

The Glu residue in the laminin γ-tail forms a bipartite integrin binding site with three globular domains of the α chain.
Abstract
Laminins regulate diverse cellular functions through interaction with integrins. Two regions of laminins—three laminin globular domains of the α chain (LG1–3) and the carboxyl-terminal tail of the γ chain (γ-tail)—are required for integrin binding, but it remains unclear how the γ-tail contributes to the binding. We determined the crystal structure of the integrin binding fragment of laminin-511, showing that the γ-tail extends to the bottom face of LG1–3. Electron microscopic imaging combined with biochemical analyses showed that integrin binds to the bottom face of LG1–3 with the γ1-tail apposed to the metal ion–dependent adhesion site (MIDAS) of integrin β1. These findings are consistent with a model in which the γ-tail coordinates the metal ion in the MIDAS through its Glu residue.
INTRODUCTION
Adhesion of cells to the basement membrane (BM) is a fundamental biological process essential for tissue development and maintenance. Laminins make a major contribution to the cell-adhesive activity of the BM and elicit a variety of cellular responses through interaction with a panel of cell surface receptors, including integrins. Laminins consist of three chains, α, β, and γ, which assemble into a cross-shaped heterotrimer. In mammals, 11 laminin chains (α1 to α5, β1 to β3, and γ1 to γ3) and 16 combinations of these have been identified (1). Laminin-511 (LM511), an isoform consisting of chains α5, β1, and γ1, is expressed in the early embryonic stage and maintains tissue homeostasis throughout life (2). Integrins are heterodimeric membrane proteins composed of noncovalently associated α and β subunits (3). Twenty-four integrins have been identified in mammals; among these, α6β1 integrin exhibits the highest binding affinity for the α5 chain–containing laminin isoforms LM511 and LM521 (4).
The integrin binding activity of laminin has been mapped within the “E8” segment, comprising the distal part of the coiled-coil domain and three laminin globular domains (LG1–3) of the α chain (Fig. 1A, middle). LG1–3, which are prerequisite for integrin binding (5–7), have been proposed to adopt a “cloverleaf” configuration based on electron microscopic observations (8), although LG1–3 alone adopted an open configuration when their structure was determined by x-ray crystallography (9). LG1–3 alone have no significant integrin binding activity (10), suggesting that β and/or γ chains facilitate the cloverleaf assembly of LG1–3 for integrin recognition.
Fig. 1. The integrin binding region of LM511.
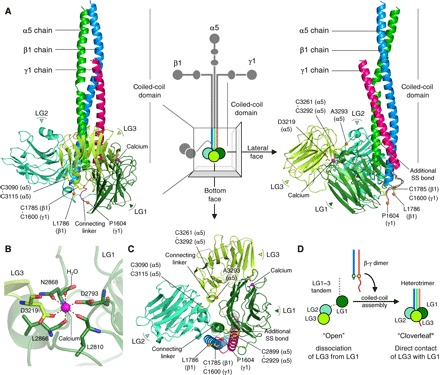
(A) Front (left) and lateral (right) faces of the integrin binding region of LM511 that consists of chains α5 (green), β1 (blue), and γ1 (red). LG1–3 of the α5 chain are colored in deep green (LG1), green-cyan (LG2), and yellow-green (LG3). A calcium ion at the LG1-LG3 interface is shown as a magenta sphere, disulfide-linked Cys residues as yellow sticks, and Cα atoms of C-terminal residues as orange spheres. (B) Octahedral calcium coordination in the LG1-LG3 interface. (C) Triangular cloverleaf configuration of LG1–3. (D) Schematic illustration of the configurational change of LG1–3 from “open” to “cloverleaf” through coiled-coil assembly.
Our previous results showed that the Glu residue at the third position from the C terminus of the γ chain is crucial for integrin binding (11, 12). However, it remains to be determined whether this Glu residue in the C-terminal region of the γ chain (designated the γ-tail) contributes to adopting an active LG1–3 conformation for integrin recognition or directly interacts with integrins by coordinating the metal ion held in the major ligand-binding site of integrins. This site is referred to as the MIDAS (metal ion–dependent adhesion site). Here, we sought to determine the role of the γ-tail in the laminin-integrin interaction by x-ray crystallography combined with electron microscopic imaging and a series of biochemical analyses.
RESULTS
Crystal structure of the integrin binding region of LM511
Through a series of truncation/deletion mutagenesis experiments coupled with evaluation of the integrin binding activity, we identified a truncated LM511E8 (tLM511E8) suitable for the structural determination, which was successfully crystallized and whose structure was determined at 1.8 Å resolution (fig. S1 and table S1). The structure of tLM511E8 exhibited a “ladle” shape with LG1–3 adopting a compact triangular cloverleaf configuration, where LG1 was in direct contact with LG3 (Fig. 1, A and C). Each LG adopts a canonical β-sandwich fold stabilized by a conserved disulfide bridge typical for this class of domain. We found one calcium ion at the LG1-LG3 interface (Fig. 1B), where its octahedrally coordinating ligands were provided by both LG1 (four ligands) and LG3 (one ligand), as well as one water molecule. The overall arrangement of LG1–3 is in sharp contrast to that seen in the same fragment of α2 chain previously reported in isolation, which adopted an open configuration with LG1 completely dissociated from LG3 (9). Thus, the cloverleaf assembly of LG1–3 in tLM511E8 should have been brought about by the heterotrimeric assembly of the α5 chain with a β1-γ1 dimer, rather than by the direct contact between LG1 and LG3 (Fig. 1D). Notably, LG1 and LG2 clamp the C-terminal region of the β1-γ1 dimer (Fig. 2A). LG1 contains a hydrophobic patch consisting of residues V2733, V2735, P2736, Y2913, F2915, L2921, and P2928 (Fig. 2B). Following the heterotrimeric assembly, the hydrophobic patch is brought into direct contact with the side chains of Y1782 of the β1 chain and L1596/P1597/F1601 of the γ1 chain. As a result, LG1 is fastened to the C-terminal region of the β1-γ1 dimer through hydrophobic side-chain interactions (Fig. 2C). LG2 faces the β1 chain pillar from the opposite side, but there are no obvious side-chain interactions at the interface. Instead, water molecules fill the gap by forming a hydrogen-bonded network and secure the contacts of LG2 with the β1-γ1 dimer (Fig. 2D). Thus, heterotrimeric assembly may impose conformational restriction on LG1 and LG2 to appose the β1 chain pillar and lead to the cloverleaf assembly of LG1–3.
