

Abstract
Identified in 1993, APOE4 is the greatest genetic risk factor for sporadic Alzheimer’s disease (AD), increasing risk up to 15-fold compared with APOE3, with APOE2 decreasing AD risk. However, the functional effects of APOE4 on AD pathology remain unclear and, in some cases, controversial. In vivo progress to understand how the human (h)-APOE genotypes affect AD pathology has been limited by the lack of a tractable familial AD-transgenic (FAD-Tg) mouse model expressing h-APOE rather than mouse (m)-APOE. The disparity between m- and h-apoE is relevant for virtually every AD-relevant pathway, including amyloid-β (Aβ) deposition and clearance, neuroinflammation, tau pathology, neural plasticity and cerebrovascular deficits. EFAD mice were designed as a temporally useful preclinical FAD-Tg-mouse model expressing the h-APOE genotypes for identifying mechanisms underlying APOE-modulated symptoms of AD pathology. From their first description in 2012, EFAD mice have enabled critical basic and therapeutic research. Here we review insights gleaned from the EFAD mice and summarize future directions.
Keywords: animal models, lipoproteins, apolipoproteins, brain, behavior, histopathology, neuronal viability, apolipoprotein E lipidation, cerebrovascular dysfunction, apolipoprotein E, EFAD
INTRODUCTION
Alzheimer’s disease: humans to transgenic mice
In humans, Alzheimer’s disease (AD) progresses over decades, resulting in synaptic dysfunction eventually leading to neuronal loss. Despite the large number of longitudinal aging studies in humans, the lack of cognitive measures coordinated with symptoms of AD pathology results in a poorly understood disease trajectory (1). Transgenic (Tg)-mice are a powerful tool to track AD pathology, address mechanistic hypothesis, and assess the activity of potential therapeutics. An overall limitation of all Tg-mouse models is that none reproduce the full spectrum of human AD symptoms and pathology. However, a number of reviews provide specific guidelines for preclinical AD studies with a consistent theme that a useful Tg-mouse model will incorporate major human AD risk factors and demonstrate dysfunction in the AD-relevant symptom of interest (2). Thus, critical to this modeling is the inclusion of the major human AD risk factors. The APOE4 genotype is the greatest genetic risk factor increasing AD risk up to 15-fold compared with the common APOE3 genotype, while APOE2 is protective (3, 4), but less frequent [estimated: ε2/2 at 0.4%, ε2/3 at 8.8%, and ε2/4 at 1.5% (5, 6)]. Although this risk was identified in 1993 and a number of hypotheses propose APOE genotype-specific functions, the mechanistic pathways that cause APOE4-induced AD risk remain unclear. Even less understood is the critical link between female (♀) sex and the APOE4-induced AD risk (6–13). As described, the sequence of pathology in human AD patients is unclear, however there are key AD-relevant outcomes that are modulated by APOE and sex in humans. AD is diagnosed by extracellular amyloid plaques and neurofibrillary tangles (NFTs) of tau. In addition, soluble oligomeric conformations of amyloid-β (oAβ) correlate with cognitive decline and disease severity (14–19). Importantly, oAβ levels in cerebrospinal fluid (CSF) are increased in AD patients versus controls; and are greater with APOE4/4 versus APOE3/3 in AD patients (20). Further pathology includes neuronal dysfunction, neuroinflammation, and cerebrovascular dysfunction (CerVD). Hence, it is essential to study the interactive role of AD risk factors: age, sex and APOE genotype in the development of human AD pathology using mouse models.
Mouse APOE versus human APOE in AD-Tg mouse models
In vivo progress to determine the effects of the human (h)-APOE genotypes on AD pathology has been limited by the lack of a tractable familial AD-Tg (FAD-Tg) mouse model expressing h-APOE rather than mouse (m)-APOE [for complete review on the introduction of h-APOE into FAD-Tg mouse models see (21)]. Why is this a critical issue? The m-apoE is structurally and functionally distinct from h-apoE. While the three human isoforms of apoE differ by a single amino acid change at residues 112 and 158 (apoE2Cys,Cys, apoE3Cys,Arg, apoE4Arg,Arg), m-apoE is expressed as a single isoform and differs from h-apoE by ∼100–300 amino acids. The disparity between m- and h-apoE is relevant for virtually all AD-relevant pathology, including Aβ deposition and clearance, neuroinflammation, tau pathology, neural plasticity, and CerVD (22–29). Further, many APOE/FAD-Tg models express h-apoE under heterologous promoters that may be activated by stressors [glial fibrillary acidic protein (GFAP) (27, 30)], or by cell types that are not the primary producers of apoE [neuron-specific enolase (31)], rather than the endogenous m-apoE promoter. Currently, the best option is the APOE-knock-in or targeted-replacement (TR) mice, developed to replace the coding domain for m-APOE with the coding domain of each of the h-APOE genotypes (32). In APOE-TR mice, glial cells express h-APOE in a native conformation at physiologically regulated levels, and in the same temporal and spatial pattern as endogenous m-APOE. Thus, it is critical to conduct mechanistic and preclinical therapeutic studies in mice that incorporate h-APOE in a relevant context.
Building an AD-Tg mouse model with h-APOE
Operationally, relative to m-APOE, the h-APOE genotypes delay AD pathology in FAD-Tg mouse models, as illustrated in Table 1. This table is designed to be a representation, using a composite of several FAD-Tg mouse models (27, 33–46) and Aβ pathology as an example of AD phenotype. First, Aβ pathology is significantly delayed when FAD-Tg mice are crossed with APOE-KO mice, eliminating m-APOE (Table 1: compare A to B) (27, 30, 39, 40, 46). Second, Aβ pathology is further delayed when h-APOE-Tg mice are crossed with FAD-Tg/APOE-KO mice (Table 1: compare B to C) (28, 30, 47, 48). Thus, incorporation of h-APOE into FAD-Tg mice that normally begin to develop pathology at 8–10 months delays the development of pathology to 12–16 months (27, 28, 30, 47, 48), extending progression of the full array of AD-related symptoms beyond the lifespan of the mouse. One approach to address this h-APOE-induced temporal delay in AD pathology is using an FAD-Tg mouse that has rapid-onset AD pathology. The 5xFAD mice exhibit accelerated development of pathology, including amyloid deposition by 2 months (Table 1: compare A to D) (49–69). The 5xFAD mice were crossed with APOE-TR mice to produce the EFAD mice2, which exhibit the anticipated delay in AD pathology, particularly with plaque development delayed to ∼6 months (Table 1: compare D to E) (20, 66, 70–75), but still >6 months before the average FAD-Tg/APOE-Tg mice (compare Table 1: C and E).
TABLE 1.
Effects of h-APOE on Aβ pathology in FAD-Tg mice
| Model | Aβ Pathology | ||||
| <4 Months | 4–8 Months | 8–12 Months | 12–16 Months | ≥16 Months | |
| (A) FAD-Tg | ↑ | ↑↑ | ↑↑↑ | ↑↑↑↑ | |
| (B) FAD-Tg×APOE-KO | ↑ | ↑↑ | ↑↑↑ | ||
| (C) FAD-Tg×APOE-Tg | ↑ | ↑↑ | |||
| (ε4 > ε3) | (ε4 > ε3) | ||||
| (D) 5xFAD+/− | ↑↑ | ↑↑↑ | ↑↑↑↑ | ↑↑↑↑↑ | ↑↑↑↑↑↑ |
| (E) 5xFAD+/−×APOE-TR = EFAD-Tg | ↑ | ↑↑ | n.m. | n.m. | |
| (ε4 > ε3) | (ε4 > ε3) | ||||
Differences in Aβ pathology (measured as Aβ levels using biochemical methods or Aβ deposition using immunohistochemistry) in FAD-Tg mice in the presence or absence of m- or h-APOE genotypes across the mouse lifespan among FAD-Tg (27, 33–35, 37–46, 327, 328) (A), FAD-Tg/APOE-KO (27, 30, 39, 40, 46) (B), FAD-Tg/APOE-Tg (27, 28, 30, 47, 48) (C), 5xFAD-Tg (50, 55, 58, 62–65, 68, 69, 222, 329–339) (D), 5xFAD/APOE-TR (EFAD-Tg mice) (20, 66, 70–75, 257) (E). Aβ pathology change: low ↑ to high ↑↑↑↑↑↑, relative to an earlier age or among FAD-Tg mouse models. If measured and significant, differences between APOE genotypes are specifically indicated within each cell (as applicable). n.m., not measured.
Development of the EFAD mouse model
The 5xFAD mice coexpress five FAD mutations [amyloid precursor protein (APP) K670N/M671L + I716V + V717I and PS1 M146L + L286V] under the control of the neuron-specific mouse thymocyte differentiation antigen 1 theta (Thy-1) promotor (50). The specificity of this Thy-1 promoter for neuronal expression in the brain has been established (76, 77). The 5xFAD+/− line, Tg6799, produces the highest amount of Aβ42 (provided by R. Vassar, Northwestern University) (50). To establish colonies of EFAD mouse lines (E2FAD, E3FAD, and E4FAD), 5xFAD mice were bred to homozygous APOE2-, APOE3-, and APOE4-TR mice (32, 78) via a contract with Taconic Biosciences2. Briefly, male (♂) APOE-TR+/+ mice on a C57/BL6 background were bred with ♀ 5xFAD+/− mice on a C57BL6/B6×SJL background. The resulting ♀ mice, APOE-TR+/+/5xFAD+/− were backcrossed to ♂ APOE-TR mice to generate APOE-TR+/+/5xFAD+/− (EFAD) mice. The EFAD mice allow for the monitoring of multiple AD-related symptoms through the mouse lifespan (Tables 2, 3). Importantly, based on the design of the breeding strategy, the EFAD mice littermates (50%) are noncarriers (EFAD-NCs) for the 5xFAD transgenes and homozygous for APOE (5xFAD−/−/APOE+/+). Thus, EFAD-NCs provide both a comparison to the EFAD mice and a complementary approach to address functional questions about APOE in the absence of FAD-induced pathology.
TABLE 2.
AD-related behavioral deficits, histopathology and loss of neuronal viability in 5xFAD and EFAD mice
| Model | Phenotype | |||||
| <4 Months | 4–8 Months | 8–12 Months | 12–16 Months | ≥16 Months | ||
| 5xFAD | Behavioral deficits | |||||
| Learning, exploration and memory | ↑ | ↑↑ | ↑↑↑ | ↑↑↑↑ | ↑↑↑↑ | |
| (MWM) | (YM, MWM, FC, NOR) | (YM, MWM, FC, CTA) | (YM, MWM, FC, CM) | (YM) | ||
| Histopathology | ||||||
| Aβ deposition | ↑↑ | ↑↑↑ | ↑↑↑↑ | ↑↑↑↑↑ | ↑↑↑↑↑↑ | |
| Plaque deposition | ↑↑ | ↑↑↑ | ↑↑↑↑ | ↑↑↑↑ | n.m. | |
| (♀ > ♂) | (♀ > ♂) | (♀ > ♂) | ||||
| CAA | ↑ | ↑↑ | ↑↑↑ | n.m. | n.m. | |
| Neuroinflammation | ↑ | ↑↑ | ↑↑↑ | ↑↑↑↑ | ↑↑↑↑ | |
| pTau | ↑ | ↑ | ↑↑ | — | n.m. | |
| Neuronal viability | ||||||
| Synaptic protein loss | ↑ | ↑↑ | ↑↑↑ | ↑↑↑ | n.m. | |
| Neuroplasticity | ↑ | ↑↑ | ↑↑↑ | n.m. | n.m. | |
| Neuronal loss | — | ↑* | ↑↑* | ↑↑↑ | n.m. | |
| EFAD | Behavioral deficits | |||||
| Learning, exploration and memory | — | ↑ | ↑ | n.m. | n.m. | |
| (MWM: ♀ε4 > ε3 ≥ ε2, YM: ♀ε4 > ε3 ≥ ε2) | (YM: ε4 > ε3; ε4 ♀ > ♂, NOR: ε4 ♀ > ♂; ♀ ε4 > ε3) | |||||
| Histopathology | ||||||
| Aβ deposition | — | ↑ | ↑↑ | n.m. | n.m. | |
| (ε4 > ε3 ≥ ε2; ♀ > ♂) | (ε4 > ε3; ♀ > ♂) | |||||
| Plaque deposition | — | ↑ | n.m. | n.m. | n.m. | |
| (ε4 > ε3 ≥ ε2; ♀ > ♂) | ||||||
| CAA | n.m. | ↑ | n.m. | n.m. | n.m. | |
| (ε4 > ε3; ♀ = ♂) | ||||||
| Neuroinflammation | n.m. | ↑ | n.m. | n.m. | n.m. | |
| (ε4 > ε3) | ||||||
| pTau | — | ↑ | n.m. | n.m. | n.m. | |
| (ε4 > ε3) | ||||||
| Neuronal viability | ||||||
| Synaptic protein loss | — | ↑ | n.m. | n.m. | n.m. | |
| (♀ε4 > ε3 ≥ ε2; ♂ε4 > ε3) | ||||||
| Neuroplasticity | n.m. | n.m. | n.m. | n.m. | n.m. | |
| Neuronal loss | n.m. | n.m. | n.m. | n.m. | n.m. | |
5xFAD mice: Behavioral deficits: MWM (49, 50, 59–61, 340), YM (49, 51, 54, 55, 58), fear conditioning (FC) (52, 56, 57), NOR (329, 330), conditioned taste aversion (CTA) (52), cross-maze (CM) (62). AD-related histopathology: Aβ deposition: immunohistochemistry (IHC)-mAb to Aβ (50, 52, 55, 58, 62–65, 68, 329–337); plaque deposition: Thio-S (50, 64, 69, 332, 334); CAA: Thio-S, methoxy-X04 (334, 341); neuroinflammation: astrogliosis or microgliosis: IHC-mAb to GFAP or ionized calcium binding adaptor molecule 1 (Iba1)/F4-80 (50, 55, 58, 61, 65, 329, 333–335); pTau: Western blot for p-sites (59, 342). Neuronal viability: Synaptic proteins: PSD95, synaptophysin, syntaxin by Western blot or IHC (50, 68, 69); neuroplasticity: basal synaptic transmission, long-term potentiation or paired-pulse facilitation (56, 57, 61, 329–331, 335, 336, 343–345); neuronal loss: cresyl violet, IHC-mAb to RNA binding protein, fox-1 homolog 3 (NeuN): *quantified by area (50, 185), or using unbiased stereology (61–63, 68, 69, 334, 338, 346). EFAD mice: Behavioral deficits: MWM, YM, NOR (70, 71). AD-related histopathology: Aβ deposition: IHC-mAb to Aβ42-specific (MOAB-2) (66, 71) or IHC-mAb to Aβ (4G8) (73); plaque deposition: Thio-S (66, 71, 257); CAA: Thio-S (73); neuroinflammation: IHC-mAb to GFAP or Iba1 (72); pTau: Western blot for p-sites (75). Neuronal viability: synaptic proteins: PSD95, synaptophysin, drebrin by Western blot (70, 172). The levels are represented as low ↑ to high ↑↑↑↑↑↑ relative to an earlier age or, if known and significant, differences between sex or APOE genotypes are specifically indicated within each cell relative to an earlier age. n.m., not measured; —, not detectable.
TABLE 3.
Differences in AD-related biochemical measures in 5xFAD and EFAD mice
| Model | Biochemical Measures | |||||
| <4 Months | 4–8 Months | 8–12 Months | 12–16 Months | ≥16 Months | ||
| 5xFAD | Aβ and apoE solubility | |||||
| Total Aβ | ↑ (♀ > ♂) | ↑↑ (♀ > ♂) | ↑↑↑ (♀ > ♂) | ↑↑↑↑ | ↑↑↑↑↑ | |
| Soluble Aβ42 | ↑ | ↑↑ | ↑↑↑ | n.m. | n.m. | |
| Total apoE | ↑ | ↑↑ | ↑↑↑ | ↑↑↑↑ | n.m. | |
| TBSX-apoE | — | — | n.m. | n.m. | n.m. | |
| APP processing | ||||||
| APP levels | ↑ | ↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | |
| BACE levels | ↑ | ↑↑ | ↑↑↑ | ↑↑↑ | ↑↑↑ | |
| C-terminal fragments | ↑ | ↑↑ | ↑↑↑ | ↑↑↑↑ | ↑↑↑↑ | |
| Neurotrophic factors | ||||||
| BDNF | ↓ | ↓↓ | n.m. | ↓↓↓ | ↓↓↓↓ | |
| pCREB | n.m. | — | ↓ | n.m. | n.m. | |
| Neuroinflammatory cytokines | ||||||
| TNF-α | ↑ | ↑ | ↑↑ | n.m. | n.m. | |
| IL-1β | ↑ | ↑↑ | n.m. | n.m. | n.m. | |
| EFAD | Aβ and apoE solubility | |||||
| Total Aβ | — | ↑ (ε4 > ε3 ≥ ε2) | n.m. | n.m. | n.m. | |
| Soluble Aβ42 | — | ↑ (ε4 > ε3 ≥ ε2; ε4 ♀ > ♂) | n.m. | n.m. | n.m. | |
| Total apoE | n.c. (ε4 < ε3 ≤ ε2) | n.c. (ε4 < ε3 ≤ ε2) | n.m. | n.m. | n.m. | |
| TBSX-apoE | n.c. (ε4 < ε3 ≤ ε2) | n.c. (ε4 < ε3 ≤ ε2) | n.m. | n.m. | n.m. | |
| APP processing | ||||||
| APP levels | n.m. | n.c. (ε4 = ε3 = ε2) | n.m. | n.m. | n.m. | |
| BACE levels | n.m. | n.m. | n.m. | n.m. | n.m. | |
| C-terminal fragments | n.m. | n.m. | n.m. | n.m. | n.m. | |
| Neurotrophic factors | ||||||
| BDNF | ↓ (♀ε4 < ε3 ≤ ε2) | ↓↓ (♀ε4 < ε3 ≤ ε2) | n.m. | n.m. | n.m. | |
| pCREB | ↓ (♀ε4 < ε3 ≤ ε2) | ↓↓ (♀ε4 < ε3 ≤ ε2) | n.m. | n.m. | n.m. | |
| Neuroinflammatory cytokines | ||||||
| TNF-α | n.m. | ↑ (ε4 > ε3) | ↑ (ε4 = ε3) | n.m. | n.m. | |
| IL-1β | n.m. | ↑ (ε4 > ε3) | ↑ (ε4 = ε3) | n.m. | n.m. | |
5xFAD mice: ApoE and Aβ solubility: total Aβ: ELISA (49, 50, 52, 53, 57, 66, 222, 348), soluble Aβ42: in TBS or PBS extraction fraction by ELISA (67, 334, 338, 348), total apoE: ELISA or Western blot (67, 348, 349); TBSX-apoE: in TBSX extraction fraction by ELISA (67). APP processing: APP, BACE, C-terminal fragments by Western blot (49, 52, 53, 58, 67, 222, 329, 336, 337, 339, 348–352). Neurotrophic factors: brain-derived neurotrophic factor (BDNF) and pCREB proteins by Western blot or mRNA by qPCR (55, 61, 336, 350, 352, 353). Neuroinflammatory cytokines: TNF-α, IL-1β by qPCR (185, 329, 342, 351, 354). EFAD mice: ApoE and Aβ solubility: total Aβ: ELISA (66, 172), soluble Aβ42: in TBS extraction fraction by ELISA (66, 73, 172), total apoE: ELISA (66, 70), TBSX-apoE: in TBSX extraction fraction by ELISA (66, 172). APP processing: APP by Western blot (66). Neurotrophic factors: BDNF and pCREB proteins by Western blot (70, 193). Neuroinflammatory cytokines: TNF-α, IL1-β mRNA by qPCR (72, 106). Change: Direction: increase ↑, decrease ↓. Extent: the number of arrows (lower-higher) relative to an earlier age. If known and significant, differences between sex or APOE genotypes are specifically indicated within each cell relative to an earlier age. n.c., no change; n.m., not measured; —, not detectable.
