

Key Points
A genetic risk profile integrating α-thalassemia and BCL11A status improves associations with hemolytic markers and stroke history.
Abstract
α-Thalassemia and the BCL11A rs1427407 T allele are commonly observed in sickle cell anemia (SCA) patients and are associated with reduced hemolysis and higher hemoglobin F levels, respectively. We investigated whether a high-risk genetic profile, defined as SCA patients who did not inherit either α-thalassemia or the BCL11A rs1427407 T allele, had stronger associations with clinical and laboratory variables than the individual genetic components in the University of Ibadan cohort (N = 249). We then replicated our findings in SCA cohorts from the University of Illinois at Chicago (UIC) (N = 260) and the Walk-Treatment of Pulmonary Hypertension and Sickle Cell Disease with Sildenafil Therapy study (Walk-PHaSST) (N = 387). A high-risk genetic profile was associated with higher reticulocytes (15.0% vs 7.8%, P = .08) and stroke history (6% vs 1%, P = .02) than standard-risk patients, and these associations were more significant than the individual genetic components in the University of Ibadan cohort. These findings were replicated in high-risk patients from UIC and Walk-PHaSST for reticulocytes (UIC: 13.5% vs 11.8%, P = .03; Walk-PHaSST: 9.6% vs 8.2%, P = .0003) and stroke history (UIC: 32% vs 22%, P = .07; Walk-PHaSST: 14% vs 7%, P = .01). On combined analysis, a high-risk genetic profile had strong associations with increased markers of hemolysis (hemoglobin β = –0.29, 95% confidence interval [CI]: −0.50 to −0.09; P=.006; reticulocyte% β = 2.29, 95% CI: 1.31-3.25; P = 1 × 10−5) and stroke history (odds ratio = 2.0, 95% CI: 1.3-3.0; P = .0002), but no association with frequent vaso-occlusive crises (≥3 per year). A high-risk genetic profile is associated with increased hemolysis and stroke history in 3 independent cohorts. This profile may help identify patients to prioritize for hydroxyurea and for closer monitoring strategies for stroke.
Visual Abstract
Introduction
Sickle cell anemia (SCA) is an inherited red blood cell disorder that affects ∼25 million people worldwide.1 This monogenic disease is caused by a single–base point mutation in the β-globin chain that results in a glutamic acid to valine substitution and leads to vaso-occlusion and chronic hemolysis. There are protean acute and chronic complications in SCA, and downstream genetic modifiers may influence the phenotype of this disease.
Complex diseases and disease-related complications may result from the joint effects of multiple genetic factors, and combining these genetic factors into a risk profile may strengthen the predictive value compared with the individual factors alone. By using common variants with strong effects, genetic risk profiles have been developed for age-related macular degeneration and for hypertriglyceridemia.2,3 Coinheritance of α-thalassemia is observed in approximately one-third of SCA patients and is associated with reduced hemolysis and protection against some SCA-related complications.4,5 The BCL11A rs1427407 T variant (minor allele frequency: 0.25) is also commonly observed in patients with SCA and may modify disease severity by increasing fetal hemoglobin (HbF) levels.6
We conducted a study to determine if a genetic risk profile based on the coinheritance of α-thalassemia and the BCL11A rs1427407 variants influences laboratory and clinical variables in a cohort of SCA patients treated at the University of Ibadan and repeated our analyses in the University of Illinois at Chicago (UIC) and in the multicenter Walk-Treatment of Pulmonary Hypertension and Sickle Cell Disease with Sildenafil Therapy study (Walk-PHaSST) cohorts. We hypothesized that a genetic risk profile would provide additional value in identifying SCA patients at higher risk for clinical complications than the individual genetic components.