Fig. 2. Association of LG1–3 with β1-γ1 dimer.
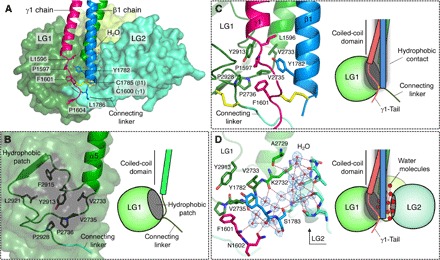
(A) Back view showing LG1-LG2 (surfaces), β1-γ1 dimer (ribbons) with side chains (sticks), and water molecules (spheres). (B) Hydrophobic patch on LG1. (C) Hydrophobic interactions between the hydrophobic patch of LG1 and the C-terminal region of the β1-γ1 dimer. (D) The interaction between LG2 and β1 chain was mediated by a layer of hydrogen-bonded water molecules. 2Fo-Fc electron density map countered at 1.0σ is shown as blue mesh around water molecules.
It has been proposed that the Glu residue in the γ-tail associates with LG1–3 to ensure their functional triangular assembly (5). However, the five C-terminal residues of the γ1 chain, including E1607 (γ1E1607), which is the critical residue for integrin binding, are disordered in our structure and have no direct contact with LG1–3 (Fig. 2 and fig. S2B), strongly arguing against its role in stabilizing the functionally active conformation of LG1–3. Recently, Pulido et al. (13) reported a high-resolution crystal structure of the integrin binding region of mouse LM111. The global structure of LM111 is essentially the same as that of LM511 shown here, with both LM111 and LM511 exhibiting a triangular configuration of LG1–3 with the β1-γ1 dimer clamped between LG1 and LG2. One interesting difference is that the LG3 in LM111 is positioned to make the transverse plane of LG1–3 orthogonal to the coiled-coil domain, whereas LG3 in LM511 forms a beveled plane of LG1–3 (fig. S2A). Nevertheless, the γ1-tail in the LM111 structure is oriented toward the bottom face of LG1/LG2 with the side chain of E1605 (equivalent to γ1E1607 in human) and the following residues disordered in the crystal structure (fig. S2B), as in our human tLM511E8 structure.
LM511 binds to α6β1 integrin in a bottom-to-head manner
The ligand-binding site of integrins has been mapped to the upper face of the integrin’s headpiece that consists of the β-propeller domain of the α subunit and the βI/hybrid domains of the β subunit (3). It is generally accepted that recognition of physiological ligands by integrins relies on an acidic residue in the ligands, coordinating a divalent metal ion in the MIDAS of the integrin β subunit (β-MIDAS) (14). Consistent with this scheme, the Glu residues in the γ1-tail (E1607) and γ2-tail (E1191) are essential for integrin binding by laminins; the γ3 chain does not have an equivalent Glu residue (11, 12). Furthermore, our exhaustive survey demonstrated that no single acidic residue in the LM511E8 fragment other than γ1E1607 is critically required for integrin binding (15), pointing toward the possibility that γ1E1607 directly coordinates with the β-MIDAS. To gain insight into the location of the integrin binding site within LM511, we examined LM511E8 complexed with recombinant α6β1 integrin by electron microscopy. A gallery of electron microscopic images revealed that the headpiece of α6β1 integrin always bound to LM511E8 at the site opposite the filament-like coiled-coil extension (Fig. 3A and fig. S3). These results indicate that LM511 binds to α6β1 integrin via the bottom face of LG1–3, where the disordered five C-terminal residues of the γ1-tail are predicted to reside (Fig. 3B).
Fig. 3. Close contact of the γ1-tail with the headpiece of α6β1 integrin.
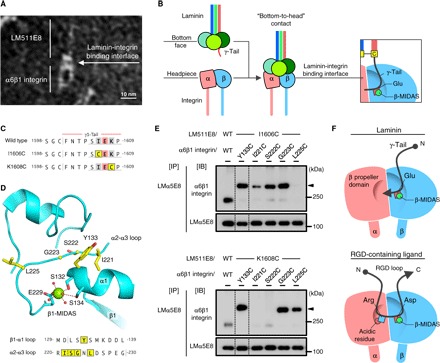
(A) Electron microscopic image of LM511E8 complexed with α6β1 integrin (see also fig. S3). (B) Schematic illustration of the interaction between LM511E8 and α6β1 integrin. (C) Amino acid sequences of wild-type (WT) and Cys-substituted γ1-tails. Cys-substituted residues are shown in yellow. (D) The residues cross-linked to the γ1-tail (yellow sticks) lie near the metal ion (green sphere) in the MIDAS site of integrin β1 (β1-MIDAS) (Protein Data Bank ID: 4WJK). The β1-MIDAS metal ion and water molecules are depicted as green and red spheres, respectively. (E) Intermolecular disulfide formation between LM511E8 having γ1-I1606C (top) or γ1-K1608C (bottom) substitution and Cys-substituted α6β1 integrins (see also fig. S6). Arrowheads indicate disulfide-linked products. (F) Distinct topologies of the γ-tail (top) and the RGD motif (bottom) on the integrin’s headpiece.
