Post-transplant lymphoproliferative disorders. Annu. Rev. Med. 2005;56:29–44. doi: 10.1146/annurev.med.56.082103.104727. [DOI] [PubMed] [Google Scholar]
- 5.Rickinson AB, Kieff E. Epstein-Barr Virus. In: Fields BN, Knipe DM, Howley PM, editors. Fields’ Virology. 5th ed Wolters Kluwer/Lippincott Williams & Wilkins; Philadelphia, Pa. ; London: 2007. pp. 2655–2700. [Google Scholar]
- 6.Pudney VA, Leese AM, Rickinson AB, Hislop AD. CD8+ immunodominance among Epstein-Barr virus lytic cycle antigens directly reflects the efficiency of antigen presentation in lytically infected cells. J. Exp. Med. 2005;201:349–360. doi: 10.1084/jem.20041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steven NM, Annels NE, Kumar A, Leese AM, Kurilla MG, Rickinson AB. Immediate early and early lytic cycle proteins are frequent targets of the Epstein-Barr virus-induced cytotoxic T cell response. J. Exp. Med. 1997;185:1605–1617. doi: 10.1084/jem.185.9.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catalina MD, Sullivan JL, Bak KR, Luzuriaga K. Differential evolution and stability of epitope-specific CD8(+) T cell responses in EBV infection. J. Immunol. 2001;167:4450–4457. doi: 10.4049/jimmunol.167.8.4450. [DOI] [PubMed] [Google Scholar]
- 9.Hislop AD, Annels NE, Gudgeon NH, Leese AM, Rickinson AB. Epitope-specific evolution of human CD8(+) T cell responses from primary to persistent phases of Epstein-Barr virus infection. J. Exp. Med. 2002;195:893–905. doi: 10.1084/jem.20011692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodberry T, Suscovich TJ, Henry LM, Davis JK, Frahm N, Walker BD, Scadden DT, Wang F, Brander C. Differential targeting and shifts in the immunodominance of Epstein-Barr virus--specific CD8 and CD4 T cell responses during acute and persistent infection. J. Infect. Dis. 2005;192:1513–1524. doi: 10.1086/491741. [DOI] [PubMed] [Google Scholar]
- 11.Croft NP, Shannon-Lowe C, Bell AI, Horst D, Kremmer E, Ressing ME, Wiertz EJ, Middeldorp JM, Rowe M, Rickinson AB, Hislop AD. Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog. 2009;5:e1000490. doi: 10.1371/journal.ppat.1000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hislop AD, Ressing ME, van Leeuwen D, Pudney VA, Horst D, Koppers-Lalic D, Croft NP, Neefjes JJ, Rickinson AB, Wiertz EJ. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J. Exp. Med. 2007;204:1863–1873. doi: 10.1084/jem.20070256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuo J, Currin A, Griffin BD, Shannon-Lowe C, Thomas WA, Ressing ME, Wiertz EJ, Rowe M. The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 2009;5:e1000255. doi: 10.1371/journal.ppat.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuo J, Quinn LL, Tamblyn J, Thomas WA, Feederle R, Delecluse HJ, Hislop AD, Rowe M. The Epstein-Barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J. Virol. 2011;85:1604–1614. doi: 10.1128/JVI.01608-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012;8:e1002704. doi: 10.1371/journal.ppat.1002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowe M, Glaunsinger B, van Leeuwen D, Zuo J, Sweetman D, Ganem D, Middeldorp J, Wiertz EJ, Ressing ME. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. U. S. A. 2007;104:3366–3371. doi: 10.1073/pnas.0611128104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeidler R, Eissner G, Meissner P, Uebel S, Tampe R, Lazis S, Hammerschmidt W. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood. 1997;90:2390–2397. [PubMed] [Google Scholar]