Methods
The protocol was approved by the institutional review board of the respective institutions before initiating the study and all subjects provided written informed consent prior to blood sample and data collection. Between 2012 and 2015, 249 patients from the University of Ibadan with SCA (defined as HbSS or HbSβ0-thalassemia) were recruited and genotyped. Clinical and laboratory data were collected by the medical health professionals and by chart review. HbF levels were only available by high-pressure liquid chromatography in 25 SCA patients who were enrolled in a low-dose hydroxyurea study (NCT02149537) at the University of Ibadan and were used to determine the association between the HbF levels and the BCL11A rs1427407 T variant. Replication analyses were conducted in 260 SCA patients from the UIC recruited between August 2010 and March 2016 and in 387 SCA patients from the Walk-PHaSST cohort, which represents 1 UK and 9 US centers, recruited between February 2008 and June 2009. The UIC was a participating site for Walk-PHaSST, and patients from UIC were excluded from this cohort.
The Taqman genotyping assay (Applied BioSystems, Foster City, CA) was used for genotyping the BCL11A rs1427407 polymorphism in the University of Ibadan and UIC subjects by using 10 ng of genomic DNA according to the manufacturer's instructions in a Bio-Rad CFX 384 Real-Time System with a C1000 Thermal Cycler. The genotyping calls were analyzed by using Bio-Rad CFX Manager version 3.1. In the Walk-PHaSST cohort, genotypes for the BCL11A rs1427407 variant were determined by imputation as previously described.7 Multiplex polymerase chain reaction (PCR) reactions on 100 to 200 ng of genomic DNA were used to detect α-thalassemia status by using methods previously described in the University of Ibadan and UIC cohorts.8 To avoid primer dimers, only primers for the HBA2 gene, α-3.7K deletion, and α-4.2K deletion were used in the PCR reaction. Genotyping for BCL11A and α-thalassemia status for the University of Ibadan and UIC subjects was performed centrally at UIC. In the Walk-PHaSST cohort, the α-3.7K deletion and α-4.2K deletion were determined by a multiplex PCR reaction assay9 performed at the University of Pittsburgh. A genetic risk profile was defined a priori as follows: high risk = absence of both α-thalassemia and the BCL11A rs1427407 T allele; and standard risk = inheritance of α-thalassemia and/or the BCL11A rs1427407 T allele.
Comparisons according to genotype or risk profile in the individual cohorts were performed by using the Kruskall-Wallis test for continuous variables and χ2 analysis for categorical variables. In a combined analysis of the 3 cohorts, we tested the associations of the genetic risk profile with continuous and categorical variables using linear and logistic regression models, respectively, adjusting for site (University of Ibadan, UIC, or Walk-PHaSST), age, sex, and hydroxyurea therapy (yes or no). Systat version 11 (Systat Software Corporation, Chicago, IL) was used for the statistical analyses. Median values and interquartile ranges (IQR) are provided for linear variables.
Results
University of Ibadan
In the University of Ibadan cohort, the median age was 19 years (IQR, 15-24 years), 53% were male, and 7 (3%) were on hydroxyurea therapy. Coinheritance of α-thalassemia was observed in 42.5% of patients (α-/αα: 33.3%, α-/α-: 9.2%) and was associated with a lower white blood cell (WBC) count (Table 1). The BCL11A rs1427407 T variant was observed in 46.6% of patients (G/T: 41.4%, T/T: 5.2%) and was associated with a trend for higher hemoglobin concentration (Table 1). The BCL11A rs1427407 T variant correlated with a higher HbF level by high-pressure liquid chromatography in the subset of patients enrolled in the low-dose hydroxyurea study at baseline (G/G: 4.1 ± 1.1%, G/T: 5.8 ± 1.1%, T/T: 7.1 ± 3.1%) and after 10 mg/kg of hydroxyurea therapy (G/G: 7.2 ± 1.7%, G/T: 10.1 ± 1.6%, T/T: 19.0 ± 4.8%) (repeated measures P = .01). Trends for a higher prevalence of vaso-occlusive crises (VOCs) ≥3 per year were observed in SCA patients who did not inherit α-thalassemia or the BCL11A rs1427407 T variant. Similarly, prevalence of stroke history was higher in SCA patients who did not inherit α-thalassemia or the BCL11A rs1427407 T variant, although the associations were not statistically significant (Table 1).