The γ1-tail is positioned close to the metal ion of the β1-MIDAS
To further clarify the possibility that the γ1-tail directly interacts with α6β1 integrin, we simultaneously introduced Cys substitutions into the γ1-tail and the βI domain of integrin β1 and then performed exhaustive screening for intermolecular disulfide formation between the Cys-substituted LM511E8 and the α6β1 integrin. We reasoned that a Cys residue introduced adjacent to γ1E1607 would become cross-linked to a Cys residue introduced near the β1-MIDAS if the γ1-tail directly interacts with integrin β1. Thus, we introduced Cys substitutions of residues I1606 and K1608 of the γ1-tail (Fig. 3C) and of 19 residues in the βI domain that are predicted to be surface-exposed near the β1-MIDAS (fig. S4). LM511E8 with γ1-I1606C and γ1-K1608C substitutions (designated LM511E8/I1606C and LM511E8/K1608C, respectively) retained integrin binding activity, whereas those having additional Gln substitutions for γ1E1607 (designated LM511E8/I1606C/EQ and LM511E8/K1608C/EQ) were almost devoid of the activity (fig. S5). Coexpression of Cys-substituted LM511E8 and α6β1 integrin in mammalian cells and subsequent immunoprecipitation of secreted LM511E8, followed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) under nonreducing conditions, identified a total of seven disulfide-linked products between LM511E8 and α6β1 integrin, depending on the position of the Cys substitution near the β1-MIDAS (Fig. 3, D and E, and fig. S6). LM511E8/I1606C was disulfide-linked to four Cys-substituted integrin β1 residues: 133, 221, 222, and 223 (Fig. 3E, top); LM511E8/K1608C was disulfide-linked to three Cys-substituted residues: 133, 223, and 225 (Fig. 3E, bottom). The ability of both laminin mutants to form disulfide bonds with integrin β1 residues 133 and 223 is consistent with the fact that these residues are closest to the MIDAS metal ion, where γ1E1607 is predicted to ligate (Fig. 3D). In contrast, residues 221 and 222, which are located further away from the α subunit, preferentially cross-linked to LM511E8/I1606C, whereas residue 225, situated closer to the α subunit, efficiently cross-linked to LM511E8/K1608C. These results suggest that the I1606-E1607-K1608 segment of the γ1-tail is aligned parallel to the segment with residues 221 to 225 of the α2-α3 loop of integrin β1, with the C-terminal end of the former pointing toward the β-propeller domain of integrin α6 when γ1E1607 coordinates with the β1-MIDAS metal ion (Fig. 3F, top). This is in sharp contrast to the common mode of Arg-Gly-Asp (RGD) motif recognition by many integrins, where a short peptide segment is docked at the α-β interface in the N→C direction, with the side chains of Arg and Asp being recognized by the α and β subunits of integrins, respectively (Fig. 3F, bottom) (16–18).
To confirm the specificity of the intermolecular disulfide formation between the Cys-substituted LM511E8 and the α6β1 integrin, we performed the disulfide cross-link assays using laminin mutants carrying the inactivating Gln mutation at γ1E1607 (fig. S7). The Gln substitution resulted in a significant reduction in the disulfide formation, suggesting that efficient disulfide bond formation requires correct steering of the γ1-tail guided by the γ1E1607-MIDAS interaction. It is also notable that intermolecular disulfide bonds, particularly involving integrin β1 residue 133, form to some extent in the absence of γ1E1607, suggesting that LM511E8 devoid of γ1E1607 has weak but appreciable binding ability to integrin.
γ1-Tail–derived peptide inhibits laminin-integrin interaction
Although these results strongly support the idea that the γ1-tail comprises the integrin binding site of LM511 and directly interacts with the β1-MIDAS of α6β1 integrin, we have been unable to show competitive inhibition of integrin-ligand interaction using synthetic peptides derived from the γ1-tail (11), which has been repeatedly observed in RGD-recognizing integrins with RGD and related peptides (19). To revisit the effect of synthetic γ1-tail peptides on laminin-integrin interactions, we performed inhibition assays using a γ1-tail–derived pentapeptide SIEKP (designated γ1C5) and its E-to-Q mutant (SIQKP) under conditions where α6β1 integrin was rendered conformationally active by the activating anti-integrin β1 antibody TS2/16. γ1C5 was only weakly inhibitory to α6β1 integrin binding to LM511E8 in the absence of TS2/16, but it exerted a significant inhibitory activity in the presence of TS2/16 [half-maximal inhibitory concentration (IC50) approximately 25 μM] (Fig. 4A). The inhibitory activity of γ1C5 was abrogated by the E-to-Q substitution, lending support to the idea of direct interaction of the γ1-tail with α6β1 integrin in a γ1E1607-dependent manner.
Fig. 4. Direct contribution of γ1E1607 to laminin-integrin interaction.
