

Abstract
The synthesis of novel series of sulfamoylbenzamides as HBV capsid assembly effector is reported. The structure was divided into five parts which were independently modified as part of our lead optimization. All synthesized compounds were evaluated for their anti-HBV activity and toxicity in human hepatocytes, lymphocytes and other cells. Additionally, we assessed their effect on HBV cccDNA formation in an HBeAg reporter cell-based assay. Among the 27 compounds reported, several analogs exhibited submicromolar activities and significant reduction of HBeAg secretion. Selected compounds were studied under negative-stain electron microscopy for their ability to disrupt the HBV capsid formation. Structures were modeled into a binding site recently identified in the HBV capsid protein for similar molecules to rationalize the structure-activity relationships for this family of compounds.
Keywords: sulfamoylbenzamide, antiviral agent, HBV, capsid assembly effector, cccDNA, HBeAg
Graphical abstract
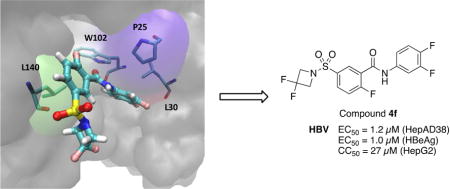
1. Introduction
Hepatitis B virus (HBV) remains a major public health concern with over 2 billion people estimated to be infected worldwide. However, while most healthy adults are capable of recovering from the infection, up to 50% of children will develop chronic infections if the infection occurs before the age of 6 [1]. If left untreated, these subjects, adults and children, will experience liver diseases such as cirrhosis and hepatocellular carcinoma. A constant suppression of the viral replication is then required with available treatments involving polymerase inhibitors such as lamivudine (3TC), entecavir (ETV) and tenofovir-diisoproxyl fumarate (TDF) or tenofovir alafenamide (TAF) [2]. Although these drugs are very effective at controlling virus infection, long-term treatments may cause drug resistance or severe side effects and more importantly, none of these drugs can cure HBV infections [3, 4].
Nucleocapsids have an important role in the HBV viral replication cycle since they play a role during genome packaging, reverse transcription, intracellular trafficking and maintenance of chronic infection [5]. Early discoveries have shown that interactions of the core proteins with small heterocyclic molecules can induce faulty assembly which leads to the formation of dysfunctional nucleocapsids [6, 7]. By acting on the encapsidation process of viral material, capsid assembly effectors (CAEs) are expected to significantly decrease the formation of cccDNA partly responsible of the viral persistence [8]. Disrupting HBV capsids allows exposure of cccDNA to degrading enzymes. In recent years, several classes of CAE targeting the HBV nucleocapsid formation/disruption have emerged. Among them, heteroarylpyrimidines (GLS-4, phase II) [9], phenylpropenamides (AT-130) [10] and sulfamoylbenzamide derivatives (DVR-23 [11], NVR 3-778 [12], phase IIa, structure not yet disclosed) exhibited promising antiviral effects and could potentially lead to a curative treatment (Figure 1). As part of our HBV research program, we turned our attention towards the sulfamoyl scaffold of DVR-23 and designed new series of derivatives. Herein, we wish to report their detailed synthesis along with their antiviral evaluation and characterization.
Fig 1.
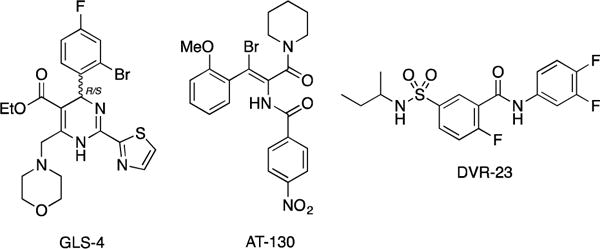
Capsid assembly effectors.
2. Results and discussion
2.1. Chemical synthesis
To simplify the structure-activity relationship (SAR) study, the sulfamoylbenzamide scaffold was divided in five parts labeled A, B, C, D and E (Figure 2).
Fig 2.
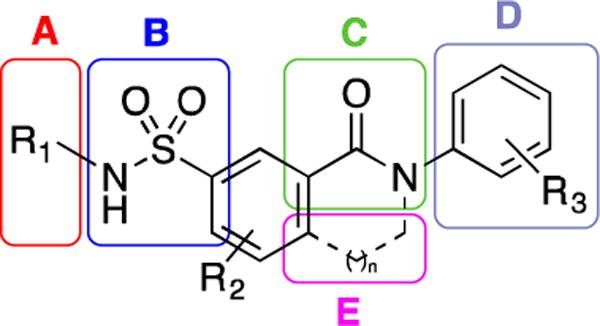
Structure of the sulfamoylbenzamide scaffold.
Part A: Various groups selected from alkyls, cycloalkyls, alkylaryls or sulfonyls were introduced at this position. Sulfamoylbenzamide derivatives 4a–s were prepared from 2-fluorobenzoic acid 1 by reaction with chlorosulfonic acid leading to sulfonyl chloride 2 in 77% yield (Scheme 1) [13]. Subsequent reaction with thionyl chloride followed by treatment with 3,4-difluoroaniline provided the amide 3 in 65% yield. This key intermediate was then reacted with various primary and secondary aliphatic and benzyl amines, amino acids and sulfonamides to afford a small library of structurally diverse sulfamoyl analogs (Compounds 4a–s).
Scheme 1.
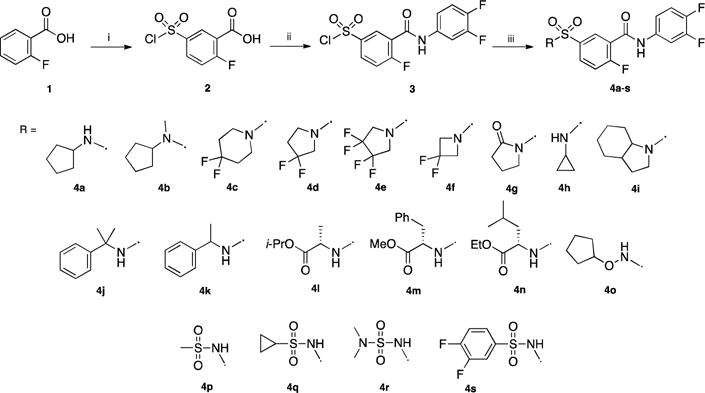
Reagents and conditions: (i) ClSO3H, 80 °C, 4 h, 77%; (ii) SOCl2, 80 °C, 16 h then 3,4-difluoroaniline, toluene, 110 °C, 2 h, 65%; (iii) amine, Et3N, CH2Cl2, 0 °C to rt (or 40 °C), 2 h, 26–94%.
Part B: Inversion of the sulfonamide portion was also investigated. The synthesis of derivatives 9–11 was achieved in four steps from commercially available 5-amino-2-fluorobenzoic acid 5 by first Boc-protection of the amino group to afford derivative 6 in 74% yield (Scheme 2). The resulting intermediate was then activated using HATU and reacted with 3,4-difluoroaniline to afford compound 7 in 90% yield. Finally, deprotection of the Boc group using TFA followed by reaction with different cycloalkylsulfonyl chlorides (cycloproyl, cyclopentyl and cyclohexyl) provided the desired analogs 9–11 in moderate yields (23–47%).
Scheme 2.
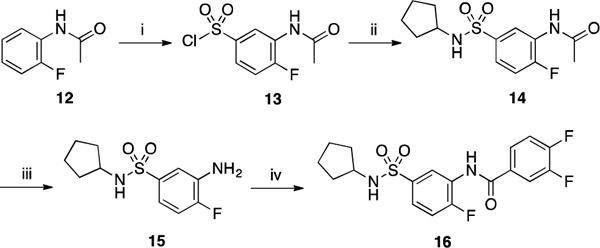
Reagents and conditions: (i) Boc2O, NaHCO3, 1,4-dioxane/H2O, 0 °C to rt, 16 h, 74%; (ii) 3,4-difluoroaniline, HATU, DIPEA, DMF, rt, 1 h, 90%; (iii) TFA, DCM, 0 °C to rt, 2 h, 60%; (iv) cycloalkylsulfonyl chloride, Et3N, CH2Cl2, DMAP, 40 °C, 3 h, 23–47%.