Table 1.
Comparison of laboratory variables by genetic variants in patients with SCA from the University of Ibadan
| α-Thalassemia | |||||
|---|---|---|---|---|---|
| N | αα/αα | N | α-/αα or α-/α- | P | |
| WBC count, × 103/µL | 143 | 12.5 (10.2-15.4) | 106 | 10.8 (9.0-14.6) | .024 |
| Hemoglobin, g/dL | 143 | 7.7 (6.9-8.5) | 106 | 7.5 (6.6-8.6) | .5 |
| Reticulocyte, % | 40 | 10.5 (3.5-17.1) | 32 | 7.9 (3.3-12.7) | .4 |
| VOCs ≥3/y, n (%) | 143 | 124 (87) | 106 | 82 (77) | .1 |
| Stroke history, n (%) | 143 | 6 (4) | 106 | 1 (1) | .1 |
| BCL11A rs1427407 | |||||
|---|---|---|---|---|---|
| N | G/G | N | G/T or T/T | P | |
| WBC count, × 103/µL | 133 | 11.4 (9.6-14.7) | 116 | 12.0 (9.8-15.4) | .7 |
| Hemoglobin, g/dL | 133 | 7.5 (6.6-8.3) | 116 | 7.8 (6.9-8.6) | .074 |
| Reticulocyte, % | 32 | 12.3 (3.5-18.2) | 40 | 7.6 (3.4-11.8) | .2 |
| VOCs ≥3/y, n (%) | 133 | 115 (86) | 116 | 93 (80) | .2 |
| Stroke history, n (%) | 133 | 5 (4) | 116 | 2 (2) | .3 |
| Genetic risk profile | |||||
|---|---|---|---|---|---|
| N | High risk | N | Standard risk | P | |
| WBC count, × 103/µL | 78 | 12.2 (10.2-15.1) | 171 | 11.4 (9.5-14.9) | .3 |
| Hemoglobin, g/dL | 78 | 7.6 (6.8-8.3) | 171 | 7.7 (6.7-8.6) | .6 |
| Reticulocyte, % | 15 | 15.0 (5.1-20.2) | 57 | 7.8 (3.0-12.9) | .076 |
| VOCs ≥ 3/y, n (%) | 78 | 70 (90) | 171 | 136 (80) | .069 |
| Stroke history, n (%) | 78 | 5 (6) | 171 | 2 (1) | .020 |
Values are expressed as medians and IQRs.
Using a genetic risk profile integrating α-thalassemia and BCL11A rs1427407 variant status, high risk (no α-thalassemia and no BCL11A rs1427407 T allele) was observed in 31.3% (78 of 249) whereas standard risk (either α-thalassemia and/or the BCL11A rs1427407 T allele) was observed in 68.7% (171 of 249) of SCA patients from the University of Ibadan cohort. This risk profile was associated with trends for higher reticulocyte percentage and increased risk for VOCs ≥3 per year and was significantly associated with a higher prevalence of stroke history (P = .02) (Table 1).
