

Cardiolipin mediates dynamic receptor-channel interactions within the mitochondrial TIM23 protein import complex.
Abstract
The phospholipid cardiolipin mediates the functional interactions of proteins that reside within energy-conserving biological membranes. However, the molecular basis by which this lipid performs this essential cellular role is not well understood. We address this role of cardiolipin using the multisubunit mitochondrial TIM23 protein transport complex as a model system. The early stages of protein import by this complex require specific interactions between the polypeptide substrate receptor, Tim50, and the membrane-bound channel-forming subunit, Tim23. Using analyses performed in vivo, in isolated mitochondria, and in reductionist nanoscale model membrane systems, we show that the soluble receptor domain of Tim50 interacts with membranes and with specific sites on the Tim23 channel in a manner that is directly modulated by cardiolipin. To obtain structural insights into the nature of these interactions, we obtained the first small-angle x-ray scattering-based structure of the soluble Tim50 receptor in its entirety. Using these structural insights, molecular dynamics simulations combined with a range of biophysical measurements confirmed the role of cardiolipin in driving the association of the Tim50 receptor with lipid bilayers with concomitant structural changes, highlighting the role of key structural elements in mediating this interaction. Together, these results show that cardiolipin is required to mediate specific receptor-channel associations in the TIM23 complex. Our results support a new working model for the dynamic structural changes that occur within the complex during transport. More broadly, this work strongly advances our understanding of how cardiolipin mediates interactions among membrane-associated proteins.
INTRODUCTION
In addition to generating most of the adenosine 5′-triphosphate (ATP) within eukaryotic cells, mitochondria regulate many essential processes, including lipid synthesis, calcium homeostasis, and apoptosis. The vast majority of proteins that reside within mitochondria (approximately 1000 in yeast; 1500 in humans) are encoded within the nuclear genome, translated on cytosolic ribosomes, and targeted to the different compartments of the organelle by mitochondria-resident translocons (1–4). Most of these polypeptides are routed via the TIM23 (translocase of the inner mitochondrial membrane 23) complex, which mediates the translocation of soluble proteins into the matrix and the integration of hydrophobic proteins into the inner membrane (IM), as well as the biogenesis of some soluble proteins in the intermembrane space (IMS) and the outer membrane (OM) (5–10). The TIM23 complex is a dynamic, multisubunit machinery for which 11 subunits have now been identified (fig. S1A). The membrane-bound core of the complex includes two essential proteins, which are the focus of the present work: the central subunit Tim23, which forms at least part of the polypeptide-conducting channel (11–14), and Tim50, the central receptor of the complex (15–20).
Polypeptide substrates of the TIM23 complex are synthesized with an N-terminal cleavable presequence that consists of an amphipathic α helix with a positively charged face (21). After binding specific receptors of the TOM (translocase of the outer mitochondrial membrane) complex, the unfolded preprotein is directed across the TOM complex channel to the IMS where the presequence then engages the TIM23 machinery. Together, the Tim23 channel and the Tim50 receptor mediate the early stages of TIM23 complex–mediated protein import. Upon interaction with the Tim23-Tim50 receptor complex, the precursor is transferred to the TIM23 import channel in a manner that depends on the membrane potential (Δψm) across the IM. The Δψm is sufficient to drive lateral integration of membrane proteins into the bilayer. By comparison, soluble matrix-directed preproteins are driven across the IM by the adenosine triphosphatase (ATPase) import motor (also termed the presequence translocase–associated motor or PAM complex) (fig. S1A).
Current models of the Tim23-Tim50 association indicate that the two subunits assemble in a highly dynamic manner through multiple potential interactive sites. Tim23 has a bipartite domain organization, consisting of a channel-forming domain at the C terminus with four predicted transmembrane helices and a region of approximately 100 residues in the IMS (Tim23IMS) at the N terminus that is intrinsically disordered (22, 23). Tim50 consists of a large globular receptor domain in the IMS (Tim50IMS) that is anchored to the IM by a single transmembrane segment. The crystal structure of the trypsin-resistant core of the Saccharomyces cerevisiae Tim50 IMS receptor (Tim50CORE) revealed a phosphatase-like fold, with conserved features including a highly basic protruding β-hairpin proximal to an acidic groove that may serve to bind presequences (24, 25). Tim50 of fungal species also contains a C-terminal presequence binding domain (PBD) (18–20), although the structure of this region is currently unknown. The Tim23-Tim50 association is mediated, at least in part, by interaction between the heptad leucine repeat region in the Tim23IMS unstructured domain (residues 68 to 85; S. cerevisiae numbering) and the IMS domain of Tim50 (15–17, 23, 26–28). This interaction is critical for TIM23 complex function and is likely stabilized as a coiled coil; however, the corresponding interactive site on Tim50IMS has yet to be unambiguously identified. It has also been shown that the conserved β-hairpin of the Tim50CORE promotes interaction with Tim23 (25). Finally, it has been shown that the IMS domain of Tim50 makes contact with the first transmembrane helix of Tim23 (26). A graphical summary of the known Tim23-Tim50 interactive sites is shown in fig. S1B.
Membrane proteins do not operate in isolation; there is an increasing awareness of the importance of specific lipids in regulating the dynamics and associations of proteins in biomembranes (29–32), including the operation of mitochondrial protein import machineries (33). Cardiolipin (CL) is the signature phospholipid of energy-transducing membranes, which include the mitochondrial IM (34, 35). CL is unusual among phospholipids in that it contains two phosphate groups and four hydrocarbon chains. This molecular architecture renders CL capable of forming inverted hexagonal structures in isolation under certain conditions (for example, low pH and with divalent cations). Hence, CL can have nonbilayer propensity in the context of biomembranes, promoting local regions of high curvature (36). CL is synthesized within the mitochondrion to yield a nascent form of the lipid with nonuniform acyl chains, followed by a remodeling transacylation process to produce mature CL with highly unsaturated acyl chains whose composition is largely species- or tissue-specific (fig. S1C) (37–42). The importance of this process is underscored by the known diseases that are linked to defects in CL biogenesis, including diabetes, cardiovascular disease, neurodegenerative disease, and the multisystem disorder Barth syndrome (43–45). It has long been known that CL mediates protein-protein interactions. Most notably, it plays a predominant role in mediating the association of respiratory complexes of the IM to form supercomplexes (46–48). This lipid has also been implicated in the functional interactions of the TIM23 complex. For example, CL promotes the interactions of Tim23IMS (49) and the peripheral membrane subunit Tim44 (50, 51) with lipid bilayers, and the reconstitution of the motor-free TIM23 complex requires CL for optimal function (52). Many studies have shown that CL is required for the functional integrity of the TIM23 complex; two enzymes now known to be involved in CL biogenesis, namely, Tam41 (53–56) and Ups1 (fig. S1C) (57–59), were originally identified on the basis of the effects that their abrogation had on the general assembly status of the TIM23 complex. Moreover, it has been shown that the lack of CL causes measureable decreases in the protein import rates of TIM23 substrates (60).
Therefore, it is accepted that CL is required to maintain the overall functional integrity of the TIM23 complex. However, the mechanistic basis that underpins this critical role for CL is poorly understood. Here, we show that CL modulates the interaction between the Tim50 receptor and the Tim23 channel in a manner that is functionally relevant for TIM23 complex operation. A confluence of site-specific fluorescence, structural analysis, and molecular dynamics (MD) simulations shows that the soluble Tim50IMS receptor domain interacts with lipid bilayers in a CL-dependent manner, providing a mechanistic basis for the receptor-channel interaction. This work provides new insights into how a specific lipid mediates functionally critical protein interactions within a multicomponent machinery.
RESULTS
CL plays a role in stabilizing the Tim23-Tim50 interaction
On the basis of several different studies, Tim23 appears to be the main subunit that recruits the Tim50 receptor to the TIM23 complex (15–17, 27). Moreover, it has been demonstrated in yeast that the soluble receptor domain of Tim50 alone (Tim50IMS) is sufficient to support the function of the full-length Tim50 receptor (27). Nonetheless, throughout all eukaryotes, Tim50 contains a transmembrane segment, which prompted us to address the potential structural or functional role that anchorage of the receptor to the IM might have. We first compared the interaction of Tim23 to full-length Tim50 and to Tim50IMS using coimmunoprecipitation (Fig. 1A). Mitochondria isolated from a wild-type (WT) strain or from a strain containing Tim50IMS as the only version of the receptor were subjected to digitonin solubilization and incubation with affinity-purified antibodies to Tim23 or Tim50 or with preimmune immunoglobulins bound to protein A–Sepharose (PAS) beads, followed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) of bound and unbound fractions and immunoblotting with antibodies against different TIM23 subunits. In WT mitochondria (Fig. 1A, left), αTim23 immunodepleted virtually all Tim23 and Tim17 and immunoprecipitated a significant fraction of Tim50, whereas αTim50 immunodepleted Tim50 and immunoprecipitated part of the Tim23 and Tim17 population, consistent with previous results (27). In samples containing Tim50IMS (Fig. 1A, right), we detected the same robust Tim23-Tim17 interaction observed with WT; however, αTim23 did not precipitate Tim50IMS, nor did αTim50 immunoprecipitate Tim23. Hence, although the IMS regions of Tim23 and Tim50 functionally interact, the lack of transmembrane attachment in Tim50 renders its association with Tim23 less stable when evaluated by this method.
Fig. 1. In vivo and in organello analysis of the CL dependence of the Tim23-Tim50 interaction.
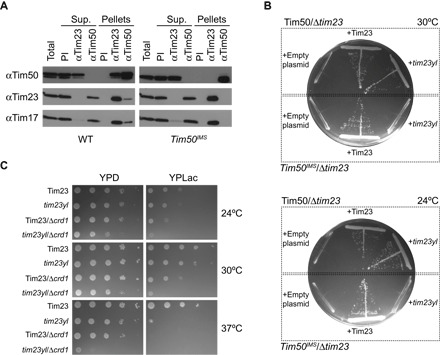
(A) Coimmunoprecipitation analysis of the Tim23-Tim50 interaction. Digitonin lysates of mitochondria from yeast strains containing full-length Tim50 (left) or Tim50IMS (right) were immunoprecipitated with affinity-purified antibodies to Tim23 (αTim23) or Tim50 (αTim50) or preimmune immunoglobulins (PI). Unbound (Sup.) and bound (Pellets) fractions were analyzed by SDS-PAGE and immunodecoration with antibodies against Tim50, Tim23, or Tim17, as indicated. Total and Sup. lanes indicate 20% of starting material; all other lanes represent undiluted samples. (B) Synthetic lethality of tim23yl and Tim50IMS. Yeast cells containing a chromosomal deletion of TIM23 complemented with a TIM23-containing URA3 plasmid were generated in a Tim50 or Tim50IMS background (delineated by dotted boxes). Cells were transformed with centromeric plasmids carrying either Tim23 or tim23yl, as indicated, and subjected to URA3 plasmid loss by plating on glucose-containing medium with 5-fluoroorotic acid (5-FOA). Cells were incubated at 30°C (upper plate) or 24°C (lower plate). (C) Genetic interaction of Δcrd1 and tim23yl. Tenfold serial dilutions were prepared from yeast cells containing WT Tim23 or the tim23yl mutant in a background of normal CL synthesis or CL synthase knockouts (Δcrd1). Cells were spotted onto YPD or YPLac medium as shown and grown at the indicated temperatures.
To obtain further evidence for the role of Tim50 membrane association in its interaction with Tim23, we reasoned that a targeted disruption of the association between the Tim23 and Tim50 IMS domains would show a stronger effect in strains containing Tim50IMS than in strains with full-length Tim50. Specific mutations in the predicted heptad leucine repeat of the Tim23 IMS domain (Y70A,L71A) have been shown to destabilize the interaction between the Tim23 and Tim50 IMS domains and to cause a temperature-sensitive growth phenotype in vivo (23). Thus, we generated strains with chromosomal deletions of Tim23 in a Tim50 or Tim50IMS background and introduced plasmid-borne copies of WT Tim23 or Tim23 Y70A,L71A (tim23yl) (Fig. 1B). We found that whereas the tim23yl double mutant was viable in the Tim50 background, we did not recover any viable cells containing the tim23yl mutant in the Tim50IMS background, either at 30°C (commonly used for yeast growth) or at 24°C (which should allow the growth of the temperature-sensitive mutants). As positive controls, we obtained viable cells when WT Tim23 was used in rescue experiments with either Tim50 or Tim50IMS. As negative controls, when empty plasmids were used, no viable cells were recovered in either of the two backgrounds, confirming the specificity of the assay. These results indicate that the anchorage of the soluble Tim50 receptor domain to the membrane can, at least partially, compensate for a destabilization in the interaction between the IMS domains of Tim23 and Tim50.
Given this apparent role of Tim50IMS membrane association, we then addressed whether the lipid composition of the IM may affect the interaction between Tim23 and Tim50. Because derangements in CL biogenesis have been shown to affect TIM23 complex stability (53–59), we asked whether tim23yl shows a genetic interaction with CRD1, the gene encoding CL synthase (fig. S1C). Thus, we analyzed the growth of yeast strains containing WT Tim23 or tim23yl in a background of normal CL biosynthesis or with a knockout of the CRD1 gene (Fig. 1C). On fermentable medium [yeast extract, peptone, and dextrose (YPD)], in comparison to either of the single mutants (tim23yl or Δcrd1), the tim23yl/Δcrd1 cells grew slower at 24° and 30°C and had a marked growth defect at 37°C. When grown on a nonfermentable carbon source [yeast extract, peptone, and lactate (YPLac)], tim23yl/Δcrd1 cells showed greatly reduced viability in comparison with either of the single mutants. This strong synthetic growth defect of tim23yl and Δcrd1 suggests that CL could play a role in the Tim23-Tim50 interaction. However, further coimmunoprecipitation tests showed that the presence of CL does not affect the association of Tim50IMS with the TIM23 complex (fig. S2A). In addition, we did not observe a synthetic growth defect between Tim50IMS and the Δcrd1 knockout (fig. S2B). This latter result is likely due, in part, to the ability of phosphatidylglycerol (PG), the biosynthetic precursor of CL, to compensate for the lack of CL in cell growth under these conditions. Thus, to more directly test the potential role of CL in the Tim23-Tim50 interaction, we proceeded using both in organello and model membrane systems.
CL modulates the interactive sites between Tim23 and Tim50
To further explore the possible CL dependence of the Tim23-Tim50 interaction, we used a strategy based on chemical cross-linking and immunoprecipitation developed previously to probe subunit interactions of Tim23 in active mitochondria (26). By this approach, radiolabeled monocysteine variants of S. cerevisiae Tim23 ([35S]Tim23) were translated in a cell-free system and imported into mitochondria isolated from different yeast strains. It has been amply demonstrated that, by this approach, in vitro–translated Tim23 can properly integrate into the mitochondrial IM and assemble with endogenous TIM23 complex subunits (26, 61). Using these samples, chemical cross-linking with homobifunctional thiol-reactive reagents was performed to create cross-linked adducts with subunits containing native cysteine sites proximal to the cysteine site within each Tim23 variant, and immunoprecipitation was performed to identify the cross-linked partner. It has been shown by this method that the single native cysteine in the S. cerevisiae Tim50 MS domain (C268, near the acidic groove and β-hairpin of Tim50CORE) is proximal to both the C-terminal part of the Tim23 IMS domain and the first predicted transmembrane segment of Tim23 (fig. S1B) (26). For the present analysis, we drew from our Tim23 monocysteine variant library those constructs having cysteines at key sites within the IMS domain and in the first two transmembrane segments of Tim23 (Fig. 2A).
Fig. 2. In organello cross-linking–detected Tim23-Tim50 interactions.