Amyloid plaque deposition is not the same as soluble Aβ peptide
Amyloid plaques (together with NFTs) are a major pathological hallmark of AD; however, there is strong debate on their significance. Understanding this issue is relevant for interpreting data obtained in EFAD mice. Recently, patience has grown thin in both the public and academic communities for the continued failure of both Aβ vaccine trials (https://endpts.com/mercks-leading-phiii-bace-drug-implodes-in-latest-alzheimers-disaster/) and amyloid imaging studies (79–81). This has led to an apparent bias against amyloid plaques as a therapeutic target and even Aβ as a measure of AD pathology, evident from publications (82–85), press releases (http://www.thetimes.co.uk/article/how-mice-immunity-is-hindering-research-n5fl2j5fj, https://www.theatlantic.com/health/archive/2017/02/alzheimers-amyloid-hypothesis/517185/), documentaries (http://www.monsterinthemind.com/), and TEDx talks (https://www.youtube.com/watch?v=6tBSFEzkk0A). One aspect of this debate is the nature of the aggregation pathways for Aβ, particularly Aβ42. As illustrated in Fig. 1, one paradigm for considering this assembly process is whether soluble oAβ is either “on” or “off” pathway to the assembly of amyloid plaques (86). Via the on pathway, oAβ can be pathogenic, but are structural precursors for the formation of the traditional insoluble fibrils that continue to assemble into the parallel β-sheet structure of amyloid plaques also considered to be pathogenic (Fig. 1A)? Alternatively, in the off pathway assembly process, Aβ can proceed via two separate processes, the pathogenic formation of oAβ or formation of the traditional insoluble fibrils that continue to assemble into amyloid plaques, considered to be benign (Fig. 1B). Whether the on or off pathway is correct has not yet been determined. However, both the vaccine trials and the imaging studies focus on amyloid plaques, not oAβ, as the pathogenic target. This distinction is critical as FAD mutations indicate only that increases in Aβ42 or the increase in the Aβ42/Aβ40 ratio cause FAD, not that the toxic assembly form of the peptide is amyloid plaques. Indeed, a novel FAD-Tg mouse model that promotes oAβ formation, rather than fibrils and amyloid plaques, develops features of AD pathology, including tau hyperphosphorylation, neuroinflammation, synaptic alteration, cognitive deficits, and neuronal loss (87). For the purposes of this review, we consider that oAβ levels and induced signaling cascades are pathogenic, and amyloid plaques are likely, at best, less neurotoxic and more likely to be benign.
Fig. 1.

Aβ assembly pathways: oAβ on or off pathways for amyloid plaque deposition. A: Via the on pathway, oAβs can be pathogenic, but are structural precursors for the formation of the traditional insoluble fibrils that continue to assemble into the parallel β-sheet structure of amyloid plaques, also considered to be pathogenic. B: Via the off pathway assembly process, Aβ can proceed via two separate processes, the pathogenic formation of oAβ or formation of the traditional insoluble fibrils that continue to assemble into amyloid plaques, considered to be benign [reproduced from (86)].
PHENOTYPIC CHARACTERIZATION OF EFAD MICE
The 5xFAD mice versus EFAD mice
The EFAD mice are a tractable mouse model to study the development of a number of components of AD pathology (Tables 2, 3). Although 5xFAD mice express m-apoE, which we argue is distinct from h-apoE in the proximal mechanistic pathways that lead to AD pathology, we have included a comparison of AD pathology in 5xFAD versus EFAD mice to demonstrate, in part, the rapid onset and accelerated development of AD symptoms in the 5xFAD mice (Tables 2, 3). In addition, pathology in 5xFAD mice can be used to predict pathology in the EFAD mice. A particularly useful example is neuronal loss, which is significant by 12 months in the 5xFAD mice, predicting that in the EFAD mice neuronal loss will likely occur at >16 months. However, APOE genotype will significantly influence most components of AD pathology and analysis of these differences will illuminate APOE-modulation of AD pathology. In general, there is an APOE genotype (E4FAD > E3FAD ≥ E2FAD), as well as an emerging sex effect (♀ > ♂) on behavioral deficits, histopathology, and neuronal viability (Table 2), as well as Aβ42 and apoE solubility, neurotrophic factors, and neuroinflammatory cytokines, but not with APP processing (Table 3). Thus, the EFAD mice allow for the monitoring of multiple AD-related symptoms through the mouse lifespan (Tables 2, 3). An important note is that the sex and APOE4 genotype interactions are recent findings, and ♂ EFAD mice are more extensively characterized than ♀ mice. Here our goal is to detail APOE-modulated AD pathology in EFAD mice and, where applicable, ♀ versus ♂ comparisons.
APOE modulated AD symptoms exhibited in EFAD mice
Behavioral deficits in ♂ and ♀ EFAD mice.
In humans, AD is characterized by progressive behavioral changes, including memory loss, a decrease in executive function, and impairments in social interaction (88–98) (Figs. 2–5). Cognitive decline in human AD patients (99) and the normal population follows ε4 > ε3 > ε2 (100), greatest in ♀ ε4 carriers (5, 101, 102). Similarly, in ♀ EFAD mice, cognitive impairment is E4FAD > E3FAD ≥ E2FAD, as measured by Morris water maze (MWM), with the deficits increasing from 2 to 6 months (Fig. 2A) (70). In 8-month-old E3FAD and E4FAD mice, cognitive impairment is E4FAD > E3FAD and ♀ > ♂, as measured by novel object recognition (NOR) and Y-maze (YM) (Fig. 2B) (71). Although cognitive testing in mice is not without drawbacks, it is striking that EFAD mice demonstrate APOE genotype- and sex-induced loss of cognitive function similar to humans.
Fig. 2.

Behavior deficits in EFAD mice. A: MWM in 2-, 4-, and 6-month-old ♀ EFAD mice, behavior performed during light cycle [adapted from (70)]. B: NOR preference index and YM spontaneous alternation in 8-month-old ♂ and ♀ EFAD mice, behavior performed during dark cycle [adapted from (71)]. P < 0.05: *versus E4FAD within sex, #versus 2 months, †♀ versus ♂ within genotype.
Fig. 5.

Aβ42 and apoE solubility in 6-month-old ♂ 5xFAD, C57BL/6, and EFAD mice. A: Total Aβ42 levels in the brain. B: Aβ42 extraction profile after sequential extraction of CX with TBS, TBSX, and formic acid [adapted from (66, 67)]. C: Total apoE levels in the brain. D: ApoE extraction profile of CX, as described for (B) [adapted from (66, 67)]. E: Soluble profile (TBS fraction): apoE, Aβ42, oAβ and apoE/Aβ complex from HP of EFAD mice [adapted from (20, 66, 67)]. *P < 0.05 significantly less than E4FAD, #P < 0.05 significantly greater than E4FAD, ¶P < 0.05 significantly less than C57BL/6. All values are represented as mean ± SE normalized per milligram tissue (A–D) or per milligram protein (E).
Extracellular amyloid and Aβ in ♂ EFAD mice.
Human data demonstrate higher levels of extracellular amyloid/Aβ with APOE4 compared with APOE3 (20, 103, 104). For our initial characterization of the pathology in the EFAD mice, we used ♂ mice aged 2–6 months (Fig. 3) (66). Both Aβ accumulation and thioflavine-S (Thio-S)-positive plaque deposition begin in the subiculum (SB), followed by the deep layers of the frontal cortex (CX) (Fig. 3A, B) then spreading to the outer layers of the CX and the thalamus (50, 66). As described above, introduction of h-APOE delayed extracellular Aβ accumulation from ∼2 to 6 months compared with the 5xFAD mice expressing m-APOE: 5xFAD > E4FAD > E3FAD ≥ E2FAD (Fig. 3A). Plaque morphology is also affected by APOE genotype, with diffuse plaques: E2FAD = E3FAD > E4FAD and compact plaques: E2FAD = E3FAD < E4FAD (66).
Fig. 3.

AD histopathology in 2- to 6-month-old ♂ 5xFAD and EFAD mice. A: Total Aβ deposition by immunohistochemistry (IHC) in 5xFAD and ♂EFAD sagittal brain sections at 2, 4, and 6 months with mAb for Aβ (MOAB-2, red) and mAb for neurons [RNA binding protein, fox-1 homolog 3 (NeuN), green]. B: Plaque deposition: Thio-S staining in 6-month-old ♂ EFAD sagittal brain sections, quantified by percent area of CX [reproduced from (66)]. *P < 0.05 versus E4FAD. C: Neuroinflammation: IHC staining in 6 month ♂ EFAD sagittal brain sections for astrocytes (GFAP) [adapted from (72)], quantified as percent area of CX; and for reactive microglia [ionized calcium binding adaptor molecule 1 (Iba1), green] and Aβ (MOAB-2, red), quantified as plaque-associated microglial density [reproduced from (72)]. *P < 0.05 versus E4FAD. D: pTau: IHC in 7 month ♂ EFAD CX for phosphorylated Tau pT205 and pS404 sites. Insets: 20× magnification (black box). pTau quantified in EFAD CX by Western blot, expressed as E4FAD/E3FAD, #P < 0.05 versus E3FAD [reproduced from (75)]. Arrows for pathology: yellow = HP, red = CX.
Neuroinflammation in ♂ EFAD mice.
Neuroinflammation is an important component of AD pathology and is modulated by APOE (105, 106). For example, with APOE4 there is evidence of greater glial activation (107), higher levels of pro-inflammatory cytokines, and lower levels of anti-inflammatory cytokines in humans (108–110) and in APOE-TR mice, both at basal levels (108, 111) and in response to an inflammatory insult (25, 106, 112–114). In E4FAD mice, there is greater astrogliosis and microgliosis compared with E3FAD and E2FAD, particularly in the SB and deep layers of the frontal CX, brain regions with high levels of extracellular Aβ deposition and a higher density of microglia associated with amyloid plaques (Fig. 3C) (72). Further, there are higher levels of the pro-inflammatory cytokine, interleukin (IL)-1β, in E4FAD mice compared with E3FAD mice. Neuroinflammation is a complex response, involving multiple receptors and mediators, which have been assessed in EFAD mice via mRNA (106), including the complement receptor 1 (115), triggering receptor expressed on myeloid cells 2 (TREM2) (116, 117), and CD33 (118–120), all AD-relevant receptors. Further, at 6 months, E4FAD mice have lower levels of interleukin-4 receptor (IL-4R)-related cytokines and higher levels of toll-like receptor 4 (TLR4)-related cytokines compared with E3FAD mice (106), consistent with studies suggesting an association of IL-4R with an anti-inflammatory/repair response (121) and TLR4 with neuronal loss (122–124). Thus, E4FAD mice exhibit a neuroinflammatory phenotype, which may be directly relevant to the human condition.
Tau pathology in ♂ EFAD mice.
In AD brains, there are higher levels of phosphorylated tau (pTau) and NFTs, the latter correlated with the severity of cognitive impairment (4, 29, 125–134). Although a general limitation is that FAD mice do not develop overt tau pathology, they do enable the assessment of subtler changes in phosphorylation. There are higher pTau levels in the SB and CA2/3 regions of the hippocampus (HP) in E4FAD mice at 7 months (Fig. 3D), not detectable at 3 months (75). Thus, data from EFAD mice demonstrate that APOE4 and Aβ interact to induce tau phosphorylation.
Neuronal viability (synaptic proteins) in ♀ EFAD mice.
Decreases in presynaptic and postsynaptic proteins in HP and CX contribute to altered functional connectivity, suggesting that synaptic deficits may precede frank neuronal loss in humans (135–139), eventually resulting in the cognitive impairments characteristic of AD. Postsynaptic density protein 95 (PSD95) and drebrin are postsynaptic intracellular scaffold proteins commonly used to assess the structural integrity of synapses. In particular, AD patients exhibit decreased levels PSD95 and drebrin (140–144). PSD95 regulates synaptic strength and plasticity (145–149) via molecular organization of the postsynaptic region (149–151). Drebrin, a spine-resident side-binding protein of filamentous actin, regulates spine morphology, size, density, and maturation (152–157) via regulation of actin cytoskeleton dynamics (158–165). In a recent publication using only ♀ EFAD mice, postsynaptic proteins (PSD95 and drebrin) were reduced in the E4FAD-HP from 2 to 6 months compared with E3FAD and E2FAD mice (Fig. 4) (70). These deficits in synaptic proteins are consistent with the behavioral deficits.
Fig. 4.
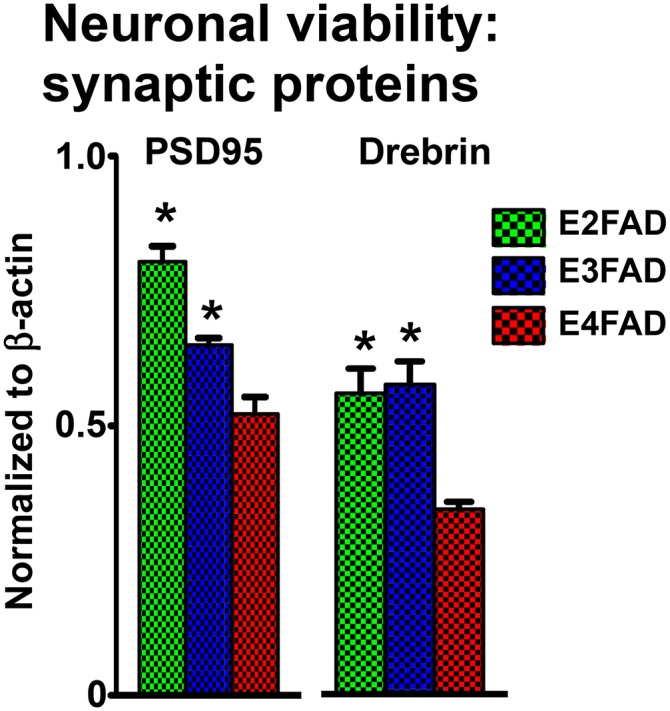
Synaptic protein levels in ♀ EFAD mice. Synaptic proteins PSD95 and drebrin measured in 6-month-old ♀ EFAD mice by Western blot and normalized to β-actin [adapted from (70)], *P < 0.05 versus E4FAD.
Solubility of apoE and Aβ42 in ♂ EFAD mice.
In brain homogenates, total Aβ42 levels are 5xFAD >> E4FAD > E3FAD = E2FAD (Fig. 5A) (66). To measure the solubility of both Aβ and apoE, particularly the apoE-lipoproteins in brain tissue, we adapted a three-step sequential protein extraction protocol using TBS, TBS + Triton X-100 (TBSX), and formic acid, producing extraction profiles for Aβ and apoE (Fig. 5B, D) (66, 67). In the Aβ extraction profile, the 5xFAD >> E4FAD > E3FAD = E2FAD pattern was observed in the TBS and formic acid extraction fractions, while the TBSX fraction contained low levels of Aβ42 that were comparable across the Tg-mouse strains [Fig. 5B, adapted from (66, 67)]. These biochemical results are consistent with the delay in Aβ deposition induced by the replacement of m-APOE with h-APOE (Fig. 3A), as are the genotype effects in levels of total and soluble Aβ42 with E4FAD > E3FAD = E2FAD (Fig. 5A, B).
The apoE extraction profile detects apoE-lipoproteins in the TBSX fraction, a nonionic detergent that releases apoE from lipoproteins without inducing the formation of new micelles, as can occur with SDS and other ionic detergents (66, 67). Thus, although total levels of apoE4 < apoE3 = apoE2 in the brains of humans (103, 166, 167), APOE-TR mice (5, 47, 166–170), and EFAD mice (Fig. 5C), this isoform-specific difference is only in the TBSX-apoE4 (Fig. 5D) (66). C57BL/6 WT mice do not express Aβ42, but do express m-apoE and, so, are included for comparison with the 5xFAD mice. For total apoE, the order is the reverse of the total Aβ levels: WT < 5xFAD ≤ E4FAD < E3FAD = E2FAD [Fig. 5D, adapted from (66, 67)]. However, in the extraction fractions, m-apoE from the WT is detected in the TBSX fraction, while the m-apoE from the 5xFAD mice is not detectable, though low levels of m-apoE from both WT and 5xFAD are detected in the TBS and formic acid fractions. Thus, the presence of Aβ42 affects m-apoE solubility, specifically eliminating TBSX-m-apoE.
In the soluble fraction, the levels of the apoE isoforms are not significantly different, while soluble Aβ42 and oAβ are significantly higher with apoE4, and apoE4/Aβ complex levels are significantly lower (Fig. 5E) (20, 66). Indeed, APOE4 is characterized by increased levels of soluble Aβ (Aβ42 and oAβ) (20, 66). Soluble Aβ correlates with cognitive decline and disease severity in humans, and memory decline in FAD-Tg mice [for review see (14)]. To address the pathways that may underlie this correlation between apoE4 and increased soluble Aβ required the development of novel reagents, including a new monoclonal antibody (mAb) specific to Aβ (MOAB-2) (171) that enabled development of the critical oAβ (66) and apoE/Aβ ELISAs (20). Importantly, lower levels of apoE lipidation and apoE/Aβ complex in APOE4 versus APOE3 negatively correlate with soluble Aβ levels (Fig. 5E) (20, 66, 172).
Lipidomic analysis in ♂ EFAD mice.