UIC and Walk-PHaSST
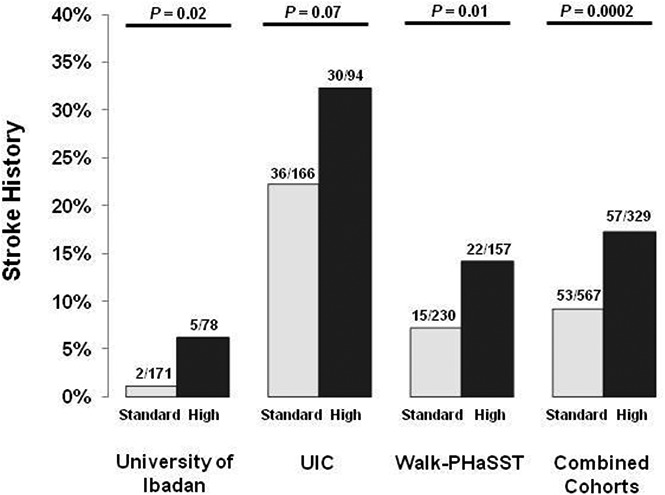
We then performed similar analyses to test for associations between the genetic risk profile and laboratory and clinical phenotypes in SCA patients from the UIC and Walk-PHaSST cohorts. In the UIC cohort, high-risk status was observed in 36.2% (94 of 260) and standard-risk status was observed in 63.8% (166 of 260) of SCA patients. The high-risk category was significantly associated with a higher reticulocyte percentage (P = .03) (Table 2). No significant association between the genetic risk profile and VOCs ≥3 per year was observed (Figure 1A), whereas a trend for a higher prevalence of stroke history (Figure 1B) was observed in high-risk vs standard-risk SCA patients. In the Walk-PHaSST cohort, high-risk status was observed in 40.6% (157 of 387) and standard-risk status in 59.4% (230 of 387) of SCA patients. The high-risk group was significantly (P < .05) associated with a lower hemoglobin concentration, a higher reticulocyte percentage, and a higher prevalence of stroke history (Figure 1B; Table 2), whereas no significant association with VOCs ≥3 per year compared with standard-risk SCA patients was observed (Figure 1A).
Table 2.
Comparison of laboratory variables by genetic risk profile in patients with SCA from the UIC and Walk-PHaSST cohorts
| N | High risk | N | Standard risk | P | |
|---|---|---|---|---|---|
| UIC | |||||
| WBC count, × 103/µL | 94 | 10.4 (7.9-13.0) | 166 | 10.1 (8.0-12.1) | .3 |
| Hemoglobin, g/dL | 94 | 8.5 (7.4-9.6) | 166 | 8.8 (8.0-9.6) | .6 |
| Reticulocyte, % | 93 | 13.5 (9.1-16.9) | 166 | 11.8 (8.3-15.6) | .027 |
| UK and US cohorts (Walk-PHaSST) | |||||
| WBC count, × 103/µL | 157 | 10.1 (8.1-13.1) | 230 | 9.6 (7.5-12.2) | .069 |
| Hemoglobin, g/dL | 157 | 8.4 (7.3-9.7) | 230 | 8.8 (7.9-9.8) | .025 |
| Reticulocyte, % | 151 | 9.6 (7.4-15.8) | 212 | 8.2 (5.8-12.3) | .0003 |
Values are expressed as medians and IQRs.
Figure 1.

A genetic risk profile predicts stroke but not VOC in SCA patients. (A) The association of a genetic risk profile with VOCs in 3 independent cohorts of SCA patients. (B) The association of a genetic risk profile with stroke in 3 independent cohorts of SCA patients.
Combined analysis
Next, we investigated the association of the genetic risk profile with the laboratory and clinical variables in a combined analysis of SCA patients from the 3 cohorts. Baseline differences between the 3 cohorts are demonstrated in Table 3. SCA patients in the high-risk category had a higher degree of hemolysis, reflected by a lower hemoglobin concentration (β = −0.29, 95% confidence interval [CI]: −0.50 to −0.09) and higher reticulocyte percentage (β = 2.29, 95% CI: 1.31-3.25), and a higher WBC count (natural log β = 0.05, 95% CI: 0.001-0.09) compared with SCA patients in the standard-risk category (Table 4). Total bilirubin concentration, another laboratory marker of hemolysis, was also available in all 3 cohorts and was elevated in SCA patients categorized as high risk (median: 2.6 mg/dL, IQR: 1.6-4.2 mg/dL) vs standard risk (median: 2.3 mg/dL, IQR: 1.6-3.4) (β = 0.51, 95% CI: 0.14-0.86; P = .0062). An increased risk for stroke history was also observed in the high- vs standard-risk SCA patients (odds ratio: 2.0, 95% CI: 1.33-2.99) (Figure 1B; Table 4). Leg ulcers and priapisms are considered hemolytic complications of SCA, and we observed a higher prevalence of leg ulcer history (20.3% vs 14.4%, respectively; odds ratio: 1.47, 95% CI: 1.01-2.15; P = .047), but no statistically significant differences for priapism history (28.2% vs 27.5%, respectively) in SCA patients categorized as high risk vs standard risk.