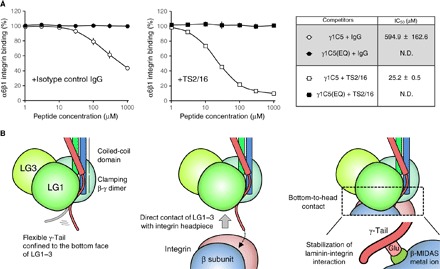
(A) Inhibition of the LM511E8–α6β1 integrin interaction by γ1-tail–derived peptides [white, γ1C5; black, γ1C5(EQ)] in the absence (circle) or presence (square) of the integrin β1 activating monoclonal antibody (mAb) TS2/16. IC50 values of peptides (means ± SD of three independent experiments) are shown in the table (right). (B) Schematic model for the mechanism by which integrin recognizes laminin.
DISCUSSION
Despite the requirement for the Glu residue within the γ-tail in integrin recognition by laminins, it has remained unsettled whether the γ-tail is required for the maintenance of a functionally active conformation of LG1–3 of the α chain or whether it directly interacts with integrin by coordinating the metal ion in the β-MIDAS. Several lines of evidence obtained in this study support the latter possibility. First, the γ1-tail of tLM511E8 was found disordered in the crystallized structure and therefore does not seem to contribute significantly to the maintenance of LG1–3 conformation. The C-terminal region of the β1-γ1 dimer is clamped between LG1 and LG2, suggesting that the disordered γ1-tail extends to the bottom face of the ladle-shaped tLM511E8. Second, electron microscopic imaging of LM511E8 complexed with α6β1 integrin revealed that LM511E8 binds to the integrin in a bottom-to-head manner, consistent with the prediction that the disordered γ1-tail lies at the bottom face of LG1–3. Third, intermolecular disulfide cross-link screening with a panel of Cys-substituted LM511E8s and α6β1 integrins demonstrated that the γ1-tail is selectively disulfide-linked to residues near the β1-MIDAS, supporting the idea of direct interaction of the γ1-tail with integrin’s β1-MIDAS. Finally, the γ1C5 peptide SIEKP inhibited the binding of LM511E8 to α6β1 integrin, but its E-to-Q mutant did not, corroborating the direct interaction of the γ1-tail with α6β1 integrin, in which γ1E1607 is prerequisite. Together, these findings lead us to conclude that the γ1-tail directly interacts with integrin’s β1-MIDAS, with γ1E1607 serving as the critical acidic residue that coordinates the metal ion in the β1-MIDAS.
There is compelling evidence that LG1–3 is required for laminin recognition by integrins. Deletion of LG1–3 or substitution of any one of the LGs nullifies the integrin binding activity of laminins (10, 20). Furthermore, the integrin binding specificity, as well as the affinity of laminin isoforms, has been shown to be primarily defined by the laminin α chains (4). In support of the involvement of LG1–3 in laminin recognition by integrins, the LM511E8 mutant lacking the five C-terminal residues of the γ1-tail competitively inhibited the binding of α6β1 integrin to LM511E8, albeit at a much higher concentration than intact E8 (fig. S8). Given that LM511E8 binds to α6β1 integrin in a bottom-to-head manner, it is conceivable that LG1–3 harbor an auxiliary integrin recognition site(s) at their bottom face that can complement the primary site involving the γ1-tail, although the detailed location of this site(s) remains to be explored. Pulido et al. (13) noted that the LG1 and LG2 surfaces on either side of the γ1-tail are more highly conserved than that of LG3, implying that the residues involved in integrin binding lie within the highly conserved surfaces. Our exhaustive disulfide cross-link screening with Cys-substituted LM511E8 and α6β1 integrin showed that the γ1-tail came into close contact with and cross-linked with integrin β1 residue 133 near the β1-MIDAS even in the absence of γ1E1607. These findings suggest that the interaction of α6β1 integrin with the bottom face of LG1–3 brings the γ1-tail into close contact with the β1-MIDAS, thereby stabilizing the laminin-integrin complex (Fig. 4B).
In conclusion, we report evidence that supports a scheme in which the γ1-tail directly interacts with integrin β1, with γ1E1607 coordinating the metal ion in the β1-MIDAS during formation of the LM511–α6β1 integrin complex. These findings contribute to a better understanding of the molecular basis of laminin-integrin interactions in diverse aspects of cellular function.
MATERIALS AND METHODS
Antibodies and reagents
Horseradish peroxidase (HRP)–conjugated mouse anti-5×His mAb was from Qiagen. Monoclonal ANTI-FLAG M2 antibody was purchased from Sigma-Aldrich. HRP-conjugated mouse anti–c-Myc mAb (clone 9E10) was purchased from Abcam. Rabbit polyclonal antibody (pAb) against the ACID/BASE coiled-coil region (designated Velcro) was produced by immunization with ACID/BASE coiled-coil peptides, as described previously (21). The anti-Velcro pAb was biotinylated using EZ-Link Sulfo-NHS-Biotin reagents (Thermo Fisher Scientific) according to the manufacturer’s instructions. mAb 5D6 against the human laminin α5 chain was generated in our laboratory, as described previously (22). mAb 4C7 against the LG of human laminin α5 chain (10, 23, 24) was from Merck Millipore. Mouse immunoglobulin G (IgG) was from Thermo Fisher Scientific. HRP-conjugated goat anti-mouse IgG pAb was purchased from Jackson ImmunoResearch. HRP-conjugated streptavidin was from Thermo Fisher Scientific. The mAb TS2/16 against human integrin β1 was purified on a Protein G Sepharose 4 Fast Flow column (GE Healthcare) from the conditioned media of hybridoma cells obtained from the American Type Culture Collection. Restriction enzymes Nhe I, Bam HI, and Not I were obtained from New England BioLabs. AcTEV protease, which is an enhanced form of tobacco etch virus (TEV) protease, was purchased from Thermo Fisher Scientific. Synthetic peptides derived from the five C-terminal residues of the human laminin γ1 chain, namely, γ1C5 and γ1C5(EQ), were purchased from Greiner Bio-One.