Part C: Inversion of the amide group was also evaluated through the synthesis of compound 16. The synthesis was initiated by a Friedel-Craft sulfonylation of 2-fluoroacetanilide 12 by treatment with chlorosulfonic acid [14]. As expected, the reaction produced a mixture of regioisomers separable by column chromatography. Structure of both isomers was established by careful analysis of coupling constant observed in 1H NMR. The regioisomer 13 was then reacted with cyclopentylamine to afford intermediate 14 in 88% yield. Finally, deacetylation under acidic conditions followed by reaction with 3,4-difluorobenzoyl chloride gave the targeted analog 16 (Scheme 3).
Scheme 3.
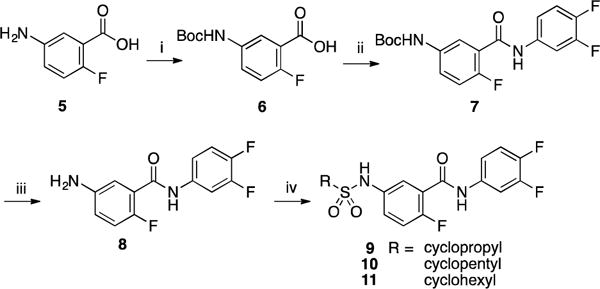
Reagents and conditions: (i) ClSO3H, 80 °C, 5 h, 62%; (ii) cyclopentylamine, Et3N, CH2Cl2, rt, 2 h, 88%; (iii) HCl 6N, 100 °C, 1 h, 74%; (iv) 3,4-difluorobenzoyl chloride, Et3N, CH2Cl2, 0 °C to rt, 2 h, 36%.
Part D: The original aniline moiety of our scaffold was replaced with various substituted benzyl amines using the chemistry described in scheme 4. Cyclopropyl-substituted benzylamine 18 was prepared by treatment of 3,4-difluorobenzonitrile 17 with ethylmagnesium bromide and titanium isopropoxide [15], while α-dimethyl substituted benzylamine 20 was prepared from 17 by treatment with methyl lithium in presence of cerium chloride [16]. Coupling of acid 2 with either 18 or 20 and subsequent reaction with cyclopentylamine afforded derivatives 19 and 21, respectively.
Scheme 4.
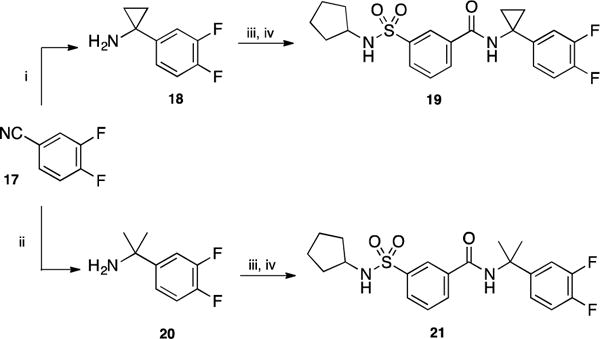
Reagents and conditions: (i) EtMgBr, Ti(Oi-Pr)4, BF3.Et2O, Et2O, −78 °C, 1 h, 56%; (ii) MeLi, CeCl3, THF, −25 °C, 1 h, 51%; (iii) 2, SOCl2, 80 °C, 16 h then 18 or 20, toluene, 110 °C, 2 h; (iv) cyclopentylamine, Et3N, CH2Cl2, rt, 2 h, 34% for 19 and 25% for 21 over two steps.
Part E: Finally, a new series of more rigid bicyclic derivatives was synthesized (Scheme 5). Commercially available phenylethylamine 22 and 3-phenyl-1-propylamine 23 were acylated using methyl chloroformate and subsequently treated with trifluoromethanesulfonic acid to form derivatives 24 and 25, respectively. These intermediates were then regioselectively sulfonylated by treatment with chlorosulfonic acid to afford the corresponding 3-sulfonyl chloride derivatives 26 and 27 in high yields. 26 and 27 were then reacted with cyclopentylamine and N-methylcyclopentylamine to afford compounds 28–30 in yields ranging from 64% to 94%. These intermediates were then N-alkylated with 3,4-difluorobromobenzene via a copper iodide catalyzed Ullmann reaction. Overall, these reactions were sluggish, requiring 12–15 hr of heating at high temperature (150 °C) and a stoichiometric amount (and sometimes an excess) of copper iodide to reach full conversion. Interestingly, the coupling was found to proceed faster with N-methyl substrates 30 and 31 under microwave irradiation after 90 minutes while no improvement was observed for the unsubstituted substrates 28 and 29. It is noteworthy that additional arylation of the sulfonamide moiety was also observed when starting from substrates 28 and 29.
Scheme 5.
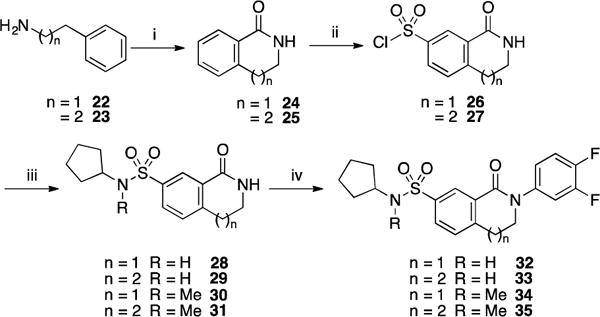
Reagents and conditions: (i) (a) methyl chloroformate, Et3N, DMF, 0 °C to rt, 1 h; b) CF3SO3H, 70 °C, 24 h, 82% for 24 and 84% for 25 over two steps; (ii) ClSO3H, 60 °C, 16 h, 83 % for 26 and 79% for 27; (iii) cyclopentylamine or N-methylcyclopentylamine, Et3N, CH2Cl2, rt, 2 h, 81% for 28, 97% for 29, 69% for 30, 94% for 31; (iv) 3,4-difluorobromobenzene, CuI, K2CO3, DMF, 150 °C, 16 h (or microwave, 150 °C, 90 min), 56% for 32, 58% for 33, 28% for 34 and 35% for 35.
2.2. Biological evaluation
The in vitro anti-HBV activity and safety profile of all new compounds were assessed by RT-PCR in HepAD38 cells as previously described by Stuyver et al. [17] The concentration of compound that inhibited HBV DNA replication by 50% (EC50) was determined by linear regression. All data were given relative to the untreated control. In addition, cytotoxicity was determined by using the CellTiter 96 non-radioactive cell proliferation colorimetric assay (Promega) in peripheral blood mononuclear (PBM) cells and in human T lymphoblast (CEM), African green monkey kidney (Vero), and human hepatocellular carcinoma (HepG2) cell lines. Toxicity levels were measured as the concentration of test compound that inhibited cell growth by 50% (CC50). Several derivatives exhibited potent activities with EC50 comprised between 0.8 and 9.2 μM while only displaying moderate toxicities in primary human lymphocytes, Vero and CEM cells (compounds 4a–s, 9–11, 16, 19, 21, 32–35, Table 1). Overall, cycloalkyls gave the best results among the first 19 synthesized analogs while benzyl, amino acid or disulfonamide substituted compounds were found to be less potent or devoid of antiviral activity (Part A). The potency of cyclopentyl sulfonamide 4a, previously reported by Campagna et al. [11] (DVR-56), was confirmed in our assay with an EC50 of 0.8 ± 0.2 μM (reported EC50 in HepDES19 cells: 0.14 ± 0.09 μM). The difluoro azetidine substituted derivative 4f exhibited a comparable anti-HBV activity (EC50 = 1.2 ± 0.6 μM) but displayed higher toxicity in CEM and Vero cells. It is noteworthy that inversion of the sulfonamide group on part B (compounds 9–11) mostly preserved the potency of the parent compound with the cyclopentyl-substituted analog 10 being the most active compound of this series (EC50 = 2.1 ± 1.3 μM). Regarding modifications on both part C and D, the inversion of the amide group (compound 16) and replacement of the 3,4-difluoroarylamide group with its α-substituted benzyl counterparts (compound 19 and 21) led to complete loss of activity. Similarly, the bicyclic analogs 32–35 did not exhibit any activity suggesting the relative importance of preserving the NH-aryl amide moiety or the flexibility of the molecule (Part E).