Recent improvements in lipidomic technology have advanced the field to a point where it is now possible to accurately identify and quantify thousands of phospholipids (PLs) in complex biological samples with relative ease. Recently, a lipidomics study revealed that the ratios of arachidonic acid (AA) and DHA increased in several major PL classes in the blood from cognitively normal ε4 carriers who converted to mild cognitive impairment/AD within 3 years (173). Longitudinal profiling of E3FAD and E4FAD mice showed that blood AA- and DHA-containing PL species were altered as early as 2.5 months of age. At 6 months, AA- and DHA-containing lysophosphatidylcholine species increased in blood, but decreased in the brains of E4FAD compared with E3FAD mice (173). Previous studies have shown that lysophosphatidylcholine-DHA is a preferred DHA carrier to the brain (174), and that the major facilitator superfamily domain-containing protein 2 (mfsd2a) transports this lipid across the blood-brain barrier (BBB) (175, 176). Hence, an increase in this lipid in the blood and a decrease in the brain suggests reduced transport of DHA to the brain in E4FAD mice compared to E3FAD and E2FAD mice. In addition, brain DHA transport is reduced in APOE4-TR mice compared with APOE2-TR mice, evidence for DHA transport deficiencies (177). Collectively, these studies suggest that an imbalance in AA and DHA may be due to transport deficiencies among the ε4 carriers, which further contribute to the neuroinflammation associated with AD pathogenesis.
EFAD mice as a model for cerebrovascular dysfunction: a detailed example
CerVD is reemerging as a key component of AD, with unresolved questions as to the extent of CerVD and its significance to cognitive decline. Additional issues include the apparent differences between CerVD outcomes in humans and mouse models caused by APOE4, Aβ, and sex. EFAD mice provide critical insight on the interactive role of APOE, ♀ sex, and Aβ in CerVD.
Cerebrovascular leakiness and vessel coverage in ♂ and ♀ EFAD mice.
Plasma protein extravasation into the brain is a key outcome of CerVD, particularly BBB capillary leakage. Here, we demonstrate that fibrinogen levels in the CX and SB in 8-month-old EFAD mice follow the order: ♀ E4FAD > ♀ E3FAD = ♂ E4FAD > ♂ E3FAD (Fig. 6A). Total vessel coverage is a complimentary outcome measure of BBB damage, and in the SB and deep CX layers follow the order: ♂ E3FAD > ♂ E4FAD ≥ ♀ E3FAD > ♀ E4FAD [Fig. 6B, C reproduced from (71)]. Thus, the combination of ♀ sex, APOE4, and Aβ induce pronounced BBB deficits that likely contribute to cognitive deficits. Indeed, high fibrinogen levels have been demonstrated in AD patients that are ε4 carriers (178) and induce glial activation and neuronal dysfunction. Angiogenic growth factors are important for maintaining BBB function. Recent data demonstrate that plasma levels of one angiogenic growth factor, the epidermal growth factor (EGF), follow the order: ♂ E3FAD > ♂ E4FAD ≥ ♀ E3FAD > ♀ E4FAD (71). Importantly, peripheral EGF administration to mice prevented cognitive decline, cerebrovascular leakiness, and vessel coverage deficits in ♀ E4FAD (71). Therefore, disruption of plasma angiogenic growth factor levels is a potential downstream pathway that contributes to BBB dysfunction in EFAD mice.
Fig. 6.

Cerebrovascular leakiness and vessel coverage in 8-month-old EFAD mice. Fibrinogen (red) levels in the CX (A) and SB (B) follow the order: ♀ E4FAD > ♀ E3FAD = ♂ E4FAD > ♂ E3FAD. Green = CD31; n = 8. Data expressed as mean ± SEM. *P < 0.05 by two-way ANOVA and Tukey’s post hoc comparisons. #P < 0.05 by two-way AVOVA followed Fisher’s LSD test (new data). C: Laminin (green) staining as a marker of total vessel coverage follows the order: ♂ E3FAD > ♂ E4FAD ≥ ♀ E3FAD > ♀ E4FAD in the deep CX and SB [adapted from (71)].
Cerebral amyloid angiopathy and microbleeds in ♂ and ♀ EFAD mice.
Cerebral amyloid angiopathy (CAA) and microbleeds are often described as linked pathology. Here, we present data that CAA in the CX is higher in E4FAD mice regardless of sex (upper layers > deep layers), but not the SB (Fig. 7A), consistent with other investigators (73). The CAA is present primarily in larger vessels, although some capillary CAA is observed. When assessed using triple staining confocal analysis of the larger vessels, Aβ is found attached to the outside of laminin, in the perivascular space between brain endothelial cells and laminin, and penetrating the vessel (Fig. 7B). These data raise the important general question of how Aβ in the brain interstitial fluid deposits as CAA. Identified interstitial fluid drainage pathways include: 1) perivascular flow along the capillaries to the arteriole/artery basement membrane and; 2) the glymphatic pathway, in which CSF enters the brain along paravascular channels surrounding small arteries, exchanges with interstitial fluid, which is then cleared along paravascular spaces of large veins. We propose that our data are consistent with apoE4-induced impaired perivascular Aβ drainage in arterioles (179, 180), which also results in capillary CAA. In human AD patients, CAA is higher with APOE4 (181). However, in APOE genotype-matched AD patients, CAA is higher in ♂ compared with ♀. While one suggestion for this apparent difference between the EFAD mice and humans is that CerVD in humans is unique (73), there are several alternative explanations. First, the ♂/♀ differences in CAA may become more apparent in aged EFAD mice. Second, apoE3 may be more protective in ♀ versus ♂ ε3/4 carriers. Third, and our hypothesis, is that peripheral AD-risk factors are greater in ♂, inducing cerebrovascular and BBB dysfunction, leading to CAA.
Fig. 7.

CAA and microbleeds in 8-month-old EFAD mice. A: Cortical CAA-like deposition is highest in ♀ E4FAD and ♂ E4FAD; n = 8. Data expressed as mean ± SEM. *P < 0.05 by two-way ANOVA and Tukey’s post hoc comparisons (new data). B: Aβ deposits are found attached to laminin, in the perivascular space and inside the vessel lumen. Examples from ♀ E4FAD mice (new data). C: Microbleeds are highest in ♀ E4FAD mice [adapted from (71)].
In EFAD mice, microbleeds partially mimic fibrinogen extravasation, rather than CAA (71, 73, 182): ♀ E4FAD > ♀ E3FAD > ♂ E4FAD = ♂ E3FAD (Fig. 7C) (71). In AD, microbleeds are associated with ♂, higher blood pressure, lower CSF Aβ42, and APOE4 (183). The same considerations for CAA may underlie the microbleed differences between EFAD mice and humans. In addition, microbleeds are not severe, even in ♀ E4FAD mice, and in our experience, are localized to the deeper layers of the CX. Therefore, at 8 months, microbleeds could be driven by capillary or postcapillary venule breakdown, rather than CAA-induced arteriole damage. Alternatively, APOE-modulated damage to different parts of the vascular tree may be APOE genotype- and sex-specific, eventually leading to microbleeds.
EFAD MICE AND THERAPEUTIC TREATMENT
EFAD mice are a vital tool for testing therapeutics
EFAD mice exhibit temporally-defined, APOE-modulated changes in outcomes for efficacy (behavior, neuronal protein levels), pharmacodynamic activity (Aβ levels, neuroinflammation, and CerVD), and indirect targeted engagement. Thus, the activity of therapies/drug-like molecules can be assessed in prevention, treatment, and reversal paradigms. Therapies can target proximal (e.g., apoE lipidation) or downstream processes (e.g., neuroinflammation) that are disrupted by APOE4 and the detrimental interaction of ♀, APOE4, and Aβ (e.g., sex hormone based). Further, whether a therapy is specific for APOE4, Aβ, or is applicable for all groups (APOE genotype, sex, ±Αβ) can be determined by incorporating E2FAD, E3FAD mice, ±FAD mutations (EFAD, EFAD-NC). Thus far, pharmacological and nonpharmacological therapies targeting apoE-lipidation, general neuroprotection, CerVD, and sex hormone pathways have been tested in EFAD mice.
Targeting apoE4 lipidation
Lower lipidation/lipoprotein-associated levels in apoE4 was targeted in ♂ E4FAD mice using retinoid X receptor (RXR) agonists. The history of RXR agonists in the context of AD has been extensively reviewed elsewhere (184–192). Briefly, key issues center on whether RXR agonists increase apoE levels or lipidation (via increasing ABCA1 levels), the effect of human apoE4 and the duration of treatment. In ♂ E4FAD mice, short-term RXR agonist treatment (5.75–6 months) increased ABCA1 levels, apoE4 lipoprotein-association/lipidation, and apoE4/Aβ complex, decreased soluble Aβ, and increased PSD95 in the HP (172). However, RXR agonists induced no beneficial effects in ♂ E4FAD using a prevention protocol (5–6 months) and actually increased soluble Aβ levels in ♂ E3FAD and ♂ E4FAD CX with the short-term protocol, possibly the result of systemic hepatomegaly. These data support RXR agonists to address the loss-of-function associated with APOE4 and exacerbated by Aβ pathology, i.e., low levels of apoE4 lipoprotein association/lipidation. However, further development is needed to address detrimental systemic effects (RXR response elements in critical hepatic enzymes) and to discern whether RXR agonists are beneficial in specific paradigms and EFAD groups (as discussed above).
Neuroprotectants
The experimental drug, 4-methyl-5-(2-(nitrooxy) ethyl) thiazol-3-ium chloride, was designed based on the anticonvulsant drug, Zendra, to activate the NO/cyclic guanosine monophosphate/phosphorylated cAMP-response element binding protein (pCREB) pathway as a multifunctional protective drug. 4-methyl-5-(2-(nitrooxy) ethyl) thiazol-3-ium chloride treatment of ♂ E4FAD mice (3.5–6 months) lowered Aβ levels (soluble and insoluble) and increased both pCREB and PSD95 (193).
Nonpharmacological treatments
Additional nonpharmacological treatments tested in EFAD mice include EGF targeting cerebrovascular dysfunction (71) and 17-β estradiol (E2) treatment of ovariectomized (OVX) ♀ EFAD mice. E2 decreased soluble Aβ42 levels in ♀ E3FAD and ♀ E4FAD mice. However, insoluble Aβ levels increased in ♀ E4FAD mice (194). Therefore, the activity of E2 may be dependent on the relative impact of extracellular and soluble Aβ on AD-induced neurodegeneration, with the results consistent with the hypothesis that soluble oAβ is toxic, while amyloid plaques are relatively benign (Fig. 1).
POTENTIAL DISADVANTAGES OF THE EFAD MOUSE MODEL
EFAD mice share weaknesses common to all FAD-Tg mice, including questions regarding the relevance of FAD transgene-induced pathology to sporadic AD, particularly during aging. The comparison of rodent to human aging is also a construct with inherent limitations based on differences in species, and strain differences among mice. Thus, it is useful to evaluate whether FAD-Tg mice can mimic aspects of aging and AD pathology. A major issue with h-APP-Tg mice is that their 2 year life-span may not be sufficient to observe the development of AD pathology (195). As with most FAD-Tg models, AD-related pathology, particularly Aβ deposition, develops prior to middle age, which does not model the human condition. These concerns are mitigated to some extent in the EFAD by two factors. First, based on the genetic background of EFAD mice [(B6SJLF1×C57BL/6) from 5xFAD (50) × (C57BL/6) from APOE-TR (32)], we estimate that 10–14 months will represent middle age and 18 months will represent old age (https://www.nia.nih.gov/research/dab/aged-rodent-colonies-handbook/strain-survival-information) (196). Specifically, with the known survival rates for the background strains of the EFAD mice: 1) 5xFAD have a ∼75% survival rate at 16 months (197); and 2) C57BL/6 and APOE-TR have 75% survival rate at 24 months for ♂ and 22 months for ♀ (196). Thus, the EFAD mice have ∼75% survival at 20 months for ♂ and 19 months for ♀, making our target “old age” 18 months. Although specific measures of AD pathology in the EFAD are significant by 6 months, pathology continues to develop until at least 18 months, the oldest EFAD mice we have examined thus far (data not shown). Second, the EFAD-NC littermates provide both a comparison to the EFAD mice and a complementary approach to the address functional questions about APOE in the absence of FAD-induced pathology.
Despite these limitations, EFAD mice are the only well-characterized FAD/h-APOE-Tg mouse model with an extensive and growing provenance. Consistent with human AD patients, E4FAD mice develop pathology in a number of APOE genotype-, sex-, and age-dependent pathways. EFAD mice are a tractable mouse model to study a number of AD-related outcomes, including changes in behavior, Aβ deposition, tau pathology, neuroinflammation, and neuronal viability (Table 2), as well as apoE lipidation and Aβ solubility (Table 3). These mice also allow for study of the interactions among AD risk factors, including age, APOE genotype, and sex.
FUTURE DIRECTIONS
Using EFAD mice as a model of aging and development of AD pathology
Understanding the interaction and dominance of APOE genotype versus sex with aging.
Identification of the interactions between APOE genotype and sex are critical to understanding both aging and the development of AD pathology. Making predictions requires identification of the dominant risk factor in a given comparison, APOE genotype or sex: 1) The levels of Aβ and amyloid deposition, as well as soluble Aβ levels are higher in ♀ versus ♂ in several FAD-Tg mice (Tg2576, APP/PS1, 3xTg-AD) (64, 198–203), as well as the EFAD mice (Table 3) (73). 2) In APOE-Tg mice, cognitive deficits are greater in ♀ APOE4 versus APOE3 [for review (21, 204–209)]. In EFAD mice, behavioral deficits are E4FAD > E3FAD and ♀ > ♂ in 6- and 8-month-old mice (Fig. 2, Table 2). 3) In humans, lifetime AD risk, cognitive decline and accumulation of Aβ is ♀ > ♂ in ε4 carriers. These data suggest that the greatest risk for AD is with ♀ APOE4 carriers (6–13, 102, 210–213). These observations introduce a reoccurring theme in this field of research: which risk factor is dominant in its effects on AD pathology: APOE genotype or sex, and does this change with age? Based on cognition and AD-related histopathology, our general predicted order for AD pathology, with the addition of heterozygous E3/4FAD, is: ♂ E3FAD < ♀ E3FAD < ♂ E3/4FAD < ♀ E3/4FAD < ♂ E4FAD < ♀ E4FAD (the effect of the APOE2 genotypes are discussed separately). However, the dominant risk factor in a given comparison, APOE genotype or sex, is unclear. In general, the key comparisons for establishing the dominant effects of APOE versus sex will be determined by heterozygous E3/4FAD mice versus homozygous E3FAD and E4FAD mice, as established by age and AD pathology. For example, Aβ deposition, neuroinflammation, and tau pathology in ♂ E4FAD versus ♀ E3/4FAD will predict a dominant risk factor: APOE4 if ♂ E4FAD shows the greatest pathology, or ♀ sex if ♀ E3/4FAD has the greater pathology. How this relative risk changes with age is critical. As well, using the EFAD-NC we can determine the effect of APOE versus sex interactions on normal aging.
Understanding trajectories, cliffs, and therapeutic windows.
Multiple measures of AD pathology during aging will inform two critical components that indicate the relative contribution of risk factors, APOE or sex, and how their contributions are altered along the trajectory of the disease: 1) “Cliffs” or tipping points suggest a clear dominant risk factor: ♀ sex or APOE. For example, while ♀ E4FAD mice exhibit the greatest behavioral deficits and Aβ pathology at both 6 and 8 months, ♀ E3FAD ≈ ♂ E4FAD at 8 months (71), suggesting a cliff or tipping point where ♀ sex is dominant compared with APOE genotype. However, unlike humans, as the ♀ EFAD mice age, they maintain 45–80% E2 levels and normal uterine weight (214–218), which may produce an interesting phenotype at older ages with the scale tipping toward the dominance of APOE genotype. This change can be compared with OVX ± E2 replacement. 2) Therapeutic windows are periods during which specific components of AD pathology are differentially affected by APOE or ♀ sex, allowing us to design and test specific therapeutic targets in preclinical studies using prevention or reversal paradigms.
Understanding the function of APOE2.
The majority of the published data on the EFAD mice have used ♂ mice ≤8 months. As ♂ and ♀ mice are aged from 10 to 14 to 18 months, sex and APOE genotype interact to induce significant differences in various components of AD pathology (data not shown). As well, all of our work thus far has been with APOE+/+/5xFAD+/− mice. As the APOE heterozygous genotypes are investigated (ε2/3, ε2/4, ε3/4), the influence of APOE genotype and sex interactions can be fully defined. In studying the APOE2 genotypes, it is important to keep in mind that if there are functional differences among ε2/2, ε2/3, and ε2/4, it will likely go unidentified in all but the largest human cohort studies. This is because most studies will be underpowered for significance because of the low frequency of the ε2 alleles [estimated: ε2/2 at 0.4%, ε2/3 at 8.8%, and ε2/4 at 1.5% (5, 6)]. This effect is exacerbated if the APOE2 genotypes are further stratified by age, AD status, and sex, resulting in the apparently contradictory literature for this field. However, heterozygous genotypes of APOE2 mice can be bred to reach significance via power analysis for any variable in comparison to heterozygous genotypes of APOE3 and APOE4. Indeed, the study of ε2/2, ε2/3, and ε2/4 is perhaps a more subtle model to study the protective effects in both a normal (EFAD-NC) and AD (EFAD) cohort of mice. These results are key for identifying how the genotypes of APOE2 may cause differential effects in the context of being protective factors, for example, does ε2/4 behave more like the risk ε4 or the protective ε2. These studies will provide new insights into how APOE2 imparts healthy brain aging and reduces AD risk, leading to diagnostic biomarkers and identification of therapeutic targets.
Using EFAD to identify environmental risk factors in AD pathology
About 98% of the human AD cases are sporadic with only half the cases linked to APOE4 and other genetic loci identified by genome-wide association study, suggesting the presence of other genetic or environmental risk factors and, thus, the potential interaction between genetic and environmental risk factors (88, 219–227). Thus, while APOE4 is the major genetic risk factor for AD, a number of environmental or lifestyle risk factors, have also been identified (228–233). Two examples are given below.
Effect of high fat diets on AD.
Epidemiological studies in humans consistently show an interaction between obesity and dementia/increased AD risk (228, 234–239), though the interaction with sex remains controversial (240–243). High fat diet-induced obesity accelerates AD pathology in FAD-Tg mice (244–248) and impairs cognition in APOE4-TR mice (249). However, the interaction among obesity, APOE genotype, and sex in modulating development of AD pathology is poorly understood (250, 251). EFAD mice are a relevant model to address this question and the importance of lifestyle risk factors and their association with APOE in a genotype- and sex-dependent manner.
Effect of particulate air pollutants.