Construction of expression vectors
Expression vectors for recombinant E8 fragments of human laminin α5, β1, γ1, and γ1 having a Gln substitution for γ1E1607 were prepared as described (11). 6×His, hemagglutinin (HA), and FLAG tags were added to the N termini of LMα5E8, LMβ1E8, and LMγ1E8, respectively. An expression vector for LMα5E8 in which the 6×His tag was replaced with a c-Myc tag was generated by extension polymerase chain reaction (PCR) using 6×His tag–conjugated LMα5E8 expression vector as the template. Expression vectors for LMγ1E8/I1606C, LMγ1E8/I1606C/EQ, LMγ1E8/K1608C, and LMγ1E8/K1608C/EQ were generated by extension PCR, using LMγ1E8 and LMγ1E8/EQ expression vectors as templates. The pcDNA3.4+MCS vector was generated by inserting the multiple cloning site sequence derived from the expression vector pSecTag2A into the TOPO cloning site of pcDNA3.4-TOPO (Thermo Fisher Scientific).
Expression vectors for individual truncated-E8 fragments of human laminin α5, β1, and γ1 chains (designated tLMα5E8, tLMβ1E8, and tLMγ1E8, respectively) were constructed as follows. Complementary DNAs encoding tLMα5E8 (E2655–A3327), tLMβ1E8 (D1714–L1786), and tLMγ1E8 (D1528–P1609) were amplified by PCR using individual E8 expression vectors as templates. The PCR products were digested with Nhe I and Not I and then ligated into the Nhe I–Not I sites, that is, between the Nhe site and the Not I site, of pcDNA3.4+MCS. 6×His, HA, and FLAG tags, followed by a TEV protease recognition sequence, were added by extension PCR. The PCR products were digested with Nhe I/Not I and inserted into the corresponding restriction sites of pcDNA3.4+MCS. tLMα5E8/I2723C and tLMγ1E8/D1585C mutants were generated by extension PCR using tLMα5E8 and tLMγ1E8 expression vectors as templates, respectively. The PCR products were digested with Nhe I/Not I and inserted into the corresponding restriction sites of pcDNA3.4+MCS.
An expression vector for the extracellular domain of human integrin α6 with C-terminal ACID peptide and FLAG tag sequences was prepared as described previously (10). An expression vector for the extracellular domain of human integrin β1 with a BASE peptide and a 6×His tag sequence at the C terminus was prepared as described previously (21). Expression vectors for integrin β1 mutants, in which 19 residues located on the I-like domain were individually Cys-substituted, were produced by extension PCR, using integrin β1 expression vector as the template. The PCR products were digested with Bam HI/Not I and cloned into the same sites of the wild-type construct. All DNA sequences were verified using ABI PRISM 3130xl Genetic Analyzer (Thermo Fisher Scientific).
Expression and purification of recombinant proteins
LM511E8 was transiently expressed in FreeStyle 293-F cells (Thermo Fisher Scientific) according to the manufacturer’s instructions. Conditioned media were collected 72 hours after transfection and loaded onto cOmplete His-Tag Purification Resin (Roche). The resin was washed with Hepes-buffered saline (HBS) (8.0) [20 mM Hepes and 137 mM NaCl (pH 8.0)], and the protein was eluted with HBS (8.0) containing 250 mM imidazole. Fractions containing LM511E8 were further loaded onto DDDDK-tagged Protein Purification Gel (MBL). The resin was washed with HBS (7.4) [20 mM Hepes and 137 mM NaCl (pH 7.4)], and the proteins were eluted with HBS (7.4) containing DDDDK peptide (100 μg/ml; MBL). Fractions containing LM511E8 were concentrated with Amicon Ultra-15 (Merck Millipore) and further purified on a Superdex 200 10/300 GL column (GE Healthcare) using HBS (7.4) as the running buffer.
tLM511E8 for crystallization was produced using FreeStyle 293-F cells and purified by affinity chromatography as for LM511E8. Fractions containing tLM511E8 were concentrated using Amicon Ultra-15 and digested with TEV protease (2000 U/ml) at 25°C for 24 hours. The reaction mixture was subjected to gel filtration on a Superdex 200 10/300 GL column using HBS (7.4) as the running buffer. Fractions containing TEV-digested tLM511E8 were concentrated with Amicon Ultra-2 (Merck Millipore) to ~30 mg/ml and stored at −80°C.
Recombinant human α6β1 integrin was prepared as described previously (10, 11). Intact α6β1 integrin for electron microscopic observation was prepared using the same expression system. The conditioned media were loaded onto ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich), and bound proteins were eluted with HBS (7.4) containing FLAG peptide (100 μg/ml). Fractions containing α6β1 integrin were concentrated with Amicon Ultra-4 (Merck Millipore) and immediately subjected to gel filtration on a Superdex 200 10/300 GL column using HBS (7.4) as the running buffer. Fractions containing α6β1 integrin were collected and stored at −80°C. Protein concentrations of all recombinant products were determined using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) using bovine serum albumin (BSA) as the standard.