Table 1.
Anti-HBV activity and cytotoxicity
| Compound | Anti-HBV activity (μM)
|
Cytotoxicity CC50 (μM)
|
||||
|---|---|---|---|---|---|---|
| EC50 | EC90 | PBM | CEM | Vero | HepG2 | |
| 4a | 0.8 ± 0.2 | 7.8 ± 0.6 | 15.6 ± 0.01 | 14.0 ± 0.01 | 32.7 ± 0.11 | 37.1 ± 0.04 |
| 4b | 7.7 ± 1.6 | ≥ 10 (87 ± 0.2) | 25.8 ± 0.03 | 14.5 ± 0.07 | 18.3 ± 0.13 | 48.6 ± 0.01 |
| 4c | 4.5 ± 0.3 | 9.4 ± 0.1 | > 100 | 10.3 ± 0.09 | > 100 | > 100 |
| 4d | 6.7 ± 0.6 | > 10 (83 ± 1.7) | > 100 | 4.8 ± 0.78 | 21.3 ± 0.06 | > 100 |
| 4e | 9.2 ± 1.3 | > 10 (59 ± 14) | > 100 | ≤ 1 | 9.0 ± 0.02 | > 100 |
| 4f | 1.2 ± 0.6 | 6.3 ± 2.9 | 69.1 ± 0.07 | 3.7 ± 0.04 | 14.7 ± 0.04 | 27.0 ± 0.07 |
| 4g | > 10 | N/A | > 100 | 37.8 ± 0.16 | 39.6 ± 0.13 | >100 |
| 4h | 3.2 ± 0.4 | 8.9 ± 0.05 | 30.1 ± 0.06 | 13.6 ± 0.12 | 79.2 ± 0.03 | 62.3 ± 0.13 |
| 4i | 4.6 ± 1.2 | 10 ± 0.01 | > 100 | 22.9 ± 0.05 | 30.7 ± 0.02 | > 100 |
| 4j | 6.8 ± 0.9 | 9.6 ± 0.1 | > 100 | 3.8 ± 0.07 | 39.4 ± 0.26 | 88.2 ± 0.08 |
| 4k | 7.7 ± 2.5 | 9.3 ± 0.03 | > 100 | 3.6 ± 0.16 | 68.0 ± 0.03 | > 100 |
| 4l | > 10 | N/A | > 100 | 31.1 ± 0.16 | 13.1 ± 0.06 | 45.5 ± 0.11 |
| 4m | > 10 | N/A | > 100 | 31.6 ± 0.08 | > 100 | 76.8 ± 0.01 |
| 4n | > 10 | N/A | 58.1 ± 0.04 | 10.8 ± 0.05 | 39.1 ± 0.09 | > 100 |
| 4o | 2.7 ± 1.1 | > 10 (82 ± 3.2) | 13.0 ± 0.05 | 6.4 ± 0.04 | 5.8 ± 0.04 | 26.4 ± 0.03 |
| 4p | > 10 | N/A | > 100 | > 100 | > 100 | > 100 |
| 4q | > 10 | N/A | > 100 | > 100 | > 100 | > 100 |
| 4r | > 10 | N/A | > 100 | > 100 | > 100 | > 100 |
| 4s | > 10 | N/A | > 100 | > 100 | > 100 | > 100 |
| 9 | 6.9 ± 0.1 | > 10 (76 ± 1.6) | 39.2 ± 0.04 | 11.8 ± 0.12 | 30.7 ± 0.04 | 61.8 ± 0.03 |
| 10 | 2.1 ± 1.3 | 9.5 ± 0.8 | > 100 | 54.2 ± 0.11 | 29.1 ± 0.08 | > 100 |
| 11 | 3.7 ± 0.1 | ≥ 10 (86 ± 2.1) | > 100 | 74.5 ± 0.07 | 6.4 ± 0.06 | 80.4 ± 0.11 |
| 16 | ≥ 10 | N/A | 81.0 ± 0.01 | 13.6 ± 0.01 | 87.5 ± 0.01 | >100 |
| 19 | > 10 | N/A | > 100 | > 100 | 12.7 ± 0.03 | > 100 |
| 21 | > 10 | N/A | > 100 | 64.0 ± 0.04 | 84.3 ± 0.04 | > 100 |
| 32 | > 10 | N/A | > 100 | > 100 | > 100 | > 100 |
| 33 | > 10 | N/A | > 100 | 50.6 ± 0.10 | > 100 | > 100 |
| 34 | > 10 | N/A | 71.2 ± 0.03 | 36.0 ± 0.07 | > 100 | > 100 |
| 35 | > 10 | N/A | > 100 | > 100 | > 100 | > 100 |
| 3TC | 0.04 ± 0.03 | 0.3 ± 0.02 | > 100 | > 100 | > 100 | > 10 |
Data are the mean of replicates of two or three ± standard deviation (SD) from 2 independent experiments with representative results. N/A: not applied. Results in parenthesis: % of inhibition ± SD at 10 μM.
To determine whether our active derivatives have an effect on the levels of cccDNA formation, secretion of HBe antigen was measured (by ELISA, Biochain) as a cccDNA-dependent marker using the HepAD38 cells and tested them in parallel with GLS4, and 3TC or entecavir [18, 19]. Potent polymerase-based inhibitors, such as 3TC or entecavir, are expected to have negligible on cccDNA formation/amplification both in vivo and in vitro. Although they suppress HBV DNA levels in serum, they do not inhibit de novo formation of viral cccDNA in infected hepatocytes [18–21]. Thus, as expected, GLS4 reduced secretion of HBeAg whereas 10 μM 3TC or ETV had no effect (Table 2). All five selected derivatives reduced secreted HBeAg, with EC50s varying from 1.0 to 9.7 μM, and with drug-induced inhibition of intra/extra viral replication. Remarkably, HBeAg secretion was reduced by 90% at 3–4 μM with compounds 4a and 4f versus 2 μM with GLS-4. The novel sulfamoyl derivatives could be negatively acting on the stabilization maturation-associated nucleocapsid, consequently interrupting the release of relaxed capsid into the nucleus, and the subsequent cccDNA formation.
Table 2.
Sulfamoyl derivatives reduce HBV cccDNA formation in an HBeAg reporter cell-based assay.
| Compound | HBeAg Secretion | HBV DNA – Extracellular | HBV DNA – Intracellular | |||
|---|---|---|---|---|---|---|
|
| ||||||
| EC50 (μM) | EC90 (μM) | EC50 (μM) | EC90 (μM) | EC50 (μM) | EC90 (μM) | |
| 4a | 1.3 ± 0.4 | 4.6 ± 2.0 | 0.8 ± 0.2 | 7.8 ± 0.6 | 0.6 ± 0.002 | 4.3 ± 0.07 |
| 4f | 1.0 ± 0.05 | 3.2 ± 0.01 | 1.2 ± 0.6 | 6.3 ± 2.9 | 0.9 ± 0.05 | 9.7 ± 0.04 |
| 9 | 5.5 ± 0.02 | > 10 (61 ± 1.7) | 6.9 ± 0.1 | > 10 (76 ± 1.6) | 0.8 ± 0.01 | 8.1 ± 0.001 |
| 10 | 6.0 ± 2.9 | > 10 (66 ± 5.9) | 2.1 ± 1.3 | 9.5 ± 0.8 | 0.3 ± 0.04 | 3.6 ± 0.14 |
| 11 | 9.7 ± 2.5 | > 10 (67 ± 15.3) | 3.7 ± 0.1 | ≥ 10 (86 ± 2.1) | < 0.1 (63 ± 2.3) | 7.9 ± 0.17 |
| 3TC | > 10 (<1 ± 0.03) | N/A | 0.04 ± 0.03 | 0.3 ± 0.02 | < 0.01 (74 ± 4.4) | 0.7 ± 0.05 |
| ETV | > 10 (<1 ± 0.2) | N/A | 0.0007 ± 0.00001 | 0.1 ± 0.03 | ≤ 0.0001 (53 ± 0.5) | 0.009 ± 0.002 |
| GLS-4 | 0.7 ± 0.2 | 1.9 ± 0.04 | 0.2 ± 0.1 | 1.0 ± 0.1 | < 0.01 (82 ± 3.4) | 0.05 ± 0.0001 |
Data are the mean of replicates of two or three ± standard deviation (SD) from 2 independent experiments with representative results. N/A: not applied. Results in parenthesis: % of inhibition ± SD at 10 μM.