The role of particulate air pollutants in accelerating cognitive impairment has been established in human (252–255) and WT mouse studies (256). Exposure to particulate air pollutants increased Aβ deposition, amyloid plaques and soluble oAβ in ♀ E4FAD compared with ♀ E3FAD mice (257). This increased susceptibility of ♀ ε4 carriers to the neurotoxicity of particulate air pollutants provides evidence for interactive effects among genetic and environmental risk factors.
Using EFAD as a therapeutic model
Repurposing cardiovascular disease drugs.
As discussed above, we previously demonstrated that in EFAD mice, induction of ABCA1/ABCG1 with RXR agonists increased apoE4 lipoprotein-association/lipidation, decreased soluble Aβ, and increased PSD95 in the HP (172). However, treatment induced severe hepatomegaly, limiting RXR agonism for AD treatment. Approaches for targeting apoE lipoprotein-association/lipidation in the brain without the use of RXR agonists emerged as a promising alternative as the major enzymatic and lipid transport activities involved in the peripheral system are also expressed in the brain (258–279). The lipoprotein-association/lipidation of apoE in the brain parenchyma is the result of intercellular lipoprotein maturation and remodeling (263, 274, 280–291). Current strategies include directly targeting ABCA1 activity with an apoE mimetic peptide in the EFAD to evaluate its effect on apoE levels or apoE4 lipoprotein-association/lipidation and reduction of AD pathology.
Cerebrovascular dysfunction (CerVD).
Many of the planned treatment strategies that target either the proximal or downstream processes modulated by APOE and sex will likely also target CerVD. Proximally, directly targeting the structural and functional deficits of apoE4 may ameliorate detrimental changes that cause CerVD (190, 292–294). Targeting downstream signaling pathways or the soluble mediators produced by APOE-modulated activated glia (astrocytes and microglia) and pericytes may ameliorate CerVD, or prevent the risk with a subsequent additional hit, such as peripheral inflammation and high fat diets (292). Further, brain endothelial cells are often overlooked as a direct therapeutic target. The advantages of this target include: 1) Brain penetration is not required; 2) Peripheral risk factors will likely initially target brain endothelial cells rather than cells in the brain and; 3) As highlighted by the EGF treatment study, as brain endothelial cells play a central role in the homeostasis of the CNS, targeting brain endothelial cells may induce a pronounced beneficial effect on cognition. Currently, the ability of EGF to reverse cognitive and cerebrovascular deficits is under evaluation.
Neuroinflammation.
Epidemiological studies targeting peripheral inflammation for AD indicate APOE-dependent lowering of AD risk due to nonsteroidal anti-inflammatory drugs (NSAIDs), with a beneficial effect for ε4 resulting in initiation of AD Anti-inflammatory Prevention Trial (ADAPT) (295–304). However, ADAPT failed and led to more criticism for evaluating the role of neuroinflammation in AD. It remains unclear whether targeting AD-relevant neuroinflammation receptor pathways is beneficial, detrimental, or not effective. For example, data from FAD-Tg mice provide evidence for beneficial (25, 305–307) and detrimental effects from TLR4 inhibition (308–311). Inflammatory receptors may function differently depending on stage of AD pathology and APOE genotype, necessitating prevention and treatment protocols. EFAD mice are an ideal model to investigate this interplay between neuroinflammation and neurodegeneration that result in cognitive behavioral impairments, and for identifying the appropriate timing and targets involved in AD-associated neuroinflammation. Currently, EFAD are being evaluated with a prevention and reversal paradigm trial with a small TLR4 antagonist to evaluate its effect on AD pathology.
Selective estrogen mimics and selective estrogen receptor modulators.
E2 is key for ♀ vulnerability to APOE4-induced AD risk and pathology: OVX-induced loss of circulating E2 in premenopausal women (312–316) and FAD-Tg mice (201, 317, 318) causes cognitive deficits that can be reversed by E2 and estrogen therapy (ET), and in FAD-Tg mice, the OVX-induced increase in amyloid deposition is also reversed with ET (319, 320). However, the timing of ET in relation to the risk of AD in naturally menopausal women is a critical factor due to the apparent opposing outcomes based on early versus late menopause treatment (321). The controversial outcomes associated with timing could be addressed with the development of safe ET alternatives for the prevention and treatment of AD, potentially specific for the APOE genotype of patient. Based on the need for ET alternatives, we plan to study selective estrogen receptor modulators (322–325), or selective estrogen mimics (326) in ±OVX ♀ EFAD mice.
SUMMARY
Given the prevalence of AD and the repeated failure of clinical trials, it is critical to develop Tg-mouse models to understand the mechanisms driving the trajectory of AD, identify early-stage biomarkers, and test preclinical therapeutic targets. EFAD mice mimic a range of AD-related pathologies, including cognitive decline, region-specific Aβ and plaque deposition, progressive neuroinflammation, reduced synaptic viability, and cerebrovascular dysfunction. EFAD mice provide insight into the specific pathways and mechanisms that underlie APOE- and sex-dependent modulation of AD pathology. A complete characterization of the EFAD mice with age will enable an understanding of how the interaction among the greatest AD risk factors modulates AD-related pathology, specifically age, APOE genotype, and sex. Consistent with the underlying principles of personalized medicine, only when we understand these interactions can we begin to design therapeutic approaches for the prevention and treatment of AD.
Supplementary Material
Acknowledgments
The authors acknowledge the histology/imaging services that were provided by the Research Resources Center, Research Histology and Tissue Imaging Core at the University of Illinois at Chicago established with the support of the Vice Chancellor of Research.
Footnotes
Abbreviations:
- AA
- arachidonic acid
- Aβ
- amyloid-β
- AD
- Alzheimer’s disease
- APP
- amyloid precursor protein
- BBB
- blood-brain barrier
- CAA
- cerebral amyloid angiopathy
- CSF
- cerebrospinal fluid
- CerVD
- cerebrovascular dysfunction
- CX
- cortex
- E2
- 17-β estradiol
- EFAD-NC
- EFAD noncarrier
- EGF
- epidermal growth factor
- ET
- estrogen therapy
- FAD
- familial Alzheimer’s disease
- GFAP
- glial fibrillary acidic protein
- h-APOE
- human APOE
- HP
- hippocampus
- IL-1β
- interleukin-1β
- IL-4R
- interleukin-4 receptor
- mAb
- monoclonal antibody
- m-APOE
- mouse APOE
- MOAB-2
- mAb specific to amyloid-β
- MWM
- Morris water maze
- NFT
- neurofibrillary tangle
- NOR
- novel object recognition
- NSAID
- nonsteroidal anti-inflammatory drug
- oAβ
- oligomeric conformation of amyloid-β
- OVX
- ovariectomized
- pCREB
- phosphorylated cAMP-response element binding protein
- PL
- phospholipid
- PSD95
- postsynaptic density protein 95
- pTau
- phosphorylated tau
- RXR
- retinoid X receptor
- SB
- subiculum
- TBSX
- TBS + Triton X-100
- Tg
- transgenic
- Thio-S
- thioflavine-S
- Thy-1
- thymocyte differentiation antigen 1 theta
- TLR4
- toll-like receptor 4
- TR
- targeted-replacement
- YM
- Y-maze
- ♀
- female
- ♂
- male
This work was supported by National Institutes of Health Grants R21 AG051233 (M.J.L.), R21 AG044682 (M.J.L.), UH2NS100127 (M.J.L.), and institutional funding from University of Illinois College of Medicine (M.J.L. and L.M.T.).
The online version of this article (available at http://www.jlr.org) contains a supplement.
REFERENCES
- 1.Erten-Lyons D., Sherbakov L. O., Piccinin A. M., Hofer S. M., Dodge H. H., Quinn J. F., Woltjer R. L., Kramer P. L., and Kaye J. A.. 2012. Review of selected databases of longitudinal aging studies. Alzheimers Dement. 8: 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shineman D. W., Basi G. S., Bizon J. L., Colton C. A., Greenberg B. D., Hollister B. A., Lincecum J., Leblanc G. G., Lee L. B., Luo F., et al. . 2011. Accelerating drug discovery for Alzheimer’s disease: best practices for preclinical animal studies. Alzheimers Res. Ther. 3: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reitz C., and Mayeux R.. 2010. Use of genetic variation as biomarkers for mild cognitive impairment and progression of mild cognitive impairment to dementia. J. Alzheimers Dis. 19: 229–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leoni V. 2011. The effect of apolipoprotein E (ApoE) genotype on biomarkers of amyloidogenesis, tau pathology and neurodegeneration in Alzheimer’s disease. Clin. Chem. Lab. Med. 49: 375–383. [DOI] [PubMed] [Google Scholar]
- 5.Shinohara M., Kanekiyo T., Yang L., Linthicum D., Shinohara M., Fu Y., Price L., Frisch-Daiello J. L., Han X., Fryer J. D., et al. . APOE2 eases cognitive decline during aging: clinical and preclinical evaluations. Ann. Neurol. Epub ahead of print. March 2, 2016; doi:10.1002/ana.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrer L. A., Cupples L. A., Haines J. L., Hyman B., Kukull W. A., Mayeux R., Myers R. H., Pericak-Vance M. A., Risch N., and van Duijn C. M.. 1997. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. J. Am. Med. Assoc. 278: 1349–1356. [PubMed] [Google Scholar]
- 7.Breitner J. C., Wyse B. W., Anthony J. C., Welsh-Bohmer K. A., Steffens D. C., Norton M. C., Tschanz J. T., Plassman B. L., Meyer M. R., Skoog I., et al. . 1999. APOE-epsilon4 count predicts age when prevalence of AD increases, then declines: the Cache County Study. Neurology. 53: 321–331. [DOI] [PubMed] [Google Scholar]
- 8.Martinez M., Campion D., Brice A., Hannequin D., Dubois B., Didierjean O., Michon A., Thomas-Anterion C., Puel M., Frebourg T., et al. . 1998. Apolipoprotein E epsilon4 allele and familial aggregation of Alzheimer disease. Arch. Neurol. 55: 810–816. [DOI] [PubMed] [Google Scholar]
- 9.Andersen K., Launer L. J., Dewey M. E., Letenneur L., Ott A., Copeland J. R., Dartigues J. F., Kragh-Sorensen P., Baldereschi M., Brayne C., et al. . 1999. Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. EURODEM Incidence Research Group. Neurology. 53: 1992–1997. [DOI] [PubMed] [Google Scholar]
- 10.Bretsky P. M., Buckwalter J. G., Seeman T. E., Miller C. A., Poirier J., Schellenberg G. D., Finch C. E., and Henderson V. W.. 1999. Evidence for an interaction between apolipoprotein E genotype, gender, and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 13: 216–221. [DOI] [PubMed] [Google Scholar]
- 11.Molero A. E., Pino-Ramirez G., and Maestre G. E.. 2001. Modulation by age and gender of risk for Alzheimer’s disease and vascular dementia associated with the apolipoprotein E-epsilon4 allele in Latin Americans: findings from the Maracaibo Aging Study. Neurosci. Lett. 307: 5–8. [DOI] [PubMed] [Google Scholar]
- 12.Corder E. H., Ghebremedhin E., Taylor M. G., Thal D. R., Ohm T. G., and Braak H.. 2004. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann. N. Y. Acad. Sci. 1019: 24–28. [DOI] [PubMed] [Google Scholar]
- 13.Altmann A., Tian L., Henderson V. W., and Greicius M. D.; Alzheimer’s Disease Neuroimaging Initiative Investigators. 2014. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 75: 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larson M. E., and Lesne S. E.. 2012. Soluble Abeta oligomer production and toxicity. J. Neurochem. 120 (Suppl. 1): 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selkoe D. J. 2008. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192: 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tu S., Okamoto S., Lipton S. A., and Xu H.. 2014. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 9: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klyubin I., Cullen W. K., Hu N. W., and Rowan M. J.. 2012. Alzheimer’s disease Abeta assemblies mediating rapid disruption of synaptic plasticity and memory. Mol. Brain. 5: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., and Masters C. L.. 1999. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46: 860–866. [DOI] [PubMed] [Google Scholar]
- 19.Tomic J. L., Pensalfini A., Head E., and Glabe C. G.. 2009. Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol. Dis. 35: 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tai L. M., Bilousova T., Jungbauer L., Roeske S. K., Youmans K. L., Yu C., Poon W. W., Cornwell L. B., Miller C. A., Vinters H. V., et al. . 2013. Levels of soluble apolipoprotein E/amyloid-beta (Abeta) complex are reduced and oligomeric Abeta increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J. Biol. Chem. 288: 5914–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tai L. M., Youmans K. L., Jungbauer L., Yu C., and Ladu M. J.. 2011. Introducing human APOE into Abeta transgenic mouse models. Int. J. Alzheimers Dis. 2011: 810981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trommer B. L., Shah C., Yun S. H., Gamkrelidze G., Pasternak E. S., Ye G. L., Sotak M., Sullivan P. M., Pasternak J. F., and LaDu M. J.. 2004. ApoE isoform affects LTP in human targeted replacement mice. Neuroreport. 15: 2655–2658. [DOI] [PubMed] [Google Scholar]
- 23.Trommer B. L., Shah C., Yun S. H., Gamkrelidze G., Pasternak E. S., Stine W. B., Manelli A., Sullivan P., Pasternak J. F., and LaDu M. J.. 2005. ApoE isoform-specific effects on LTP: blockade by oligomeric amyloid-beta1–42. Neurobiol. Dis. 18: 75–82. [DOI] [PubMed] [Google Scholar]
- 24.Bien-Ly N., Andrews-Zwilling Y., Xu Q., Bernardo A., Wang C., and Huang Y.. 2011. C-terminal-truncated apolipoprotein apoE4 inefficiently clears amyloid-β (Aβ) and acts in concert with Aβ to elicit neuronal and behavioral deficits in mice. Proc. Natl. Acad. Sci. USA. 108: 4236–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y., Nwabuisi-Heath E., Dumanis S. B., Tai L. M., Yu C., Rebeck G. W., and LaDu M. J.. 2012. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 60: 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao F., Zhang T. J., Jiang H., Lefton K. B., Robinson G. O., Vassar R., Sullivan P. M., and Holtzman D. M.. 2015. Murine versus human apolipoprotein E4: differential facilitation of and co-localization in cerebral amyloid angiopathy and amyloid plaques in APP transgenic mouse models. Acta Neuropathol. Commun. 3: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fagan A. M., Watson M., Parsadanian M., Bales K. R., Paul S. M., and Holtzman D. M.. 2002. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 9: 305–318. [DOI] [PubMed] [Google Scholar]
- 28.Fryer J. D., Simmons K., Parsadanian M., Bales K. R., Paul S. M., Sullivan P. M., and Holtzman D. M.. 2005. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J. Neurosci. 25: 2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oddo S., Caccamo A., Cheng D., and LaFerla F. M.. 2009. Genetically altering Abeta distribution from the brain to the vasculature ameliorates tau pathology. Brain Pathol. 19: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holtzman D. M., Bales K. R., Tenkova T., Fagan A. M., Parsadanian M., Sartorius L. J., Mackey B., Olney J., McKeel D., Wozniak D., et al. . 2000. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 97: 2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buttini M., Yu G. Q., Shockley K., Huang Y., Jones B., Masliah E., Mallory M., Yeo T., Longo F. M., and Mucke L.. 2002. Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid beta peptides but not on plaque formation. J. Neurosci. 22: 10539–10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sullivan P. M., Mezdour H., Aratani Y., Knouff C., Najib J., Reddick R. L., Quarfordt S. H., and Maeda N.. 1997. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 272: 17972–17980. [DOI] [PubMed] [Google Scholar]
- 33.Dodart J. C., Meziane H., Mathis C., Bales K. R., Paul S. M., and Ungerer A.. 1999. Behavioral disturbances in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Behav. Neurosci. 113: 982–990. [DOI] [PubMed] [Google Scholar]
- 34.Hartman R. E., Izumi Y., Bales K. R., Paul S. M., Wozniak D. F., and Holtzman D. M.. 2005. Treatment with an amyloid-beta antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer’s disease. J. Neurosci. 25: 6213–6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen G., Chen K. S., Knox J., Inglis J., Bernard A., Martin S. J., Justice A., McConlogue L., Games D., Freedman S. B., et al. . 2000. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Nature. 408: 975–979. [DOI] [PubMed] [Google Scholar]
- 36.Nilsson L. N., Arendash G. W., Leighty R. E., Costa D. A., Low M. A., Garcia M. F., Cracciolo J. R., Rojiani A., Wu X., Bales K. R., Paul S. M., and Potter H.. 2004. Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol Aging. 25: 1153–1167. [DOI] [PubMed] [Google Scholar]
- 37.Games D., Adams D., Alessandrini R., Barbour R., Berthelette P., Blackwell C., Carr T., Clemens J., Donaldson T., Gillespie F., et al. . 1995. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 373: 523–527. [DOI] [PubMed] [Google Scholar]
- 38.Johnson-Wood K., Lee M., Motter R., Hu K., Gordon G., Barbour R., Khan K., Gordon M., Tan H., Games D., et al. . 1997. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl. Acad. Sci. USA. 94: 1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bales K. R., Verina T., Cummins D. J., Du Y., Dodel R. C., Saura J., Fishman C. E., DeLong C. A., Piccardo P., Petegnief V., et al. . 1999. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 96: 15233–15238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dodart J. C., Mathis C., Bales K. R., Paul S. M., and Ungerer A.. 2000. Behavioral deficits in APP(V717F) transgenic mice deficient for the apolipoprotein E gene. Neuroreport. 11: 603–607. [DOI] [PubMed] [Google Scholar]
- 41.Irizarry M. C., Soriano F., McNamara M., Page K. J., Schenk D., Games D., and Hyman B. T.. 1997. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J. Neurosci. 17: 7053–7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masliah E., Sisk A., Mallory M., and Games D.. 2001. Neurofibrillary pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. J. Neuropathol. Exp. Neurol. 60: 357–368. [DOI] [PubMed] [Google Scholar]
- 43.Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Youkin S., Yang F., and Cole G.. 1996. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 274: 99–102. [DOI] [PubMed] [Google Scholar]