(A) Topology diagram of Tim23 and Tim50 in the mitochondrial IM. Tim23 (yellow) contains four predicted transmembrane segments (TMS1 to TMS4) and an intrinsically disordered IMS region with a heptad leucine repeat (thickened line). Sites in white depict residues changed to cysteine for thiol-based cross-linking. Sites in red depict the region of the Y70A,L71A mutations. Tim50 (cyan) contains a single native cysteine site (C268) in the IMS domain. (B) Cross-linking and immunoprecipitation (IP)–detected interaction of Tim50 with Tim23IMS. [35S]Tim23(S80C) (“23”) or [35S]Tim23(S80C) with the Y70A,L71A mutations (“yl”) was imported into mitochondria isolated from the strains indicated (WT, Δcrd1, Δtaz1, and Δcld1) and subjected to chemical cross-linking with BMOE. One subset of samples was retained for analysis of the cross-linking reaction (“Total”), and the other was subjected to immunoprecipitation with antibodies to Tim50 to identify Tim23-Tim50 adducts (“Immunoprecipitation”). The band corresponding to non–cross-linked [35S]Tim23(S80C) (or [35S]Tim23(S80C)yl) is indicated by an open arrowhead on the Total gel; cross-linked adducts between radiolabeled Tim23 constructs and Tim50 [used for quantitation in (C)] are indicated by the closed arrowheads on the Immunoprecipitation gel. (C) Quantitation of cross-linking efficiency between Tim50 and Tim23IMS. Means represent values from three independent experiments (normalized relative to [35S]Tim23 in WT mitochondria); error bars represent SDs. Dots indicate significant differences in comparison to results from control ([35S]Tim23(S80C) in WT mitochondria) by Student’s t test (•P < 0.05; ••P < 0.01; •••P < 0.0001). (D) Interaction between Tim50 and Tim23 transmembrane segments. Left: Representative cross-linking and IP-detected interaction between Tim50 and the indicated sites on TMS1 and TMS2, showing the higher–molecular weight immunoprecipitated band. Right: Helical wheel projections of TMS1 and TMS2 indicating sites used for cross-linking. Sites in yellow indicate those with highest cross-linking to Tim50. The red arc on TMS2 indicates the aqueous channel–facing, substrate-interactive face of the helix. (E) IP-detected interaction of Tim50 with Tim23 TMS1. [35S]Tim23(F114C) (“23”) or [35S]Tim23(F114C)yl (“yl”) was subjected to chemical cross-linking and IP in mitochondria from the indicated strains exactly as in (B). Cross-linked adducts between radiolabeled Tim23 constructs and Tim50 [used for quantitation in (F)] are indicated by the closed arrowheads. (F) Quantitation of cross-linking efficiency between Tim50 and Tim23 TMS1. Means represent values from three independent experiments (normalized relative to [35S]Tim23 in WT mitochondria); error bars represent SDs. Dots indicate significant differences in comparison to results from control ([35S]Tim23(F114C) in WT mitochondria) by Student’s t test (••P < 0.01; •••P < 0.0001).
Because the goal of this work was to evaluate the effect of CL on the Tim23-Tim50 interaction, we used mitochondria that were isolated from yeast strains deficient in specific steps of the CL biosynthesis and remodeling pathway (fig. S1C), which have been described and characterized elsewhere (62). Mitochondria from yeast lacking CL synthase (Δcrd1) are devoid of CL and have increased concentration of its biosynthetic precursor, PG. Mitochondria from yeast lacking the CL-specific deacylase (Δcld1) contain unremodeled CL with acyl chains that are more saturated and heterogeneous than those of remodeled CL. In addition, mitochondria from yeast lacking the transacylase tafazzin (Δtaz1) contain CL that is at a decreased concentration and contain unremodeled acyl chain composition, as well as an accumulation of monolysocardiolipin (MLCL). To confirm that cell-free translated [35S]Tim23 can be imported into mitochondria from each of these strains, we performed diagnostic proteolysis of intact mitochondria and of mitochondria subject to osmotic rupture of the OM to expose the IMS (fig. S3) (63). [35S]Tim23 was imported with roughly equal efficiency into mitochondria from all four strains, judging by the protease protection of intact mitochondria. When integration efficiency was evaluated by the ~14-kDa proteolytic fragment following osmotic swelling, mitochondria from the CL biogenesis mutants showed slightly decreased efficiencies to different extents. We note that blocking CL synthesis has been previously shown to result in measurable decreases in mitochondrial protein import, for instance, in the decreased import of TIM23 substrates in mitochondria from Δcrd1 yeast (60).
We first evaluated the cross-linking–detected interaction between the Tim50IMS receptor and the Tim23IMS domain in different CL backgrounds using Tim23 constructs with a single cysteine at position 80 (Fig. 2B, with cross-linking band intensities quantified in Fig. 2C). In mitochondria with a WT CL background, [35S]Tim23(S80C) generated adducts to Tim50, consistent with previous work showing the interaction between the IMS region of Tim50 and the C-terminal half of the Tim23 IMS domain (Fig. 2B, lanes 1 and 9) (15–17, 26). In addition, as shown previously (17, 26), two immunoprecipitated adducts are observed: a prominent band at ~97 kDa and a weaker band at ~70 kDa that likely represents a proteolysis product. Control samples in which the [35S]Tim23(S80C) construct contained the Y70A,L71A mutations did not yield cross-linked adducts to Tim50 (Fig. 2B, lanes 2 and 10), confirming that our cross-linking–based assay was a good reporter of the native interaction between the IMS domains of these subunits (23). When we performed the same analysis in mitochondria lacking CL (Δcrd1), we found that [35S]Tim23(S80C) cross-linked Tim50 with significantly higher efficiency (Fig. 2B, compare lanes 1 and 3 and lanes 9 and 11). In mitochondria synthesizing CL but defective in remodeling at the transacylase (Δtaz1) or phospholipase (Δcld1) steps, the degree of cross-linking was still higher (Fig. 2B, compare lane 1 with lanes 5 and 7 and lane 9 with lanes 13 and 15). As observed with the WT lipid complement, in all of the CL knockout backgrounds, negligible Tim50 cross-linking of Tim23 with the yl mutations was observed (Fig. 2B, lanes 4, 6, 8, 12, 14, and 16). These results suggest that when the Tim23-Tim50 coiled-coil interaction is intact, defects in CL biogenesis may actually promote the interaction between the soluble IMS domain of Tim23 and the Tim50 receptor region in the IMS.
We extended our cross-linking–based analysis to test Tim50IMS interactions with the two N-terminal transmembrane segments of Tim23 (Fig. 2A, TMS1 and TMS2). Notably, the Tim50IMS domain was previously shown to contact residues within Tim23 TMS1 (26). This suggests that the Tim50 receptor domain might protrude into the lipid bilayer and/or that Tim23 has the conformational freedom to move this transmembrane segment into the aqueous IMS. Consistent with these observations, we observed cross-linking between Tim50IMS and Tim23 TMS1 sites near the center of the predicted transmembrane helix, with the highest efficiency on the helical face in which F114 resides (Fig. 2D). Continuing this analysis for Tim23 TMS2, which has not been addressed before, we again observed Tim50IMS site-specific cross-links that are highest on a particular helical face, the side in which A152 and A156 reside (Fig. 2D). These observations are notable for two reasons. First, Tim23 TMS1 and TMS2 have both been shown to be in contact with the polypeptide substrate in transit across the aqueous channel of the TIM23 complex (11). Second, both TMS1 (26) and TMS2 (13) gain exposure to the aqueous IMS in a manner coupled to a reduction in Δψm across the IM. Together with our cross-linking data, these observations suggest potential mechanisms for the Tim50-Tim23 interaction (see Discussion). For the remainder of this study, we focused on the Tim50IMS–Tim23 TMS1 interaction because this has been more extensively characterized.
We then extended our analysis to address the CL dependence of the interaction between Tim50IMS and Tim23 TMS1 (Fig. 2E, with cross-linking band intensities quantified in Fig. 2F). Because Tim50IMS showed greatest proximity to F114, we used [35S]Tim23(F114C) as a test construct. This analysis yielded two surprising findings. First, whereas the Tim23 Y70A,L71A mutations (in which the IMS coiled-coil interaction is obstructed) almost completely blocked the cross-linking–detected interaction between Tim23IMS and Tim50IMS (Fig. 2, B and C), these mutations had a much less marked effect on the interaction between Tim50IMS and Tim23 TMS1 in mitochondria with a normal CL composition (Fig. 2E, compare lanes 1 and 2). Hence, the interaction between Tim50IMS and the Tim23 transmembrane segment appears to be stabilized, at least in part, by factors other than the IMS coiled-coil interaction. Second, mitochondria lacking CL (from the Δcrd1 strains) supported much reduced interaction between Tim50IMS and TMS1 compared with the WT strain (Fig. 2E, compare lanes 1 and 3 and lanes 2 and 4), in stark contrast to the increased Tim50IMS-Tim23IMS interaction observed in the absence of CL (Fig. 2, B and C). Strains synthesizing CL but with defects in remodeling (Δtaz1 and Δcld1) showed varying degrees of recovery of the Tim50IMS–Tim23 TMS1 interaction to WT levels (Fig. 2E, lanes 5 to 8).
Notably, we have previously shown that the magnitude of the Δψm affects the cross-linking–detected Tim23-Tim50 interaction. Specifically, a decrease in the Δψm was shown to increase the interaction of the IMS regions of Tim23 and Tim50 (26), consistent with the enhanced interactions of the IMS domains with CL biogenesis defects (Fig. 2, B and C). Thus, in principle, a lowered Δψm accompanying a block in CL synthesis (64) could, in part, account for our observed cross-linking profiles. However, we also observe a progressive increase in the interaction between the Tim50 IMS domain and Tim23 TMS1 when the Δψm is reduced by titration of the protonophore CCCP (carbonyl cyanide m-chlorophenyl hydrazone) (fig. S4), which is in contrast to the respective interaction observed in the presence of CL biogenesis defects (Fig. 2, E and F). Hence, whereas decreasing Δψm causes a general, site-independent increase in Tim50 interaction with Tim23, the alteration in cross-linking profiles with defective CL biogenesis is site-dependent (increasing the interaction of the Tim50 receptor domain with the Tim23 IMS domain and decreasing its interaction with the Tim23 channel region). These results support the idea that our observed cross-linking patterns result from CL-mediated Tim23-Tim50 interaction and do not result from an indirect effect of different Δψm among the strains used, although it cannot be ruled out that the Δψm could play a minor role in our observed interactions.
Together, this cross-linking analysis suggests that CL modulates the site-specific interactions between the Tim23 and Tim50 subunits. Whereas the association between Tim50IMS and Tim23IMS is stabilized by the predicted coiled-coil interaction, the association between Tim50IMS and Tim23 TMS1 is additionally stabilized by the presence of CL. This finding is consistent with a model in which defective CL synthesis reduces the interaction of Tim50IMS with Tim23 TMS1, thereby concurrently increasing its interaction with Tim23IMS. However, given the complexities of this in organello experimental system (for example, differences in [35S]Tim23 import efficiencies among strains and indirect effects of genetic knockouts, including possible effects of Δψm), we next more directly addressed CL-mediated Tim23-Tim50 interactions using a reductionist approach with model membrane systems.
Analysis of Tim23-Tim50 interactions in nanoscale model bilayers
Nanodiscs are discoidal patches of lipid bilayer that are bound by annuli of membrane scaffold protein (MSP) (65–67). As platforms for the reconstitution of membrane proteins in a lamellar bilayer of defined lipid composition, they are excellent experimental systems for probing the lipid dependence of membrane protein interactions. Thus, to investigate interactions between Tim50IMS and full-length Tim23 in this reductionist system, we incorporated [35S]Tim23 monocysteine constructs into nanodiscs and performed site-directed cross-linking with purified soluble Tim50IMS (Fig. 3A). To prepare Tim23-containing nanodiscs, [35S]Tim23 variants were translated in a cell-free system and added to a reaction containing the MSP1E3D1 scaffold protein (66), mixed micelles of cholate, and the desired phospholipid(s), and disc self-assembly was initiated by the addition of hydrophobic adsorbants. [35S]Tim23-containing nanodiscs ([35S]Tim23-ND) were purified from the rest of the assembly mixture on a Ni–nitrilotriacetic acid (NTA) matrix that bound the 6×His tag on the MSP1E3D1 (fig. S5A). The fraction of [35S]Tim23 that did not assemble into nanodiscs came out in the flow-through; the fraction of [35S]Tim23 assembled into nanodiscs coeluted with the MSP1E3D1 protein (fig. S5A, compare upper and lower panels). To confirm that nonincorporated [35S]Tim23 did not nonspecifically bind to the Ni-NTA matrix, we performed mock assembly reactions in the absence of MSP1E3D1 and found that the radiolabeled protein eluted in early fractions (fig. S5B). To address whether Tim23 was incorporated into nanodiscs with proper topology, we cotranslationally labeled cysteine positions in the soluble domain (Tim23 T24C) or in TMS1 (Tim23 I111C) with the environment-sensitive fluorescent probe NBD exactly as previously described (11, 13) before incorporation into nanodiscs (yielding NBD-Tim23-ND). Samples with the NBD probe in the transmembrane helix had a higher emission yield and a blue-shifted λmax value compared with samples containing the probe in the soluble domain (Fig. 3B). These differences in fluorescence parameters are nearly identical to a similar comparison for NBD-Tim23 integrated into the mitochondrial IM (11, 13), supporting the proper integration of the transmembrane region into nanodiscs. Finally, we conducted several controls to confirm that our nanodisc system could be used to evaluate physiologically relevant cross-linking patterns (Fig. 3C). In the presence of the chemical cross-linker, [35S]Tim23(T94C)-ND formed cross-links with the single cysteine in Tim50IMS (lane 1), revealing the same type of Tim23–Tim50IMS domain interaction observed in organello. However, no such cross-linked adduct was generated in the absence of Tim50IMS (lane 2) or the cross-linker (lane 3) or when the [35S]Tim23 construct lacked a Cys site (lane 4) or contained the Y70A,L71A mutations (lane 5). On the basis of these controls, we conclude that our [35S]Tim23-ND preparations contain Tim23 assembled into nanodiscs with the correct topology, suitable for direct probing of Tim50IMS interactions by site-directed cross-linking.
Fig. 3. Interactions between Tim50IMS and full-length Tim23 in nanodiscs.
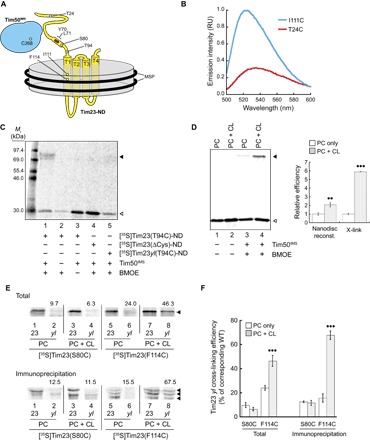
(A) Diagram of the experimental design. Full-length Tim23 (yellow) was reconstituted into nanodiscs (gray disc) containing an experimentally defined lipid composition and bound by MSP (black rings). Sites in white depict residues changed to cysteine for thiol-based cross-linking or fluorescence analysis. Sites in red depict the region of the Y70A,L71A mutation. Purified soluble Tim50IMS (cyan) contains a single cysteine site (C268). (B) Fluorescence analysis of NBD-Tim23-ND. NBD was cotranslationally incorporated into the Tim23 IMS region (Tim23 T24C) or TMS1 (Tim23 I111C) during in vitro translation, assembled into nanodiscs, and purified for spectral analyses. On the basis of emission scans, samples with the NBD probe in TMS1 [NBD-Tim23(I111C)-ND, cyan] had a 3.1-fold higher fluorescence yield and a 10-nm blue-shifted λmax relative to samples with the probe in the IMS domain [NBD-Tim23(T24C)-ND, red]. RU, relative units. (C) Quality control tests with the nanodisc-based cross-linking system. Purified nanodiscs containing [35S]Tim23(T94C) (lanes 1 to 3), [35S]Tim23ΔCys (lane 4), or [35S]Tim23yl(T94C) (lane 5) were subjected to reactions in the presence or absence of a cross-linking agent [bis-maleimidoethane (BMOE)] and Tim50IMS, as indicated, followed by IP with αTim50. The band corresponding to the non–cross-linked [35S]Tim23(T94C)-ND is indicated by the open arrowhead, and the cross-linked adduct between [35S]Tim23(T94C)-ND and Tim50IMS is indicated by the closed arrowhead. (D) Relative efficiency of nanodisc incorporation and Tim50IMS-[35S]Tim23-ND cross-linking. [35S]Tim23(F114C) was reconstituted into nanodiscs containing only POPC (“PC”) or a binary lipid composition containing 20 mole percent (mol %) TOCL (“PC + CL”), as indicated. Samples were resolved by SDS-PAGE in the absence or presence of cross-linking with Tim50IMS, as shown. The bands corresponding to the non–cross-linked [35S]Tim23(F114C)-ND are indicated by the open arrowhead, and the cross-linked adducts between [35S]Tim23(F114C)-ND and Tim50IMS are indicated by the closed arrowhead. The panel to the right shows the quantitation (means with SDs) of relative incorporation efficiencies of [35S]Tim23 into nanodiscs (“Nanodisc reconst.”) and Tim50IMS cross-linking (“X-link”) for both types of nanodisc lipid compositions (means of three independent experiments with SDs are shown). Dots indicate significant differences between PC + CL and the cognate and PC-only samples by Student’s t test (••P < 0.01; •••P < 0.0001). (E) Cross-linking and IP-detected interaction between Tim50IMS and [35S]Tim23-ND. [35S]Tim23(S80C) or [35S]Tim23(F114C) in the absence (“23”) or presence (“yl”) of the Y70A,L71A mutations was reconstituted into nanodiscs containing only POPC (“PC”) or with a binary lipid composition containing 20 mol % TOCL (“PC + CL”), as indicated. Samples from cross-linking reactions with Tim50IMS were resolved directly by SDS-PAGE (Total; top) or subjected to IP with αTim50 and resolved by SDS-PAGE (Immunoprecipitation; bottom). Cross-linked adducts between radiolabeled Tim23 constructs and Tim50 (used for quantitation) are indicated by the closed arrowheads. Numbers above each “yl” lane indicate the percent cross-linking efficiency relative to the corresponding experiment with WT Tim23 (lane to the immediate left), shown graphically in (F). (F) Quantitation of cross-linking efficiency between Tim50IMS and [35S]Tim23-ND. Means represent values from three independent experiments; error bars represent SDs. Dots indicate significant differences between PC + CL and the cognate and PC-only samples by Student’s t test (•••P < 0.0001).