2.3. Electron microscopy
Recently published results show that a compound chemically similar to 4f inhibits replication by disrupting the HBV capsid protein [22]. To determine if our compounds act by this mechanism, HBV capsid formation was monitored by negative-stain electron microscopy. In the absence of drug, the HBV Cp149 dimers assemble upon addition of NaCl into regular hollow spheres with a diameter of approximately 40 nm (Figure 3A) [23]. Three compounds including 4a, 4f and the inactive compound 21 were tested at 20 μM in a single-blind series of experiments. HBV Cp149 dimers were incubated with the agents for 1 hr, and assembly was induced with the addition of NaCl overnight. One sample yielded properly formed HBV Cp149 capsids of similar shape, size and yield as the drug-lacking images (Figure 3B and 3C). The other two samples exhibited morphology dissimilar to the wild-type capsids correctly identifying 4a and 4f as the active agents. Compound 4a induces Cp149 to form greater numbers of disfigured circular assemblies (Figure 3D). Pre-incubation with compound 4f yielded a large number of incomplete, but typically spherical capsids aggregated into clusters (Figure 3E and 3F). These results support that sulfamoyl agents like 4f inhibit HBV replication by binding to the capsid protein and affecting assembly.
Fig 3.
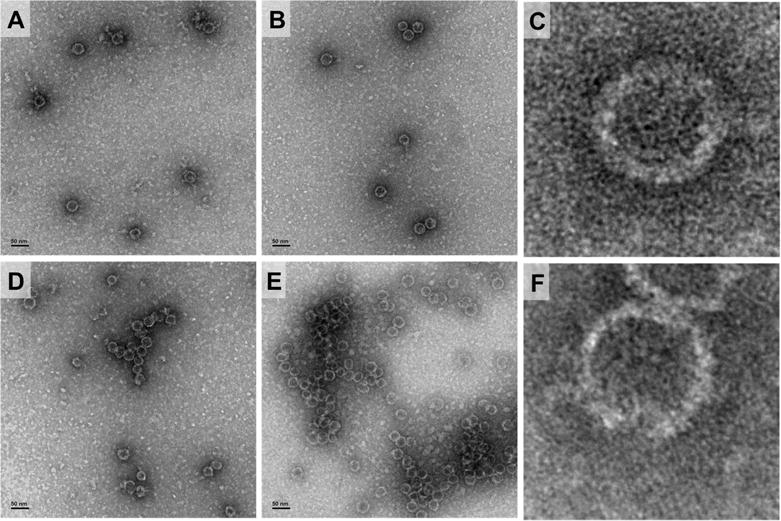
Perturbation of HBV Cp149 capsid formation by sulfamoyl compounds at 20 μM determined by negative-stain TEM. A) HBV Cp149 capsids formed in the absence of drug, B) in the presence of 21, C) properly formed capsid D) in the presence of 4a, E) in the presence of 4f and F) incomplete and aggregated capsid.
2.4. Molecular modeling
A recent crystal structure revealed the binding site for sulfamoyl-based HBV capsid effectors at the Cp149 chain B-C dimer-dimer interface (Figure 4A) [22]. Chain B formed a “concave” that contributes most protein-ligand interactions while chain C acted as a “lid”. The compound 4f was modeled into this pocket at the interface of chain B and chain C using Glide Docking. To refine the docked model and identify transient interactions, the entire HBV Cp149 hexamer was simulated for 10 ns in explicit solvent using Desmond MD (Figure 4A). The simulation rapidly equilibrated in ~2.5 ns, and protein RMSF agreed with the experimentally observed thermal factors (see Supporting Information). The resulting protein-ligand contacts from the molecular dynamics simulation provided insight into the structure-activity relationships observed for this series (Figure 4B, 4C). In most frames, the amide oxygen accepted a hydrogen bond from Trp102 NH while also engaging in a minor water-mediated bridge with the Phe23 backbone (observed in 5% of frames). The amide nitrogen acted as a hydrogen bond donor to the Thr128 side chain alcohol on the other interfacial partner. These amide noncovalent bonds bridged the two proteins comprising the binding pocket. Compounds 32–35 are cyclized at the amide N thereby lacking the hydrogen-bond donating functionality, and these agents do not inhibit HBV replication supporting the necessity of the hydrogen bonding interactions. The difluorobenzyl ring positioned into a hydrophobic pocket formed by Pro25, Leu30, Trp102, and Val124 from the interfacial partner. The central monofluorobenzyl ring falls into a hydrophobic pocket lined by Phe110, Leu140, and Thr128 from chain C. The sulfonamide group formed transient water bridges with Ser141. The Ser141 water bridge must be weak and dynamic since the “flipped” sulfonamide compounds 9–11 retain modest anti-HBV activity. The difluoroazetidine group positions in a narrow, hydrophobic solvent-exposed tunnel. Since this group does not form specific interactions with the HBV capsid protein, several small, hydrophobic substitutions are tolerated (compounds 4a, 4b, 4c, 4d, 4e, 4h, 4i, 4j, 4k, and 4o). Bulkier substitutions on the sulfonamide (compounds 4l, 4m, and 4n) are predicted to sterically clash with the HBV protein resulting in inactivity. This model and subsequent dynamical observations help explain the structure-activity relationships for this series and will be useful in optimizing future sulfamoyl agents as HBV inhibitors.
Fig 4.
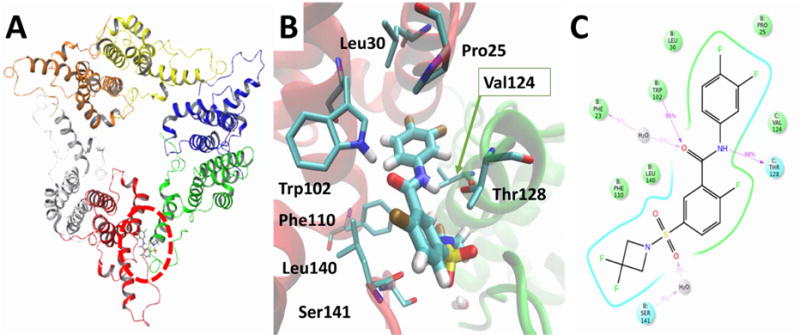
Model of sulfonamide compound 4f bound to HBV capsid protein from dynamic simulation. The model was generated by docking compound 4f into HBV Cp149 Y132A hexamer crystal structure (PDBID 5T2P) followed by 10ns dynamic simulation. A) Binding locus for compound 4f in HBV capsid hexamer. B) The binding pocket lies at the interface of two HBV Cp149 proteins (chain B in red and chain C in green). Residues that interact with compound 4f from the simulation are shown. C) Simulation interaction diagram of compound 4f bound to HBV Cp149 Y132A hexamer from the productive 7.5 ns of a 10 ns simulation. Green residues are hydrophobic, hydrogen bond interactions are indicated by purple arrows, polar residues are shown in cyan, and water bridges are shown in grey. The values in purple indicate the percentage of frames from the production phase in which these interactions were observed.