- 44.Irizarry M. C., McNamara M., Fedorchak K., Hsiao K., and Hyman B. T.. 1997. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J. Neuropathol. Exp. Neurol. 56: 965–973. [DOI] [PubMed] [Google Scholar]
- 45.Yan P., Zhu A., Liao F., Xiao Q., Kraft A. W., Gonzales E., Perez R., Greenberg S. M., Holtzman D. M., and Lee J. M.. 2015. Minocycline reduces spontaneous hemorrhage in mouse models of cerebral amyloid angiopathy. Stroke. 46: 1633–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irizarry M. C., Rebeck G. W., Cheung B., Bales K., Paul S. M., Holzman D., and Hyman B. T.. 2000. Modulation of A beta deposition in APP transgenic mice by an apolipoprotein E null background. Ann. N. Y. Acad. Sci. 920: 171–178. [DOI] [PubMed] [Google Scholar]
- 47.Bales K. R., Liu F., Wu S., Lin S., Koger D., DeLong C., Hansen J. C., Sullivan P. M., and Paul S. M.. 2009. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29: 6771–6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R. B., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., et al. . 2011. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 3: 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohno M., Chang L., Tseng W., Oakley H., Citron M., Klein W. L., Vassar R., and Disterhoft J. F.. 2006. Temporal memory deficits in Alzheimer’s mouse models: rescue by genetic deletion of BACE1. Eur. J. Neurosci. 23: 251–260. [DOI] [PubMed] [Google Scholar]
- 50.Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., et al. . 2006. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26: 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shukla V., Zheng Y. L., Mishra S. K., Amin N. D., Steiner J., Grant P., Kesavapany S., and Pant H. C.. 2013. A truncated peptide from p35, a Cdk5 activator, prevents Alzheimer’s disease phenotypes in model mice. FASEB J. 27: 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devi L., and Ohno M.. 2010. Genetic reductions of beta-site amyloid precursor protein-cleaving enzyme 1 and amyloid-beta ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer’s disease model mice. Eur. J. Neurosci. 31: 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Devi L., and Ohno M.. 2010. Phospho-eIF2alpha level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS One. 5: e12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Devi L., and Ohno M.. 2012. Mitochondrial dysfunction and accumulation of the beta-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol. Dis. 45: 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hüttenrauch M., Walter S., Kaufmann M., Weggen S., and Wirths O.. Limited effects of prolonged environmental enrichment on the pathology of 5XFAD mice. Mol. Neurobiol. Epub ahead of print. October 12, 2016; doi:10.1007/s12035-016-0167-x. [DOI] [PubMed] [Google Scholar]
- 56.Kimura R., and Ohno M.. 2009. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol. Dis. 33: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaczorowski C. C., Sametsky E., Shah S., Vassar R., and Disterhoft J. F.. 2011. Mechanisms underlying basal and learning-related intrinsic excitability in a mouse model of Alzheimer’s disease. Neurobiol. Aging. 32: 1452–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohno M., Cole S. L., Yasvoina M., Zhao J., Citron M., Berry R., Disterhoft J. F., and Vassar R.. 2007. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol. Dis. 26: 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kanno T., Tsuchiya A., and Nishizaki T.. 2014. Hyperphosphorylation of Tau at Ser396 occurs in the much earlier stage than appearance of learning and memory disorders in 5XFAD mice. Behav. Brain Res. 274: 302–306. [DOI] [PubMed] [Google Scholar]
- 60.Urano T., and Tohda C.. 2010. Icariin improves memory impairment in Alzheimer’s disease model mice (5xFAD) and attenuates amyloid beta-induced neurite atrophy. Phytother. Res. 24: 1658–1663. [DOI] [PubMed] [Google Scholar]
- 61.Kalinin S., Polak P. E., Lin S. X., Sakharkar A. J., Pandey S. C., and Feinstein D. L.. 2012. The noradrenaline precursor L-DOPS reduces pathology in a mouse model of Alzheimer’s disease. Neurobiol. Aging. 33: 1651–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jawhar S., Trawicka A., Jenneckens C., Bayer T. A., and Wirths O.. 2012. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging. 33: 196.e29–196.e40. [DOI] [PubMed] [Google Scholar]
- 63.Eimer W. A., and Vassar R.. 2013. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol. Neurodegener. 8: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhattacharya S., Haertel C., Maelicke A., and Montag D.. 2014. Galantamine slows down plaque formation and behavioral decline in the 5XFAD mouse model of Alzheimer’s disease. PLoS One. 9: e89454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spencer N. G., Lovell D. P., Elderfield K., Austen B., and Howe F. A.. 2017. Can MRI T1 be used to detect early changes in 5xFAD Alzheimer’s mouse brain? MAGMA. 30: 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Youmans K. L., Tai L. M., Nwabuisi-Heath E., Jungbauer L., Kanekiyo T., Gan M., Kim J., Eimer W. A., Estus S., Rebeck G. W., et al. . 2012. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem. 287: 41774–41786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Youmans K. L., Leung S., Zhang J., Maus E., Baysac K., Bu G., Vassar R., Yu C., and Ladu M. J.. 2011. Amyloid-beta42 alters apolipoprotein E solubility in brains of mice with five familial AD mutations. J. Neurosci. Methods. 196: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shao C. Y., Mirra S. S., Sait H. B., Sacktor T. C., and Sigurdsson E. M.. 2011. Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced Abeta and tau pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol. 122: 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neuman K. M., Molina-Campos E., Musial T. F., Price A. L., Oh K. J., Wolke M. L., Buss E. W., Scheff S. W., Mufson E. J., and Nicholson D. A.. 2015. Evidence for Alzheimer’s disease-linked synapse loss and compensation in mouse and human hippocampal CA1 pyramidal neurons. Brain Struct. Funct. 220: 3143–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu D. S., Pan X. D., Zhang J., Shen H., Collins N. C., Cole A. M., Koster K. P., Ben Aissa M., Dai X. M., Zhou M., et al. . 2015. APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol. Neurodegener. 10: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas R., Zuchowska P., Morris A. W., Marottoli F. M., Sunny S., Deaton R., Gann P. H., and Tai L. M.. 2016. Epidermal growth factor prevents APOE4 and amyloid-beta-induced cognitive and cerebrovascular deficits in female mice. Acta Neuropathol. Commun. 4: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodriguez G. A., Tai L. M., LaDu M. J., and Rebeck G. W.. 2014. Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J. Neuroinflammation. 11: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cacciottolo M., Christensen A., Moser A., Liu J., Pike C. J., Smith C., LaDu M. J., Sullivan P. M., Morgan T. E., Dolzhenko E., et al. . 2016. The APOE4 allele shows opposite sex bias in microbleeds and Alzheimer’s disease of humans and mice. Neurobiol. Aging. 37: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tai L. M., Mehra S., Shete V., Estus S., Rebeck G. W., Bu G., and Ladu M. J.. 2014. Soluble apoE/Abeta complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol. Neurodegener. 9: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou M., Huang T., Collins N., Zhang J., Shen H., Dai X., Xiao N., Wu X., Wei Z., York J., et al. . 2016. APOE4 induces site-specific tau phosphorylation through calpain-CDK5 signaling pathway in EFAD-Tg mice. Curr. Alzheimer Res. 13: 1048–1055. [DOI] [PubMed] [Google Scholar]
- 76.Moechars D., Lorent K., De Strooper B., Dewachter I., and Van Leuven F.. 1996. Expression in brain of amyloid precursor protein mutated in the alpha- secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 15: 1265–1274. [PMC free article] [PubMed] [Google Scholar]
- 77.Vidal M., Morris R., Grosveld F., and Spanopoulou E.. 1990. Tissue-specific control elements of the Thy-1 gene. EMBO J. 9: 833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sullivan P. M., Mezdour H., Quarfordt S. H., and Maeda N.. 1998. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J. Clin. Invest. 102: 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vandenberghe R., Rinne J. O., Boada M., Katayama S., Scheltens P., Vellas B., Tuchman M., Gass A., Fiebach J. B., Hill D., et al. . 2016. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 8: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carlson C., Siemers E., Hake A., Case M., Hayduk R., Suhy J., Oh J., and Barakos J.. 2016. Amyloid-related imaging abnormalities from trials of solanezumab for Alzheimer’s disease. Alzheimers Dement (Amst). 2: 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doody R. S., Thomas R. G., Farlow M., Iwatsubo T., Vellas B., Joffe S., Kieburtz K., Raman R., Sun X., Aisen P. S., et al. . 2014. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370: 311–321. [DOI] [PubMed] [Google Scholar]
- 82.The Lancet Neurology. 2017. Solanezumab: too late in mild Alzheimer’s disease? Lancet Neurol. 16: 97. [DOI] [PubMed] [Google Scholar]
- 83.Le Couteur D. G., Hunter S., and Brayne C.. 2016. Solanezumab and the amyloid hypothesis for Alzheimer’s disease. BMJ. 355: i6771. [DOI] [PubMed] [Google Scholar]
- 84.Piazza F., and Winblad B.. 2016. Amyloid-related imaging abnormalities (ARIA) in immunotherapy trials for Alzheimer’s disease: need for prognostic biomarkers? J. Alzheimers Dis. 52: 417–420. [DOI] [PubMed] [Google Scholar]
- 85.Abbott A., and Dolgin E.. 2016. Failed Alzheimer’s trial does not kill leading theory of disease. Nature. 540: 15–16. [DOI] [PubMed] [Google Scholar]
- 86.Pimplikar S. W. 2009. Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int. J. Biochem. Cell Biol. 41: 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tomiyama T., Matsuyama S., Iso H., Umeda T., Takuma H., Ohnishi K., Ishibashi K., Teraoka R., Sakama N., Yamashita T., et al. . 2010. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 30: 4845–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lazarov O., and Marr R. A.. 2013. Of mice and men: neurogenesis, cognition and Alzheimer’s disease. Front. Aging Neurosci. 5: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reid A. T., and Evans A. C.. 2013. Structural networks in Alzheimer’s disease. Eur. Neuropsychopharmacol. 23: 63–77. [DOI] [PubMed] [Google Scholar]
- 90.Risacher S. L., Shen L., West J. D., Kim S., McDonald B. C., Beckett L. A., Harvey D. J., Jack C. R. Jr., Weiner M. W., and Saykin A. J.. 2010. Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol. Aging. 31: 1401–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hsiung G. Y., Sadovnick A. D., and Feldman H.. 2004. Apolipoprotein E epsilon4 genotype as a risk factor for cognitive decline and dementia: data from the Canadian Study of Health and Aging. CMAJ. 171: 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grossberg G. T. 2003. Diagnosis and treatment of Alzheimer’s disease. J. Clin. Psychiatry. 64(Suppl. 9): 3–6. [PubMed] [Google Scholar]
- 93.Reiman E. M., Caselli R. J., Chen K., Alexander G. E., Bandy D., and Frost J.. 2001. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: a foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 98: 3334–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Budson A. E., Desikan R., Daffner K. R., and Schacter D. L.. 2001. Perceptual false recognition in Alzheimer’s disease. Neuropsychology. 15: 230–243. [PubMed] [Google Scholar]
- 95.Devanand D. P., Sano M., Tang M. X., Taylor S., Gurland B. J., Wilder D., Stern Y., and Mayeux R.. 1996. Depressed mood and the incidence of Alzheimer’s disease in the elderly living in the community. Arch. Gen. Psychiatry. 53: 175–182. [DOI] [PubMed] [Google Scholar]
- 96.Braak H., and Braak E.. 1994. Morphological criteria for the recognition of Alzheimer’s disease and the distribution pattern of cortical changes related to this disorder. Neurobiol. Aging. 15: 355–356; discussion 379–380. [DOI] [PubMed] [Google Scholar]
- 97.Mirra S. S., Heyman A., McKeel D., Sumi S. M., Crain B. J., Brownlee L. M., Vogel F. S., Hughes J. P., van Belle G., and Berg L.. 1991. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 41: 479–486. [DOI] [PubMed] [Google Scholar]
- 98.Evans D. A., Funkenstein H. H., Albert M. S., Scherr P. A., Cook N. R., Chown M. J., Hebert L. E., Hennekens C. H., and Taylor J. O.. 1989. Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. J. Am. Med. Assoc. 262: 2551–2556. [PubMed] [Google Scholar]
- 99.Berlau D. J., Corrada M. M., Head E., and Kawas C. H.. 2009. APOE epsilon2 is associated with intact cognition but increased Alzheimer pathology in the oldest old. Neurology. 72: 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wilson R. S., Bienias J. L., Berry-Kravis E., Evans D. A., and Bennett D. A.. 2002. The apolipoprotein E epsilon 2 allele and decline in episodic memory. J. Neurol. Neurosurg. Psychiatry. 73: 672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Corrada M. M., Paganini-Hill A., Berlau D. J., and Kawas C. H.. 2013. Apolipoprotein E genotype, dementia, and mortality in the oldest old: the 90+ Study. Alzheimers Dement. 9: 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hyman B. T., Gomez-Isla T., Briggs M., Chung H., Nichols S., Kohout F., and Wallace R.. 1996. Apolipoprotein E and cognitive change in an elderly population. Ann. Neurol. 40: 55–66. [DOI] [PubMed] [Google Scholar]
- 103.Beffert U., Cohn J. S., Petit-Turcotte C., Tremblay M., Aumont N., Ramassamy C., Davignon J., and Poirier J.. 1999. Apolipoprotein E and beta-amyloid levels in the hippocampus and frontal cortex of Alzheimer’s disease subjects are disease-related and apolipoprotein E genotype dependent. Brain Res. 843: 87–94. [DOI] [PubMed] [Google Scholar]
- 104.Nagy Z., Esiri M. M., Jobst K. A., Johnston C., Litchfield S., Sim E., and Smith A. D.. 1995. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience. 69: 757–761. [DOI] [PubMed] [Google Scholar]
- 105.Keene C. D., Cudaback E., Li X., Montine K. S., and Montine T. J.. 2011. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr. Opin. Neurobiol. 21: 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tai L. M., Ghura S., Koster K. P., Liakaite V., Maienschein-Cline M., Kanabar P., Collins N., Ben-Aissa M., Lei A. Z., Bahroos N., et al. . 2015. APOE-modulated Abeta-induced neuroinflammation in Alzheimer’s disease: current landscape, novel data, and future perspective. J. Neurochem. 133: 465–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Egensperger R., Kosel S., von Eitzen U., and Graeber M. B.. 1998. Microglial activation in Alzheimer disease: association with APOE genotype. Brain Pathol. 8: 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gale S. C., Gao L., Mikacenic C., Coyle S. M., Rafaels N., Murray Dudenkov T., Madenspacher J. H., Draper D. W., Ge W., Aloor J. J., et al. . 2014. APOepsilon4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol. 134: 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tziakas D. N., Chalikias G. K., Antonoglou C. O., Veletza S., Tentes I. K., Kortsaris A. X., Hatseras D. I., and Kaski J. C.. 2006. Apolipoprotein E genotype and circulating interleukin-10 levels in patients with stable and unstable coronary artery disease. J. Am. Coll. Cardiol. 48: 2471–2481. [DOI] [PubMed] [Google Scholar]
- 110.Licastro F., Porcellini E., Caruso C., Lio D., and Corder E. H.. 2007. Genetic risk profiles for Alzheimer’s disease: integration of APOE genotype and variants that up-regulate inflammation. Neurobiol. Aging. 28: 1637–1643. [DOI] [PubMed] [Google Scholar]
- 111.Lynch J. R., Tang W., Wang H., Vitek M. P., Bennett E. R., Sullivan P. M., Warner D. S., and Laskowitz D. T.. 2003. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 278: 48529–48533. [DOI] [PubMed] [Google Scholar]
- 112.Ophir G., Amariglio N., Jacob-Hirsch J., Elkon R., Rechavi G., and Michaelson D. M.. 2005. Apolipoprotein E4 enhances brain inflammation by modulation of the NF-kappaB signaling cascade. Neurobiol. Dis. 20: 709–718. [DOI] [PubMed] [Google Scholar]
- 113.Maezawa I., Nivison M., Montine K. S., Maeda N., and Montine T. J.. 2006. Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J. 20: 797–799. [DOI] [PubMed] [Google Scholar]
- 114.Ghura S., Tai L., Zhao M., Collins N., Che C. T., Warpeha K. M., and LaDu M. J.. 2016. Arabidopsis thaliana extracts optimized for polyphenols production as potential therapeutics for the APOE-modulated neuroinflammation characteristic of Alzheimer’s disease in vitro. Sci. Rep. 6: 29364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brouwers N., Van Cauwenberghe C., Engelborghs S., Lambert J. C., Bettens K., Le Bastard N., Pasquier F., Montoya A. G., Peeters K., Mattheijssens M., et al. . 2012. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol. Psychiatry. 17: 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J. S., Younkin S., et al. . 2013. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P. V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A. I., Lah J. J., et al. . 2013. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368: 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Green A. E., Gray J. R., Deyoung C. G., Mhyre T. R., Padilla R., Dibattista A. M., and William Rebeck G.. 2014. A combined effect of two Alzheimer’s risk genes on medial temporal activity during executive attention in young adults. Neuropsychologia. 56: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Naj A. C., Jun G., Beecham G. W., Wang L. S., Vardarajan B. N., Buros J., Gallins P. J., Buxbaum J. D., Jarvik G. P., Crane P. K., et al. . 2011. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43: 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hollingworth P., Harold D., Sims R., Gerrish A., Lambert J. C., Carrasquillo M. M., Abraham R., Hamshere M. L., Pahwa J. S., Moskvina V., et al. . 2011. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43: 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gadani S. P., Cronk J. C., Norris G. T., and Kipnis J.. 2012. IL-4 in the brain: a cytokine to remember. J. Immunol. 189: 4213–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lehnardt S. 2010. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 58: 253–263. [DOI] [PubMed] [Google Scholar]