- 18.Bogedain C, Wolf H, Modrow S, Stuber G, Jilg W. Specific cytotoxic T lymphocytes recognize the immediate-early transactivator Zta of Epstein-Barr virus. J. Virol. 1995;69:4872–4879. doi: 10.1128/jvi.69.8.4872-4879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pepperl S, Benninger-Doring G, Modrow S, Wolf H, Jilg W. Immediate-early transactivator Rta of Epstein-Barr virus (EBV) shows multiple epitopes recognized by EBV-specific cytotoxic T lymphocytes. J. Virol. 1998;72:8644–8649. doi: 10.1128/jvi.72.11.8644-8649.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saulquin X, Ibisch C, Peyrat MA, Scotet E, Hourmant M, Vie H, Bonneville M, Houssaint E. A global appraisal of immunodominant CD8 T cell responses to Epstein-Barr virus and cytomegalovirus by bulk screening. Eur. J. Immunol. 2000;30:2531–2539. doi: 10.1002/1521-4141(200009)30:9<2531::AID-IMMU2531>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 21.Scotet E, David-Ameline J, Peyrat MA, Moreau-Aubry A, Pinczon D, Lim A, Even J, Semana G, Berthelot JM, Breathnach R, Bonneville M, Houssaint E. T cell response to Epstein-Barr virus transactivators in chronic rheumatoid arthritis. J. Exp. Med. 1996;184:1791–1800. doi: 10.1084/jem.184.5.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bharadwaj M, Sherritt M, Khanna R, Moss DJ. Contrasting Epstein-Barr virus-specific cytotoxic T cell responses to HLA A2-restricted epitopes in humans and HLA transgenic mice: implications for vaccine design. Vaccine. 2001;19:3769–3777. doi: 10.1016/s0264-410x(01)00085-8. [DOI] [PubMed] [Google Scholar]
- 23.Khanna R, Sherritt M, Burrows SR. EBV structural antigens, gp350 and gp85, as targets for ex vivo virus-specific CTL during acute infectious mononucleosis: potential use of gp350/gp85 CTL epitopes for vaccine design. J. Immunol. 1999;162:3063–3069. [PubMed] [Google Scholar]
- 24.Saulquin X, Bodinier M, Peyrat MA, Hislop A, Scotet E, Lang F, Bonneville M, Houssaint E. Frequent recognition of BCRF1, a late lytic cycle protein of Epstein-Barr virus, in the HLA-B*2705 context: evidence for a TAP-independent processing. Eur. J. Immunol. 2001;31:708–715. doi: 10.1002/1521-4141(200103)31:3<708::aid-immu708>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 25.Orlova N, Wang F, Fogg MH. Persistent infection drives the development of CD8+ T cells specific for late lytic infection antigens in lymphocryptovirus-infected macaques and Epstein-Barr virus-infected humans. J. Virol. 2011;85:12821–12824. doi: 10.1128/JVI.05742-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan LC, Gudgeon N, Annels NE, Hansasuta P, O’Callaghan CA, Rowland-Jones S, McMichael AJ, Rickinson AB, Callan MF. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J. Immunol. 1999;162:1827–1835. [PubMed] [Google Scholar]
- 27.Feederle R, Kost M, Baumann M, Janz A, Drouet E, Hammerschmidt W, Delecluse HJ. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000;19:3080–3089. doi: 10.1093/emboj/19.12.3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rist MJ, Theodossis A, Croft NP, Neller MA, Welland A, Chen Z, Sullivan LC, Burrows JM, Miles JJ, Brennan RM, Gras S, Khanna R, Brooks AG, McCluskey J, Purcell AW, Rossjohn J, Burrows SR. HLA Peptide Length Preferences Control CD8+ T Cell Responses. J. Immunol. 2013;191:561–571. doi: 10.4049/jimmunol.1300292. [DOI] [PubMed] [Google Scholar]
- 29.Couedel C, Bodinier M, Peyrat MA, Bonneville M, Davodeau F, Lang F. Selection and long-term persistence of reactive CTL clones during an EBV chronic response are determined by avidity, CD8 variable contribution compensating for differences in TCR affinities. J. Immunol. 1999;162:6351–6358. [PubMed] [Google Scholar]
- 30.Sauce D, Larsen M, Abbott RJ, Hislop AD, Leese AM, Khan N, Papagno L, Freeman GJ, Rickinson AB. Upregulation of interleukin 7 receptor alpha and programmed death 1 marks an epitope-specific CD8+ T-cell response that disappears following primary Epstein-Barr virus infection. J. Virol. 2009;83:9068–9078. doi: 10.1128/JVI.00141-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Hara GA, Welten SP, Klenerman P, Arens R. Memory T cell inflation: understanding cause and effect. Trends Immunol. 2012;33:84–90. doi: 10.1016/j.it.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 32.Weekes MP, Wills MR, Mynard K, Hicks R, Sissons JG, Carmichael AJ. Large clonal expansions of human virus-specific memory cytotoxic T lymphocytes within the CD57+ CD28− CD8+ T-cell population. Immunology. 1999;98:443–449. doi: 10.1046/j.1365-2567.1999.00901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chapman AL, Rickinson AB, Thomas WA, Jarrett RF, Crocker J, Lee SP. Epstein-Barr virus-specific cytotoxic T lymphocyte responses in the blood and tumor site of Hodgkin’s disease patients: implications for a T-cell-based therapy. Cancer Res. 2001;61:6219–6226. [PubMed] [Google Scholar]