Table 3.
Clinical and laboratory variables in the University of Ibadan, UIC, and Walk-PHaSST cohorts
| University of Ibadan, N = 249 | UIC, N = 260 | Walk-PHaSST, N = 387 | |
|---|---|---|---|
| Age, y | 19 (15-24) | 31 (23-42) | 35 (25-46) |
| Sex (female:male), % | 47:53 | 58:42 | 52:48 |
| Hydroxyurea therapy, n (%) | 7 (3) | 135 (52) | 160 (41) |
| BMI, kg/m2 | 19 (16-20) | 23 (21-26) | 23 (21-25) |
| Mean arterial pressure, mm Hg | 76 (70-82) | 86 (80-93) | 83 (78-90) |
| α-Thalassemia (α-/αα or α-/α-), n (%) | 106 (43) | 96 (37) | 125 (32) |
| BCL11A rs1427407 (G/T or T/T), n (%) | 116 (47) | 116 (45) | 159 (41) |
Values are expressed as medians and IQRs.
BMI, body mass index.
Table 4.
Analysis of laboratory and clinical outcomes in combined data from the University of Ibadan, UIC, and Walk-PHaSST cohorts
| N | High risk | N | Standard risk | P* | |
|---|---|---|---|---|---|
| WBC count, × 103/µL | 329 | 10.7 (8.5-13.5) | 567 | 10.2 (8.2-12.6) | .049 |
| Hemoglobin, g/dL | 329 | 8.2 (7.2-9.3) | 567 | 8.5 (7.5-9.5) | .0055 |
| Reticulocyte, % | 259 | 11.5 (7.9-16.7) | 434 | 9.6 (6.2-13.9) | 1.0 × 10−5 |
| VOCs ≥3/y, n (%) | 329 | 181 (55) | 567 | 306 (54) | .3 |
| Stroke history, n (%) | 329 | 57 (17) | 567 | 53 (9) | .0002 |
Values are expressed as medians and IQRs.
P value adjusted for age, sex, hydroxyurea therapy, and site by using linear and logistic regression analysis.
Discussion
α-Thalassemia and the BCL11A rs1427407 T variant were both commonly observed in SCA patients from the University of Ibadan. Consistent with the literature, α-thalassemia and the BCL11A rs1427407 T variant were associated with lower WBC counts10 and a trend for higher hemoglobin concentrations,11 respectively. When combining the absence of α-thalassemia with the absence of the BCL11A rs1427407 T variant to define a high-risk group of SCA patients, we observed a replicated association of the genetic risk profile with higher reticulocyte percentage and stroke risk in 3 independent cohorts.
Coinheritance of α-thalassemia has been associated with protection against some SCA-related complications, such as acute chest syndrome, leg ulcers, and chronic kidney disease,4,5 whereas similar rates of VOC and higher rates of avascular necrosis and retinal disease have been observed compared with SCA patients without α-thalassemia.4,12 Furthermore, the effects of α-thalassemia on cerebral vasculopathy and stroke risk in SCA patients have been mixed, with some studies showing a protective benefit,13-18 whereas others have not.10,19-21 In the University of Ibadan cohort of SCA patients, α-thalassemia was commonly observed, but was not associated with a statistically significant reduction in the frequency of SCA-related complications.