- 123.Lehnardt S., Lachance C., Patrizi S., Lefebvre S., Follett P. L., Jensen F. E., Rosenberg P. A., Volpe J. J., and Vartanian T.. 2002. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J. Neurosci. 22: 2478–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lehnardt S., Massillon L., Follett P., Jensen F. E., Ratan R., Rosenberg P. A., Volpe J. J., and Vartanian T.. 2003. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA. 100: 8514–8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fortea J., Vilaplana E., Alcolea D., Carmona-Iragui M., Sánchez-Saudinos M. B., Sala I., Antón-Aguirre S., González S., Medrano S., Pegueroles J., et al. . 2014. Cerebrospinal fluid beta-amyloid and phospho-tau biomarker interactions affecting brain structure in preclinical Alzheimer disease. Ann. Neurol. 76: 223–230. [DOI] [PubMed] [Google Scholar]
- 126.Hampel H., Blennow K., Shaw L. M., Hoessler Y. C., Zetterberg H., and Trojanowski J. Q.. 2010. Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp. Gerontol. 45: 30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ibach B., Wittmann M., Pfannenschmid F., Poljansky S., Haen E., and Hajak G.. 2004. Genetic tau-variants in patients with frontotemporal dementia [article in German]. Psychiatr. Prax. 31(Suppl. 1): S55–S57. [DOI] [PubMed] [Google Scholar]
- 128.Jagust W. J., Landau S. M., Shaw L. M., Trojanowski J. Q., Koeppe R. A., Reiman E. M., Foster N. L., Petersen R. C., Weiner M. W., Price J. C., et al. . 2009. Relationships between biomarkers in aging and dementia. Neurology. 73: 1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.King M. E., Ahuja V., Binder L. I., and Kuret J.. 1999. Ligand-dependent tau filament formation: implications for Alzheimer’s disease progression. Biochemistry. 38: 14851–14859. [DOI] [PubMed] [Google Scholar]
- 130.López-González I., Schlüter A., Aso E., Garcia-Esparcia P., Ansoleaga B., LLorens F., Carmona M., Moreno J., Fuso A., Portero-Otin M., et al. . 2015. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J. Neuropathol. Exp. Neurol. 74: 319–344. [DOI] [PubMed] [Google Scholar]
- 131.Morris J. C., and Selkoe D. J.. 2011. Recommendations for the incorporation of biomarkers into Alzheimer clinical trials: an overview. Neurobiol. Aging. 32(Suppl. 1): S1–S3. [DOI] [PubMed] [Google Scholar]
- 132.Papasozomenos S. C. 1989. Tau protein immunoreactivity in dementia of the Alzheimer type. I. Morphology, evolution, distribution, and pathogenetic implications. Lab. Invest. 60: 123–137. [PubMed] [Google Scholar]
- 133.Patrick G. N., Zukerberg L., Nikolic M., de la Monte S., Dikkes P., and Tsai L. H.. 1999. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 402: 615–622. [DOI] [PubMed] [Google Scholar]
- 134.Peskind E. R. 1996. Neurobiology of Alzheimer’s disease. J. Clin. Psychiatry. 57(Suppl. 14): 5–8. [PubMed] [Google Scholar]
- 135.Davies C. A., Mann D. M., Sumpter P. Q., and Yates P. O.. 1987. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J. Neurol. Sci. 78: 151–164. [DOI] [PubMed] [Google Scholar]
- 136.Masliah E., Mallory M., Alford M., DeTeresa R., Hansen L. A., McKeel D. W. Jr., and Morris J. C.. 2001. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 56: 127–129. [DOI] [PubMed] [Google Scholar]
- 137.Scheff S. W., Price D. A., Schmitt F. A., DeKosky S. T., and Mufson E. J.. 2007. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 68: 1501–1508. [DOI] [PubMed] [Google Scholar]
- 138.Bertoni-Freddari C., Fattoretti P., Solazzi M., Giorgetti B., Di Stefano G., Casoli T., and Meier-Ruge W.. 2003. Neuronal death versus synaptic pathology in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1010: 635–638. [DOI] [PubMed] [Google Scholar]
- 139.DeKosky S. T., and Scheff S. W.. 1990. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27: 457–464. [DOI] [PubMed] [Google Scholar]
- 140.Danysz W., and Parsons C. G.. 2012. Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine–searching for the connections. Br. J. Pharmacol. 167: 324–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gong Y., and Lippa C. F.. 2010. Review: disruption of the postsynaptic density in Alzheimer’s disease and other neurodegenerative dementias. Am. J. Alzheimers Dis. Other Demen. 25: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stranahan A. M., and Mattson M. P.. 2010. Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer’s disease. Neural Plast. 2010: 108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kojima N., and Shirao T.. 2007. Synaptic dysfunction and disruption of postsynaptic drebrin-actin complex: a study of neurological disorders accompanied by cognitive deficits. Neurosci. Res. 58: 1–5. [DOI] [PubMed] [Google Scholar]
- 144.Counts S. E., He B., Nadeem M., Wuu J., Scheff S. W., and Mufson E. J.. 2012. Hippocampal drebrin loss in mild cognitive impairment. Neurodegener. Dis. 10: 216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cheng D., Hoogenraad C. C., Rush J., Ramm E., Schlager M. A., Duong D. M., Xu P., Wijayawardana S. R., Hanfelt J., Nakagawa T., et al. . 2006. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteomics. 5: 1158–1170. [DOI] [PubMed] [Google Scholar]
- 146.Chen L., Chetkovich D. M., Petralia R. S., Sweeney N. T., Kawasaki Y., Wenthold R. J., Bredt D. S., and Nicoll R. A.. 2000. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 408: 936–943. [DOI] [PubMed] [Google Scholar]
- 147.Bats C., Groc L., and Choquet D.. 2007. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 53: 719–734. [DOI] [PubMed] [Google Scholar]
- 148.Kim E., and Sheng M.. 2004. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 5: 771–781. [DOI] [PubMed] [Google Scholar]
- 149.Elias G. M., and Nicoll R. A.. 2007. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 17: 343–352. [DOI] [PubMed] [Google Scholar]
- 150.Sheng M., and Hoogenraad C. C.. 2007. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 76: 823–847. [DOI] [PubMed] [Google Scholar]
- 151.Feng W., and Zhang M.. 2009. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. Neurosci. 10: 87–99. [DOI] [PubMed] [Google Scholar]
- 152.Keller A. 2002. Use-dependent inhibition of dendritic spines. Trends Neurosci. 25: 541–543; discussion 543–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Marrs G. S., Green S. H., and Dailey M. E.. 2001. Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat. Neurosci. 4: 1006–1013. [DOI] [PubMed] [Google Scholar]
- 154.Okabe S., Miwa A., and Okado H.. 2001. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J. Neurosci. 21: 6105–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shirao T., and Sekino Y.. 2001. Clustering and anchoring mechanisms of molecular constituents of postsynaptic scaffolds in dendritic spines. Neurosci. Res. 40: 1–7. [DOI] [PubMed] [Google Scholar]
- 156.Mizui T., Takahashi H., Sekino Y., and Shirao T.. 2005. Overexpression of drebrin A in immature neurons induces the accumulation of F-actin and PSD-95 into dendritic filopodia, and the formation of large abnormal protrusions. Mol. Cell. Neurosci. 30: 149–157. [DOI] [PubMed] [Google Scholar]
- 157.Ivanov A., Esclapez M., and Ferhat L.. 2009. Role of drebrin A in dendritic spine plasticity and synaptic function: implications in neurological disorders. Commun. Integr. Biol. 2: 268–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shirao T. 1995. The roles of microfilament-associated proteins, drebrins, in brain morphogenesis: a review. J. Biochem. 117: 231–236. [DOI] [PubMed] [Google Scholar]
- 159.Shirao T., Kojima N., and Obata K.. 1992. Cloning of drebrin A and induction of neurite-like processes in drebrin-transfected cells. Neuroreport. 3: 109–112. [DOI] [PubMed] [Google Scholar]
- 160.Ishikawa R., Hayashi K., Shirao T., Xue Y., Takagi T., Sasaki Y., and Kohama K.. 1994. Drebrin, a development-associated brain protein from rat embryo, causes the dissociation of tropomyosin from actin filaments. J. Biol. Chem. 269: 29928–29933. [PubMed] [Google Scholar]
- 161.Shirao T., Hayashi K., Ishikawa R., Isa K., Asada H., Ikeda K., and Uyemura K.. 1994. Formation of thick, curving bundles of actin by drebrin A expressed in fibroblasts. Exp. Cell Res. 215: 145–153. [DOI] [PubMed] [Google Scholar]
- 162.Hayashi K., Ishikawa R., Ye L. H., He X. L., Takata K., Kohama K., and Shirao T.. 1996. Modulatory role of drebrin on the cytoskeleton within dendritic spines in the rat cerebral cortex. J. Neurosci. 16: 7161–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shirao T., Hanamura K., Koganezawa N., Ishizuka Y., Yamazaki H., and Sekino Y.. The role of drebrin in neurons. J. Neurochem. Epub ahead of print. February 15, 2017; doi:10.1111/jnc.13988. [DOI] [PubMed] [Google Scholar]
- 164.Koganezawa N., Hanamura K., Sekino Y., and Shirao T.. The role of drebrin in dendritic spines. Mol. Cell. Neurosci. Epub ahead of print. February 1, 2017; doi:10.1016/j.mcn.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 165.Ma L., Li Y., and Wang R.. 2015. Drebrin and cognitive impairment. Clin. Chim. Acta. 451: 121–124. [DOI] [PubMed] [Google Scholar]
- 166.Sullivan P. M., Mace B. E., Maeda N., and Schmechel D. E.. 2004. Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience. 124: 725–733. [DOI] [PubMed] [Google Scholar]
- 167.Sullivan P. M., Han B., Liu F., Mace B. E., Ervin J. F., Wu S., Koger D., Paul S., and Bales K. R.. 2011. Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol. Aging. 32: 791–801. [DOI] [PubMed] [Google Scholar]
- 168.Riddell D. R., Zhou H., Atchison K., Warwick H. K., Atkinson P. J., Jefferson J., Xu L., Aschmies S., Kirksey Y., Hu Y., et al. . 2008. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci. 28: 11445–11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Vitek M. P., Brown C. M., and Colton C. A.. 2009. APOE genotype-specific differences in the innate immune response. Neurobiol. Aging. 30: 1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yong S. M., Lim M. L., Low C. M., and Wong B. S.. 2014. Reduced neuronal signaling in the ageing apolipoprotein-E4 targeted replacement female mice. Sci. Rep. 4: 6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Youmans K. L., Tai L. M., Kanekiyo T., Stine W. B. Jr., Michon S. C., Nwabuisi-Heath E., Manelli A., Fu Y., Riordan S., Eimer W. A., et al. . 2012. Intraneuronal Abeta detection in 5xFAD mice by a new Abeta-specific antibody. Mol. Neurodegener. 7: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tai L. M., Koster K. P., Luo J., Lee S. H., Wang Y. T., Collins N. C., Ben Aissa M., Thatcher G. R., and LaDu M. J.. 2014. Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem. 289: 30538–30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Abdullah L., Evans J. E., Emmerich T., Crynen G., Shackleton B., Keegan A. P., Luis C., Tai L., LaDu M. J., Mullan M., et al. . 2017. APOE epsilon4 specific imbalance of arachidonic acid and docosahexaenoic acid in serum phospholipids identifies individuals with preclinical mild cognitive impairment/Alzheimer’s disease. Aging (Albany NY). 9: 964–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Picq M., Chen P., Perez M., Michaud M., Vericel E., Guichardant M., and Lagarde M.. 2010. DHA metabolism: targeting the brain and lipoxygenation. Mol. Neurobiol. 42: 48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nguyen L. N., Ma D., Shui G., Wong P., Cazenave-Gassiot A., Zhang X., Wenk M. R., Goh E. L., and Silver D. L.. 2014. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature. 509: 503–506. [DOI] [PubMed] [Google Scholar]
- 176.Guemez-Gamboa A., Nguyen L. N., Yang H., Zaki M. S., Kara M., Ben-Omran T., Akizu N., Rosti R. O., Rosti B., Scott E., et al. . 2015. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 47: 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vandal M., Alata W., Tremblay C., Rioux-Perreault C., Salem N. Jr., Calon F., and Plourde M.. 2014. Reduction in DHA transport to the brain of mice expressing human APOE4 compared to APOE2. J. Neurochem. 129: 516–526. [DOI] [PubMed] [Google Scholar]
- 178.Hultman K., Strickland S., and Norris E. H.. 2013. The APOE varepsilon4/varepsilon4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 33: 1251–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zekonyte J., Sakai K., Nicoll J. A., Weller R. O., and Carare R. O.. 2016. Quantification of molecular interactions between ApoE, amyloid-beta (Abeta) and laminin: relevance to accumulation of Abeta in Alzheimer’s disease. Biochim. Biophys. Acta. 1862: 1047–1053. [DOI] [PubMed] [Google Scholar]
- 180.Bakker E. N., Bacskai B. J., Arbel-Ornath M., Aldea R., Bedussi B., Morris A. W., Weller R. O., and Carare R. O.. 2016. Lymphatic clearance of the brain: perivascular, paravascular and significance for neurodegenerative diseases. Cell. Mol. Neurobiol. 36: 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shinohara M., Murray M. E., Frank R. D., Shinohara M., DeTure M., Yamazaki Y., Tachibana M., Atagi Y., Davis M. D., Liu C. C., et al. . 2016. Impact of sex and APOE4 on cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol. 132: 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Finch C. E., and Shams S.. 2016. Apolipoprotein E and sex bias in cerebrovascular aging of men and mice. Trends Neurosci. 39: 625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Benedictus M. R., Goos J. D., Binnewijzend M. A., Muller M., Barkhof F., Scheltens P., Prins N. D., and van der Flier W. M.. 2013. Specific risk factors for microbleeds and white matter hyperintensities in Alzheimer’s disease. Neurobiol. Aging. 34: 2488–2494. [DOI] [PubMed] [Google Scholar]
- 184.Koster K. P., Smith C., Valencia-Olvera A. C., Thatcher G. R., Tai L. M., and LaDu M. J.. 2017. Rexinoids as therapeutics for Alzheimer’s disease: role of APOE. Curr. Top. Med. Chem. 17: 708–720. [DOI] [PubMed] [Google Scholar]
- 185.Mariani M. M., Malm T., Lamb R., Jay T. R., Neilson L., Casali B., Medarametla L., and Landreth G. E.. 2017. Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Sci. Rep. 7: 42270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Moutinho M., and Landreth G. E.. Therapeutic potential of nuclear receptor agonists in Alzheimer’s disease. J. Lipid Res. Epub ahead of print. March 6, 2017; doi:10.1194/jlr.R075556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Skerrett R., Malm T., and Landreth G.. 2014. Nuclear receptors in neurodegenerative diseases. Neurobiol. Dis. 72: 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sodhi R. K., and Singh N.. 2014. Retinoids as potential targets for Alzheimer’s disease. Pharmacol. Biochem. Behav. 120: 117–123. [DOI] [PubMed] [Google Scholar]
- 189.Lee J. H., Jiang Y., Han D. H., Shin S. K., Choi W. H., and Lee M. J.. 2014. Targeting estrogen receptors for the treatment of Alzheimer’s disease. Mol. Neurobiol. 49: 39–49. [DOI] [PubMed] [Google Scholar]
- 190.Zolezzi J. M., and Inestrosa N. C.. 2013. Peroxisome proliferator-activated receptors and Alzheimer’s disease: hitting the blood-brain barrier. Mol. Neurobiol. 48: 438–451. [DOI] [PubMed] [Google Scholar]
- 191.Sodhi R. K., and Singh N.. 2013. Liver X receptors: emerging therapeutic targets for Alzheimer’s disease. Pharmacol. Res. 72: 45–51. [DOI] [PubMed] [Google Scholar]
- 192.Mandrekar-Colucci S., and Landreth G. E.. 2011. Nuclear receptors as therapeutic targets for Alzheimer’s disease. Expert Opin. Ther. Targets. 15: 1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Luo J., Lee S. H., VandeVrede L., Qin Z., Ben Aissa M., Larson J., Teich A. F., Arancio O., D’Souza Y., Elharram A., et al. . 2016. A multifunctional therapeutic approach to disease modification in multiple familial mouse models and a novel sporadic model of Alzheimer’s disease. Mol. Neurodegener. 11: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kunzler J., Youmans K. L., Yu C., Ladu M. J., and Tai L. M.. 2014. APOE modulates the effect of estrogen therapy on Abeta accumulation EFAD-Tg mice. Neurosci. Lett. 560: 131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Morrissette D. A., Parachikova A., Green K. N., and LaFerla F. M.. 2009. Relevance of transgenic mouse models to human Alzheimer disease. J. Biol. Chem. 284: 6033–6037. [DOI] [PubMed] [Google Scholar]
- 196.Turturro A., Witt W. W., Lewis S., Hass B. S., Lipman R. D., and Hart R. W.. 1999. Growth curves and survival characteristics of the animals used in the Biomarkers of Aging Program. J. Gerontol. A Biol. Sci. Med. Sci. 54: B492–B501. [DOI] [PubMed] [Google Scholar]
- 197.Rae E. A., and Brown R. E.. 2015. The problem of genotype and sex differences in life expectancy in transgenic AD mice. Neurosci. Biobehav. Rev. 57: 238–251. [DOI] [PubMed] [Google Scholar]
- 198.Bories C., Guitton M. J., Julien C., Tremblay C., Vandal M., Msaid M., De Koninck Y., and Calon F.. 2012. Sex-dependent alterations in social behaviour and cortical synaptic activity coincide at different ages in a model of Alzheimer’s disease. PLoS One. 7: e46111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Overk C. R., Perez S. E., Ma C., Taves M. D., Soma K. K., and Mufson E. J.. 2013. Sex steroid levels and AD-like pathology in 3xTgAD mice. J. Neuroendocrinol. 25: 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Overk C. R., Lu P. Y., Wang Y. T., Choi J., Shaw J. W., Thatcher G. R., and Mufson E. J.. 2012. Effects of aromatase inhibition versus gonadectomy on hippocampal complex amyloid pathology in triple transgenic mice. Neurobiol. Dis. 45: 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Carroll J. C., Rosario E. R., Villamagna A., and Pike C. J.. 2010. Continuous and cyclic progesterone differentially interact with estradiol in the regulation of Alzheimer-like pathology in female 3xTransgenic-Alzheimer’s disease mice. Endocrinology. 151: 2713–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hirata-Fukae C., Li H. F., Hoe H. S., Gray A. J., Minami S. S., Hamada K., Niikura T., Hua F., Tsukagoshi-Nagai H., Horikoshi-Sakuraba Y., et al. . 2008. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 1216: 92–103. [DOI] [PubMed] [Google Scholar]
- 203.Wang J., Tanila H., Puolivali J., Kadish I., and van Groen T.. 2003. Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol. Dis. 14: 318–327. [DOI] [PubMed] [Google Scholar]