- 35.Maecker HT, Dunn HS, Suni MA, Khatamzas E, Pitcher CJ, Bunde T, Persaud N, Trigona W, Fu TM, Sinclair E, Bredt BM, McCune JM, Maino VC, Kern F, Picker LJ. Use of overlapping peptide mixtures as antigens for cytokine flow cytometry. J. Immunol. Methods. 2001;255:27–40. doi: 10.1016/s0022-1759(01)00416-1. [DOI] [PubMed] [Google Scholar]
- 36.Draenert R, Altfeld M, Brander C, Basgoz N, Corcoran C, Wurcel AG, Stone DR, Kalams SA, Trocha A, Addo MM, Goulder PJ, Walker BD. Comparison of overlapping peptide sets for detection of antiviral CD8 and CD4 T cell responses. J. Immunol. Methods. 2003;275:19–29. doi: 10.1016/s0022-1759(02)00541-0. [DOI] [PubMed] [Google Scholar]
- 37.Long HM, Leese AM, Chagoury OL, Connerty SR, Quarcoopome J, Quinn LL, Shannon-Lowe C, Rickinson AB. Cytotoxic CD4+ T cell responses to EBV contrast with CD8 responses in breadth of lytic cycle antigen choice and in lytic cycle recognition. J. Immunol. 2011;187:92–101. doi: 10.4049/jimmunol.1100590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rowe M, Zuo J. Immune responses to Epstein-Barr virus: molecular interactions in the virus evasion of CD8+ T cell immunity. Microbes Infect. 2010;12:173–181. doi: 10.1016/j.micinf.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montone KT, Hodinka RL, Salhany KE, Lavi E, Rostami A, Tomaszewski JE. Identification of Epstein-Barr virus lytic activity in post-transplantation lymphoproliferative disease. Mod. Pathol. 1996;9:621–630. [PubMed] [Google Scholar]
- 40.Rea D, Delecluse HJ, Hamilton-Dutoit SJ, Marelle L, Joab I, Edelman L, Finet JF, Raphael M. Epstein-Barr virus latent and replicative gene expression in post-transplant lymphoproliferative disorders and AIDS-related non-Hodgkin’s lymphomas. French Study Group of Pathology for HIV-associated Tumors. Ann. Oncol. 1994;5(Suppl 1):113–116. doi: 10.1093/annonc/5.suppl_1.s113. [DOI] [PubMed] [Google Scholar]
- 41.Tanner JE, Alfieri C. The Epstein-Barr virus and post-transplant lymphoproliferative disease: interplay of immunosuppression, EBV, and the immune system in disease pathogenesis. Transpl. Infect. Dis. 2001;3:60–69. doi: 10.1034/j.1399-3062.2001.003002060.x. [DOI] [PubMed] [Google Scholar]
- 42.Feederle R, Neuhierl B, Baldwin G, Bannert H, Hub B, Mautner J, Behrends U, Delecluse HJ. Epstein-Barr virus BNRF1 protein allows efficient transfer from the endosomal compartment to the nucleus of primary B lymphocytes. J. Virol. 2006;80:9435–9443. doi: 10.1128/JVI.00473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsai K, Thikmyanova N, Wojcechowskyj JA, Delecluse HJ, Lieberman PM. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011;7:e1002376. doi: 10.1371/journal.ppat.1002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Callan MF, Fazou C, Yang H, Rostron T, Poon K, Hatton C, McMichael AJ. CD8(+) T-cell selection, function, and death in the primary immune response in vivo. J. Clin. Invest. 2000;106:1251–1261. doi: 10.1172/JCI10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adhikary D, Behrends U, Moosmann A, Witter K, Bornkamm GW, Mautner J. Control of Epstein-Barr virus infection in vitro by T helper cells specific for virion glycoproteins. J. Exp. Med. 2006;203:995–1006. doi: 10.1084/jem.20051287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mautner J, Bornkamm GW. The role of virus-specific CD4+ T cells in the control of Epstein-Barr virus infection. Eur. J. Cell Biol. 2012;91:31–35. doi: 10.1016/j.ejcb.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 47.Norbury CC, Sigal LJ. Cross priming or direct priming: is that really the question? Curr. Opin. Immunol. 2003;15:82–88. doi: 10.1016/s0952791502000031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.