The BCL11A rs1427407 T variant has been associated with higher hemoglobin concentrations11,22,23 and may ameliorate SCA-related complications based on composite endpoints, such as all-cause hospitalizations11,24 or disease severity scores.25,26 However, associations between the BCL11A variants and specific SCA-related complications, such as VOC frequency11,27 and stroke,11,25 have shown conflicting results. We did not observe a statistically significant reduction in ≥3 VOCs per year or stroke history with inheritance of the BCL11A T variant in the University of Ibadan cohort.
When we combined α-thalassemia and BCL11A rs1427407 T variant status into a genetic risk profile, we observed a significantly higher proportion of SCA patients with stroke history and trends for higher reticulocyte percentage and VOCs ≥3 per year in SCA patients categorized as high risk vs standard risk. When testing these associations in 2 other independent cohorts of SCA patients, one representing an urban center in the United States (UIC) and the other representing a multicenter cohort of SCA patients selected for suspicion of having pulmonary hypertension from the United States and the United Kingdom (Walk-PHaSST), we were able to replicate the association of this genetic risk profile with reticulocyte percentage and stroke history. We could not replicate the association with VOCs ≥3 per year observed in the University of Ibadan cohort in the UIC or Walk-PHaSST cohort, and this may be due to differences in environmental factors or prevalence of the use of hydroxyurea therapy. In combined analysis of the 3 cohorts, the genetic risk profile had a strong association with markers of hemolysis and identified SCA patients at a twofold increased risk of stroke.
The association of a hemolytic genetic risk profile with stroke history emphasizes the hemolytic role in the pathophysiologic process of cerebral vasculopathy. Mechanisms for how increased hemolysis may lead to cerebral vasculopathy and stroke include reduced nitric oxide bioavailability, leading to increased vascular resistance28 and upregulation of vascular adhesion molecules (eg, VCAM1, E-selectin),29 more severe anemia in the setting of impaired cerebrovascular reserve capacity,30 and increased coagulation activation.31,32 Our findings are also consistent with other investigators that have demonstrated that a lower hemoglobin concentration is a risk factor for stroke33 and that elevated markers of hemolysis are independently associated with increased transcranial Doppler velocities13 and silent strokes.34
There are several limitations to our study. First, this was a cross-sectional study, and differences in stroke history may have been subject to survival bias. Stroke history was determined by clinical history and was not confirmed by a clinical assessment tool or by magnetic resonance imaging or magnetic resonance angiography findings and, this may have led to a detection bias and excluded silent infarcts from this analysis.
In conclusion, a genetic risk profile incorporating 2 common variants that influence the degree of hemolysis (α-thalassemia) and HbF levels (BCL11A rs1427407 T variant) may improve our ability to identify SCA patients who are at elevated risk for stroke. Our findings are a first attempt at a proof-of-concept for a genetic risk profile in SCA and demonstrate the potential to be important clinically. Our cohorts included older adolescents and adults, and future studies of children that investigate the association of this genetic profile with transcranial Doppler velocities and integrate this risk profile with transcranial Doppler velocities to predict stroke risk may help focus screening practices. Future studies prospectively evaluating whether this genetic risk profile can detect SCA patients with a higher risk of stroke may also help prioritize hydroxyurea therapy and transfusion strategies, particularly in low-resource areas.
Acknowledgments
This work was supported by Doris Duke Charitable Foundation grant 2013140 for clinical trial registration NCT02149537 (V.R.G. and B.O.T.) and by National Institutes of Health, National Heart, Lung, and Blood Institute grants K23HL125984 (S.L.S.), R01HL111656, and R01HL127342 (R.F.M.). The Walk-PHaSST project was supported by federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health under contract HHSN268200617182C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding institutions.