- 204.Hartman R. E., Wozniak D. F., Nardi A., Olney J. W., Sartorius L., and Holtzman D. M.. 2001. Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: apoE4 mice show profound working memory impairments in the absence of Alzheimer’s-like neuropathology. Exp. Neurol. 170: 326–344. [DOI] [PubMed] [Google Scholar]
- 205.van Meer P., Acevedo S., and Raber J.. 2007. Impairments in spatial memory retention of GFAP-apoE4 female mice. Behav. Brain Res. 176: 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Buttini M., Orth M., Bellosta S., Akeefe H., Pitas R. E., Wyss-Coray T., Mucke L., and Mahley R. W.. 1999. Expression of human apolipoprotein E3 or E4 in the brains of Apoe-/- mice: isoform-specific effects on neurodegeneration. J. Neurosci. 19: 4867–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bour A., Grootendorst J., Vogel E., Kelche C., Dodart J. C., Bales K., Moreau P. H., Sullivan P. M., and Mathis C.. 2008. Middle-aged human apoE4 targeted-replacement mice show retention deficits on a wide range of spatial memory tasks. Behav. Brain Res. 193: 174–182. [DOI] [PubMed] [Google Scholar]
- 208.Grootendorst J., Bour A., Vogel E., Kelche C., Sullivan P. M., Dodart J. C., Bales K., and Mathis C.. 2005. Human apoE targeted replacement mouse lines: h-apoE4 and h-apoE3 mice differ on spatial memory performance and avoidance behavior. Behav. Brain Res. 159: 1–14. [DOI] [PubMed] [Google Scholar]
- 209.Raber J., Wong D., Buttini M., Orth M., Bellosta S., Pitas R. E., Mahley R. W., and Mucke L.. 1998. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc. Natl. Acad. Sci. USA. 95: 10914–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Heise V., Filippini N., Trachtenberg A. J., Suri S., Ebmeier K. P., and Mackay C. E.. 2014. Apolipoprotein E genotype, gender and age modulate connectivity of the hippocampus in healthy adults. Neuroimage. 98: 23–30. [DOI] [PubMed] [Google Scholar]
- 211.Mortensen E. L., and Hogh P.. 2001. A gender difference in the association between APOE genotype and age-related cognitive decline. Neurology. 57: 89–95. [DOI] [PubMed] [Google Scholar]
- 212.Holland D., Desikan R. S., Dale A. M., and McEvoy L. K.; Alzheimer’s Disease Neuroimaging Initiative. 2013. Higher rates of decline for women and apolipoprotein E epsilon4 carriers. AJNR Am. J. Neuroradiol. 34: 2287–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fleisher A. S., Houston W. S., Eyler L. T., Frye S., Jenkins C., Thal L. J., and Bondi M. W.. 2005. Identification of Alzheimer disease risk by functional magnetic resonance imaging. Arch. Neurol. 62: 1881–1888. [DOI] [PubMed] [Google Scholar]
- 214.Frick K. M., Burlingame L. A., Arters J. A., and Berger-Sweeney J.. 2000. Reference memory, anxiety and estrous cyclicity in C57BL/6NIA mice are affected by age and sex. Neuroscience. 95: 293–307. [DOI] [PubMed] [Google Scholar]
- 215.Frick K. M. 2009. Estrogens and age-related memory decline in rodents: what have we learned and where do we go from here? Horm. Behav. 55: 2–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Nelson J. F., Felicio L. S., Randall P. K., Sims C., and Finch C. E.. 1982. A longitudinal study of estrous cyclicity in aging C57BL/6J mice: I. Cycle frequency, length and vaginal cytology. Biol. Reprod. 27: 327–339. [DOI] [PubMed] [Google Scholar]
- 217.Finch C. E., Felicio L. S., Mobbs C. V., and Nelson J. F.. 1984. Ovarian and steroidal influences on neuroendocrine aging processes in female rodents. Endocr. Rev. 5: 467–497. [DOI] [PubMed] [Google Scholar]
- 218.Jilka R. L. 2013. The relevance of mouse models for investigating age-related bone loss in humans. J. Gerontol. A Biol. Sci. Med. Sci. 68: 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Blázquez G., Cañete T., Tobeña A., Giménez-Llort L., and Fernández-Teruel A.. 2014. Cognitive and emotional profiles of aged Alzheimer’s disease (3xTgAD) mice: effects of environmental enrichment and sexual dimorphism. Behav. Brain Res. 268: 185–201. [DOI] [PubMed] [Google Scholar]
- 220.Lambert J. C., Ibrahim-Verbaas C. A., Harold D., Naj A. C., Sims R., Bellenguez C., DeStafano A. L., Bis J. C., Beecham G. W., Grenier-Boley B., et al. . 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45: 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Arranz L., De Castro N. M., Baeza I., Gimenez-Llort L., and De la Fuente M.. 2011. Effect of environmental enrichment on the immunoendocrine aging of male and female triple-transgenic 3xTg-AD mice for Alzheimer’s disease. J. Alzheimers Dis. 25: 727–737. [DOI] [PubMed] [Google Scholar]
- 222.Devi L., Alldred M. J., Ginsberg S. D., and Ohno M.. 2010. Sex- and brain region-specific acceleration of beta-amyloidogenesis following behavioral stress in a mouse model of Alzheimer’s disease. Mol. Brain. 3: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Levi O., and Michaelson D. M.. 2007. Environmental enrichment stimulates neurogenesis in apolipoprotein E3 and neuronal apoptosis in apolipoprotein E4 transgenic mice. J. Neurochem. 100: 202–210. [DOI] [PubMed] [Google Scholar]
- 224.Szczygielski J., Mautes A., Steudel W. I., Falkai P., Bayer T. A., and Wirths O.. 2005. Traumatic brain injury: cause or risk of Alzheimer’s disease? A review of experimental studies. J. Neural Transm. 112: 1547–1564. [DOI] [PubMed] [Google Scholar]
- 225.Mucke L., and Pitas R. E.. 2004. Food for thought: essential fatty acid protects against neuronal deficits in transgenic mouse model of AD. Neuron. 43: 596–599. [DOI] [PubMed] [Google Scholar]
- 226.Grootendorst J., de Kloet E. R., Dalm S., and Oitzl M. S.. 2001. Reversal of cognitive deficit of apolipoprotein E knockout mice after repeated exposure to a common environmental experience. Neuroscience. 108: 237–247. [DOI] [PubMed] [Google Scholar]
- 227.Nicoll J. A., Roberts G. W., and Graham D. I.. 1996. Amyloid beta-protein, APOE genotype and head injury. Ann. N. Y. Acad. Sci. 777: 271–275. [DOI] [PubMed] [Google Scholar]
- 228.Emmerzaal T. L., Kiliaan A. J., and Gustafson D. R.. 2015. 2003-2013: a decade of body mass index, Alzheimer’s disease, and dementia. J. Alzheimers Dis. 43: 739–755. [DOI] [PubMed] [Google Scholar]
- 229.Ferrari C., Nacmias B., Bagnoli S., Piaceri I., Lombardi G., Pradella S., Tedde A., and Sorbi S.. 2014. Imaging and cognitive reserve studies predict dementia in presymptomatic Alzheimer’s disease subjects. Neurodegener. Dis. 13: 157–159. [DOI] [PubMed] [Google Scholar]
- 230.Breunig J. J., Guillot-Sestier M. V., and Town T.. 2013. Brain injury, neuroinflammation and Alzheimer’s disease. Front. Aging Neurosci. 5: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Calderón-Garcidueñas L., Kavanaugh M., Block M., D’Angiulli A., Delgado-Chávez R., Torres-Jardón R., González-Maciel A., Reynoso-Robles R., Osnaya N., Villarreal-Calderon R., et al. . 2012. Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J. Alzheimers Dis. 28: 93–107. [DOI] [PubMed] [Google Scholar]
- 232.Brown B. M., Peiffer J. J., and Martins R. N.. 2013. Multiple effects of physical activity on molecular and cognitive signs of brain aging: can exercise slow neurodegeneration and delay Alzheimer’s disease? Mol. Psychiatry. 18: 864–874. [DOI] [PubMed] [Google Scholar]
- 233.Tolppanen A. M., Solomon A., Kulmala J., Kareholt I., Ngandu T., Rusanen M., Laatikainen T., Soininen H., and Kivipelto M.. 2015. Leisure-time physical activity from mid- to late life, body mass index, and risk of dementia. Alzheimers Dement. 11: 434–443.e6. [DOI] [PubMed] [Google Scholar]
- 234.Fitzpatrick A. L., Kuller L. H., Lopez O. L., Diehr P., O’Meara E. S., Longstreth W. T. Jr., and Luchsinger J. A.. 2009. Midlife and late-life obesity and the risk of dementia: cardiovascular health study. Arch. Neurol. 66: 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gustafson D., Rothenberg E., Blennow K., Steen B., and Skoog I.. 2003. An 18-year follow-up of overweight and risk of Alzheimer disease. Arch. Intern. Med. 163: 1524–1528. [DOI] [PubMed] [Google Scholar]
- 236.Gustafson D. R., Backman K., Waern M., Ostling S., Guo X., Zandi P., Mielke M. M., Bengtsson C., and Skoog I.. 2009. Adiposity indicators and dementia over 32 years in Sweden. Neurology. 73: 1559–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Luchsinger J. A., Cheng D., Tang M. X., Schupf N., and Mayeux R.. 2012. Central obesity in the elderly is related to late-onset Alzheimer disease. Alzheimer Dis. Assoc. Disord. 26: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Meng X. F., Yu J. T., Wang H. F., Tan M. S., Wang C., Tan C. C., and Tan L.. 2014. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J. Alzheimers Dis. 42: 1295–1310. [DOI] [PubMed] [Google Scholar]
- 239.Profenno L. A., Porsteinsson A. P., and Faraone S. V.. 2010. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol. Psychiatry. 67: 505–512. [DOI] [PubMed] [Google Scholar]
- 240.Hayden K. M., Zandi P. P., Lyketsos C. G., Khachaturian A. S., Bastian L. A., Charoonruk G., Tschanz J. T., Norton M. C., Pieper C. F., Munger R. G., et al. . 2006. Vascular risk factors for incident Alzheimer disease and vascular dementia: the Cache County study. Alzheimer Dis. Assoc. Disord. 20: 93–100. [DOI] [PubMed] [Google Scholar]
- 241.Whitmer R. A., Gunderson E. P., Quesenberry C. P. Jr., Zhou J., and Yaffe K.. 2007. Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Curr. Alzheimer Res. 4: 103–109. [DOI] [PubMed] [Google Scholar]
- 242.Exalto L. G., Quesenberry C. P., Barnes D., Kivipelto M., Biessels G. J., and Whitmer R. A.. 2014. Midlife risk score for the prediction of dementia four decades later. Alzheimers Dement. 10: 562–570. [DOI] [PubMed] [Google Scholar]
- 243.Isaac V., Sim S., Zheng H., Zagorodnov V., Tai E. S., and Chee M.. 2011. Adverse associations between visceral adiposity, brain structure, and cognitive performance in healthy elderly. Front. Aging Neurosci. 3: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jayaraman A., and Pike C. J.. 2014. Alzheimer’s disease and type 2 diabetes: multiple mechanisms contribute to interactions. Curr. Diab. Rep. 14: 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Barron A. M., Rosario E. R., Elteriefi R., and Pike C. J.. 2013. Sex-specific effects of high fat diet on indices of metabolic syndrome in 3xTg-AD mice: implications for Alzheimer’s disease. PLoS One. 8: e78554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ho L., Qin W., Pompl P. N., Xiang Z., Wang J., Zhao Z., Peng Y., Cambareri G., Rocher A., Mobbs C. V., et al. . 2004. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 18: 902–904. [DOI] [PubMed] [Google Scholar]
- 247.Julien C., Tremblay C., Phivilay A., Berthiaume L., Emond V., Julien P., and Calon F.. 2010. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol. Aging. 31: 1516–1531. [DOI] [PubMed] [Google Scholar]
- 248.Kohjima M., Sun Y., and Chan L.. 2010. Increased food intake leads to obesity and insulin resistance in the tg2576 Alzheimer’s disease mouse model. Endocrinology. 151: 1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Johnson L. A., Torres E. R., Impey S., Stevens J. F., and Raber J.. 2017. Apolipoprotein E4 and insulin resistance interact to impair cognition and alter the epigenome and metabolome. Sci. Rep. 7: 43701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Janssen C. I., Jansen D., Mutsaers M. P., Dederen P. J., Geenen B., Mulder M. T., and Kiliaan A. J.. 2016. The effect of a high-fat diet on brain plasticity, inflammation and cognition in female apoE4-knockin and apoE-knockout mice. PLoS One. 11: e0155307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Hanson A. J., Bayer-Carter J. L., Green P. S., Montine T. J., Wilkinson C. W., Baker L. D., Watson G. S., Bonner L. M., Callaghan M., Leverenz J. B., et al. . 2013. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol. 70: 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Weuve J., Kaufman J. D., Szpiro A. A., Curl C., Puett R. C., Beck T., Evans D. A., and Mendes de Leon C. F.. 2016. Exposure to traffic-related air pollution in relation to progression in physical disability among older adults. Environ. Health Perspect. 124: 1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Clifford A., Lang L., Chen R., Anstey K. J., and Seaton A.. 2016. Exposure to air pollution and cognitive functioning across the life course–a systematic literature review. Environ. Res. 147: 383–398. [DOI] [PubMed] [Google Scholar]
- 254.Sunyer J., Esnaola M., Alvarez-Pedrerol M., Forns J., Rivas I., Lopez-Vicente M., Suades-Gonzalez E., Foraster M., Garcia-Esteban R., Basagana X., et al. . 2015. Association between traffic-related air pollution in schools and cognitive development in primary school children: a prospective cohort study. PLoS Med. 12: e1001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Calderón-Garcidueñas L., San Juan Chávez V., Vacaseydel-Aceves N. B., Calderón-Sánchez R., Macías-Escobedo E., Frías C., Giacometto M., Velasquez L., Félix-Villarreal R., Martin J. D., et al. . 2016. Chocolate, air pollution and children’s neuroprotection: what cognition tools should be at hand to evaluate interventions? Front. Pharmacol. 7: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Fonken L. K., Xu X., Weil Z. M., Chen G., Sun Q., Rajagopalan S., and Nelson R. J.. 2011. Air pollution impairs cognition, provokes depressive-like behaviors and alters hippocampal cytokine expression and morphology. Mol. Psychiatry. 16: 987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Cacciottolo M., Wang X., Driscoll I., Woodward N., Saffari A., Reyes J., Serre M. L., Vizuete W., Sioutas C., Morgan T. E., et al. . 2017. Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl. Psychiatry. 7: e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Tarr P. T., and Edwards P. A.. 2008. ABCG1 and ABCG4 are coexpressed in neurons and astrocytes of the CNS and regulate cholesterol homeostasis through SREBP-2. J. Lipid Res. 49: 169–182. [DOI] [PubMed] [Google Scholar]
- 259.Wang N., Yvan-Charvet L., Lutjohann D., Mulder M., Vanmierlo T., Kim T. W., Tall A. R., Wang N., Yvan-Charvet L., Lutjohann D., et al. . 2008. ATP-binding cassette transporters G1 and G4 mediate cholesterol and desmosterol efflux to HDL and regulate sterol accumulation in the brain. FASEB J. 22: 1073–1082. [DOI] [PubMed] [Google Scholar]
- 260.Hirsch-Reinshagen V., Donkin J., Stukas S., Chan J., Wilkinson A., Fan J., Parks J. S., Kuivenhoven J. A., Lutjohann D., Pritchard H., et al. . 2009. LCAT synthesized by primary astrocytes esterifies cholesterol on glia-derived lipoproteins. J. Lipid Res. 50: 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Karasinska J. M., Rinninger F., Lutjohann D., Ruddle P., Franciosi S., Kruit J. K., Singaraja R. R., Hirsch-Reinshagen V., Fan J., Brunham L. R., et al. . 2009. Specific loss of brain ABCA1 increases brain cholesterol uptake and influences neuronal structure and function. J. Neurosci. 29: 3579–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hirsch-Reinshagen V., Zhou S., Burgess B. L., Bernier L., McIsaac S. A., Chan J. Y., Tansley G. H., Cohn J. S., Hayden M. R., and Wellington C. L.. 2004. Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 279: 41197–41207. [DOI] [PubMed] [Google Scholar]
- 263.Tachikawa M., Watanabe M., Hori S., Fukaya M., Ohtsuki S., Asashima T., and Terasaki T.. 2005. Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J. Neurochem. 95: 294–304. [DOI] [PubMed] [Google Scholar]
- 264.McLean J., Wion K., Drayna D., Fielding C., and Lawn R.. 1986. Human lecithin-cholesterol acyltransferase gene: complete gene sequence and sites of expression. Nucleic Acids Res. 14: 9397–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Warden C. H., Langner C. A., Gordon J. I., Taylor B. A., McLean J. W., and Lusis A. J.. 1989. Tissue-specific expression, developmental regulation, and chromosomal mapping of the lecithin: cholesterol acyltransferase gene. Evidence for expression in brain and testes as well as liver. J. Biol. Chem. 264: 21573–21581. [PubMed] [Google Scholar]
- 266.Albers J. J., Marcovina S. M., and Christenson R. H.. 1992. Lecithin cholesterol acyltransferase in human cerebrospinal fluid: reduced level in patients with multiple sclerosis and evidence of direct synthesis in the brain. Int. J. Clin. Lab. Res. 22: 169–172. [DOI] [PubMed] [Google Scholar]
- 267.Smith K. M., Lawn R. M., and Wilcox J. N.. 1990. Cellular localization of apolipoprotein D and lecithin:cholesterol acyltransferase mRNA in rhesus monkey tissues by in situ hybridization. J. Lipid Res. 31: 995–1004. [PubMed] [Google Scholar]
- 268.Rigotti A., Miettinen H. E., and Krieger M.. 2003. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr. Rev. 24: 357–387. [DOI] [PubMed] [Google Scholar]
- 269.Husemann J., Loike J. D., Anankov R., Febbraio M., Silverstein S. C., Husemann J., Loike J. D., Anankov R., Febbraio M., and Silverstein S. C.. 2002. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia. 40: 195–205. [DOI] [PubMed] [Google Scholar]
- 270.Husemann J., and Silverstein S. C.. 2001. Expression of scavenger receptor class B, type I, by astrocytes and vascular smooth muscle cells in normal adult mouse and human brain and in Alzheimer’s disease brain. Am. J. Pathol. 158: 825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Holtzman D. M., Pitas R. E., Kilbridge J., Nathan B., Mahley R. W., Bu G., and Schwartz A. L.. 1995. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc. Natl. Acad. Sci. USA. 92: 9480–9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Clatworthy A. E., Stockinger W., Christie R. H., Schneider W. J., Nimpf J., Hyman B. T., and Rebeck G. W.. 1999. Expression and alternate splicing of apolipoprotein E receptor 2 in brain. Neuroscience. 90: 903–911. [DOI] [PubMed] [Google Scholar]
- 273.Rebeck G. W., Reiter J. S., Strickland D. K., and Hyman B. T.. 1993. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 11: 575–580. [DOI] [PubMed] [Google Scholar]
- 274.Pitas R. E., Boyles J. K., Lee S. H., Hui D., and Weisgraber K. H.. 1987. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J. Biol. Chem. 262: 14352–14360. [PubMed] [Google Scholar]
- 275.Rebeck G. W., Harr S. D., Strickland D. K., and Hyman B. T.. 1995. Multiple, diverse senile plaque-associated proteins are ligands of an apolipoprotein E receptor, the alpha 2-macroglobulin receptor/low-density-lipoprotein receptor-related protein. Ann. Neurol. 37: 211–217. [DOI] [PubMed] [Google Scholar]
- 276.Bu G., Maksymovitch E. A., Nerbonne J. M., and Schwartz A. L.. 1994. Expression and function of the low density lipoprotein receptor-related protein (LRP) in mammalian central neurons. J. Biol. Chem. 269: 18521–18528. [PubMed] [Google Scholar]