3. Conclusion
Among the 27 sulfamoylbenzamides prepared, several analogs exhibited potent anti-HBV activities in the low micromolar range. Antiviral activity was correlated with reduction in cccDNA level by measuring HBeAg secretion. These sulfamoyl derivatives were found to inhibit HBV replication by disrupting the capsid assembly leading to misshaped unviable assemblies. Results from this work validate this novel class of small molecule as CAE and the model used to rationalize our SAR could lead to the discovery of more potent and effective analogs. Ultimately these sulfamoyl derivatives, when combined with other modalities (e.g., nucleoside analogs), could lead to a novel therapeutic strategy with reduced treatment duration and a functional cure for HBV infection.
4. Experimental section
4.1. Chemistry
Commercially available chemicals were of reagent grade and used as received. Nuclear magnetic resonance (NMR) spectra (1H, 13C and 19F) were recorded on a Bruker Ascend™ 400 MHz Fourier transform spectrometer at rt, with tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm) and signals are quoted as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), bs (broad signal), dd (doublet of doublets) or ddd (doublet of doublets of doublets). 13C NMR data is reported as observed, that is, some carbon signals overlap with solvent signals. High-resolution mass spectra (HRMS) were recorded on a ThermoFisher Q exactive Plus high-resolution mass spectrometer with electrospray ionization (ESI). Thin-layer chromatography (TLC) was performed on 0.25 mm silica gel. Purifications were performed on silica gel column chromatography (60 Å, 63–200 or 40–75 μm). Reactions under microwave irradiation were performed on a CEM Discovery SP Microwave Synthesizer using 5 mL sealed tubes.
4.1.1. 5-Chlorosulfonyl-2-fluorobenzoic acid (2)
To chlorosulfonic acid (23.8 mL, 0.35 mol, 10 equiv.) cooled to 0 °C was added portion wise 2-fluorobenzoic acid 1 (5.0 g, 35 mmol). After addition, the yellow solution was allowed to warm to room temperature and heated at 75 °C for 16 h. The reaction mixture was cooled to room temperature and carefully added dropwise into crushed ice. The white precipitate was filtered, washed with water and dried in vacuo to afford compound 2 as a white solid (6.4 g, 77%). Spectral data was consistent with that previously reported. CAS: 37098-75-2.
4.1.2. 3-((3,4-Difluorophenyl)carbamoyl)-4-fluorobenzenesulfonyl chloride (3)
A solution of 2 (3 g, 12.6 mmol) in SOCl2 (20 mL) was heated at 80 °C for 16 h. The mixture was concentrated under reduced pressure and co-evaporated with toluene. The crude residue was dissolved in toluene (25 mL) and 3,4-difluoroaniline (1.24 mL, 12.6 mmol) was added. The mixture was heated at 110 °C for 2 h and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (8:2) to afford 3 (2.87 g, 65% over two steps). 1H NMR (400 MHz, Acetone-d6) δ 9.99 (s, 1H), 8.54 (dd, J = 6.1, 2.6 Hz, 1H), 8.43 – 8.32 (m, 1H), 7.98 – 7.87 (m, 1H), 7.73 (dd, J = 9.7, 8.9 Hz, 1H), 7.54 – 7.45 (m, 1H), 7.33 (dt, J = 10.5, 9.0 Hz, 1H). 13C NMR (101 MHz, Acetone-d6) δ 163.4 (d, J = 262.3 Hz), 160.2, 149.6 (dd, J = 244.5, 13.3 Hz), 146.8 (dd, J = 243.6, 12.8 Hz), 140.2 (d, J = 3.2 Hz), 135.3 (dd, J = 8.9, 3.2 Hz), 132.2 (d, J = 11.2 Hz), 130.1 (d, J = 4.9 Hz), 125.9 (d, J = 16.7 Hz), 118.9 (d, J = 25.0 Hz), 117.3 (d, J = 18.3 Hz), 116.5 (dd, J = 6.1, 3.5 Hz), 109.6 (d, J = 22.1 Hz). 19F NMR (377 MHz, Acetone-d6) δ −103.6, −139.3 – −139.4 (m), −145.6 – −145.7 (m).
4.1.3. General procedure to compounds 4a–n
To a solution of sulfonyl chloride derivative 3 (100 mg, 0.286 mmol) in CH2Cl2 (3 mL) were added the appropriate amine (0.286 mmol) and Et3N (0.315 mmol) at 0 °C. The mixture was stirred 2 h at room temperature, diluted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc to afford the desired sulfonamide derivatives.
4.1.3.1. 5-(N-Cyclopentylsulfamoyl)-N-(3,4-difluorophenyl)-2-fluorobenzamide (4a)
Yield 81%. 1H NMR (400 MHz, Acetone-d6) δ 9.89 (s, 1H), 8.29 (dd, J = 6.6, 2.4 Hz, 1H), 8.12 – 8.05 (m, 1H), 8.01 – 7.92 (m, 1H), 7.58 – 7.47 (m, 2H), 7.43 – 7.28 (m, 1H), 6.72 (d, J = 7.3 Hz, 1H), 3.72 – 3.57 (m, 1H), 1.83 – 1.71 (m, 2H), 1.68 – 1.58 (m, 2H), 1.55 – 1.37 (m, 4H). 13C NMR (101 MHz, Acetone-d6) δ 161.4 (d, J = 255.9 Hz), 161.2, 149.6 (dd, J = 244.2, 13.3 Hz), 146.6 (dd, J = 243.4, 12.7 Hz), 138.8 (d, J = 3.5 Hz), 135.5 (dd, J = 9.0, 3.2 Hz), 131.9 (d, J = 10.0 Hz), 129.7 (d, J = 3.8 Hz), 124.5 (d, J = 15.7 Hz), 117.4 (d, J = 6.1 Hz), 117.2, 116.3 (dd, J = 6.0, 3.5 Hz), 109.4 (d, J = 22.1 Hz), 78.3, 55.1, 54.1, 32.9, 23.0. 19F NMR (377 MHz, Acetone-d6) δ −110.5 – −110.6 (m), −139.5 – −139.6 (m), −146.0 – −146.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C18H18F3N2O3S: 399.0990, found: 399.0985.
4.1.3.2. 5-(N-Cyclopentyl-N-methylsulfamoyl)-N-(3,4-difluorophenyl)-2-fluorobenzamide (4b)
Yield 84%. 1H NMR (400 MHz, Acetone-d6) δ 9.88 (s, 1H), 8.24 (dd, J = 6.5, 2.5 Hz, 1H), 8.10 – 8.02 (m, 1H), 8.02 – 7.94 (m, 1H), 7.61 – 7.49 (m, 2H), 7.35 (dt, J = 10.5, 9.0 Hz, 1H), 4.38 (p, J = 8.1 Hz, 1H), 2.77 (s, 3H), 1.71 – 1.54 (m, 4H), 1.52 – 1.35 (m, 4H). 13C NMR (101 MHz, Acetone-d6) δ 161.6 (d, J = 256.4 Hz), 161.1 (d, J = 1.7 Hz), 149.6 (dd, J = 244.2, 13.3 Hz), 146.6 (dd, J = 243.3, 12.7 Hz), 136.4 (d, J = 3.7 Hz), 135.5 (dd, J = 9.0, 3.2 Hz), 132.1 (d, J = 10.0 Hz), 129.9 (d, J = 3.8 Hz), 124.7 (d, J = 15.8 Hz), 117.5 (d, J = 24.4 Hz), 117.3 (d, J = 18.5 Hz), 116.3 (dd, J = 6.1, 3.6 Hz), 109.4 (d, J = 22.2 Hz), 58.3, 28.2, 27.6, 23.8. 19F NMR (377 MHz, Acetone-d6) δ −110.1 – −110.2 (m), −139.4 – −139.6 (m), −146.0 – −146.1 (m). HRMS (ESI): m/z [M+H]+ calcd for C19H19F3N2O3S: 413.1147, found: 413.1142.