Authorship
Contribution: S.L.S., T.S.A., B.N.S., C.A.E., O.S., X.Z., V.R.G., and B.O.T. designed and performed the research, analyzed the data, and wrote the paper; and L.L.H., M.T.G., R.F.M., and R.S.C. designed the research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Santosh L. Saraf, Division of Hematology and Oncology, Department of Medicine, University of Illinois at Chicago, 820 South Wood St, MC 712, Chicago, IL 60612; e-mail: ssaraf@uic.edu; and Bamidele O. Tayo, Department of Public Health Sciences, Stritch School of Medicine, Loyola University Chicago, 2160 South First Ave, Maywood, IL 60153; e-mail: btayo@luc.edu.
References
- 1.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maller J, George S, Purcell S, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38(9):1055-1059. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Ban MR, Zou GY, et al. Polygenic determinants of severe hypertriglyceridemia. Hum Mol Genet. 2008;17(18):2894-2899. [DOI] [PubMed] [Google Scholar]
- 4.Higgs DR, Aldridge BE, Lamb J, et al. The interaction of alpha-thalassemia and homozygous sickle-cell disease. N Engl J Med. 1982;306(24):1441-1446. [DOI] [PubMed] [Google Scholar]
- 5.Guasch A, Zayas CF, Eckman JR, Muralidharan K, Zhang W, Elsas LJ. Evidence that microdeletions in the alpha globin gene protect against the development of sickle cell glomerulopathy in humans. J Am Soc Nephrol. 1999;10(5):1014-1019. [DOI] [PubMed] [Google Scholar]
- 6.Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342(6155):253-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saraf SL, Zhang X, Shah B, et al. Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica. 2015;100(10):1275-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kidd JL, Azimi M, Lubin B, Vichinsky E, Hoppe C. Application of an expanded multiplex genotyping assay for the simultaneous detection of Hemoglobin Constant Spring and common deletional alpha-thalassemia mutations. Int J Lab Hematol. 2010;32(4):373-380. [DOI] [PubMed] [Google Scholar]
- 9.Tan AS, Quah TC, Low PS, Chong SS. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood. 2001;98(1):250-251. [DOI] [PubMed] [Google Scholar]
- 10.Rumaney MB, Ngo Bitoungui VJ, Vorster AA, et al. The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLoS One. 2014;9(6):e100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wonkam A, Ngo Bitoungui VJ, Vorster AA, et al. Association of variants at BCL11A and HBS1L-MYB with hemoglobin F and hospitalization rates among sickle cell patients in Cameroon. PLoS One. 2014;9(3):e92506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagel RL, Fabry ME, Kaul DK, et al. Known and potential sources for epistatic effects in sickle cell anemia. Ann N Y Acad Sci. 1989;565:228-238. [DOI] [PubMed] [Google Scholar]
- 13.Bernaudin F, Verlhac S, Chevret S, et al. G6PD deficiency, absence of alpha-thalassemia, and hemolytic rate at baseline are significant independent risk factors for abnormally high cerebral velocities in patients with sickle cell anemia [published correction appears in Blood 2010;116(23):5079]. Blood. 2008;112(10):4314-4317. [DOI] [PubMed] [Google Scholar]
- 14.Belisário AR, Rodrigues CV, Martins ML, Silva CM, Viana MB. Coinheritance of α-thalassemia decreases the risk of cerebrovascular disease in a cohort of children with sickle cell anemia. Hemoglobin. 2010;34(6):516-529. [DOI] [PubMed] [Google Scholar]
- 15.Hsu LL, Miller ST, Wright E, et al. ; Stroke Prevention Trial (STOP) and the Cooperative Study of Sickle Cell Disease (CSSCD). Alpha thalassemia is associated with decreased risk of abnormal transcranial Doppler ultrasonography in children with sickle cell anemia. J Pediatr Hematol Oncol. 2003;25(8):622-628. [DOI] [PubMed] [Google Scholar]