- 277.Christie R. H., Chung H., Rebeck G. W., Strickland D., and Hyman B. T.. 1996. Expression of the very low-density lipoprotein receptor (VLDL-r), an apolipoprotein-E receptor, in the central nervous system and in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 55: 491–498. [DOI] [PubMed] [Google Scholar]
- 278.Kim D. H., Iijima H., Goto K., Sakai J., Ishii H., Kim H. J., Suzuki H., Kondo H., Saeki S., and Yamamoto T.. 1996. Human apolipoprotein E receptor 2. A novel lipoprotein receptor of the low density lipoprotein receptor family predominantly expressed in brain. J. Biol. Chem. 271: 8373–8380. [DOI] [PubMed] [Google Scholar]
- 279.Marzolo M. P., von Bernhardi R., Bu G., and Inestrosa N. C.. 2000. Expression of alpha(2)-macroglobulin receptor/low density lipoprotein receptor-related protein (LRP) in rat microglial cells. J. Neurosci. Res. 60: 401–411. [DOI] [PubMed] [Google Scholar]
- 280.Ladu M. J., Reardon C., Van Eldik L., Fagan A. M., Bu G., Holtzman D., and Getz G. S.. 2000. Lipoproteins in the central nervous system. Ann. N. Y. Acad. Sci. 903: 167–175. [DOI] [PubMed] [Google Scholar]
- 281.LaDu M. J., Gilligan S. M., Lukens J. R., Cabana V. G., Reardon C. A., Van Eldik L. J., and Holtzman D. M.. 1998. Nascent astrocyte particles differ from lipoproteins in CSF. J. Neurochem. 70: 2070–2081. [DOI] [PubMed] [Google Scholar]
- 282.Weisgraber K. H., and Mahley R. W.. 1996. Human apolipoprotein E: the Alzheimer’s disease connection. FASEB J. 10: 1485–1494. [DOI] [PubMed] [Google Scholar]
- 283.Weisgraber K. H. 1994. Apolipoprotein E: structure-function relationships. Adv. Protein Chem. 45: 249–302. [DOI] [PubMed] [Google Scholar]
- 284.Roheim P. S., Carey M., Forte T., and Vega G. L.. 1979. Apolipoproteins in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA. 76: 4646–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Koudinov A. R., Koudinova N. V., Kumar A., Beavis R. C., and Ghiso J.. 1996. Biochemical characterization of Alzheimer’s soluble amyloid beta protein in human cerebrospinal fluid: association with high density lipoproteins. Biochem. Biophys. Res. Commun. 223: 592–597. [DOI] [PubMed] [Google Scholar]
- 286.Borghini I., Barja F., Pometta D., and James R. W.. 1995. Characterization of subpopulations of lipoprotein particles isolated from human cerebrospinal fluid. Biochim. Biophys. Acta. 1255: 192–200. [DOI] [PubMed] [Google Scholar]
- 287.Koch S., Donarski N., Goetze K., Kreckel M., Stuerenburg H. J., Buhmann C., and Beisiegel U.. 2001. Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 42: 1143–1151. [PubMed] [Google Scholar]
- 288.Francis G. A., Knopp R. H., and Oram J. F.. 1995. Defective removal of cellular cholesterol and phospholipids by apolipoprotein A-I in Tangier disease. J. Clin. Invest. 96: 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Remaley A. T., Stonik J. A., Demosky S. J., Neufeld E. B., Bocharov A. V., Vishnyakova T. G., Eggerman T. L., Patterson A. P., Duverger N. J., Santamarina-Fojo S., et al. . 2001. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem. Biophys. Res. Commun. 280: 818–823. [DOI] [PubMed] [Google Scholar]
- 290.Jonas A. 1998. Regulation of lecithin cholesterol acyltransferase activity. Prog. Lipid Res. 37: 209–234. [DOI] [PubMed] [Google Scholar]
- 291.Wolf A., Bauer B., and Hartz A. M.. 2012. ABC transporters and the Alzheimer’s disease enigma. Front. Psychiatry. 3: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Tai L. M., Thomas R., Marottoli F. M., Koster K. P., Kanekiyo T., Morris A. W., and Bu G.. 2016. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 131: 709–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Liu C. C., Kanekiyo T., Xu H., and Bu G.. 2013. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9: 106–118. [Erratum. 2013. Nat. Rev. Neurol.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Hu J., Liu C. C., Chen X. F., Zhang Y. W., Xu H., and Bu G.. 2015. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol. Neurodegener. 10: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Gorelick P. B. 2010. Role of inflammation in cognitive impairment: results of observational epidemiological studies and clinical trials. Ann. N. Y. Acad. Sci. 1207: 155–162. [DOI] [PubMed] [Google Scholar]
- 296.Imbimbo B. P., Solfrizzi V., and Panza F.. 2010. Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front. Aging Neurosci. 21: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.McGeer P. L., and McGeer E. G.. 2007. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol. Aging. 28: 639–647. [DOI] [PubMed] [Google Scholar]
- 298.Meraz-Ríos M. A., Toral-Rios D., Franco-Bocanegra D., Villeda-Hernández J., and Campos-Peña V.. 2013. Inflammatory process in Alzheimer’s disease. Front. Integr. Nuerosci. 7: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Rich J. B., Rasmusson D. X., Folstein M. F., Carson K. A., Kawas C., and Brandt J.. 1995. Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology. 45: 51–55. [DOI] [PubMed] [Google Scholar]
- 300.Rubio-Perez J. M., and Morillas-Ruiz J. M.. 2012. A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal. 2012: 756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Stewart W. F., Kawas C., Corrada M., and Metter E. J.. 1997. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 48: 626–632. [DOI] [PubMed] [Google Scholar]
- 302.Vlad S. C., Miller D. R., Kowall N. W., and Felson D. T.. 2008. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 70: 1672–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Weggen S., Eriksen J. L., Das P., Sagi S. A., Wang R., Pietrzik C. U., Findlay K. A., Smith T. E., Murphy M. P., Bulter T., et al. . 2001. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 414: 212–216. [DOI] [PubMed] [Google Scholar]
- 304.Yip A. G., Green R. C., Huyck M., Cupples L. A., and Farrer L. A.. 2005. Nonsteroidal anti-inflammatory drug use and Alzheimer’s disease risk: the MIRAGE Study. BMC Geriatr. 5: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Minoretti P., Gazzaruso C., Vito C. D., Emanuele E., Bianchi M., Coen E., Reino M., and Geroldi D.. 2006. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci. Lett. 391: 147–149. [DOI] [PubMed] [Google Scholar]
- 306.Walter S., Letiembre M., Liu Y., Heine H., Penke B., Hao W., Bode B., Manietta N., Walter J., Schulz-Schuffer W., et al. . 2007. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell. Physiol. Biochem. 20: 947–956. [DOI] [PubMed] [Google Scholar]
- 307.Tang S. C., Lathia J. D., Selvaraj P. K., Jo D. G., Mughal M. R., Cheng A., Siler D. A., Markesbery W. R., Arumugam T. V., and Mattson M. P.. 2008. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp. Neurol. 213: 114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Yu J. T., Miao D., Cui W. Z., Ou J. R., Tian Y., Wu Z. C., Zhang W., and Tan L.. 2012. Common variants in toll-like receptor 4 confer susceptibility to Alzheimer’s disease in a Han Chinese population. Curr. Alzheimer Res. 9: 458–466. [DOI] [PubMed] [Google Scholar]
- 309.Wang L. Z., Yu J. T., Miao D., Wu Z. C., Zong Y., Wen C. Q., and Tan L.. 2011. Genetic association of TLR4/11367 polymorphism with late-onset Alzheimer’s disease in a Han Chinese population. Brain Res. 1381: 202–207. [DOI] [PubMed] [Google Scholar]
- 310.Song M., Jin J., Lim J. E., Kou J., Pattanayak A., Rehman J. A., Kim H. D., Tahara K., Lalonde R., and Fukuchi K.. 2011. TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflammation. 8: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Tahara K., Kim H. D., Jin J. J., Maxwell J. A., Li L., and Fukuchi K.. 2006. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 129: 3006–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Rocca W. A., Bower J. H., Maraganore D. M., Ahlskog J. E., Grossardt B. R., de Andrade M., and Melton L. J. III. 2007. Increased risk of cognitive impairment or dementia in women who underwent oophorectomy before menopause. Neurology. 69: 1074–1083. [DOI] [PubMed] [Google Scholar]
- 313.Phillips S. M., and Sherwin B. B.. 1992. Effects of estrogen on memory function in surgically menopausal women. Psychoneuroendocrinology. 17: 485–495. [DOI] [PubMed] [Google Scholar]
- 314.Sherwin B. B. 1988. Estrogen and/or androgen replacement therapy and cognitive functioning in surgically menopausal women. Psychoneuroendocrinology. 13: 345–357. [DOI] [PubMed] [Google Scholar]
- 315.Sherwin B. B. 1988. Affective changes with estrogen and androgen replacement therapy in surgically menopausal women. J. Affect. Disord. 14: 177–187. [DOI] [PubMed] [Google Scholar]
- 316.Sherwin B. B. 2009. Estrogen therapy: is time of initiation critical for neuroprotection? Nat. Rev. Endocrinol. 5: 620–627. [DOI] [PubMed] [Google Scholar]
- 317.Heikkinen T., Kalesnykas G., Rissanen A., Tapiola T., Iivonen S., Wang J., Chaudhuri J., Tanila H., Miettinen R., and Puolivali J.. 2004. Estrogen treatment improves spatial learning in APP + PS1 mice but does not affect beta amyloid accumulation and plaque formation. Exp. Neurol. 187: 105–117. [DOI] [PubMed] [Google Scholar]
- 318.Carroll J. C., Rosario E. R., Chang L., Stanczyk F. Z., Oddo S., LaFerla F. M., and Pike C. J.. 2007. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. 27: 13357–13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Zheng H., Xu H., Uljon S. N., Gross R., Hardy K., Gaynor J., Lafrancois J., Simpkins J., Refolo L. M., Petanceska S., et al. . 2002. Modulation of A(beta) peptides by estrogen in mouse models. J. Neurochem. 80: 191–196. [DOI] [PubMed] [Google Scholar]
- 320.Li R., He P., Cui J., Staufenbiel M., Harada N., and Shen Y.. 2013. Brain endogenous estrogen levels determine responses to estrogen replacement therapy via regulation of BACE1 and NEP in female Alzheimer’s transgenic mice. Mol. Neurobiol. 47: 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Maki P. M. 2013. Critical window hypothesis of hormone therapy and cognition: a scientific update on clinical studies. Menopause. 20: 695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Yaffe K., Krueger K., Cummings S. R., Blackwell T., Henderson V. W., Sarkar S., Ensrud K., and Grady D.. 2005. Effect of raloxifene on prevention of dementia and cognitive impairment in older women: the Multiple Outcomes of Raloxifene Evaluation (MORE) randomized trial. Am. J. Psychiatry. 162: 683–690. [DOI] [PubMed] [Google Scholar]
- 323.Yaffe K., Krueger K., Sarkar S., Grady D., Barrett-Connor E., Cox D. A., and Nickelsen T.; Multiple Outcomes of Raloxifene Evaluation Investigators. 2001. Cognitive function in postmenopausal women treated with raloxifene. N. Engl. J. Med. 344: 1207–1213. [DOI] [PubMed] [Google Scholar]
- 324.Jacobsen D. E., Samson M. M., Emmelot-Vonk M. H., and Verhaar H. J.. 2010. Raloxifene improves verbal memory in late postmenopausal women: a randomized, double-blind, placebo-controlled trial. Menopause. 17: 309–314. [DOI] [PubMed] [Google Scholar]
- 325.Agnusdei D., and Iori N.. 2000. Raloxifene: results from the MORE study. J. Musculoskelet. Neuronal Interact. 1: 127–132. [PubMed] [Google Scholar]
- 326.Molloy M. E., White B. E., Gherezghiher T., Michalsen B. T., Xiong R., Patel H., Zhao H., Maximov P. Y., Jordan V. C., Thatcher G. R., et al. . 2014. Novel selective estrogen mimics for the treatment of tamoxifen-resistant breast cancer. Mol. Cancer Ther. 13: 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Daumas S., Sandin J., Chen K. S., Kobayashi D., Tulloch J., Martin S. J., Games D., and Morris R. G.. 2008. Faster forgetting contributes to impaired spatial memory in the PDAPP mouse: deficit in memory retrieval associated with increased sensitivity to interference? Learn. Mem. 15: 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Ishii M., Wang G., Racchumi G. , Dyke J. P., and Iadecola C.. 2014. Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J. Neurosci. 34: 9096–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Colie S., Sarroca S., Palenzuela R., Garcia I., Matheu A., Corpas R., Dotti C. G., Esteban J. A., Sanfeliu C., and Nebreda A. R.. 2017. Neuronal p38alpha mediates synaptic and cognitive dysfunction in an Alzheimer’s mouse model by controlling beta-amyloid production. Sci. Rep. 7: 45306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Tohda C., Urano T., Umezaki M., Nemere I., and Kuboyama T.. 2012. Diosgenin is an exogenous activator of 1,25D(3)-MARRS/Pdia3/ERp57 and improves Alzheimer’s disease pathologies in 5XFAD mice. Sci. Rep. 2: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Crouzin N., Baranger K., Cavalier M., Marchalant Y., Cohen-Solal C., Roman F. S., Khrestchatisky M., Rivera S., Feron F., and Vignes M.. 2013. Area-specific alterations of synaptic plasticity in the 5XFAD mouse model of Alzheimer’s disease: dissociation between somatosensory cortex and hippocampus. PLoS One. 8: e74667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Fragkouli A., Tsilibary E. C., and Tzinia A. K.. 2014. Neuroprotective role of MMP-9 overexpression in the brain of Alzheimer’s 5xFAD mice. Neurobiol. Dis. 70: 179–189. [DOI] [PubMed] [Google Scholar]
- 333.Moon M., Jeong I., Kim C. H., Kim J., Lee P. K., Mook-Jung I., Leblanc P., and Kim K. S.. 2015. Correlation between orphan nuclear receptor Nurr1 expression and amyloid deposition in 5XFAD mice, an animal model of Alzheimer’s disease. J. Neurochem. 132: 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Xu F., Kotarba A. E., Ou-Yang M. H., Fu Z., Davis J., Smith S. O., and Van Nostrand W. E.. 2014. Early-onset formation of parenchymal plaque amyloid abrogates cerebral microvascular amyloid accumulation in transgenic mice. J. Biol. Chem. 289: 17895–17908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Wu Z., Guo Z., Gearing M., and Chen G.. 2014. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat. Commun. 5: 4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Kimura R., Devi L., and Ohno M.. 2010. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J. Neurochem. 113: 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Zhao J., Fu Y., Yasvoina M., Shao P., Hitt B., O’Connor T., Logan S., Maus E., Citron M., Berry R., et al. . 2007. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J. Neurosci. 27: 3639–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Zhang Z., Liu X., Schroeder J. P., Chan C. B., Song M., Yu S. P., Weinshenker D., and Ye K.. 2014. 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 39: 638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Devi L., and Ohno M.. 2015. A combination Alzheimer’s therapy targeting BACE1 and neprilysin in 5XFAD transgenic mice. Mol. Brain. 8: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Tang X., Wu D., Gu L. H., Nie B. B., Qi X. Y., Wang Y. J., Wu F. F., Li X. L., Bai F., Chen X. C., et al. . 2016. Spatial learning and memory impairments are associated with increased neuronal activity in 5XFAD mouse as measured by manganese-enhanced magnetic resonance imaging. Oncotarget. 7: 57556–57570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Giannoni P., Arango-Lievano M., Neves I. D., Rousset M. C., Baranger K., Rivera S., Jeanneteau F., Claeysen S., and Marchi N.. 2016. Cerebrovascular pathology during the progression of experimental Alzheimer’s disease. Neurobiol. Dis. 88: 107–117. [DOI] [PubMed] [Google Scholar]
- 342.Griiñán-Ferré C., Sarroca S., Ivanova A., Puigoriol-Illamola D., Aguado F., Camins A., Sanfeliu C., and Pallàs M.. 2016. Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging (Albany NY). 8: 664–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Kandalepas P. C., Sadleir K. R., Eimer W. A., Zhao J., Nicholson D. A., and Vassar R.. 2013. The Alzheimer’s beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 126: 329–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Buskila Y., Crowe S. E., and Ellis-Davies G. C.. 2013. Synaptic deficits in layer 5 neurons precede overt structural decay in 5xFAD mice. Neuroscience. 254: 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Lee K., Kim H., An K., Kwon O. B., Park S., Cha J. H., Kim M. H., Lee Y., Kim J. H., Cho K., et al. . 2016. Replenishment of microRNA-188–5p restores the synaptic and cognitive deficits in 5XFAD mouse model of Alzheimer’s disease. Sci. Rep. 6: 34433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Spangenberg E. E., Lee R. J., Najafi A. R., Rice R. A., Elmore M. R., Blurton-Jones M., West B. L., and Green K. N.. 2016. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain. 139: 1265–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Deleted in proof. [Google Scholar]
- 348.Maarouf C. L., Kokjohn T. A., Whiteside C. M., Macias M. P., Kalback W. M., Sabbagh M. N., Beach T. G., Vassar R., and Roher A. E.. 2013. Molecular differences and similarities between Alzheimer’s disease and the 5XFAD transgenic mouse model of amyloidosis. Biochem. Insights. 6: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Saul A., and Wirths O.. 2017. Endogenous apolipoprotein E (ApoE) fragmentation is linked to amyloid pathology in transgenic mouse models of Alzheimer’s disease. Mol. Neurobiol. 54: 319–327. [DOI] [PubMed] [Google Scholar]
- 350.Devi L., and Ohno M.. 2012. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology. 37: 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Py N. A., Bonnet A. E., Bernard A., Marchalant Y., Charrat E., Checler F., Khrestchatisky M., Baranger K., and Rivera S.. 2014. Differential spatio-temporal regulation of MMPs in the 5xFAD mouse model of Alzheimer’s disease: evidence for a pro-amyloidogenic role of MT1-MMP. Front. Aging Neurosci. 6: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Devi L., and Ohno M.. 2014. PERK mediates eIF2alpha phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging. 35: 2272–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Devi L., and Ohno M.. 2015. TrkB reduction exacerbates Alzheimer’s disease-like signaling aberrations and memory deficits without affecting beta-amyloidosis in 5XFAD mice. Transl. Psychiatry. 5: e562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Hillmann A., Hahn S., Schilling S., Hoffmann T., Demuth H. U., Bulic B., Schneider-Axmann T., Bayer T. A., Weggen S., and Wirths O.. 2012. No improvement after chronic ibuprofen treatment in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging. 33: 833.e39–833.e50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.