Leveraging the ability to reconstitute Tim23 into nanodiscs containing or lacking CL, we used this experimental system to directly analyze the effect of CL on stabilizing the interaction between Tim50IMS and nanodisc-bound Tim23, both at the Tim23 IMS domain (Cys80) and at Tim23 TMS1 (Cys114). We compared the cross-linking efficiency of Tim50IMS to [35S]Tim23(F114C)-ND in nanodiscs with and without CL (Fig. 3D). We found that the efficiency of reconstitution of Tim23 into nanodiscs containing 1′,3′-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol (tetraoleoyl CL, or TOCL) was approximately twice the efficiency of reconstitution into nanodiscs containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) only (Fig. 3D, open arrowhead), consistent with our previous observations that CL enhances incorporation of in vitro–translated proteins into model membranes (68). However, we also found that the cross-linking–detected interaction of Tim50IMS with the Tim23 transmembrane region was approximately sixfold higher in the presence of TOCL than in its absence (Fig. 3D, closed arrowhead), supporting a CL-enhanced interaction between the Tim50 receptor and the Tim23 channel region. To further investigate the site-specific effects of CL on the Tim23-Tim50 interaction, we developed a strategy to factor out any lipid-dependent effects of Tim23 reconstitution into nanodiscs. Namely, [35S]Tim23(S80C) and [35S]Tim23(F114C) constructs with or without the Y70A,L71A mutations were reconstituted into nanodiscs with or without TOCL, and the dependence of the Tim50IMS interaction on the yl mutations was measured (Fig. 3E and quantitation in Fig. 3F). As expected, the yl mutations obliterated the cross-linking–detected interaction between the IMS domains of Tim50 and Tim23 (Fig. 3E, lane 2), and this interaction was not enhanced by the presence of TOCL in the nanodiscs (lane 4). By contrast, whereas the yl mutations strongly reduced the interaction between Tim50IMS and Tim23 TMS1 in the presence of POPC only (lane 6), this interaction was significantly enhanced by the presence of TOCL (lane 8). Hence, in agreement with our in organello work, this reductionist system shows that the presence of CL in the bilayer specifically promotes the interaction between the Tim50 receptor domain and TMS1 of Tim23.
Analysis and modeling of the Tim50IMS solution structure
The S. cerevisiae Tim50IMS receptor (residues 133 to 476) contains a highly conserved core (Tim50CORE; residues 164 to 361) and a C-terminal PBD (residues 395 to 476) (fig. S6A) (15–20). Several regions within Tim50IMS are predicted to mediate coiled-coil interactions (fig. S6B), and mutation of key sites within heptad leucine repeat regions of Tim50CORE has been shown to inhibit its interaction with Tim23IMS (28). Advances in our understanding of the structure of S. cerevisiae Tim50 have focused on the highly ordered and protease-resistant “conserved core” (Tim50CC; residues 176 to 361). The crystal structure of this region revealed a structure homologous to C-terminal repeat domain phosphatases, specifically the Scp1 transcriptional repressor (24, 25, 69). Key features of this structure include a highly exposed basic β-hairpin that is required for its interaction with Tim23 and a proximal pocket with negative charge density that may serve as a binding groove for positively charged presequences. In addition, nuclear magnetic resonance (NMR) analysis of a small segment of the C-terminal PBD (sPBD; residues 400 to 450) confirmed that this site functions to bind presequences and also binds the Tim50CORE (19). Obtaining structural information of the Tim50IMS receptor domain in its entirety could provide further insights into the mechanism by which Tim50IMS might interact with CL-containing bilayers. Measurements of the Tim50IMS secondary structure in solution confirm that it is highly structured (23); however, in comparison with the core domain, Tim50IMS may contain intrinsically disordered regions (fig. S6C) (70), consistent with its role in mediating protein-protein interactions (71). The predicted conformational flexibility and molecular mass (~41 kDa) of Tim50IMS would make it recalcitrant to x-ray crystallography or NMR spectroscopy, respectively. We therefore used small-angle x-ray scattering (SAXS) to obtain structural information on the entire Tim50IMS in solution.
A series of SAXS scattering curves was collected for three concentrations of purified Tim50IMS (fig. S6D). Guinier analyses of scattering data showed linear fits, confirming that the protein was monodisperse at all concentrations (fig. S6D, inset). On the basis of Guinier plots, the calculated radii of gyration (Rg) were concentration-independent, with an average value of 32.7 ± 1.0 Å, and the average molecular mass was 42.0 ± 1.4 kDa (table S1), which agreed closely with the value calculated from the primary sequence of Tim50IMS (40.3 kDa). The pair distance distribution function P(r) of Tim50IMS revealed a maximum at ~30 Å with a tail toward longer distances, suggesting the presence of flexible regions and/or an elongated structure with a maximum dimension Dmax of ~120 Å (fig. S6E). The envelope of the Tim50IMS structure generated by ab initio reconstructions demonstrated its elongated rodlike shape (Fig. 4A). The normalized spatial discrepancy (NSD) for the 10 independent reconstructions from DAMMIF was calculated to be 0.658 for Tim50IMS (0.5 mg/ml), suggesting high quantitative similarity among the different bead models.
Fig. 4. Ab initio SAXS reconstruction of Tim50IMS.
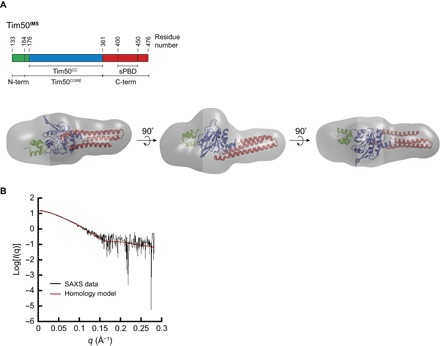
(A) Tim50IMS SAXS molecular envelope aligned with the homology model. The ab initio envelope generated by DAMAVER analysis of SAXS data from Tim50IMS (0.5 mg/ml; gray) is shown in comparison with the Tim50IMS homology model (ribbon structure), aligned using SITUS. The domains of the Tim50IMS homology model are color-coded with respect to the linear domain representation shown above. (B) Analysis of the fit between the envelope and the homology model. The experimental SAXS scattering data for Tim50IMS (0.5 mg/ml; black) and theoretical scattering data for the Tim50IMS homology model (red) were fit using the CRYSOL package, yielding a χ2 value of 1.12.
Next, a model for the full-length Tim50IMS was constructed by combining the known coordinates of the conserved core region [Protein Data Bank (PDB) 3QLE] with homology-modeled structures of the flanking regions (Fig. 4A). The structures of the N-terminal (residues 133 to 175) and C-terminal (residues 362 to 476) regions were predicted by the I-TASSER (Iterative Threading ASSEmbly Refinement) web server. As described in Materials and Methods, the structure was energy-minimized in the CHARMM (Chemistry at HARvard Macromolecular Mechanics) program (72) using the CHARMM22 force field (73) with CMAP correction (74) to relieve local steric strains. The minimized structure was then rigidly docked in the SAXS-derived density map using the Situs package (75). The theoretical scattering profile for the homology model was compared with the experimental scattering values of Tim50IMS using the program CRYSOL (76). The model displayed an excellent fit to the experimental scattering data (χ2 = 1.12) (Fig. 4B). Moreover, the homology model contained a coiled-coil conformation at the C terminus of Tim50IMS, in agreement with the predicted coiled-coil propensity of that region (fig. S6B), as well as the previously reported NMR and circular dichroism spectra of the sPBD, which indicated a domain rich in α-helical content (19).
To further validate our SAXS model for Tim50IMS, we extended our SAXS analysis to include just the Tim50CORE domain (residues 165 to 361) in comparison with the known crystal structure of Tim50CC (25). The distance distribution function from Tim50CORE SAXS scattering data revealed a parabolic profile (fig. S7A), consistent with the globular structure of Tim50CC. On the basis of Tim50CORE SAXS data, 10 independent reconstructions in DAMMIF were highly consistent (with a low NSD of 0.468), and the resulting ab initio model of the Tim50CORE molecular envelope confirmed its globular shape, consistent with the crystal structure (fig. S7B). As confirmation, the theoretical scattering profile of Tim50CC showed excellent agreement with the experimental SAXS scatter of Tim50CORE (fig. S7C).
This SAXS analysis has therefore provided the first low-resolution structure of the Tim50IMS receptor in its entirety, which was subsequently used to inform a Tim50IMS homology model consistent with the SAXS envelope. With these structural insights, we then sought to understand our observed CL dependence of the Tim50-Tim23 interaction, which could arise from an effect of CL on Tim23 conformation and/or an effect of CL on Tim50IMS-bilayer interactions. We proceeded to directly investigate the latter using both in silico and experimental approaches.
MD analysis of the interaction between Tim50IMS and lipid bilayers
We next sought to characterize Tim50IMS-bilayer interactions via MD simulations using a coarse-grained (CG) model to generate structural models for the orientation of Tim50IMS bound to membranes and the role of CL in mediating these interactions. We initiated our investigation of the membrane binding properties of Tim50IMS by performing spontaneous membrane binding simulations. In these simulations, our Tim50IMS structural model (Fig. 4) was placed in the aqueous phase and allowed to spontaneously adsorb to a bilayer. We initiated the simulations with five different random orientations of Tim50IMS and performed MD simulations in the presence of a pure POPC bilayer and in the presence of a TOCL-containing bilayer (POPC/TOCL molar ratio, 80:20). When the bilayer contained only POPC lipids, we did not observe a stable association between Tim50IMS and the bilayer over any of the 4-μs simulations (Fig. 5A and movie S1). However, when TOCL was present in the bilayer, a stable interaction between Tim50IMS and the bilayer was observed under 3 μs for all five simulations (Fig. 5B and movie S2). These results suggest a role for CL in mediating the rapid and stable interaction of Tim50IMS with lipid bilayers.
Fig. 5. Analysis of Tim50IMS-bilayer interactions from CG MD simulations.
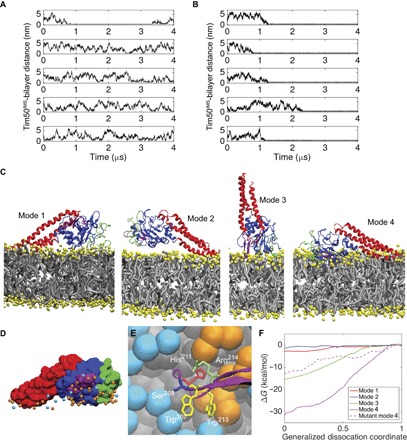
(A and B) Time course profiles of the Tim50IMS-bilayer distance. The minimum distance between Tim50IMS and the bilayers composed of pure POPC (A) or POPC with 20 mol % TOCL (B) is shown for five MD simulations each. (C) Four unique modes of Tim50IMS binding to TOCL-containing bilayers. N-terminal region, green; Tim50CORE, blue; β-hairpin, purple; C-terminal region, red; lipid headgroups, yellow; lipid hydrocarbon chains, gray. (D) Lipid headgroup phosphate contacts with Tim50IMS in binding mode 4. Tim50IMS is shown in space-filling representation with protein coloring the same as that in (C). POPC phosphates, cyan; TOCL phosphates, orange. (E) Tim50IMS β-hairpin–bilayer interactions in binding mode 4. Detailed image of key β-hairpin side chains shown to stabilize the Tim50IMS-bilayer interaction (W207, H211, W213, and R214) and the site of extrinsic fluorescent probe attachment (S208; see Fig. 7). POPC headgroup beads, cyan; TOCL headgroup beads, orange; lipid hydrocarbon tails, gray. (F) Free energy binding curves. Umbrella sampling results are shown for the four unique binding modes as well as for the four-point mutant of mode 4. The abscissa is a normalized coordinate such that x = 0 when Tim50IMS is bound and x = 1 when Tim50IMS is fully dissociated. This coordinate was used for clarity because the different modes required different absolute displacements from the bilayer to become fully dissociated.
The binding orientation (mode) of Tim50IMS to CL-containing bilayers differed among the simulations, although one orientation was observed in two of the simulations. The four unique binding orientations are shown in Fig. 5C and can be described as follows: mode 1 involved the binding of the C-terminal, N-terminal, and core regions, but the β-hairpin was not bound; mode 2 involved only the C-terminal region; mode 3 involved the N-terminal region and the core, including the β-hairpin; and mode 4 involved the N-terminal and C-terminal regions with the core, including the β-hairpin. With these binding modes defined, we first addressed whether there was an enrichment of TOCL in the vicinity of Tim50IMS. On the basis of the molar concentration of TOCL in the bilayer and the fact that CL has a two-phosphate headgroup, a random distribution of TOCL in the area of Tim50IMS contact would produce a TOCL/POPC phosphate contact ratio of 0.5. However, when we analyzed the ratio of TOCL/POPC headgroup phosphate contacts to Tim50IMS (averaged over the final 800 ns of each simulation), the ratios ranged from 1.6 to 3.8, indicating a significant enrichment of CL at the region of Tim50IMS binding. A snapshot of mode 4 with the surrounding POPC and TOCL phosphates is shown in Fig. 5D, which illustrates the enrichment of TOCL at the bilayer binding interface of Tim50IMS. A detailed view of the β-hairpin in proximity to the interfacial region for binding mode 4 is shown in Fig. 5E.
The binding strengths of the four unique binding modes were also evaluated by performing umbrella sampling unbinding calculations to determine the ΔG of binding (Fig. 5F). The binding modes involving the β-hairpin (modes 3 and 4) showed significantly stronger binding than those not involving the β-hairpin (modes 1 and 2). The rationale for the observation of multiple binding modes is that the weaker binding modes are metastable orientations that are kinetically trapped, and given the limited time scales of the simulations, we do not observe unbinding to adopt a lower free energy state (for example, mode 4). We then endeavored to identify residues of Tim50IMS that are critical in mediating bilayer interaction using rational mutation of key sites. Given that the β-hairpin is implicated in the strongest binding modes, we mutated residues W207, H211, W213, and R214 of this structural element to alanine residues and performed umbrella sampling unbinding of mode 4, as described above. The introduction of these mutations reduced the ΔG of Tim50IMS binding by more than half (Fig. 5F), underscoring the role of β-hairpin residues in stabilizing the bilayer interaction. Together, the results of our CG MD analysis indicate that Tim50IMS binds to lipid bilayers in a CL-dependent manner and with a binding mode that is strongly stabilized by the protruding β-hairpin. We next tested these computational predictions using in vitro binding and mutational studies.
Empirical characterization of the CL dependence of the Tim50IMS-bilayer interaction
The results of our in silico analyses prompted us to empirically test the CL dependence of the interaction between the soluble receptor domain of Tim50 and model membrane systems. To this end, we first used an established cosedimentation approach (77), which has been used to assay the binding of peripheral membrane proteins to lipid bilayers (Fig. 6). In this technique, soluble proteins are added to oligosaccharide-loaded liposomes of specific lipid composition, followed by an ultracentrifugation step in which vesicle-bound proteins are separated from unbound proteins in the supernatant. Cosedimentation analysis of Tim50IMS with sucrose-loaded large unilamellar vesicles (LUVs) composed solely of POPC showed negligible binding with increasing liposome concentration (Fig. 6A, “PC”). However, when LUVs contained 20 mol % TOCL, Tim50IMS showed a robust interaction with the vesicles (Fig. 6A, “PC + CL”), consistent with our MD analyses (Fig. 5). We then performed cosedimentation experiments to test whether lipids with properties similar to CL might also recruit Tim50IMS to the bilayer. To test the general requirement for a negatively charged headgroup, we repeated the cosedimentation analysis with LUVs containing 20 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG), which, like CL, is an anionic phospholipid. The presence of POPG enhanced the binding of Tim50IMS to LUVs relative to those with POPC only but not to the level observed in the presence of CL (Fig. 6A, “PC + PG”). To test whether the presence of a nonbilayer preferring lipid might affect Tim50IMS binding, we performed the cosedimentation analysis with LUVs containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), which, like CL, has the propensity to form hexagonal II (HII) structures in isolation under certain conditions (78). However, even when the POPE concentration was as high as 50 mol %, we still did not observe an increase in Tim50IMS binding relative to that in the presence of POPC only (Fig. 6A, “PC + PE”). Together, these results indicate that Tim50IMS binds lipid bilayers in a CL-dependent manner and suggest that the mode of binding depends, at least in part, on electrostatic interactions with negatively charged headgroups.
Fig. 6. Cosedimentation analysis of the CL-dependent association of Tim50IMS with lipid bilayers.