4.1.3.3. N-(3,4-Difluorophenyl)-5-((4,4-difluoropiperidin-1-yl)sulfonyl)-2-fluorobenzamide (4c)
Yield 86%. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 8.07 (dd, J = 6.2, 2.4 Hz, 1H), 8.06 – 7.97 (m, 1H), 7.92 – 7.83 (m, 1H), 7.68 (t, J = 9.1 Hz, 1H), 7.52 – 7.41 (m, 2H), 3.18 – 3.09 (m, 4H), 2.18 – 1.99 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 161.8 (d, J = 257.5 Hz), 161.7, 149.4 (dd, J = 243.7, 13.2 Hz), 146.3 (dd, J = 242.4, 12.7 Hz), 135.9 (d, J = 8.9 Hz), 132.6 (d, J = 10.5 Hz), 130.0 (d, J = 4.3 Hz), 126.1 (d, J = 17.0 Hz), 124.5, 122.1, 119.7, 118.5 (d, J = 23.5 Hz), 118.1 (d, J = 17.8 Hz), 116.8, 109.4 (d, J = 21.7 Hz), 43.8 (t, J = 5.7 Hz), 33.2 (t, J = 23.6 Hz). 19F NMR (377 MHz, DMSO-d6) δ −97.5, −107.0, −136.9 (d, J = 23.0 Hz), −143.5 (d, J = 23.0 Hz). HRMS (ESI): m/z [M+H]+ calcd for C18H15F5N2O3S: 435.0802, found: 435.0796.
4.1.3.4. N-(3,4-Difluorophenyl)-5-((3,3-difluoropyrrolidin-1-yl)sulfonyl)-2-fluorobenzamide (4d)
Yield 79%. 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1H), 8.22 – 8.05 (m, 2H), 7.98 – 7.83 (m, 1H), 7.68 (t, J = 9.2 Hz, 1H), 7.52 – 7.41 (m, 2H), 3.68 (t, J = 12.9 Hz, 2H), 3.42 (t, J = 7.4 Hz, 2H), 2.43 – 2.25 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 162.0 (d, J = 257.7 Hz), 161.7, 149.4 (dd, J = 243.8, 13.2 Hz), 146.3 (dd, J = 242.6, 12.6 Hz), 135.9 (dd, J = 9.0, 3.0 Hz), 132.8 (d, J = 10.1 Hz), 132.4 (d, J = 3.3 Hz), 130.4, 130.3 (d, J = 4.3 Hz), 128.0, 125.9 (d, J = 16.5 Hz), 125.5, 118.4 (d, J = 23.5 Hz), 118.1 (d, J = 18.0 Hz), 117.0 – 116.4 (m), 109.5 (d, J = 21.6 Hz), 54.1 (t, J = 32.0 Hz), 46.1, 33.9 (t, J = 23.9 Hz). 19F NMR (377 MHz, Acetone-d6) δ −101.5 – −101.6 (m), −109.0 – −109.1 (m), −139.6 – −139.7 (m), −146.1 – −146.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C17H13F5N2O3S: 421.0645, found: 421.0639.
4.1.3.5. N-(3,4-Difluorophenyl)-2-fluoro-5-((3,3,4,4-tetrafluoropyrrolidin-1-yl)sulfonyl)benzamide (4e)
Yield 82%. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 8.22 (dd, J = 6.3, 2.5 Hz, 1H), 8.21 – 8.12 (m, 1H), 7.97 – 7.83 (m, 1H), 7.70 (t, J = 9.2 Hz, 1H), 7.56 – 7.42 (m, 2H), 4.19 – 3.92 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.3 (d, J = 258.4 Hz), 161.6, 149.4 (dd, J = 243.8, 13.3 Hz), 146.3 (dd, J = 242.6, 12.6 Hz), 136.0 (dd, J = 8.9, 2.9 Hz), 133.0 (d, J = 10.3 Hz), 132.1, 130.8 (d, J = 4.4 Hz), 126.0 (d, J = 16.6 Hz), 121.0 (t, J = 24.8 Hz), 118.6 (d, J = 23.3 Hz), 118.1 (d, J = 17.7 Hz), 117.2 – 116.3 (m), 115.8 (t, J = 24.9 Hz), 109.4 (d, J = 21.6 Hz), 51.1 (t, J = 30.7 Hz). 19F NMR (377 MHz, Acetone-d6) δ −107.97 – −108.05 (m), −124.1 – −124.2 (m), −139.5 – −139.7 (m), −146.1 – −146.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C17H11F7N2O3S: 457.0457, found: 457.0456.
4.1.3.6. 5-((3,3-Difluoroazetidin-1-yl)sulfonyl)-N-(3,4-difluorophenyl)-2-fluorobenzamide (4f)
Yield 79%. 1H NMR (400 MHz, DMSO-d6) δ 10.92 (s, 1H), 8.21 (dd, J = 6.3, 2.5 Hz, 1H), 8.20 – 8.11 (m, 1H), 7.93 – 7.86 (m, 1H), 7.73 (t, J = 9.2 Hz, 1H), 7.52 – 7.42 (m, 2H), 4.36 (t, J = 12.7 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.4 (d, J = 258.7 Hz), 161.6, 149.4 (dd, J = 243.8, 13.2 Hz), 146.3 (dd, J = 242.7, 12.6 Hz), 135.9 (dd, J = 9.1, 2.9 Hz), 133.6 (d, J = 10.2 Hz), 131.3 (d, J = 4.4 Hz), 130.4 (d, J = 3.2 Hz), 126.1 (d, J = 16.5 Hz), 118.6 (d, J = 23.7 Hz), 118.1 (d, J = 17.8 Hz), 117.5, 116.9 (dd, J = 6.0, 3.4 Hz), 114.8, 112.1, 109.5 (d, J = 21.5 Hz), 62.4 (t, J = 27.6 Hz), 46.1. 19F NMR (377 MHz, DMSO-d6) δ −98.3, −105.8, −136.9 (d, J = 22.8 Hz), −143.5 (d, J = 23.1 Hz). HRMS (ESI): m/z [M+H]+ calcd for C16H11F5N2O3S: 407.0489, found: 407.0484.
4.1.3.7. N-(3,4-Difluorophenyl)-2-fluoro-5-((2-oxopyrrolidin-1-yl)sulfonyl)benzamide (4g)
Note: 2-pyrolidinone (24 mg, 0.286 mmol) was treated with sodium hydride (12 mg, 0.286 mmol, 60% dispersion in mineral oil) in THF (3 mL) prior to the addition of 3. Yield 37%. 1H NMR (400 MHz, Acetone-d6) δ 9.93 (s, 1H), 8.41 (dd, J = 6.5, 2.5 Hz, 1H), 8.30 – 8.20 (m, 1H), 8.03 – 7.92 (m, 1H), 7.57 (dd, J = 10.0, 8.8 Hz, 1H), 7.55 – 7.49 (m, 1H), 7.36 (dt, J = 10.5, 9.0 Hz, 1H), 4.01 (t, J = 7.0 Hz, 2H), 2.46 (dd, J = 8.4, 7.6 Hz, 2H), 2.20 – 2.11 (m, 2H). 13C NMR (101 MHz, Acetone-d6) δ 13C NMR (101 MHz, Acetone-d6) δ 173.5, 162.5 (d, J = 258.3 Hz), 160.9, 149.6 (dd, J = 244.3, 13.3 Hz), 146.6 (dd, J = 243.4, 12.9 Hz), 135.5 (dd, J = 8.9, 3.2 Hz), 135.2, 133.3 (d, J = 10.6 Hz), 130.8 (d, J = 4.4 Hz), 124.7 (d, J = 16.2 Hz), 117.5 (d, J = 5.5 Hz), 117.2, 116.6 – 115.9 (m), 109.4 (d, J = 22.2 Hz), 47.4, 31.6, 18.1. 19F NMR (377 MHz, Acetone-d6) δ −107.9, −139.5 – −139.7 (m), −146.0 – −146.2 (m). HRMS (ESI): m/z [M+H]+ calcd for C17H13F3N2O4S: 399.0626, found: 399.0619.