- 16.Cox SE, Makani J, Soka D, et al. Haptoglobin, alpha-thalassaemia and glucose-6-phosphate dehydrogenase polymorphisms and risk of abnormal transcranial Doppler among patients with sickle cell anaemia in Tanzania. Br J Haematol. 2014;165(5):699-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flanagan JM, Frohlich DM, Howard TA, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011;117(24):6681-6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joly P, Garnier N, Kebaili K, et al. G6PD deficiency and absence of α-thalassemia increase the risk for cerebral vasculopathy in children with sickle cell anemia. Eur J Haematol. 2016;96(4):404-408. [DOI] [PubMed] [Google Scholar]
- 19.Domingos IF, Falcão DA, Hatzlhofer BL, et al. Influence of the βs haplotype and α-thalassemia on stroke development in a Brazilian population with sickle cell anaemia. Ann Hematol. 2014;93(7):1123-1129. [DOI] [PubMed] [Google Scholar]
- 20.Sommet J, Alberti C, Couque N, et al. Clinical and haematological risk factors for cerebral macrovasculopathy in a sickle cell disease newborn cohort: a prospective study. Br J Haematol. 2016;172(6):966-977. [DOI] [PubMed] [Google Scholar]
- 21.Filho IL, Leite AC, Moura PG, et al. Genetic polymorphisms and cerebrovascular disease in children with sickle cell anemia from Rio de Janeiro, Brazil. Arq Neuropsiquiatr. 2011;69(3):431-435. [DOI] [PubMed] [Google Scholar]
- 22.Sheehan VA, Luo Z, Flanagan JM, et al. ; BABY HUG Investigators. Genetic modifiers of sickle cell anemia in the BABY HUG cohort: influence on laboratory and clinical phenotypes. Am J Hematol. 2013;88(7):571-576. [DOI] [PubMed] [Google Scholar]
- 23.Mtatiro SN, Makani J, Mmbando B, Thein SL, Menzel S, Cox SE. Genetic variants at HbF-modifier loci moderate anemia and leukocytosis in sickle cell disease in Tanzania. Am J Hematol. 2015;90(1):E1-E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muszlak M, Pissard S, Badens C, Chamouine A, Maillard O, Thuret I. Genetic modifiers of sickle cell disease: a genotype-phenotype relationship study in a cohort of 82 children on Mayotte Island. Hemoglobin. 2015;39(3):156-161. [DOI] [PubMed] [Google Scholar]
- 25.Leonardo FC, Brugnerotto AF, Domingos IF, et al. Reduced rate of sickle-related complications in Brazilian patients carrying HbF-promoting alleles at the BCL11A and HMIP-2 loci. Br J Haematol. 2016;173(3):456-460. [DOI] [PubMed] [Google Scholar]
- 26.Upadhye D, Jain D, Trivedi Y, Nadkarni A, Ghosh K, Colah R. Influence of single nucleotide polymorphisms in the BCL11A and HBS1L-MYB gene on the HbF levels and clinical severity of sickle cell anaemia patients. Ann Hematol. 2016;95(7):1201-1203. [DOI] [PubMed] [Google Scholar]
- 27.Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA. 2008;105(33):11869-11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Caterina R, Libby P, Peng HB, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96(1):60-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nur E, Kim YS, Truijen J, et al. Cerebrovascular reserve capacity is impaired in patients with sickle cell disease. Blood. 2009;114(16):3473-3478. [DOI] [PubMed] [Google Scholar]
- 31.Ataga KI, Brittain JE, Desai P, et al. Association of coagulation activation with clinical complications in sickle cell disease. PLoS One. 2012;7(1):e29786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Connes P, Verlhac S, Bernaudin F. Advances in understanding the pathogenesis of cerebrovascular vasculopathy in sickle cell anaemia. Br J Haematol. 2013;161(4):484-498. [DOI] [PubMed] [Google Scholar]
- 33.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288-294. [PubMed] [Google Scholar]
- 34.Kwiatkowski JL, Zimmerman RA, Pollock AN, et al. Silent infarcts in young children with sickle cell disease. Br J Haematol. 2009;146(3):300-305. [DOI] [PMC free article] [PubMed] [Google Scholar]