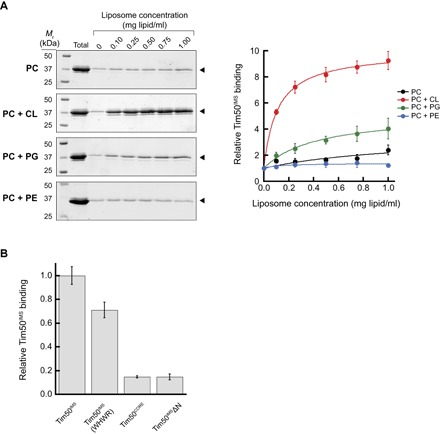
(A) Liposome cosedimentation analysis of Tim50IMS. Left: SDS-PAGE/SYPRO Orange stain profiles of Tim50IMS (1 μM) cosedimented with sucrose-loaded LUVs of defined lipid composition and increasing lipid concentration (up to 1.0 mg lipid/ml). Lipid compositions include PC (100 mol % POPC), PC + CL (80 mol % POPC and 20 mol % TOCL), PC + PG (80 mol % POPC and 20 mol % POPG), and PC + PE (50 mol % POPC and 50 mol % POPE). Bands used for quantitation are shown by the closed arrowheads. Right: Quantitation of relative Tim50IMS liposome cosedimentation for each lipid composition. Means represent values from three independent experiments (normalized with respect to band intensity in the absence of added liposomes); error bars represent SDs. (B) Cosedimentation analysis of Tim50IMS variants. Cosedimentation experiments conducted as above for 1 μM Tim50IMS, Tim50IMS W207A H211A W213A R214A (Tim50IMS WHWR), Tim50CORE (residues 165 to 361), and Tim50IMSΔN (residues 165 to 476) in the presence of PC + CL LUVs (final concentration, 0.5 mg lipid/ml). Means are normalized relative to Tim50IMS binding for three independent experiments; error bars represent SDs.
The membrane affinity of Tim50IMS is reminiscent of another TIM23 complex subunit, the peripheral membrane protein Tim44, which also binds bilayers in a CL-dependent manner (50, 51). Using a Tim44 truncation series, it was shown that deleting α helices A1 and A2 of the C-terminal domain abolished binding to CL-containing bilayers, a result corroborated by MD analysis (50). Informed by our own MD analyses of Tim50IMS, we sought to empirically analyze by cosedimentation which regions may be important for bilayer interactions (Fig. 6B). Compared with intact Tim50IMS, the core region of Tim50 (Tim50CORE) displayed only about 15% binding efficiency, and this binding was not improved by the presence of the C-terminal region (Tim50IMSΔN). This result is consistent with the role of the N-terminal domain in Tim50IMS-bilayer interactions indicated by MD analysis (Fig. 5). Unfortunately, we were unable to test the binding of the Tim50 core with only the N-terminal region (Tim50IMSΔC) because this protein was very unstable, degrading soon after purification. However, we did successfully generate a construct with alanine substitutions at sites within the β-hairpin that were identified by MD to contribute significantly to the binding energy (Tim50IMS W207A, H211A, W213A, and R214A), and this mutant showed a significantly reduced cosedimentation compared with WT Tim50IMS (Fig. 6B). These results not only confirm the importance of the β-hairpin in mediating the Tim50IMS-bilayer interaction but also point to an important role of the N-terminal region. In line with our MD analyses, this empirical analysis indicates that Tim50IMS likely has multiple surfaces that bind to CL-containing bilayers.
Finally, to analyze the importance of Coulombic attraction in the binding of Tim50IMS to CL-containing bilayers, we then examined the effect of ionic strength on the cosedimentation-detected interaction. Taking binding in the absence of added salt as the baseline, we observed a slight increase in Tim50IMS binding at moderate salt concentrations ([NaCl] = 50 to 100 mM), followed by a significant decline in binding efficiency because ionic strength was increased further ([NaCl] up to 1 M) (fig. S8). The effect of ionic strength on the binding of basic peripheral membrane proteins to bilayers with acidic lipids has been well characterized and likely results from electrostatic screening of the Coulombic interaction (79). Yet, the fact that significant Tim50IMS binding remained even at the highest salt concentration suggests that other binding modes (for example, hydrophobic interactions) play a role in Tim50IMS recruitment to CL-containing bilayers. In summary, these results provide empirical evidence that Tim50IMS binds lipid bilayers in a CL-dependent manner; that specific regions, including the β-hairpin, contribute significantly to the strength of the interaction; and that the protein-lipid association is mediated, at least in part, by electrostatic interactions.
CL-mediated structural changes and points of bilayer contact in Tim50IMS
Having demonstrated the CL dependence of the Tim50IMS-bilayer association using both CG MD and experimental approaches, we next sought to characterize the nature of this protein-lipid interaction in more detail. We first performed limited proteolysis assays to ascertain whether the binding of Tim50IMS to CL-containing bilayers might result in altered proteolytic profiles of the protein (Fig. 7A). In the absence of lipid vesicles, Tim50IMS revealed a number of protease-resistant bands (Fig. 7A, lanes 1 to 5), consistent with previous results (23), indicating the presence of tightly folded domains. The highest protease concentration yielded a band of approximately 27 kDa (marked by “*”), slightly larger than the 23-kDa Tim50CORE generated by trypsin digestion (25). The proteolysis profile of Tim50IMS preincubated with POPC vesicles (Fig. 7A, lanes 6 to 10) was identical to that of the protein in the absence of liposomes. However, proteolysis in the presence of vesicles containing 20 mol % TOCL (Fig. 7A, lanes 11 to 15) revealed a markedly different profile, with a predominant protected band (marked by “**”), which was larger than the predominant band of Tim50IMS alone or with POPC vesicles. These data show that upon binding to CL-containing bilayers, Tim50IMS has different solvent accessibility of proteinase K sites, which may result from a structural change and/or from burial of specific regions of the protein in the membrane.
Fig. 7. Analysis of CL-mediated changes in Tim50IMS proteolysis, labeling, and bilayer exposure.
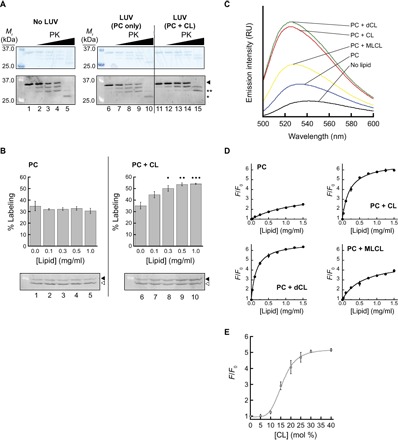
(A) Limited proteolysis of Tim50IMS. Tim50IMS (1 μM) was preincubated without lipid vesicles (lanes 1 to 5) or with vesicles composed of POPC only (“PC”; lanes 6 to 10) or POPC with 20 mol % TOCL (“PC + CL”; lanes 11 to 15). Samples were incubated in the absence of protease (lanes 1, 6, and 11) or in the presence of increasing proteinase K (“PK”; 0.67 nM, lanes 2, 7, and 12; 2.0 nM, lanes 3, 8, and 13; 5.0 nM, lanes 4, 9, and 14; 100 nM, lanes 5, 10, and 15) and resolved by SDS-PAGE. Intact Tim50IMS is indicated by the arrowhead, the smallest product detected with Tim50IMS alone or with POPC vesicles is indicated by “*,” and the predominant band protected in the presence of POPC + TOCL vesicles is indicated by “**.” (B) Lipid-dependent cysteine accessibility of Tim50IMS. Tim50IMS (1 μM) was incubated in the presence of vesicles composed of POPC only (“PC”) or POPC with 20 mol % TOCL (“PC + CL”) at the indicated lipid concentrations and subjected to labeling with the thiol-reactive reagent TMM(PEG)12. Representative gels (below) show unlabeled (open arrowhead) and labeled (closed arrowhead) Tim50IMS. Quantification of the labeling efficiency (percent labeled relative to total labeled and unlabeled; means of three independent samples with SDs) is shown for each lipid concentration in plots above. Dots indicate significant differences in labeling compared to results from control (no liposomes) by Student’s t test (•P < 0.05; ••P < 0.01; •••P < 0.0001). (C) Fluorescence-detected interaction between Tim50IMS and lipid bilayers. NBD-Tim50IMS(S208C) (500 nM) was incubated in the absence of lipid vesicles or in the presence of liposomes (1 mg lipid/ml) with differing lipid composition [POPC only (“PC”) or POPC with 20 mol % TOCL (“PC + CL”), dCL (“PC + dCL”), or MLCL (“PC + MLCL”), as indicated] and samples were subjected to fluorescence emission scans. (D) Tim50IMS-bilayer interactions with different CL variants. NBD-Tim50IMS(S208C) was incubated with varying concentrations of vesicles containing the indicated lipid compositions [described in (C)] and used for spectral analysis. The extent of interaction is reported as the fractional increase in emission intensity at the λmax (F/F0). (E) Tim50IMS-bilayer interaction with increasing TOCL concentration. NBD-Tim50IMS(S208C) was incubated with liposomes containing up to 40 mol % TOCL, and the fractional increase in emission intensity was measured.
To further address whether Tim50IMS might undergo conformational changes upon binding to CL-containing bilayers, we performed accessibility measurements of the single native cysteine at position 268 by labeling with the thiol-reactive reagent TMM(PEG)12 (Fig. 7B). In the presence of increasing concentrations of liposomes composed of POPC only (Fig. 7B, lanes 1 to 5), the accessibility of Cys268 remained moderate, with a consistent labeling efficiency of approximately 30%. However, in the presence of increasing concentrations of vesicles with 20 mol % TOCL (Fig. 7B, lanes 6 to 10), the degree of labeling increased significantly, suggesting that a structural alteration may have increased the aqueous exposure of Cys268 to the labeling reagent. Together, the proteolysis and thiol labeling results point to the possibility that Tim50IMS may undergo a significant conformational change upon binding to CL-containing bilayers.
We next addressed the possibility that a part of Tim50IMS may become exposed to a low dielectric environment upon binding to CL-containing bilayers. Given the strong contribution of the protruding β-hairpin to the bilayer binding energy revealed by our CG MD simulations (Fig. 5), we reasoned that this structural element may insert into the membrane in a CL-dependent manner. We therefore generated Tim50IMSS208C, a construct with a single cysteine at position 208 near the tip of the β-hairpin (Fig. 5E), and labeled this site with NBD to generate NBD-Tim50IMSS208C as a fluorescent reporter of the microenvironment of this region (Fig. 7C). As expected, in the absence of liposomes, the spectrum of the β-hairpin probe revealed a low emission yield and red-shifted λmax (centered at 542 nm), characteristic of NBD exposed to an aqueous environment (Fig. 7C, black trace). The presence of POPC vesicles (Fig. 7C, blue trace) caused a 7-nm blue shift in λmax and a modest (twofold) increase in fluorescence yield. By contrast, the presence of vesicles with 20 mol % TOCL (Fig. 7C, red trace) caused an 18-nm blue shift in λmax and a much larger (5.7-fold) increase in the fluorescence yield. Therefore, these results show that the propensity of the Tim50IMS β-hairpin to become exposed to a more hydrophobic environment in the presence of lipid bilayers is strongly potentiated by the presence of CL. In addition, we used this assay to address the physicochemical properties of CL that might promote Tim50IMS interaction with bilayers. The 2′-deoxycardiolipin (dCL) variant, which lacks the secondary hydroxyl of the central glycerol (the only hydrogen bond donor of the CL headgroup), consistently promoted the shift of the β-hairpin to a slightly more nonpolar environment than CL (Fig. 7C, green trace). More significantly, the monolysocardiolipin (MLCL) variant, which lacks one acyl chain, promoted this β-hairpin nonpolar exposure to a much lower extent than intact TOCL (Fig. 7C, yellow trace).
To evaluate the changes in the exposure of the Tim50IMS β-hairpin caused by these different CL variants with increasing amounts of liposomes, we titrated liposomes of different lipid composition in the presence of a constant concentration of NBD-Tim50IMSS208C and quantified the probe environment change as the fractional increase in NBD emission (Fig. 7D). Increasing amounts of liposomes composed solely of POPC caused a slight increase in fluorescence intensity that did not saturate up to 1.5 mg lipid/ml. However, vesicles with 20 mol % TOCL or dCL both showed robust increases in NBD emission that began to saturate at higher lipid concentrations, suggesting the near-complete binding of Tim50IMS to the bilayers. In contrast, vesicles containing 20 mol % MLCL showed a much weaker shift to a nonpolar environment over the same concentration range. Together, these results demonstrate that CL strongly promotes the exposure of the Tim50IMS β-hairpin to a nonpolar environment and that the lyso form of CL, which becomes abundant with defects in the tafazzin (62), promotes this interaction much more weakly. Finally, to directly evaluate the CL dependence of the Tim50IMS β-hairpin exposure to a hydrophobic environment, we measured the fractional increase of NBD-Tim50IMSS208C in the presence of liposomes at a single lipid concentration but with increasing molar amounts of TOCL (Fig. 7E). These results reveal a sigmoidal binding curve, indicating a threshold CL concentration of about 10 mol % required to promote Tim50IMS interaction, and a cooperative effect of CL on binding.
To determine the binding parameters of the Tim50IMS membrane interaction, we fit our cosedimentation (Fig. 6A) and fluorescence (Fig. 7, D and E) data to binding isotherms (fig. S9). To specifically quantify the interaction of Tim50IMS with CL, we analyzed binding efficiency as a function of the molar concentration of CL accessible on the outer leaflet. Binding curves involving increases in the total lipid (liposome) concentration without any change in the surface density of CL (fig. S9, A and B) resulted in a hyperbolic saturation curve, whose fit to a Langmuir isotherm yielded TOCL dissociation constants (Kd) of 10.5 μM (from cosedimentation data; fig. S9A) and 15.1 μM (fluorescence data; fig. S9B). As expected, the affinity for dCL was similar (Kd = 9.7 μM), whereas the affinity for MLCL was significantly diminished (Kd = 33.5 μM). Binding curves involving increases in the surface density of TOCL (fig. S9C) showed a sigmoidal curve whose fit using the Hill equation revealed a Hill coefficient of n ≈ 5. This positive cooperativity is consistent with the interaction of Tim50IMS with multiple CL molecules (Fig. 5D). This value, which should be considered an apparent cooperativity, represents a minimal stoichiometry that can be subject to nonallosteric phenomena (for example, dimensionality effects), as described in Discussion.
CL affects Tim23-Tim50IMS interactions during substrate transport
Our results show that CL promotes the strong interaction of the Tim50 receptor with the lipid bilayer, which is critical for its interaction with the membrane domain of the Tim23 channel. What implications does this have for protein transport by the TIM23 complex? To address this question, we performed our cross-linking–based analysis in the presence of translocation intermediates. To this end, mitochondria isolated from WT or Δcrd1 yeast strains were incubated with the TIM23 substrate pSu9-DHFR, a recombinant protein that contains an N-terminal presequence fused to dihydrofolate reductase (DHFR). In the presence of methotrexate, DHFR remains tightly folded and cannot traverse the TOM complex, thereby forming a translocation intermediate engaged with the TIM23 complex (80, 81). Site-directed cross-linking carried out with increasing concentrations of TIM23 translocation intermediate allowed us to address how the presence of CL (WT mitochondria) versus the absence of CL (Δcrd1 mitochondria) affected interactions between Tim50 and different sites of Tim23 during active import (Fig. 8).
Fig. 8. Substrate-dependent changes in the Tim50-Tim23 interaction with and without CL.
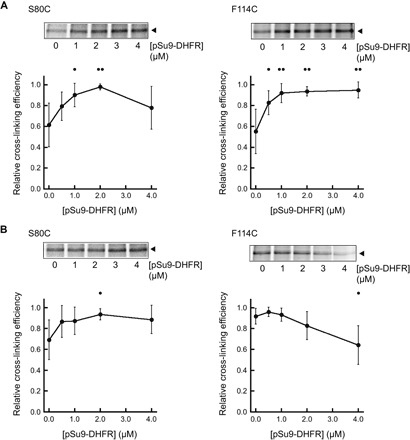
Analysis of in organello cross-linking between Tim50 and [35S]Tim23(S80C) (left) or between Tim50 and [35S]Tim23(F114C) (right) was conducted as above (Fig. 2), except in the presence of increasing pSu9-DHFR concentration. Panels above each graph show a representative cross-linked adduct between Tim50 and the relevant [35S]Tim23 construct (closed arrowhead). Relative cross-linking efficiency means represent relative efficiencies for each substrate titration type (relative to the highest efficiency), taken from a minimum of four independent experiments, and error bars represent SDs. Dots indicate significant differences in comparison to results from the respective no-substrate control by Student’s t test (•P < 0.05; ••P < 0.01). (A) Cross-linking in the presence of CL. Experiments were conducted using mitochondria isolated from WT yeast strains. (B) Cross-linking in the absence of CL. Experiments were conducted using mitochondria isolated from the Δcrd1 yeast strain.