4.1.3.8. 5-(N-Cyclopropylsulfamoyl)-N-(3,4-difluorophenyl)-2-fluorobenzamide (4h)
Yield 77%. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 8.09 (s, 2H), 8.07 – 7.98 (m, 1H), 7.91 – 7.84 (m, 1H), 7.65 (t, J = 9.2 Hz, 1H), 7.48 – 7.42 (m, 2H), 2.21 – 2.08 (m, 1H), 0.58 – 0.47 (m, 2H), 0.45 – 0.36 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 162.0, 161.2 (d, J = 256.1 Hz), 149.4 (dd, J = 243.7, 13.2 Hz), 146.3 (dd, J = 242.7, 12.9 Hz), 137.3 (d, J = 3.2 Hz), 135.9 (d, J = 6.1 Hz), 132.0 (d, J = 9.9 Hz), 129.4 (d, J = 4.0 Hz), 125.6 (d, J = 16.7 Hz), 118.2, 118.0 (d, J = 4.6 Hz), 116.8 (dd, J = 5.7, 3.4 Hz), 109.5 (d, J = 21.7 Hz), 24.6, 5.6. 19F NMR (377 MHz, DMSO-d6) δ −108.2, −136.9 (d, J = 23.0 Hz), −143.5 (d, J = 23.0 Hz). HRMS (ESI): m/z [M+H]+ calcd for C16H13F3N2O3S: 371.0677, found: 371.0672.
4.1.3.9. N-(3,4-Difluorophenyl)-2-fluoro-5-((octahydro-1H-indol-1-yl)sulfonyl)benzamide (4i)
Yield 26%. 1H NMR (400 MHz, Acetone-d6) δ 9.89 (s, 1H), 8.26 (dd, J = 6.6, 2.4 Hz, 1H), 8.14 – 8.06 (m, 1H), 8.02 – 7.94 (m, 1H), 7.58 – 7.50 (m, 2H), 7.36 (dt, J = 10.5, 9.0 Hz, 1H), 3.68 – 3.60 (m, 1H), 3.57 – 3.49 (m, 1H), 3.30 – 3.20 (m, 1H), 1.96 – 1.79 (m, 3H), 1.72 – 1.53 (m, 5H), 1.44 – 1.34 (m, 2H). 13C NMR (101 MHz, Acetone-d6) δ 161.7 (d, J = 256.4 Hz), 161.2, 149.6 (dd, J = 244.1, 13.2 Hz), 146.6 (dd, J = 243.1, 12.6 Hz), 135.6 (dd, J = 9.0, 3.0 Hz), 135.4 (d, J = 3.4 Hz), 132.2 (d, J = 10.0 Hz), 129.9 (d, J = 3.9 Hz), 124.6 (d, J = 15.7 Hz), 117.5 (d, J = 24.2 Hz), 117.5 – 117.1 (m), 116.5 – 116.1 (m), 109.3 (d, J = 22.2 Hz), 59.6, 47.3, 37.7, 29.7, 27.3, 25.9, 22.9, 21.0. 19F NMR (377 MHz, Acetone-d6) δ −110.3 – −110.4 (m), −139.6 – −139.7 (m), −146.2 – −146.3 (m). HRMS (ESI): m/z [M+H]+ calcd for C21H21F3N2O3S: 439.1303, found: 439.1297.
4.1.3.10. N-(3,4-Difluorophenyl)-2-fluoro-5-(N-(2-phenylpropan-2-yl)sulfamoyl)benzamide (4j)
Yield 69%. 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 8.25 (s, 1H), 8.05 – 7.82 (m, 1H), 7.75 (dd, J = 6.5, 2.4 Hz, 1H), 7.74 – 7.62 (m, 1H), 7.52 – 7.44 (m, 2H), 7.44 – 7.35 (m, 1H), 7.30 – 7.23 (m, 2H), 7.18 – 7.04 (m, 3H), 1.53 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 161.9, 160.7 (d, J = 255.4 Hz), 149.4 (dd, J = 243.6, 13.2 Hz), 146.3 (dd, J = 242.5, 12.6 Hz), 145.5, 140.2 (d, J = 3.3 Hz), 136.0 (dd, J = 8.8, 2.9 Hz), 131.3 (d, J = 9.9 Hz), 129.0 (d, J = 3.9 Hz), 128.1, 126.9, 126.1, 124.7 (d, J = 16.3 Hz), 118.0 (d, J = 17.8 Hz), 117.4 (d, J = 23.7 Hz), 116.9 (dd, J = 6.2, 3.3 Hz), 109.5 (d, J = 21.6 Hz), 58.1, 30.2. 19F NMR (377 MHz, Acetone-d6) δ −111.4 – −111.5 (m), −139.7 – −139.8 (m), −146.3 – −146.4 (m). HRMS (ESI): m/z [M+H]+ calcd for C22H19F3N2O3S: 449.1147, found: 449.1142.
4.1.3.11. N-(3,4-Difluorophenyl)-2-fluoro-5-(N-(1-phenylethyl)sulfamoyl)benzamide (4k)
Yield 75%. 1H NMR (400 MHz, Acetone-d6) δ 9.73 (s, 1H), 8.06 (dd, J = 6.7, 2.5 Hz, 1H), 8.02 – 7.93 (m, 1H), 7.89 – 7.77 (m, 1H), 7.59 – 7.47 (m, 1H), 7.45 – 7.27 (m, 2H), 7.24 – 7.09 (m, 6H), 4.67 – 4.51 (m, 1H), 1.41 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, Acetone-d6) δ 161.2 (d, J = 255.8 Hz), 161.0 (d, J = 9.6 Hz), 149.6 (dd, J = 244.1, 13.3 Hz), 146.6 (dd, J = 243.3, 12.8 Hz), 142.7, 138.7 (t, J = 3.9 Hz), 136.0 – 135.0 (m), 131.7 (d, J = 10.2 Hz), 129.8 (d, J = 4.0 Hz), 128.2, 127.1, 126.2, 123.9 (dd, J = 15.5, 4.6 Hz), 117.3 (d, J = 18.1 Hz), 117.0 (d, J = 24.5 Hz), 116.6 – 116.0 (m), 109.4 (dd, J = 22.2, 8.6 Hz), 53.8 (d, J = 9.6 Hz), 23.1 (d, J = 3.6 Hz). 19F NMR (377 MHz, Acetone-d6) δ −109.4 – −109.5 (m), −138.3 – −138.4 (m), −144.8 – −145.0 (m). HRMS (ESI): m/z [M+H]+ calcd for C21H17F3N2O3S: 435.0990, found: 435.0986.
4.1.3.12. Isopropyl ((3-((3,4-difluorophenyl)carbamoyl)-4-fluorophenyl)sulfonyl)-L-alaninate (4l)
Yield 94%. 1H NMR (400 MHz, Chloroform-d) δ 8.61 (d, J = 10.4 Hz, 1H), 8.48 (dd, J = 6.9, 2.5 Hz, 1H), 8.16 – 7.93 (m, 1H), 7.82 – 7.69 (m, 1H), 7.32 – 7.26 (m, 1H), 7.25 – 7.20 (m, 1H), 7.18 – 7.04 (m, 1H), 5.89 (d, J = 7.4 Hz, 1H), 4.99 – 4.71 (m, 1H), 4.00 (t, J = 7.4 Hz, 1H), 1.38 (d, J = 7.2 Hz, 3H), 1.14 (d, J = 6.3 Hz, 3H), 1.11 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 171.6, 162.1 (d, J = 256.4 Hz), 160.1 (d, J = 2.9 Hz), 150.0 (dd, J = 247.3, 13.2 Hz), 147.4 (dd, J = 246.6, 12.8 Hz), 137.5 (d, J = 3.0 Hz), 133.8 (dd, J = 8.8, 3.3 Hz), 132.5 (d, J = 10.6 Hz), 131.1 (d, J = 3.4 Hz), 122.7 (d, J = 13.4 Hz), 117.4 (d, J = 41.9 Hz), 117.4, 116.3 (dd, J = 6.1, 3.6 Hz), 110.4 (d, J = 21.9 Hz), 69.9, 51.8, 21.4, 21.4, 19.6. 19F NMR (377 MHz, Chloroform-d) δ −106.5, −135.4 (d, J = 21.2 Hz), −141.4 (d, J = 21.7 Hz). HRMS (ESI): m/z [M+H]+ calcd for C19H19F3N2O5S: 445.1045, found: 445.1038.