Solid-phase integrin binding assay
Binding activities of α6β1 integrin to wild-type and mutant LM511E8s, including tLM511E8, were measured by solid-phase binding assays as follows. LM511E8s were adsorbed onto 96-well microtiter plates (Thermo Fisher Scientific) at 10 nM overnight at 4°C and then blocked with 3% BSA at room temperature for 1 hour. The amounts of LM511E8s adsorbed on the plates were quantified with mAbs 5D6 and 4C7 to confirm the equality of the amounts of adsorbed proteins. After washing once with tris-buffered saline (TBS) [20 mM tris and 137 mM NaCl (pH 7.4)] containing 1 mM MnCl2, 0.3% BSA, and 0.02% Tween 20 (Mn2+ buffer), the plates were incubated with serially diluted α6β1 integrin solution at room temperature for 3 hours in the presence of 1 mM MnCl2 or 10 mM EDTA. After three washes with Mn2+ buffer or TBS containing 10 mM EDTA, 0.3% BSA, and 0.02% Tween 20, the plates were incubated with biotinylated rabbit anti-Velcro pAb (1.5 μg/ml) in Mn2+ buffer at room temperature for 30 min. After three washes with Mn2+ buffer, the plates were incubated with streptavidin-HRP (0.53 μg/ml) for 15 min. After three washes with Mn2+ buffer, the amount of bound α6β1 integrin was quantified by measuring the absorbance at 490 nm after incubation with o-phenylenediamine. The apparent dissociation constants were determined as described previously (25).
Preparation of laminin-integrin complex
The laminin-integrin complex was formed by mixing 200 pmol of purified LM511E8 and the same amount of purified α6β1 integrin at room temperature for 30 min in HBS (7.4) containing 1 mM MnCl2. The complex was subjected to gel filtration on a Superose 6 10/300 GL column (GE Healthcare) preequilibrated with HBS containing 1 mM MnCl2. Fractions containing the complex were identified by SDS-PAGE followed by Coomassie Brilliant Blue staining.
Electron microscopy and image processing
The fractionated complex was incubated for 1 min at room temperature on glow-discharged carbon-coated grids (Nisshin EM). Samples were washed three times in ultrapure water containing 1 mM MnCl2 and stained three times with 2% uranyl acetate for 30 s. After vacuum drying, grids were inspected with a Hitachi H-7650 transmission electron microscope operated at 80 kV.
Intermolecular disulfide bond formation between LM511E8 and α6β1 integrin
c-Myc–tagged LMα5E8 was used throughout the intermolecular disulfide formation assays. LM511E8/wild type, LM511E8/I1606C, LM511E8/I1606C/EQ, LM511E8/K1608C, and LM511E8/K1608C/EQ were transiently coexpressed with wild-type or Cys-introduced α6β1 integrin using the FreeStyle 293 Expression System. Seventy-two hours after transfection, conditioned media were incubated with mAb 5D6 (1 μg/ml) at 4°C for 1 hour. Secreted laminin-integrin complexes were precipitated with Protein G Sepharose 4 Fast Flow (GE Healthcare). Immunoprecipitates were analyzed by SDS-PAGE under nonreducing conditions, followed by immunoblotting with anti–c-Myc mAb and anti-Velcro pAb.
Inhibition of α6β1 integrin binding to LM511E8
Wild-type LM511E8 was adsorbed onto 96-well microtiter plates at 10 nM overnight at 4°C and then blocked with 3% BSA at room temperature for 1 hour. Serially diluted synthetic peptide [γ1C5 or γ1C5(EQ)] or LM511E8 (wild type or Δγ1C5) was incubated with 1 nM α6β1 integrin in Mn2+ buffer containing 3 nM isotype control IgG or integrin β1 activating mAb TS2/16 at room temperature for 1 hour, then added to LM511E8-coated plates, and allowed to bind to LM511E8 at room temperature for 1 hour. After three washes with Mn2+ buffer, the plates were incubated with biotinylated rabbit anti-Velcro pAb (1.5 μg/ml) in Mn2+ buffer at room temperature for 30 min. After three washes with Mn2+ buffer, the plates were incubated with streptavidin-HRP (0.53 μg/ml) for 15 min. After three washes with Mn2+ buffer, the amount of bound α6β1 integrin was quantified by measuring the absorbance at 490 nm after incubation with o-phenylenediamine.
IC50 values were calculated using the following equation
where x is peptide or LM511E8 concentration (in micromolars per nanomolar), “y” is activity (in percent) in the presence of inhibitors relative to the activity in the absence of inhibitors, “A” corresponds to the relative activity on the top plateau region of the curve, “B” corresponds to the slope, “C” corresponds to the inflection point of the curve, and “D” corresponds to the relative activity on the bottom plateau region of the curve. To obtain these four parameters, experimental raw data points were fitted to this equation using the “Curve Fitter” tool of ImageJ software (26). IC50 values were determined by substituting “y = 50” in the abovementioned equation.
Crystallization and diffraction data collection
Crystallization was performed at 20°C. Initial screening of crystallization conditions was performed using The Classics Neo Suite (Qiagen). For this screen, a mosquito crystallization robot (TTP LabTech) was used to dispense 0.5 μl of protein solution mixed in a 1:1 ratio with the reservoir solution. Drops were equilibrated over 80 μl of reservoir solution using the sitting drop vapor diffusion method. The initial crystallization condition [0.2 M ammonium sulfate, 0.1 M sodium acetate (pH 4.6), and 25% polyethylene glycol 4000 at room temperature] was optimized using a 24-well crystallization plate with the hanging drop vapor diffusion method. Each well contained 500 μl of reservoir solution, and the drop volume was a mixture of 0.5 μl of protein solution and 0.5 μl of reservoir solution. Diffraction quality crystals were obtained under conditions of 0.2 M ammonium sulfate, 0.1 M sodium acetate adjusted to pH 4.2 with acetic acid, and 19% polyethylene glycol 4000. Before x-ray diffraction experiments, crystals were soaked in reservoir solution containing an additional 20% glycerol and flash-cooled in liquid nitrogen.