When we analyzed the interaction between Tim50 and the Tim23 IMS region (using the Tim23 S80C variant), we found that increasing pSu9-DHFR concentration tended to increase cross-linking efficiency in both WT mitochondria (Fig. 8A, left) and mitochondria lacking CL (Fig. 8B, left) within each given titration experiment. A general trend toward enhancing Tim23-Tim50 interaction with increasing substrate may be expected, given that translocation promotes active remodeling of the TIM23 complex (82) and/or leads to increases in the Tim50-Tim23 association (18, 20). However, when we analyzed the substrate-dependent interaction between Tim50 and the channel region of Tim23 (using the Tim23 F114C variant), we observed a marked effect of CL. In WT mitochondria, increasing substrate significantly promoted the cross-linking–detected interaction between Tim50 and the Tim23 channel region (Fig. 8A, right). However, in the absence of CL, increasing substrate had the opposite effect of further decreasing the interaction of Tim50 with the Tim23 channel (Fig. 8B, right). These results suggest that the translocation-active form of the TIM23 complex promotes the interaction of the Tim50 receptor with the Tim23 channel region in a manner that is facilitated by CL.
DISCUSSION
The key outcome of this study is the discovery that the soluble receptor domain of Tim50 interacts with membranes in a CL-dependent manner and that this interaction promotes the association between the Tim50 receptor and the Tim23 channel. It has been previously shown that Tim50IMS makes several points of contact with Tim23, both (i) at the Tim23 IMS domain, likely via a coiled-coil interaction (15–17, 23, 26–28), and (ii) with Tim23 TMS1 (26). It has also been shown that CL is important for the functional integrity of the TIM23 complex (53–59). Our results here provide a conceptual framework to bridge these two phenomena.
In the first phase of this work, both biochemical and in vivo genetic evidence indicated that membrane anchorage plays a role in stabilizing the interaction of Tim50 with other TIM23 complex subunits and that the presence of CL is a requirement for this anchorage (Fig. 1). These analyses were predicated on the previous observation that, in yeast, the soluble Tim50 receptor domain can substitute for full-length Tim50 (27). We found that mutations in the Tim23 IMS domain that destabilize the Tim50-Tim23 interaction (tim23yl) not only were synthetically lethal with a version of Tim50 lacking the transmembrane domain (Tim50IMS) but also had a genetic interaction with the CL synthase gene (Δcrd). From these results, we concluded that, in addition to the established Tim23-Tim50 interaction through their respective IMS domains, there are additional stabilizing interactions between Tim50IMS and a membrane-bound component of the import machinery. This conclusion is consistent with a previous cross-linking–based analysis, which revealed an interaction between Tim50IMS and a specific helical face of Tim23 TMS1 (26). Here, site-directed cross-linking performed in organello (Fig. 2) and in model membranes (Fig. 3) confirmed that this interaction occurs in a CL-dependent manner.
The next phase of this work focused on the structural analysis of Tim50IMS. To date, high-resolution structural information on S. cerevisiae Tim50 has been limited to the crystal structure of a region of Tim50CORE (residues 176 to 361) (24, 25). However, the structure of Tim50IMS in its entirety has remained elusive. Understanding the structure of the C-terminal PBD of yeast Tim50 is particularly important because its deletion blocks TIM23-mediated protein import, resulting in a lethal phenotype (20). We used SAXS to generate an ab initio envelope of Tim50IMS, which was then used to produce a homology model of the entire receptor domain (Fig. 4). In this model, Tim50IMS forms an elongated structure with Tim50CORE at the center of the N-terminal and C-terminal domains. The C terminus forms a protruding three-helix bundle, consistent with the high α-helical content detected for the sPBD region (residues 400 to 450) (19). Informed by structural characterization, the current model for substrate recognition by yeast Tim50 includes both Tim50CORE and sPBD domains. Specifically, incoming presequences bind to the N-terminal half of sPBD, and then, the affinity of the sPBD for the core region allows the transfer of substrate to Tim50CORE, followed by the transfer to the Tim23 channel along a substrate binding affinity gradient (18–20). The putative acidic binding pocket at the base of the β-hairpin in Tim50CORE appears to be structurally malleable (24), enabling interaction with both presequences and other sites on Tim50. Consistent with this model of substrate interaction, NMR-detected sites in sPBD that interact with presequences and Tim50CORE all reside on the second α helix of our homology model, proximal to the core region (closest distances ranging from ca. 9 to 17 Å); therefore, a ternary complex of Tim50CORE, sPBD, and the presequence could be formed with a relatively minor structural adjustment.
The third phase of the study was aimed at investigating the nature of the CL-dependent membrane interactions of Tim50IMS. MD simulations based on our homology model indicated that Tim50IMS interaction with lipid bilayers requires CL and that the most stabilized mode of interaction occurs via the face of Tim50IMS that contains the protruding β-hairpin (Fig. 5). Biochemical and fluorescence-based experiments then corroborated the results of the computational simulations, showing that (i) Tim50IMS binds to model membranes in a CL-dependent manner, compared with PG- and PE-containing bilayers; (ii) the β-hairpin becomes exposed to the nonpolar bilayer core in a CL-dependent manner, with MLCL supporting much less interaction and dCL supporting slightly greater interaction; and (iii) the membrane binding surface of Tim50IMS is composed of multiple sites, including the β-hairpin of Tim50CORE and the C-terminal domain (Figs. 6 and 7).
Our analyses of the interaction between Tim50IMS and membranes of many different lipid compositions have provided insights into the molecular basis of this protein-bilayer association. Our results indicate that like other membrane-interactive amphitropic proteins, the binding of Tim50IMS entails both an electrostatic and a hydrophobic component (Fig. 9A) (83). Although yeast Tim50IMS has a negative net charge of −7e (pI = 5.76), we propose that the electrostatic docking occurs at an interface that is predominantly positively charged, including the β-hairpin and other basic surfaces (fig. S10A). The ability of overall acidic peripheral proteins to bind negatively charged bilayer surfaces via basic patches is not uncommon, as illustrated by the transducin Gβγ subunit, which also has a highly polarized electric surface potential and binds acidic bilayers (84). The role of an electrostatic interaction between Tim50IMS and the membrane was revealed by the fact that all anionic lipids resulted in some degree of Tim50IMS-bilayer interaction: PG and MLCL effected relatively weak attraction, whereas CL and dCL attracted the protein strongly. The weaker attraction of PG can be rationalized on the basis of the fact that it is a monoanionic lipid in comparison to CL, which we (85) and others (86, 87) have demonstrated to be dianionic at physiological pH. Hence, at a given molar concentration, PG establishes a lower surface charge density and, therefore, a weaker attractive surface potential and electric field than does CL. Moreover, the observation that CL induces a several-fold increase in Tim50IMS interaction over PG can be explained on theoretical grounds that with increasing negative surface charge density, there is a linear increase in binding energy and an exponential increase in polypeptide partition coefficients (88, 89). Upon electrostatic docking, we propose that there is an additional hydrophobic mode of interaction that is related to the propensity of CL to assume an HII structure and effect negative curvature (36). The binding of a polybasic surface of the protein could reduce interfacial electrostatic repulsion among CL molecules, which could promote local nonlamellar structures and lead to lipid packing alterations that expose acyl chains to bound Tim50IMS nonpolar residues (Fig. 9A). The evidence of a requirement of HII propensity comes from our experiments with MLCL, which does not form nonlamellar structures (90) and is shown here to bind Tim50IMS much more weakly than TOCL, although the two lipids have identical interfacial anionic charge (85). We also note that the enhanced binding of Tim50IMS to bilayers containing dCL may be related to a greater propensity of this lipid to support HII structures relative to CL. Finally, the lipid-dependent cross-linking profiles of our in organello experiments are notable (Fig. 2). Whereas the cross-linking–detected interaction between the Tim50 receptor and the Tim23 channel domain was more severely altered by the absence of CL (Δcrd1) than by defects in CL remodeling (Δtaz1 and Δcld1) (Fig. 2, E and F), the remodeling mutants resulted in a stronger deviation from WT for the interaction between Tim50 and the Tim23 IMS domain (Fig. 2, B and C). These disparate responses illustrate the complex nature of the relationship between the acyl chain composition of CL and protein interactions in the mitochondrial membrane. For example, the accumulation of unremodeled CL or MLCL resulting from the Δcld1 and Δtaz1 yeast mutants, respectively (62), may produce a stronger effect on the particular Tim50IMS–Tim23 TMS1 interaction than does the accumulation of PG resulting from the Δcrd1 mutant.
Fig. 9. Working models for Tim50IMS-bilayer and Tim50-Tim23 interactions.
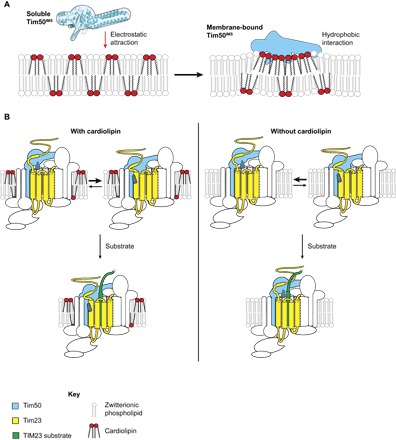
(A) Interaction between Tim50IMS and CL-containing bilayers. In solution, Tim50IMS is initially attracted to the negatively charged bilayer by long-range Coulombic forces. The electric field orients a basic face of the protein, including the β-hairpin, toward the bilayer. Upon electrostatic docking, hydrophobic interactions between the nonpolar residues at the binding interface and the nonpolar bilayer core stabilize the interaction in a manner stimulated by the HII propensity of CL. Coupled to membrane association are changes in Tim50IMS conformation relative to its water-solvated state. Membrane association of Tim50IMS will be promoted if these Coulombic and hydrophobic interactions are favorable enough to offset the energetic penalty of desolvation. Note that the left panel displays the Tim50IMS homology model of this study; subsequent panels show hypothetical outlines of the Tim50IMS structure in cyan, either in the membrane bound state or in the presence of the TIM23 complex. (B) CL- and substrate-dependent Tim50 dynamics in the context of the TIM23 complex. The presence of CL (left) favors the membrane-associated state of Tim50IMS, which facilitates interaction with the channel domain of Tim23. In the presence of substrate, Tim50IMS retains its interaction with both the Tim23IMS and Tim23 channel regions. The absence of CL (right) favors the membrane-dissociated state of Tim50IMS, which blocks its interaction with the Tim23 channel region. The presence of substrate further promotes the membrane-dissociated state of Tim50IMS. Note that the stippled region of Tim50 depicts the β-hairpin.
Our results indicate important roles of the conserved β-hairpin and N-terminal region of the Tim50 receptor in promoting membrane association. Specifically, β-hairpin structures are known to facilitate bilayer penetration. Highly basic antimicrobial peptides are known to assume β-hairpin conformations and penetrate bilayers in a manner dependent on anionic lipids (91), with some also containing conserved Trp residues for cation-π interactions, which promote membrane interaction (92), consistent with the conserved sequence of the Tim50IMS β-hairpin. Our data show that the β-hairpin of Tim50 is exposed to a nonpolar milieu upon binding to CL-containing membranes; knowing exactly how deeply it may penetrate the nonpolar core awaits future experimentation. Moreover, the C-terminal region of our homology model contains multiple segments predicted to be amphipathic, burying hydrophobic faces in the protein interior (fig. S10B). Such a helix bundle fold is a common motif for lipid-binding proteins to sequester nonpolar residues while in solution and to undergo dynamic conformational changes that expose these residues upon encountering a lipid surface to promote nonpolar interactions (93). It is possible that the Tim50IMS region undergoes a similar structural rearrangement upon membrane binding that orients these nonpolar faces toward the bilayer. Our limited proteolysis and site-directed labeling analyses (Fig. 7) strongly suggest that Tim50IMS undergoes a structural change upon binding to CL-containing bilayers. Exploring which specific regions undergo these membrane-coupled dynamics is the subject of an ongoing investigation.
Having established the role of CL in promoting the interaction of the Tim50 receptor with the bilayer and also with the channel region of Tim23, we propose a working model for CL-dependent receptor-channel dynamics (Fig. 9B). In a translocon not actively transporting a substrate, the presence of CL favors the association of the Tim50 receptor domain with the membrane-bound region of Tim23. In the absence of CL, the association between the Tim50 receptor and the IMS region of Tim23 is favored (Fig. 9B, top). Although CL plays a critical role in mitochondrial energetics (38), the CL-modulated interaction between Tim50 and Tim23 does not appear to be directly dependent on the Δψm because it is also observed in model membranes lacking a trans-bilayer electric field (Fig. 3). Further, although the CL-dependent membrane association of Tim50IMS seems to be a major determinant in directing its interaction with Tim23, we cannot currently discount the likely possibility that CL also modulates the conformation of the Tim23 channel region (not shown in the model), which may contribute to our observations. On the basis of our in organello experiments with the TIM23 substrate (Fig. 8), in CL-containing membranes, the presence of a substrate further stabilizes the interaction between the Tim50 receptor and the Tim23 channel region, whereas in membranes devoid of CL, the presence of a substrate further suppresses this interaction (Fig. 9B, bottom). One of the key functions of CL is to maintain the functional associations of membrane proteins. Our model suggests a mechanism by which CL could promote TIM23-mediated import: by stabilizing the receptor-channel interactions that would otherwise be destabilized by a substrate in the absence of CL.
Some central features of this model warrant further consideration. First, why would a receptor domain with inherent membrane affinity also be anchored to the bilayer by a transmembrane segment? One possibility is that this tethering by a transmembrane element could maintain the proximity of the receptor region to the bilayer, effectively serving as a mechanism for dimensionality reduction in its diffusion space to enhance the kinetics of membrane association (94). Another possibility is that the transmembrane segment is required to impart specificity for assembly with other subunits of the TIM23 complex. Finally, as a structural link between Tim50IMS and the as-yet poorly characterized matrix-facing domain of Tim50, the transmembrane segment could transmit signals across the topologically distinct domains of this receptor during different stages of the import cycle. By comparison, we note that Fis1 (mitochondrial fission 1 protein) of the mitochondrial OM is a tail-anchored protein with a soluble cytosolic domain that also interacts with bilayers and undergoes conformational changes in a lipid-specific manner (95). Second, what could be the physiological relevance for the receptor domain to interact with the membrane in a manner promoted by a specific lipid, in this case, CL? One possibility is that spatiotemporal variation of CL concentration in the IM could modulate the activity of the receptor. If the distribution of CL across the IM were homogeneous, then according to this model, Tim50IMS would assume a constant membrane-interactive pose regardless of its location. However, given that CL appears to be heterogeneously distributed, for example, more enriched at membrane contact sites (96), these regions of high local [CL] could promote Tim50IMS-bilayer association and the resulting conformational alterations at regions near active translocons. By contrast, when the local concentration of free CL is lower, this could promote a more membrane-dissociated state of Tim50IMS, perhaps down-regulating complex activity. We note in this respect that the observed role of Tim50 in regulating the gating of the Tim23 channel (97) or in recruiting Pam17 to transport Δψ-hypersensitive substrates (98) could also be modulated by CL.
The implications that this model has for mitochondrial protein transport could come on several fronts that could be verified experimentally. First, structural studies investigating CL-dependent conformational dynamics will elucidate exactly which regions of the Tim50 receptor are membrane-interactive and which domains may reveal altered substrate or subunit binding sites, work that could be carried out in parallel with atomistic MD simulations. Second, binding studies will reveal how the affinity of Tim50IMS for substrates may be altered in the presence of CL-containing bilayers and how CL may alter the affinity of Tim50IMS for subunits other than the Tim23 channel. Finally, comprehensive analysis of substrate import conducted in organello or in reconstituted systems could test how disruptions in CL metabolism could affect the transmembrane transport versus the lateral integration functions of the TIM23 complex.
MATERIALS AND METHODS
Yeast strains and growth conditions and isolation of mitochondria
The yeast strain containing the Tim50IMS truncation mutant was generated by transforming a haploid YPH499-based TIM50 deletion strain containing a WT copy of Tim50 on a URA plasmid with the centromeric yeast plasmid pRS315 encoding TIM50IMS and subsequently selecting for the cells lacking the URA plasmid with 5-FOA, as previously described (27). To generate yeast strains containing WT Tim23 or Tim23 Y70A,L71A in a Tim50 or Tim50IMS background, cells were transformed with pVT-U plasmids containing TIM23, and the chromosomal copy of TIM23 was deleted by homologous recombination, as previously described (23). Cells were subsequently transformed with empty pRS314 or pRS314 encoding Tim23 or Tim23 Y70,L71 under the control of the endogenous promoter and 3′ untranslated region and selected for loss of URA-containing plasmids with 5-FOA, as described above. Yeast strains containing genetic knockouts Δcrd1, Δcld1, and Δtaz1 were generated by replacing the open reading frame of the gene using polymerase chain reaction (PCR)–based gene replacement. Yeast cells were grown at 24°C in lactate medium containing 0.1% glucose, unless indicated otherwise. For the preparation of active mitochondria, yeast cells were cultivated to mid-log phase, and the mitochondria were isolated by lysis of spheroplasts and differential centrifugation, as previously described (26).
Coimmunoprecipitation
Coimmunoprecipitation experiments using detergent-solubilized mitochondria were performed as previously described (27). Briefly, mitochondria isolated from yeast strains containing full-length Tim50 or Tim50IMS were incubated at 2 mg/ml in solubilization buffer [20 mM tris-HCl (pH 8.0), 80 mM KCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1% (w/v) digitonin] at 4°C. Following a clarifying spin, solubilized material was added to protein A–Sepharose beads precoupled with antibodies (αTim23 or αTim50) and incubated under agitation for an additional 45 min. Following centrifugation, supernatants were collected, beads were washed three times, and bound proteins were eluted with nonreducing Laemmli buffer for 3 min at 95°C. Both eluted proteins and supernatants were resolved by SDS-PAGE and analyzed by immunoblotting with antibodies against Tim50, Tim23, and Tim17.
Cell-free transcription and protein synthesis
The SP6-based pSP65 plasmid library of Tim23 monocysteine variants has been described previously (11, 26). mRNA was transcribed from PCR-generated fragments using 5′ and 3′ oligonucleotides complementary to the plasmid SP6 promoter and Tim23 sequence downstream of the stop codon, respectively. Purified PCR products were transcribed in vitro with SP6 polymerase at 37°C for 1.5 hours in reactions containing 100 mM Hepes-KOH (pH 7.5), 20 mM MgCl2, 2.5 mM spermidine, 12 mM dithiothreitol (DTT), 4 mM ATP, 4 mM CTP (cytidine 5′-triphosphate), 4 mM UTP (uridine 5′-triphosphate), 0.4 mM GTP (guanosine 5′-triphosphate) G(5′)ppp(5′)G RNA cap analog (0.013 U/μl; New England Biolabs), RNasin (0.5 U/μl), and pyrophosphatase (6 U/nl); supplemented with 4 mM GTP; and allowed to incubate for an additional 30 min. mRNA was precipitated overnight at −20°C in ethanol and 90 mM sodium acetate (pH 5.2), washed in 70% (v/v) ethanol, and reconstituted in TE buffer [10 mM tris-HCl and 1 mM EDTA (pH 7.5)].
In vitro translation reactions were programmed with purified mRNA transcripts encoding Tim23 variants. Translation reactions {20 mM Hepes-KOH (pH 7.5), 100 mM potassium acetate (pH 7.5), 1 mM DTT, 200 μM spermidine, 8 μM S-adenosylmethionine, protease inhibitors [0.25 μg/ml each of leupeptin, chymostatin, antipain, and peptatin A and 0.025% (v/v) aprotinin], RNasin (0.2 U/μl), 1.2 mM ATP, 1.2 mM GTP, 64 mM creatine phosphate, creatine phosphokinase (9.6 U/nl), 30 μM amino acids (but lacking methionine for incorporation of [35S]methionine and lacking cysteine for incorporation of NBD-[14C]Cys), and 20% (v/v) wheat germ extract} were incubated at 26°C for 5 min before the addition of mRNA and either [35S]methionine (0.25 μCi/μl; for synthesis of [35S]Tim23), 0.6 μM [14C]Cys-tRNACys (for synthesis of blank samples in fluorescence experiments), or 0.6 μM NBD-[14C]Cys-tRNACys (for synthesis of NBD-labeled samples in fluorescence experiments) and then continued for 40 min at 26°C. The synthesis of the aminoacyl transfer RNA (tRNA) analog NBD-[14C]Cys-tRNACys, designed to recognize single in-frame 5′-UGC-3′ codons of Tim23 monocysteine variants for site-specific NBD probe incorporation, has been previously described (11).
Heterologously expressed proteins: Expression, purification, and labeling
For the expression of soluble domains of Tim50, sequences encoding Tim50IMS (residues 133 to 476), Tim50CORE (residues 165 to 361), Tim50IMSΔN (residues 165 to 476), and Tim50IMSΔC (residues 133 to 361) were PCR-amplified using either S. cerevisiae genomic DNA or existing plasmids as a template and subcloned into pET28a vectors, introducing a tobacco etch virus (TEV) protease cleavage site between an N-terminal 6×His tag and the coding region. Site-directed mutagenesis was performed by PCR using primers designed with mutations. The plasmid for the expression of pSu9-DHFR [the presequence of F0-ATPase subunit 9 fused with DHFR with a 6×His tag in pET30a(+)] has been previously described (26). The plasmid for the expression of membrane scaffolding protein (MSP1E3D1 in pET28a vector with a TEV-cleavable N-terminal 7×His tag) was obtained from Addgene. All plasmid sequences were verified by sequencing.
For expression of Tim50 and pSu9-DHFR constructs, competent BL21(DE3) cells (Invitrogen) were transformed with the relevant plasmid by heat shock, and cells were grown in 2 liters of YT medium supplemented with kanamycin (50 μg/ml) at 37°C to an A600 (absorbance at 600 nm) of ~0.8. Expression was induced by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside and incubation at 26°C for 2.5 hours. For purification, cells were collected by centrifugation (4000g for 10 min at 4°C); resuspended in resuspension buffer [50 mM monobasic sodium phosphate (pH 7.5), 300 mM NaCl, and 5 mM imidazole] supplemented with lysozyme (1.25 mg/ml), deoxyribonuclease (0.025 mg/ml), and 0.1 mM PMSF; incubated on ice for 30 min; and disrupted by probe sonication. After a clarifying spin (30,000g for 30 min at 4°C) to remove insoluble material, the soluble fraction was batch-incubated with Ni2+-NTA agarose (Qiagen) pre-equilibrated with resuspension buffer and loaded onto a column, followed by addition of column buffer [50 mM monobasic sodium phosphate (pH 7.5) and 300 mM NaCl] with step gradients of imidazole (up to 400 mM). Fractions enriched in protein were pooled and dialyzed against dialysis buffer [20 mM tris-HCl (pH 7.4), 100 mM NaCl, and 0.5 mM EDTA], followed by treatment with His-tagged TEV protease (0.07 U/μl) overnight at 16°C. The cleaved protein was passed over a second Ni2+-NTA column to remove the TEV protease and uncleaved protein. Protein-enriched fractions were then subjected to size exclusion chromatography on a Superdex 200 10/300 column (Amersham Biosciences) equilibrated with protein buffer [10 mM Hepes-KOH (pH 7.4), 100 mM NaCl, and 100 mM KCl] and collected at a flow rate of 0.5 ml/min. The expression and purification of MSP1E3D1 were similar, except for the fact that cells were incubated with 1% (v/v) Triton X-100 before sonication; column buffers contained 40 mM tris-HCl (pH 8.0) with 1% (v/v) Triton X-100 and 50 mM Na-cholate in the column wash steps; and following affinity purification, protein was dialyzed directly into MSP buffer [20 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 0.5 mM EDTA], based on described protocols (99, 100), and the 7×His tag was not cleaved to allow for nanodisc purification.
For the site-specific labeling of Tim50IMS with NBD, a 500-μl reaction with purified Tim50IMS (S208C) in protein buffer with a 100× molar excess of IANBD (Invitrogen) was incubated for 2 hours at room temperature (RT) with continuous rotation, and labeling reaction was quenched by the addition of 20 mM DTT. Labeled protein was separated from free dye by gel filtration with Sephadex G25 resin, and labeling efficiency was quantified as
where 23,700 M−1 cm−1 and 52,830 M−1 cm−1. For all preparations, IANBD labeling efficiency was ~60%.
Assembly of liposomes and nanodiscs
Synthetic phospholipids were purchased as chloroform stocks from Avanti Polar Lipids, including POPC, POPE, POPG, and variants of CL with 18:1 acyl chains, including TOCL, dCL, and MLCL. All stocks were stored at −20°C until use.
Unilamellar liposomes of specific lipid composition and loaded with sucrose were prepared as previously described (77). Chloroform stocks of lipids blended at the desired molar ratios were dried under a nitrogen stream in a Pyrex tube, followed by overnight evaporation in a vacuum desiccator to remove all traces of organic solvent. Dried lipid films were hydrated in buffer A [10 mM Na-Hepes (pH 7.4) and 500 mM sucrose] to a concentration of 6 mg lipids/ml, and the resulting multilamellar liposomes were passed 17 times through a Mini-Extruder (Avanti) with 0.1-μm polycarbonate membranes to produce LUVs. The liposome stock was diluted to 4 ml in buffer B [10 mM Na-Hepes (pH 7.4), 100 mM NaCl, and 100 mM KCl], aliquoted proportionally into four 1.5-ml polycarbonate centrifuge tubes, and centrifuged (137,000g for 1 hour at 22°C) in a S120-AT2 rotor (Beckman). LUV pellets were each reconstituted in 100 μl of buffer B, pooled, and quantified for lipid concentration using the ammonium ferrothiocyanate colorimetric analysis (101).
To prepare nanodiscs containing [35S]Tim23, chloroform stocks of lipid blends at the desired molar ratios were dried under nitrogen and vacuum-desiccated as above. The lipid film was solubilized in MSP buffer [20 mM tris-HCl (pH 7.4), 100 mM NaCl, and 0.5 mM EDTA] containing 59 mM Na-cholate at a molar lipid concentration that varied depending on the lipid blend used. The 2-ml assembly mixture that included MSP buffer with solubilized lipids, 50 μM purified MSP1E3D1 (at a molar ratio of 120:1), [35S]Tim23 at a final concentration of 12.5% (v/v), and protease inhibitor cocktail (Sigma-Aldrich) was then prepared. Following a 30-min incubation at RT, prehydrated hydrophobic adsorbent Bio-Beads SM (Bio-Rad) were added to remove detergent and allow nanodisc assembly to proceed for an additional 2 hours. Assembled nanodiscs were then purified by Ni2+-NTA chromatography on a column equilibrated with MSP buffer with step gradients of imidazole (up to 400 mM). The first two elution fraction columns were combined and dialyzed into ND buffer [20 mM Na-Hepes (pH 7.5), 200 mM NaCl, 50 mM KOAc, 5 mM Mg(OAc)2, and 5% (v/v) glycerol] and concentrated in Amicon Ultra centrifugal filter units (3 ml; molecular weight cutoff, 3000).
Chemical cross-linking, thiol modification, and immunoprecipitation
Cross-linking reactions with isolated mitochondria containing [35S]Tim23 were conducted exactly as previously described (26). Briefly, in vitro–synthesized [35S]Tim23 was imported into mitochondria by performing reactions (26°C for 20 min) in import buffer [20 mM Hepes-KOH (pH 7.5), 250 mM sucrose, 80 mM KCl, 5 mM MgCl2, 2 mM potassium phosphate (pH 7.5), fatty acid–free bovine serum albumin (BSA; 3 mg/ml), 3 mM NADH, and 1.5 mM ATP] containing isolated mitochondria (0.25 mg/ml) and 8% (v/v) translation reaction. Mitochondria were isolated by sedimentation (12,500g for 5 min) and resuspended in cross-linking buffer (CB; import buffer lacking BSA). Where indicated, pSu9-DHFR was added to a final concentration of up to 4 μM in the presence of methotrexate. Chemical cross-linking reactions contained mitochondria (final concentration, 0.13 mg/ml) incubated in CB with the homobifunctional thiol-reactive reagent BMOE from dimethyl sulfoxide (DMSO) stocks to a final concentration of 50 μM {final [DMSO] = 2.5% (v/v)}. Following incubation at 26°C for 20 min, the reaction was quenched by the addition of 40 mM DTT. For immunoprecipitation, mitochondria from cross-linking reactions (25 μg) were sedimented; solubilized in 50 mM tris-HCl (pH 7.5), 1 mM EDTA, and 1% (v/v) SDS; and then diluted 10-fold in lysis buffer [10 mM tris-HCl (pH 7.5), 15 mM NaCl, 5 mM EDTA, 1 mM PMSF, and 1% (v/v) Triton X-100], followed by a clarifying spin (18,000g for 10 min). Supernatants were incubated with PAS beads precoupled to αTim50, rocked for 2 hours at RT, washed three times in 1 ml of lysis buffer and once in 1 ml of 10 mM tris-HCl (pH 7.5), and eluted with 30 μl of 2× SDS sample buffer before SDS-PAGE.
Cross-linking reactions with nanodiscs containing [35S]Tim23 ([35S]Tim23-ND) were conducted in ND buffer containing 46% (v/v) [35S]Tim23-ND and 50 μM BMOE. Following incubation at 26°C for 60 min, the reaction was quenched as described above. For immunoprecipitation, reactions were transferred to a new tube and PAS beads precoupled to αTim50 were added and incubated at RT for 2 hours. Beads were then washed three times with 1 ml of radioimmunoprecipitation assay buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% (v/v) NP-40, and 0.25 (v/v) Na-deoxycholate] and once in 1 ml of 10 mM tris-HCl (pH 7.5) and eluted with 30 μl of 2× SDS sample buffer before SDS-PAGE.
For site-specific sulfhydryl modification of Tim50IMS, reactions with 1 μM Tim50IMS, 5 mM TMM(PEG)12 (Thermo Fisher Scientific), and variable concentrations of liposomes in CB were incubated at 26°C for 5 min before quenching with 200 mM DTT. Control (prequench) samples were incubated with 200 mM DTT before the addition of TMM(PEG)12. An equal volume of 2× SDS sample buffer was added before SDS-PAGE.
Cross-linking reactions (“Total” and “Immunoprecipitation”) and thiol modification reactions were resolved on 12% SDS-PAGE gels, which were subsequently fixed, dried, and placed on phosphor imaging K-screens (Kodak). Samples were visualized by radioisotope scan on a Pharos FX Plus Molecular Imager (Bio-Rad), and bands were quantified using Image Lab v3.0 software (Bio-Rad). Cross-linking efficiency was calculated as the ratio of the intensities of the cross-linked [35S]Tim23 to the non–cross-linked [35S]Tim23 bands for each experiment. Samples from CL biosynthesis mutants were normalized relative to the bands corresponding to [35S]Tim23 in WT mitochondria.
Limited proteolysis
Tim50IMS (1 μM) was incubated in buffer B with or without LUVs (1 mg lipid/ml) with the specified lipid composition for 30 min at RT in a total volume of 180 μl. Following incubation, 25 μl of the sample was added to microfuge tubes containing proteinase K at the indicated final concentrations, samples were incubated on ice for 30 min, and proteolysis was quenched by the addition of 2.8 mM PMSF. Samples were resolved by SDS-PAGE and visualized by SYPRO Orange staining and scanning on a Pharos FX Plus Molecular Imager.
Liposome cosedimentation analysis
Reactions (150 μl) with buffer B containing purified Tim50 constructs (final concentration, 1 μM) and varying concentrations of sucrose-loaded LUVs were incubated at RT for 60 min, followed by centrifugation (137,000g at 22°C for 60 min). The supernatant was discarded, and pellets were resuspended in 40 μl of 2× SDS sample buffer. Samples were resolved on 12% SDS-PAGE gels, and Tim50 was visualized by staining with SYPRO Orange protein stain (Invitrogen). Bands were visualized on a Pharos FX Plus Molecular Imager (protein stain, SYPRO Orange setting), and bands were quantified using Image Lab v3.0 software.
Fluorescence spectroscopy
Steady-state fluorescence measurements were performed with a Fluorolog 3-22 spectrofluorometer (HORIBA Jobin-Yvon) equipped with photon-counting electronics, double-grating excitation and emission monochromators, and a 450-W xenon lamp. In all cases, fluorescent samples were measured in parallel with blank samples (lacking dye but otherwise identical to the fluorescent sample) for signal subtraction to obtain the emission originating from the probe. Emission scans (5-nm bandpass) to measure NBD fluorescence were conducted with λex = 470 nm and λem = 490 to 590 nm (2-s integration times with 1-nm steps). For all sample types, spectra were measured from a minimum of three independently prepared samples.
Spectral measurements of nanodisc samples (recorded in 4-mm × 4-mm microcells) included separate readings of NBD-Tim23(T24C)-ND and NBD-Tim23(I111C)-ND (synthesized in the presence of NBD-[14C]Cys-tRNACys), as well as the corresponding blank samples (synthesized in the presence of [14C]Cys-tRNACys). Measurements of NBD-Tim50IMS in the presence of liposomes (recorded in 1-cm quartz cuvettes with continuous stirring) contained NBD-Tim50IMS (final concentration, 0.5 μM) and the indicated concentration of LUVs of defined lipid composition. Fractional changes in maximum fluorescence intensity (F/F0) for a given concentration of lipid were calculated by averaging the fluorescence intensity (F) centered about the λmax (530 to 535 nm) relative to the intensity in the absence of LUVs (F0). All data points were fit using KaleidaGraph (Synergy Software).
Quantitative analysis of binding
On the basis of Tim50IMS-liposome binding data generated by cosedimentation analysis (Fig. 6A) and fluorescence-detected binding (Fig. 7, D and E), binding parameters were evaluated using fits to Langmuir binding isotherms or Hill expansions thereof. The generalized equation
was used to fit hyperbolic binding curves (n = 1) and sigmoidal binding curves (n > 1). Here, the variable P represents Tim50IMS, the subscript B represents bound, and the subscript T represents total; therefore, the ratio [P]B/[P]T is the fraction of Tim50IMS bound. [CLacc] is the Tim50IMS-accessible CL concentration, where it is assumed, to a first approximation, that all CL of the liposomal outer leaflet is accessible. Kd is the apparent dissociation constant with respect to CL, and n represents the Hill coefficient. To analyze cooperative binding, a linear form of this equation was used
where Θ, the fraction of bound Tim50IMS, plotted as a function of log[CLacc], yields a straight line with a slope equal to the Hill coefficient. To evaluate binding data specific for the Tim50IMS-CL interaction, the background (nonspecific) binding to liposomes containing only POPC was subtracted out from the binding data for liposomes with CL or CL variants.