The diffraction data set for phasing was collected at BL-1A, Photon Factory (Tsukuba, Japan), and processed using the XDS package (27). In this data collection, three data sets of 720° each with an oscillation angle of 0.2° were merged. A higher-resolution data set for refinement was collected at BL44XU, SPring-8 (Hyogo, Japan), and processed using the HKL2000 package (28). The diffraction data statistics are shown in table S1.
Structure determination
The crystal structure of tLM511E8 was solved by the single-wavelength anomalous dispersion method using native crystals. The coordinates of the substructure including the sulfur atoms and the calcium ion were determined using SHELXC and SHELXD (29). The phase calculation using these coordinates with phase improvement followed by automated initial model building was performed using the PHENIX program package (30). This initial model was then extended and refined by manual editing using COOT (31), with the iterated implementation of refmac5 (32) on the higher-resolution data set. Finally, the crystal structure was refined to the R/Rfree factors of 0.202/0.237 at a resolution of 1.80 Å, validated with MOLPROBIDY (33). Refinement statistics are described in table S1. All figures of the tLM511E8 model in this article were produced using PyMOL (www.pymol.org/).
Supplementary Material
Acknowledgments
We thank J. Toga and E. Yagi for preparation of recombinant proteins, K. Iwasaki for his help with electron microscopic observations, and the staff in BL-1A (Photon Factory) and BL44XU (SPring-8) for their help with x-ray data collection. Funding: This work was supported by Grants-in-aid for Scientific Research #22122006 (to K.S.) and #24111006 (to J.T.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), by the “Platform for Drug Discovery, Informatics, and Structural Life Science” grant from the MEXT (to J.T.), and by the “X-ray Free Electron Laser Priority Strategy Program” grant from the MEXT (to J.T.). Author contributions: M.T., T.A., Y.T., Y.K., J.T., and K.S. contributed to the writing of the manuscript. M.T., T.A., and Y.K. crystallized the tLM511E8, collected the data, and solved the structure. Y.T. prepared the materials and performed the electron microscopic observations. M.T. and E.Y. performed disulfide cross-link assays. M.T. prepared the materials and performed the peptide competition experiments. J.T. and K.S. conceived and directed the study. All authors read and commented on the manuscript. Competing interests: K.S. is a founder and shareholder of Matrixome Inc. Y.T. is a project leader of Matrixome Inc. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Coordinates and structure factors of the crystal structure of the integrin binding fragment of LM511 have been deposited in the Protein Data Bank with accession code 5XAU.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1701497/DC1
fig. S1. Preparation and crystallization of tLM511E8.
fig. S2. Comparison of the crystal structures of tLM511E8 and mini-E8 of LM111.
fig. S3. Electron microscopic imaging of the LM511E8–α6β1 integrin complex.
fig. S4. Cys-substituted residues on βI domain.
fig. S5. Integrin binding activity of wild-type and Cys-substituted LM511E8s.
fig. S6. Disulfide formation between Cys-substituted LM511E8 and α6β1 integrin.
fig. S7. Disulfide cross-link assays using LM511E8/I1606C/EQ and LM511E8/K1608C/EQ.
fig. S8. Inhibition of the LM511E8–α6β1 integrin interaction by wild-type and Δγ1C5 LM511E8.
table S1. Data collection and refinement statistics.
REFERENCES AND NOTES
- 1.Aumailley M., Bruckner-Tuderman L., Carter W. G., Deutzmann R., Edgar D., Ekblom P., Engel J., Engvall E., Hohenester E., Jones J. C. R., Kleinman H. K., Marinkovich M. P., Martin G. R., Mayer U., Meneguzzi G., Miner J. H., Miyazaki K., Patarroyo M., Paulsson M., Quaranta V., Sanes J. R., Sasaki T., Sekiguchi K., Sorokin L. M., Talts J. F., Tryggvason K., Uitto J., Virtanen I., von der Mark K., Wewer U. M., Yamada Y., Yurchenco P. D., A simplified laminin nomenclature. Matrix Biol. 24, 326–332 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Domogatskaya A., Rodin S., Tryggvason K., Functional diversity of laminins. Annu. Rev. Cell Dev. Biol. 28, 523–553 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Hynes R. O., Integrins: Bidirectional, allosteric signaling machines. Cell 110, 673–687 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Nishiuchi R., Takagi J., Hayashi M., Ido H., Yagi Y., Sanzen N., Tsuji T., Yamada M., Sekiguchi K., Ligand-binding specificities of laminin-binding integrins: A comprehensive survey of laminin-integrin interactions using recombinant α3β1, α6β1, α7β1 and α6β4 integrins. Matrix Biol. 25, 189–197 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Hohenester E., Yurchenco P. D., Laminins in basement membrane assembly. Cell Adh. Migr. 7, 56–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deutzmann R., Aumailley M., Wiedemann H., Pysny W., Timpl R., Edgar D., Cell adhesion, spreading and neurite stimulation by laminin fragment E8 depends on maintenance of secondary and tertiary structure in its rod and globular domain. Eur. J. Biochem. 191, 513–522 (1990). [DOI] [PubMed] [Google Scholar]
- 7.Ido H., Harada K., Futaki S., Hayashi Y., Nishiuchi R., Natsuka Y., Li S., Wada Y., Combs A. C., Ervasti J. M., Sekiguchi K., Molecular dissection of the α-dystroglycan- and integrin-binding sites within the globular domain of human laminin-10. J. Biol. Chem. 279, 10946–10954 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Beck K., Hunter I., Engel J., Structure and function of laminin: Anatomy of a multidomain glycoprotein. FASEB J. 4, 148–160 (1990). [DOI] [PubMed] [Google Scholar]