SAXS data collection and analysis
SAXS scattering data were acquired at the Cornell High Energy Synchrotron source G1 station, with the beamline operating at 11.75 keV. Purified protein samples were centrifuged at 14,000 rpm for 10 min before data collection and protected from radiation damage by oscillating the x-ray beam using a computer-controlled syringe pump. The x-ray beam (wavelength, 1.055 to 1.244 Å) was collimated to 250 μm × 500 μm, and images were collected at 4°C with a Dual Pilatus 100 K-S detector with a momentum transfer interval of 0.007 Å−1 < q < 0.7 Å−1 (where the scattering vector q = 4sinθ/λ, with θ being the scattering angle and λ being the wavelength). Sample-to-detector distance was calibrated using silver behenate powder (Gem Dugout). Ten exposures of 2 s each were averaged for both protein-containing samples and background (buffer B only) samples. Buffer profiles were subtracted from the averaged protein solution profiles and reduced using BioXTAS RAW software (102). Guinier analysis was performed using PRIMUS (103, 104), and the pair-distance distribution function P(r) and values for I0, Rg, and Dmax were obtained using the program GNOM (105, 106). Rg values calculated from GNOM were consistent with those estimated from the Guinier region (table S1). The intensity of a standard (lysozyme, 4.14 mg/ml) at zero scattering angle I0 was determined by Guinier analysis and used to estimate sample molecular weight by comparison (107). The theoretical scattering data from the homology model and the crystal structure for comparison with the experimental scattering data were calculated using the CRYSOL program (76). To generate ab initio envelopes, 10 models were constructed in DAMMIF (108), which were aligned and averaged in DAMAVER (109). Final refinement was performed using DAMMIN (110).
Tim50IMS homology model generation
A model for the full-length Tim50IMS domain was constructed by combining the known coordinates of the core region residues (Tim50CC, PDB 3QLE), with homology-modeled structures of the N-terminal and C-terminal regions. Given that the conserved core structure (residues 176 to 361) was solved by x-ray crystallography (25), we chose to model the terminal regions independently, rather than generate a complete homology model for the entire Tim50IMS. The sequence of the N-terminal region (residues 133 to 195) and the C-terminal region (residues 343 to 476) was supplied to the I-TASSER web server (111) to generate models of those regions. The terminal region models contained overlapping regions with the core to facilitate structural alignments when constructing the full-length Tim50IMS model. The top-ranking models for both the N terminus and C terminus were built onto the crystal structure coordinates of Tim50CC. The model building was carried out in CHARMM (72) by performing a local alignment of residues 176 to 180 between the core and the N terminus and then copying in the coordinates of residues 132 to 175. Similarly, the C-terminal coordinates were generated by aligning residues 355 to 361 and then copying in the coordinates of residues 362 to 476 into the full model. Hydrogen atoms were constructed with the HBuild command, and steepest descent minimization was performed in the GBSW implicit solvent model (112). During the minimization, the backbone atoms of residues 132 to 360 were harmonically restrained with a force constant of 50 kcal mol−1 Å−2 for 500 steps, followed by 500 steps of unrestrained minimization.
When this initial model was compared with the Tim50IMS SAXS reconstruction, it was evident that the model (which predicted a dimeric coiled-coil structure of the C-terminal region) was too elongated, extending outside the envelope. To generate a model consistent with the experimentally derived SAXS envelope, we replaced the C-terminal region with the second highest ranked homology model from I-TASSER, which predicted a trimeric coiled-coil structure. The trimeric C-terminal model was minimized in the GBSW implicit solvent model while the backbone atoms of residues 132 to 360 were harmonically restrained. The restraint force constant was set to 200 kcal mol−1 Å−2, and 500 steepest descent steps were performed, followed by 500 steps of adopted basis Newton-Raphson (ABNR) minimization. The restraint force was lowered to 100 kcal mol−1 Å−2, and another 500 steps of ABNR were performed. This model displayed an angular architecture, which was not consistent with the linear SAXS envelope. To improve the agreement between the model and the SAXS envelope, the model was manually manipulated with the UCSF Chimera Program (113) to alter the backbone angles in residues 359 to 360 to generate a more linear conformation. Following the modifications in Chimera, the model was again minimized in CHARMM using GBSW solvent while restraining backbone atoms in residues 332 to 358 with an initial force constant of 200 kcal mol−1 Å−2 for 500 steepest descent steps, followed by 500 ABNR steps. The restraints were reduced to 100 kcal mol−1 Å−2, and 500 steps of steepest descent and ABNR were performed. Finally, all restraints were released, and 500 steps of steepest descent and ABNR minimization were performed.
CG MD simulations
CG MD simulations were used to characterize Tim50IMS-bilayer interactions and to elucidate the influence of CL on Tim50 membrane binding properties. All simulations were performed using the MARTINI CG model (114) using force field version 2.2P (115, 116) and with the polarizable water model (117). Bilayers were constructed using the insane.py python script (118) to generate a homogeneous POPC bilayer and an 80:20 POPC/CL molar ratio bilayer. The pure POPC bilayer contained 722 lipids, whereas the mixed bilayer contained 576 POPC and 144 TOCL lipids. The charge distribution and all other parameters for CL were consistent with the previous study by Dahlberg and Maliniak (119), assigning each phosphate a −0.5 charge to model proton equilibration between the phosphates. The bilayers were solvated with polarizable water CG particles, and CG Na+ and Cl− ions were added to neutralize the system and to bring the ionic concentration to 0.1 M. The bilayer systems were energy-minimized using the steepest-descents algorithm for 1500 steps. The bilayer systems were then equilibrated in the NPT ensemble for 1.6 μs.
Tim50IMS was modeled on the basis of the homology model that was generated and found to be consistent with the SAXS envelope. The martinize.py script was used to generate a CG model of Tim50. The tertiary structure of the protein was maintained through the application of an elastic network on top of the force field. In this method (ElNeDyn) (120), the distance between pairs of nonbonded backbone beads, separated by less than 0.9 nm, is harmonically restrained with a force constant of 500 kJ mol−1 nm−2. However, no elastic restraints were placed on the N-terminal region (residues 133 to 195) of Tim50, given its nature to adopt disordered structures and undergo conformational changes.
Protein-bilayer systems were constructed by taking the equilibrated bilayers and placing the protein ~10 nm above the bilayer and solvating and ionizing to neutralize and bring the ion concentration to 0.1 M. The sizes of the systems were ~115,000 CG particles for the POPC systems and ~140,000 CG particles for the POPC/CL system. The systems were energy-minimized for 1500 steepest descent steps, followed by 800-ns NPT equilibration. We performed five simulations using different orientations of Tim50IMS for both the POPC and mixed bilayer systems. The same five orientations were used for the POPC and mixed bilayer systems. Production simulations were run for 4 μs.
All simulations were performed using GROMACS v4.6 (121, 122). Electrostatic and Lennard-Jones (LJ) interactions were cut off at 1.2 nm, with electrostatics shifted from 0 to the cutoff, and LJ interactions were shifted from 0.9 nm to the cutoff. The relative dielectric was set to 2.5, as recommended when using polarizable water (117). All production runs were simulated in the NPT ensemble using the Berendsen weak coupling scheme, with the temperature maintained at 323 K and pressure kept at 1.0 bar with semi-isotropic coupling. The time constants for the pressure and temperature couplings were 3.0 and 1.0 ps, respectively, and the compressibility value was 3 × 10−5 bar−1. All simulation runs were performed using periodic boundary conditions in the x,y and z directions. The simulation time step was 0.020 ps. All reported times (with the exception of the time step) were multiplied by the standard MARTINI speed-up factor (4×) to account for the faster dynamics in the CG model. System configurations were visualized, and images were generated using VMD software (123).
To compute the potential of mean force (PMF) of Tim50IMS-bilayer binding, we performed umbrella sampling (124) simulations along the center-of-mass (com) separation coordinate between the bilayer and the protein. From the bound configurations, the protein was pulled off the bilayer during a steered MD (SMD) simulation. The SMD pull rate was 0.001 nm/ps, and the force constant was 1000 kJ mol−1 nm−2; intermediates were collected at 0.1-nm spacing intervals along the z axis, perpendicular to the bilayer plane. Umbrella sampling was performed by simulating each intermediate state for 1.2 μs with a harmonic restraint on the com separation distance; the restraining potential had a force constant of 2000 kJ mol−1 nm−2. The last 800 ns of each umbrella simulation was used to calculate the PMF using the g_wham tool available in GROMACS.
Supplementary Material
Acknowledgments
We thank S. Claypool for providing CL biosynthesis–defective yeast strains and J. Landin, P. Robisch, and Z. Stanic for expert technical assistance. Funding: This work was supported by the NIH [grants 1R01GM113092 (to N.N.A.) and K22-A1099163 (to E.R.M.)], the NSF (grant MCB-1330695 to N.N.A.), and the Deutsche Forschungsgemeinschaft (grants MO1944/1-1 and MO1944/1-2 to D.M.). Computational resources were provided on the HPC Hornet cluster located at the University of Connecticut at Storrs. Author contributions: K.M. and N.N.A. conceived the original idea of the project. K.M. and A.M. carried out cross-linking and immunoprecipitation analyses. K.M. carried out membrane interaction analyses. D.M. and U.G. generated yeast strains and carried out coimmunoprecipitation and yeast growth experiments. K.M., T.H.D., and V.L.R. carried out SAXS measurements and data interpretation. E.R.M. and S.N. carried out MD simulations and homology modeling. N.N.A. supervised the research. N.N.A., K.M., D.M., and E.R.M. wrote the manuscript. All authors contributed to the editing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1700532/DC1
fig. S1. The TIM23 complex and CL biogenesis.
fig. S2. The effect of CL synthase knockout on Tim50IMS functionality and association with the TIM23 complex.
fig. S3. Import and integration of [35S]Tim23 into isolated mitochondria.
fig. S4. Response of [35S]Tim23 cross-linking to decreased Δψm.
fig. S5. Incorporation of [35S]Tim23 into nanodiscs: control tests.
fig. S6. Tim50IMS structural predictions and SAXS analyses.
fig. S7. SAXS analysis of Tim50CORE.
fig. S8. Effect of ionic strength on the Tim50IMS-bilayer interaction.
fig. S9. Quantitative analysis of Tim50IMS binding.
fig. S10. Structural features of the Tim50IMS homology model.
table S1. SAXS structural parameters.
movie S1. Tim50IMS-bilayer no CL.
movie S2. Tim50IMS-bilayer with CL.
REFERENCES AND NOTES
- 1.Chacinska A., Koehler C. M., Milenkovic D., Lithgow T., Pfanner N., Importing mitochondrial proteins: Machineries and mechanisms. Cell 138, 628–644 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neupert W., Herrmann J. M., Translocation of proteins into mitochondria. Annu. Rev. Biochem. 76, 723–749 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Wenz L. S., Opaliński Ł., Wiedemann N., Becker T., Cooperation of protein machineries in mitochondrial protein sorting. Biochim. Biophys. Acta 1853, 1119–1129 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Stojanovski D., Bohnert M., Pfanner N., van der Laan M., Mechanisms of protein sorting in mitochondria. Cold Spring Harb. Perspect. Biol. 4, a011320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marom M., Azem A., Mokranjac D., Understanding the molecular mechanism of protein translocation across the mitochondrial inner membrane: Still a long way to go. Biochim. Biophys. Acta 1808, 990–1001 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Song J., Tamura Y., Yoshihisa T., Endo T., A novel import route for an N-anchor mitochondrial outer membrane protein aided by the TIM23 complex. EMBO Rep. 15, 670–677 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulz C., Schendzielorz A., Rehling P., Unlocking the presequence import pathway. Trends Cell Biol. 25, 265–275 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Dolezal P., Likic V., Tachezy J., Lithgow T., Evolution of the molecular machines for protein import into mitochondria. Science 313, 314–318 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Endo T., Yamano K., Kawano S., Structural insight into the mitochondrial protein import system. Biochim. Biophys. Acta 1808, 955–970 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Wenz L.-S., Opaliński Ł., Schuler M.-H., Ellenrieder L., Ieva R., Böttinger L., Qiu J., van der Laan M., Wiedemann N., Guiard B., Pfanner N., Becker T., The presequence pathway is involved in protein sorting to the mitochondrial outer membrane. EMBO Rep. 15, 678–685 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alder N. N., Jensen R. E., Johnson A. E., Fluorescence mapping of mitochondrial TIM23 complex reveals a water-facing, substrate-interacting helix surface. Cell 134, 439–450 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Lohret T. A., Jensen R. E., Kinnally K. W., Tim23, a protein import component of the mitochondrial inner membrane, is required for normal activity of the multiple conductance channel, MCC. J. Cell Biol. 137, 377–386 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malhotra K., Sathappa M., Landin J. S., Johnson A. E., Alder N. N., Structural changes in the mitochondrial Tim23 channel are coupled to the proton-motive force. Nat. Struct. Mol. Biol. 20, 965–972 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Truscott K. N., Kovermann P., Geissler A., Merlin A., Meijer M., Driessen A. J. M., Rassow J., Pfanner N., Wagner R., A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat. Struct. Biol. 8, 1074–1082 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Geissler A., Chacinska A., Truscott K. N., Wiedemann N., Brandner K., Sickmann A., Meyer H. E., Meisinger C., Pfanner N., Rehling P., The mitochondrial presequence translocase: An essential role of Tim50 in directing preproteins to the import channel. Cell 111, 507–518 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Mokranjac D., Paschen S. A., Kozany C., Prokisch H., Hoppins S. C., Nargang F. E., Neupert W., Hell K., Tim50, a novel component of the TIM23 preprotein translocase of mitochondria. EMBO J. 22, 816–825 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamamoto H., Esaki M., Kanamori T., Tamura Y., Nishikawa S.-i., Endo T., Tim50 is a subunit of the TIM23 complex that links protein translocation across the outer and inner mitochondrial membranes. Cell 111, 519–528 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Lytovchenko O., Melin J., Schulz C., Kilisch M., Hutu D. P., Rehling P., Signal recognition initiates reorganization of the presequence translocase during protein import. EMBO J. 32, 886–898 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rahman B., Kawano S., Yunoki-Esaki K., Anzai T., Endo T., NMR analyses on the interactions of the yeast Tim50 C-terminal region with the presequence and Tim50 core domain. FEBS Lett. 588, 678–684 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Schulz C., Lytovchenko O., Melin J., Chacinska A., Guiard B., Neumann P., Ficner R., Jahn O., Schmidt B., Rehling P., Tim50’s presequence receptor domain is essential for signal driven transport across the TIM23 complex. J. Cell Biol. 195, 643–656 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roise D., Schatz G., Mitochondrial presequences. J. Biol. Chem. 263, 4509–4511 (1988). [PubMed] [Google Scholar]
- 22.de la Cruz L., Bajaj R., Becker S., Zweckstetter M., The intermembrane space domain of Tim23 is intrinsically disordered with a distinct binding region for presequences. Protein Sci. 19, 2045–2054 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gevorkyan-Airapetov L., Zohary K., Popov-Čeleketić D., Mapa K., Hell K., Neupert W., Azem A., Mokranjac D., Interaction of Tim23 with Tim50 is essential for protein translocation by the mitochondrial TIM23 complex. J. Biol. Chem. 284, 4865–4872 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Li J., Sha B., The structure of Tim50(164–361) suggests the mechanism by which Tim50 receives mitochondrial presequences. Acta Crystallogr. F Struct. Biol. Commun. 71 (Pt. 9), 1146–1151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian X., Gebert M., Höpker J., Yan M., Li J., Wiedemann N., van der Laan M., Pfanner N., Sha B., Structural basis for the function of Tim50 in the mitochondrial presequence translocase. J. Mol. Biol. 411, 513–519 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alder N. N., Sutherland J., Buhring A. I., Jensen R. E., Johnson A. E., Quaternary structure of the mitochondrial TIM23 complex reveals dynamic association between Tim23p and other subunits. Mol. Biol. Cell 19, 159–170 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mokranjac D., Sichting M., Popov-Čeleketić D., Mapa K., Gevorkyan-Airapetov L., Zohary K., Hell K., Azem A., Neupert W., Role of Tim50 in the transfer of precursor proteins from the outer to the inner membrane of mitochondria. Mol. Biol. Cell 20, 1400–1407 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamura Y., Harada Y., Shiota T., Yamano K., Watanabe K., Yokota M., Yamamoto H., Sesaki H., Endo T., Tim23–Tim50 pair coordinates functions of translocators and motor proteins in mitochondrial protein import. J. Cell Biol. 184, 129–141 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersen O. S., Koeppe R. E., Bilayer thickness and membrane protein function: An energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 36, 107–130 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Cross T. A., Murray D. T., Watts A., Helical membrane protein conformations and their environment. Eur. Biophys. J. 42, 731–755 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee A. G., Lipid–protein interactions in biological membranes: A structural perspective. Biochim. Biophys. Acta 1612, 1–40 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Lee A. G., How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta 1666, 62–87 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Böttinger L., Ellenrieder L., Becker T., How lipids modulate mitochondrial protein import. J. Bioenerg. Biomembr. 48, 125–135 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Claypool S. M., Koehler C. M., The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 37, 32–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joshi A. S., Zhou J., Gohil V. M., Chen S., Greenberg M. L., Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta 1793, 212–218 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis R. N. A. H., McElhaney R. N., The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim. Biophys. Acta 1788, 2069–2079 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Baile M. G., Lu Y.-W., Claypool S. M., The topology and regulation of cardiolipin biosynthesis and remodeling in yeast. Chem. Phys. Lipids 179, 25–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren M., Phoon C. K. L., Schlame M., Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 55, 1–16 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Schlame M., Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 49, 1607–1620 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlame M., Cardiolipin remodeling and the function of tafazzin. Biochim. Biophys. Acta 1831, 582–588 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Schlame M., Ren M., Xu Y., Greenberg M. L., Haller I., Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 138, 38–49 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Ye C., Shen Z., Greenberg M. L., Cardiolipin remodeling: A regulatory hub for modulating cardiolipin metabolism and function. J. Bioenerg. Biomembr. 48, 113–123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaspard G. J., McMaster C. R., Cardiolipin metabolism and its causal role in the etiology of the inherited cardiomyopathy Barth syndrome. Chem. Phys. Lipids 193, 1–10 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Lu Y.-W., Claypool S. M., Disorders of phospholipid metabolism: An emerging class of mitochondrial disease due to defects in nuclear genes. Front. Genet. 6, 3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen Z., Ye C., McCain K., Greenberg M. L., The role of cardiolipin in cardiovascular health. Biomed. Res. Int. 2015, 891707 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arias-Cartin R., Grimaldi S., Arnoux P., Guigliarelli B., Magalon A., Cardiolipin binding in bacterial respiratory complexes: Structural and functional implications. Biochim. Biophys. Acta 1817, 1937–1949 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Chaban Y., Boekema E. J., Dudkina N. V., Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 1837, 418–426 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Mileykovskaya E., Dowhan W., Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids 179, 42–48 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bajaj R., Munari F., Becker S., Zweckstetter M., Interaction of the intermembrane space domain of Tim23 protein with mitochondrial membranes. J. Biol. Chem. 289, 34620–34626 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marom M., Safonov R., Amram S., Avneon Y., Nachliel E., Gutman M., Zohary K., Azem A., Tsfadia Y., Interaction of the Tim44 C-terminal domain with negatively charged phospholipids. Biochemistry 48, 11185–11195 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Weiss C., Oppliger W., Vergères G., Demel R., Jenö P., Horst M., de Kruijff B., Schatz G., Azem A., Domain structure and lipid interaction of recombinant yeast Tim44. Proc. Natl. Acad. Sci. U.S.A. 96, 8890–8894 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Laan M., Meinecke M., Dudek J., Hutu D. P., Lind M., Perschil I., Guiard B., Wagner R., Pfanner N., Rehling P., Motor-free mitochondrial presequence translocase drives membrane integration of preproteins. Nat. Cell Biol. 9, 1152–1159 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Gallas M. R., Dienhart M. K., Stuart R. A., Long R. M., Characterization of Mmp37p, a Saccharomyces cerevisiae mitochondrial matrix protein with a role in mitochondrial protein import. Mol. Biol. Cell 17, 4051–4062 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kutik S., Rissler M., Li Guan X., Guiard B., Shui G., Gebert N., Heacock P. N., Rehling P., Dowhan W., Wenk M. R., Pfanner N., Wiedemann N., The translocator maintenance protein Tam41 is required for mitochondrial cardiolipin biosynthesis. J. Cell Biol. 183, 1213–1221 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tamura Y., Harada Y., Nishikawa S.-i., Yamano K., Kamiya M., Shiota T., Kuroda T., Kuge O., Sesaki H., Imai K., Tomii K., Endo T., Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 17, 709–718 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tamura Y., Harada Y., Yamano K., Watanabe K., Ishikawa D., Ohshima C., Nishikawa S.-i., Yamamoto H., Endo T., Identification of Tam41 maintaining integrity of the TIM23 protein translocator complex in mitochondria. J. Cell Biol. 174, 631–637 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Connerth M., Tatsuta T., Haag M., Klecker T., Westermann B., Langer T., Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 338, 815–818 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Potting C., Wilmes C., Engmann T., Osman C., Langer T., Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 29, 2888–2898 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tamura Y., Endo T., Iijima M., Sesaki H., Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J. Cell Biol. 185, 1029–1045 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang F., Ryan M. T., Schlame M., Zhao M., Gu Z., Klingenberg M., Pfanner N., Greenberg M. L., Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Bömer U., Meijer M., Maarse A. C., Hönlinger A., Dekker P. J. T., Pfanner N., Rassow J., Multiple interactions of components mediating preprotein translocation across the inner mitochondrial membrane. EMBO J. 16, 2205–2216 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baile M. G., Sathappa M., Lu Y.-W., Pryce E., Whited K., McCaffery J. M., Han X., Alder N. N., Claypool S. M., Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J. Biol. Chem. 289, 1768–1778 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davis A. J., Ryan K. R., Jensen R. E., Tim23p contains separate and distinct signals for targeting to mitochondria and insertion into the inner membrane. Mol. Biol. Cell 9, 2577–2593 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koshkin V., Greenberg M. L., Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem. J. 347 (Pt. 3), 687–691 (2000). [PMC free article] [PubMed] [Google Scholar]
- 65.Malhotra K., Alder N. N., Advances in the use of nanoscale bilayers to study membrane protein structure and function. Biotechnol. Genet. Eng. Rev. 30, 79–93 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Schuler M. A., Denisov I. G., Sligar S. G., Nanodiscs as a new tool to examine lipid–protein interactions. Methods Mol. Biol. 974, 415–433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Denisov I. G., Sligar S. G., Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol. 23, 481–486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Long A. R., O’Brien C. C., Alder N. N., The cell-free integration of a polytopic mitochondrial membrane protein into liposomes occurs cotranslationally and in a lipid-dependent manner. PLOS ONE 7, e46332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang M., Liu J., Kim Y., Dixon J. E., Pfaff S. L., Gill G. N., Noel J. P., Zhang Y., Structural and functional analysis of the phosphoryl transfer reaction mediated by the human small C-terminal domain phosphatase, Scp1. Protein Sci. 19, 974–986 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oldfield C. J., Cheng Y., Cortese M. S., Brown C. J., Uversky V. N., Dunker A. K., Comparing and combining predictors of mostly disordered proteins. Biochemistry 44, 1989–2000 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Tompa P., Schad E., Tantos A., Kalmar L., Intrinsically disordered proteins: Emerging interaction specialists. Curr. Opin. Struct. Biol. 35, 49–59 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Brooks B. R., Brooks C. L. III, Mackerell A. D. Jr, Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M., CHARMM: The biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.MacKerell A. D. Jr, Bashford D., Bellott M., Dunbrack R. L. Jr, Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M., All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998). [DOI] [PubMed] [Google Scholar]
- 74.Mackerell A. D. Jr, Feig M., Brooks C. L. III, Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Wriggers W., Chacón P., Using Situs for the registration of protein structures with low-resolution bead models from X-ray solution scattering. J. Appl. Cryst. 34, 773–776 (2001). [Google Scholar]
- 76.Svergun D. I., Koch M. H. J., Small-angle scattering studies of biological macromolecules in solution. Rep. Prog. Phys. 66, 1735–1782 (2003). [Google Scholar]
- 77.Marom M., Azem A., The use of cardiolipin-containing liposomes as a model system to study the interaction between proteins and the inner mitochondrial membrane. Methods Mol. Biol. 1033, 147–155 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Siegel D. P., Epand R. M., The mechanism of lamellar-to-inverted hexagonal phase transitions in phosphatidylethanolamine: Implications for membrane fusion mechanisms. Biophys. J. 73, 3089–3111 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heimburg T., Marsh D., Protein surface-distribution and protein-protein interactions in the binding of peripheral proteins to charged lipid membranes. Biophys. J. 68, 536–546 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eilers M., Schatz G., Binding of a specific ligand inhibits import of a purified precursor protein into mitochondria. Nature 322, 228–232 (1986). [DOI] [PubMed] [Google Scholar]
- 81.Rapaport D., Künkele K.-P., Dembowski M., Ahting U., Nargang F. E., Neupert W., Lill R., Dynamics of the TOM complex of mitochondria during binding and translocation of preproteins. Mol. Cell. Biol. 18, 5256–5262 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Popov-Čeleketić D., Mapa K., Neupert W., Mokranjac D., Active remodelling of the TIM23 complex during translocation of preproteins into mitochondria. EMBO J. 27, 1469–1480 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mulgrew-Nesbitt A., Diraviyam K., Wang J., Singh S., Murray P., Li Z., Rogers L., Mirkovic N., Murray D., The role of electrostatics in protein–membrane interactions. Biochim. Biophys. Acta 1761, 812–826 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Murray D., McLaughlin S., Honig B., The role of electrostatic interactions in the regulation of the membrane association of G protein βγ heterodimers. J. Biol. Chem. 276, 45153–45159 (2001). [DOI] [PubMed] [Google Scholar]
- 85.Sathappa M., Alder N. N., The ionization properties of cardiolipin and its variants in model bilayers. Biochim. Biophys. Acta 1858, 1362–1372 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kooijman E. E., Swim L. A., Graber Z. T., Tyurina Y. Y., Bayır H., Kagan V. E., Magic angle spinning 31P NMR spectroscopy reveals two essentially identical ionization states for the cardiolipin phosphates in phospholipid liposomes. Biochim. Biophys. Acta 1859, 61–68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olofsson G., Sparr E., Ionization constants pKa of cardiolipin. PLOS ONE 8, e73040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arbuzova A., Wang L., Wang J., Hangyás-Mihályné G., Murray D., Honig B., McLaughlin S., Membrane binding of peptides containing both basic and aromatic residues. Experimental studies with peptides corresponding to the scaffolding region of caveolin and the effector region of MARCKS. Biochemistry 39, 10330–10339 (2000). [DOI] [PubMed] [Google Scholar]
- 89.McLaughlin S., Smith S. O., Hayman M. J., Murray D., An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. J. Gen. Physiol. 126, 41–53 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Powell G. L., Marsh D., Polymorphic phase behavior of cardiolipin derivatives studied by phosphorus-31 NMR and x-ray diffraction. Biochemistry 24, 2902–2908 (1985). [DOI] [PubMed] [Google Scholar]
- 91.Tang M., Hong M., Structure and mechanism of β-hairpin antimicrobial peptides in lipid bilayers from solid-state NMR spectroscopy. Mol. Biosyst. 5, 317–322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chan D. I., Prenner E. J., Vogel H. J., Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta 1758, 1184–1202 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Narayanaswami V., Kiss R. S., Weers P. M. M., The helix bundle: A reversible lipid binding motif. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 155, 123–133 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nag A., Dinner A. R., Enhancement of diffusion-controlled reaction rates by surface-induced orientational restriction. Biophys. J. 90, 896–902 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wells R. C., Hill R. B., The cytosolic domain of Fis1 binds and reversibly clusters lipid vesicles. PLOS ONE 6, e21384 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ardail D., Privat J.-P., Egret-Charlier M., Levrat C., Lerme F., Louisot P., Mitochondrial contact sites. Lipid composition and dynamics. J. Biol. Chem. 265, 18797–18802 (1990). [PubMed] [Google Scholar]
- 97.Meinecke M., Wagner R., Kovermann P., Guiard B., Mick D. U., Hutu D. P., Voos W., Truscott K. N., Chacinska A., Pfanner N., Rehling P., Tim50 maintains the permeability barrier of the mitochondrial inner membrane. Science 312, 1523–1526 (2006). [DOI] [PubMed] [Google Scholar]
- 98.Schendzielorz A. B., Schulz C., Lytovchenko O., Clancy A., Guiard B., Ieva R., van der Laan M., Rehling P., Two distinct membrane potential–dependent steps drive mitochondrial matrix protein translocation. J. Cell Biol. 216, 83–92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., Sligar S. G., Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schwall C. T., Greenwood V. L., Alder N. N., The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim. Biophys. Acta 1817, 1588–1596 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Stewart J. C. M., Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 104, 10–14 (1980). [DOI] [PubMed] [Google Scholar]
- 102.Nielsen S. S., Toft K. N., Snakenborg D., Jeppesen M. G., Jacobsen J. K., Vestergaard B., Kutter J. P., Arleth L., BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. J. Appl. Cryst. 42, 959–964 (2009). [Google Scholar]
- 103.A. Guinier, G. Fournet, Small-Angle Scattering of X-rays (Structure of Matter Series) (John Wiley & Sons, Inc., 1955). [Google Scholar]
- 104.Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I., PRIMUS: A windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 36, 1277–1282 (2003). [Google Scholar]
- 105.Semenyuk A. V., Svergun D. I., GNOM—A program package for small-angle scattering data processing. J. Appl. Cryst. 24, 537–540 (1991). [Google Scholar]
- 106.Svergun D. I., Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 25, 495–503 (1992). [Google Scholar]
- 107.Mylonas E., Svergun D. I., Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Cryst. 40, s245–s249 (2007). [Google Scholar]
- 108.Franke D., Svergun D. I., DAMIFF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 42, 342–346 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Volkov V. V., Svergun D. I., Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 36, 860–864 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Svergun D. I., Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang Y., I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Im W., Lee M. S., Brooks C. L. III, Generalized born model with a simple smoothing function. J. Comput. Chem. 24, 1691–1702 (2003). [DOI] [PubMed] [Google Scholar]
- 113.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 114.Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., de Vries A. H., The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 (2007). [DOI] [PubMed] [Google Scholar]
- 115.de Jong D. H., Singh G., Bennett W. F. D., Arnarez C., Wassenaar T. A., Schäfer L. V., Periole X., Tieleman D. P., Marrink S. J., Improved parameters for the Martini coarse-grained protein force field. J. Chem. Theory Comput. 9, 687–697 (2013). [DOI] [PubMed] [Google Scholar]
- 116.Monticelli L., Kandasamy S. K., Periole X., Larson R. G., Tieleman D. P., Marrink S.-J., The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 4, 819–834 (2008). [DOI] [PubMed] [Google Scholar]
- 117.Yesylevskyy S. O., Schäfer L. V., Sengupta D., Marrink S. J., Polarizable water model for the coarse-grained MARTINI force field. PLOS Comput. Biol. 6, e1000810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wassenaar T. A., Ingólfsson H. I., Böckmann R. A., Tieleman D. P., Marrink S. J., Computational lipidomics with insane: A versatile tool for generating custom membranes for molecular simulations. J. Chem. Theory Comput. 11, 2144–2155 (2015). [DOI] [PubMed] [Google Scholar]
- 119.Dahlberg M., Maliniak A., Mechanical properties of coarse-grained bilayers formed by cardiolipin and zwitterionic lipids. J. Chem. Theory Comput. 6, 1638–1649 (2010). [DOI] [PubMed] [Google Scholar]
- 120.Periole X., Cavalli M., Marrink S.-J., Ceruso M. A., Combining an elastic network with a coarse-grained molecular force field: Structure, dynamics, and intermolecular recognition. J. Chem. Theory Comput. 5, 2531–2543 (2009). [DOI] [PubMed] [Google Scholar]
- 121.Hess B., Kutzner C., van der Spoel D., Lindahl E., GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008). [DOI] [PubMed] [Google Scholar]
- 122.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. C., GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005). [DOI] [PubMed] [Google Scholar]
- 123.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 124.Torrie G. M., Valleau J. P., Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comp. Phys. 23, 187–199 (1977). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1700532/DC1
fig. S1. The TIM23 complex and CL biogenesis.
fig. S2. The effect of CL synthase knockout on Tim50IMS functionality and association with the TIM23 complex.
fig. S3. Import and integration of [35S]Tim23 into isolated mitochondria.
fig. S4. Response of [35S]Tim23 cross-linking to decreased Δψm.
fig. S5. Incorporation of [35S]Tim23 into nanodiscs: control tests.
fig. S6. Tim50IMS structural predictions and SAXS analyses.
fig. S7. SAXS analysis of Tim50CORE.
fig. S8. Effect of ionic strength on the Tim50IMS-bilayer interaction.
fig. S9. Quantitative analysis of Tim50IMS binding.
fig. S10. Structural features of the Tim50IMS homology model.
table S1. SAXS structural parameters.
movie S1. Tim50IMS-bilayer no CL.
movie S2. Tim50IMS-bilayer with CL.