- 9.Carafoli F., Clout N. J., Hohenester E., Crystal structure of the LG1-3 region of the laminin α2 chain. J. Biol. Chem. 284, 22786–22792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ido H., Harada K., Yagi Y., Sekiguchi K., Probing the integrin-binding site within the globular domain of laminin-511 with the function-blocking monoclonal antibody 4C7. Matrix Biol. 25, 112–117 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Ido H., Nakamura A., Kobayashi R., Ito S., Li S., Futaki S., Sekiguchi K., The requirement of the glutamic acid residue at the third position from the carboxyl termini of the laminin γ chains in integrin binding by laminins. J. Biol. Chem. 282, 11144–11154 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Ido H., Ito S., Taniguchi Y., Hayashi M., Sato-Nishiuchi R., Sanzen N., Hayashi Y., Futaki S., Sekiguchi K., Laminin isoforms containing the γ3 chain are unable to bind to integrins due to the absence of the glutamic acid residue conserved in the C-terminal regions of the γ1 and γ2 chains. J. Biol. Chem. 283, 28149–28157 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pulido D., Hussain S.-A., Hohenester E., Crystal structure of the heterotrimeric integrin-binding region of laminin-111. Structure 25, 530–535 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takagi J., Structural basis for ligand recognition by integrins. Curr. Opin. Cell Biol. 19, 557–564 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Taniguchi Y., Li S., Takizawa M., Oonishi E., Toga J., Yagi E., Sekiguchi K., Probing the acidic residue within the integrin binding site of laminin-511 that interacts with the metal ion-dependent adhesion site of α6β1 integrin. Biochem. Biophys. Res. Commun. 487, 525–531 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Xia W., Springer T. A., Metal ion and ligand binding of integrin α5β1. Proc. Natl. Acad. Sci. U.S.A. 111, 17863–17868 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Agthoven J. F., Xiong J.-P., Alonso J. L., Rui X., Adair B. D., Goodman S. L., Arnaout M. A., Structural basis for pure antagonism of integrin αVβ3 by a high-affinity form of fibronectin. Nat. Struct. Mol. Biol. 21, 383–388 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong X., Zhao B., Iacob R. E., Zhu J., Koksal A. C., Lu C., Engen J. R., Springer T. A., Force interacts with macromolecular structure in activation of TGF-β. Nature 542, 55–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kapp T. G., Rechenmacher F., Neubauer S., Maltsev O. V., Cavalcanti-Adam E. A., Zarka R., Reuning U., Notni J., Wester H.-J., Mas-Moruno C., Spatz J., Geiger B., Kessler H., A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins. Sci. Rep. 7, 39805 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kikkawa Y., Sasaki T., Nguyen M. T., Nomizu M., Mitaka T., Miner J. H., The LG1-3 tandem of laminin α5 harbors the binding sites of Lutheran/basal cell adhesion molecule and α3β1/α6β1 integrins. J. Biol. Chem. 282, 14853–14860 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Takagi J., Erickson H. P., Springer T. A., C-terminal opening mimics ‘inside-out’ activation of integrin α5β1. Nat. Struct. Biol. 8, 412–416 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Fujiwara H., Kikkawa Y., Sanzen N., Sekiguchi K., Purification and characterization of human laminin-8. Laminin-8 stimulates cell adhesion and migration through α3β1 and α6β1 integrins. J. Biol. Chem. 276, 17550–17558 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Engvall E., Davis G. E., Dickerson K., Ruoslahti E., Varon S., Manthorpe M., Mapping of domains in human laminin using monoclonal antibodies: Localization of the neurite-promoting site. J. Cell Biol. 103, 2457–2465 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiger C.-F., Champliaud M.-F., Pedrosa-Domellof F., Thornell L.-E., Ekblom P., Gullberg D., Presence of laminin α5 chain and lack of laminin α1 chain during human muscle development and in muscular dystrophies. J. Biol. Chem. 272, 28590–28595 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Nishiuchi R., Murayama O., Fujiwara H., Gu J., Kawakami T., Aimoto S., Wada Y., Sekiguchi K., Characterization of the ligand-binding specificities of integrin α3β1 and α6β1 using a panel of purified laminin isoforms containing distinct α chains. J. Biochem. 134, 497–504 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabsch W., XDS. Acta Crystallogr. Sect. D Struct. Biol. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otwinowski Z., Minor W., Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Sheldrick G. M., A short history of SHELX. Acta Crystallogr. A 64, 112–122 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Struct. Biol. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. Sect. D Struct. Biol. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Struct. Biol. 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Struct. Biol. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1701497/DC1
fig. S1. Preparation and crystallization of tLM511E8.
fig. S2. Comparison of the crystal structures of tLM511E8 and mini-E8 of LM111.
fig. S3. Electron microscopic imaging of the LM511E8–α6β1 integrin complex.
fig. S4. Cys-substituted residues on βI domain.
fig. S5. Integrin binding activity of wild-type and Cys-substituted LM511E8s.
fig. S6. Disulfide formation between Cys-substituted LM511E8 and α6β1 integrin.
fig. S7. Disulfide cross-link assays using LM511E8/I1606C/EQ and LM511E8/K1608C/EQ.
fig. S8. Inhibition of the LM511E8–α6β1 integrin interaction by wild-type and Δγ1C5 LM511E8.
table S1. Data collection and refinement statistics.