4.1.3.13. Methyl ((3-((3,4-difluorophenyl)carbamoyl)-4-fluorophenyl)sulfonyl)-L-phenylalaninate (4m)
Yield 91%. 1H NMR (400 MHz, Chloroform-d) δ 8.44 (d, J = 10.8 Hz, 1H), 8.23 (dd, J = 6.9, 2.5 Hz, 1H), 7.78 – 7.51 (m, 2H), 7.16 – 7.00 (m, 6H), 6.99 – 6.93 (m, 2H), 5.81 (d, J = 9.0 Hz, 1H), 4.26 – 4.10 (m, 1H), 3.49 (s, 3H), 2.98 (dd, J = 13.8, 5.5 Hz, 1H), 2.86 (dd, J = 13.8, 7.5 Hz, 1H). 13C NMR (101 MHz, Chloroform-d) δ 171.5, 162.0 (d, J = 256.2 Hz), 160.0 (d, J = 3.0 Hz), 150.0 (dd, J = 247.4, 13.3 Hz), 147.4 (dd, J = 246.6, 12.7 Hz), 137.2 (d, J = 2.9 Hz), 135.1, 133.8 (dd, J = 8.8, 3.3 Hz), 132.3 (d, J = 10.7 Hz), 131.0 (d, J = 3.4 Hz), 129.3, 128.6, 127.2, 122.4 (d, J = 13.3 Hz), 117.5 (d, J = 7.5 Hz), 117.2, 116.3 (dd, J = 6.0, 3.6 Hz), 110.4 (d, J = 21.9 Hz), 57.2, 52.7, 39.0. 19F NMR (377 MHz, Chloroform-d) δ −106.5, −135.3 (d, J = 21.8 Hz), −141.3 (d, J = 21.6 Hz). HRMS (ESI): m/z [M+H]+ calcd for C23H19F3N2O5S: 493.1045, found: 493.1038.
4.1.3.14. Ethyl ((3-((3,4-difluorophenyl)carbamoyl)-4-fluorophenyl)sulfonyl)-L-leucinate (4n)
Yield 89%. 1H NMR (400 MHz, Chloroform-d) δ 8.51 (d, J = 10.3 Hz, 1H), 8.37 (dd, J = 6.8, 2.5 Hz, 1H), 7.93 – 7.81 (m, 1H), 7.77 – 7.59 (m, 1H), 7.25 – 7.12 (m, 2H), 7.10 – 6.97 (m, 1H), 5.64 (d, J = 9.9 Hz, 1H), 3.95 – 3.88 (m, 1H), 3.84 (q, J = 7.1 Hz, 2H), 1.76 – 1.60 (m, 1H), 1.43 (t, J = 7.2 Hz, 2H), 1.03 (t, J = 7.1 Hz, 3H), 0.81 (t, J = 6.6 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 171.2, 161.1 (d, J = 256.6 Hz), 159.0 (d, J = 2.9 Hz), 149.0 (dd, J = 247.4, 13.3 Hz), 146.4 (dd, J = 246.6, 12.8 Hz), 136.2 (d, J = 3.2 Hz), 132.8 (dd, J = 8.8, 3.2 Hz), 131.5 (d, J = 10.6 Hz), 130.2 (d, J = 3.3 Hz), 121.6 (d, J = 13.4 Hz), 116.7 – 116.3 (m), 116.2 (d, J = 5.1 Hz), 115.2 (dd, J = 5.9, 3.6 Hz), 109.3 (d, J = 21.9 Hz), 60.8, 53.6, 41.0, 23.3, 21.6, 20.3, 12.9. 19F NMR (377 MHz, Chloroform-d) δ −106.4, −135.4 (d, J = 21.7 Hz), −141.4 (d, J = 21.4 Hz). HRMS (ESI): m/z [M+H]+ calcd for C21H23F3N2O5S: 473.1358, found: 473.1351.
4.1.3.15. 5-(N-(Cyclopentyloxy)sulfamoyl)-N-(3,4-difluorophenyl)-2-fluorobenzamide (4o)
To a solution of 3 (100 mg, 0.3 mmol) in acetonitrile (5 mL) were added O-cyclopentylhydroxylamine hydrochloride (0.04 mg, 0.3 mmol) and Et3N (0.2 mL, 1.5 mmol). The mixture was stirred 4 h at 65 ºC. After completion (checked by LC-MS), the reaction mixture was absorbed on silica and purified by flash chromatography using hexanes/EtOAc (7:3) to afford 4o (48 mg, 39%) as a white solid. 1H NMR (400 MHz, Acetone-d6) δ 9.88 (s, 1H), 9.36 (s, 1H), 8.29 (dd, J = 6.5, 2.5 Hz, 1H), 8.19 – 8.06 (m, 1H), 8.05 – 7.91 (m, 1H), 7.64 – 7.48 (m, 2H), 7.37 (dt, J = 10.5, 9.0 Hz, 1H), 4.70 – 4.55 (m, 1H), 1.87 – 1.76 (m, 2H), 1.76 – 1.66 (m, 2H), 1.65 – 1.47 (m, 4H). 13C NMR (101 MHz, Acetone-d6) δ 162.2 (d, J = 257.3 Hz), 161.0 (d, J = 1.7 Hz), 149.7 (dd, J = 244.3, 13.2 Hz), 146.7 (dd, J = 243.4, 12.7 Hz), 135.5 (dd, J = 9.0, 3.2 Hz), 134.5 (d, J = 3.4 Hz), 133.6 (d, J = 10.4 Hz), 131.2 (d, J = 4.2 Hz), 124.6 (d, J = 16.1 Hz), 117.3 (d, J = 18.0 Hz), 117.3 (d, J = 24.6 Hz), 116.3 (dd, J = 6.1, 3.5 Hz), 109.4 (d, J = 22.1 Hz), 88.1, 30.9, 23.2. 19F NMR (377 MHz, Acetone-d6) δ −108.6 – −108.7 (m), −139.5 – −139.7 (m), −146.0 – −146.1 (m). HRMS (ESI): m/z [M+H]+ calcd for C18H18F3N2O4S: 415.0939, found: 415.0936.
4.1.3.16. N-(3,4-Difluorophenyl)-2-fluoro-5-(N-(methylsulfonyl)sulfamoyl)benzamide (4p)
Title compound 4p was obtained from 3 using the same procedure as for compound 4o. Yield 42%, triethylamine salt. 1H NMR (400 MHz, Acetone-d6) δ 9.89 (s, 1H), 8.78 (s, 1H), 8.24 (dd, J = 6.8, 2.4 Hz, 1H), 8.09 – 8.03 (m, 1H), 8.03 – 7.94 (m, 1H), 7.57 – 7.48 (m, 1H), 7.38 – 7.28 (m, 2H), 3.32 (q, J = 7.3 Hz, 4H), 2.89 (s, 3H), 1.34 (t, J = 7.3 Hz, 6H). 13C NMR (101 MHz, Acetone-d6) δ 162.0 (d, J = 1.7 Hz), 160.5 (d, J = 253.4 Hz), 149.6 (dd, J = 243.8, 13.2 Hz), 146.5 (dd, J = 242.9, 12.8 Hz), 142.8 (d, J = 3.5 Hz), 135.8 (dd, J = 9.0, 3.1 Hz), 131.9 (d, J = 9.6 Hz), 129.2 (d, J = 3.5 Hz), 123.4 (d, J = 15.3 Hz), 117.2 (d, J = 18.1 Hz), 116.3 (dd, J = 6.1, 3.5 Hz), 116.0 (d, J = 23.8 Hz), 109.3 (d, J = 22.2 Hz), 46.2, 42.3, 8.1. 19F NMR (377 MHz, Acetone-d6) δ −113.6 – −113.7 (m), −139.7 – −139.8 (m), −146.4 – −146.5 (m). HRMS (ESI): m/z [M+H]+ calcd for C14H12F3N2O5S2: 409.0140, found: 409.0133.
4.1.3.17. 5-(N-(Cyclopropylsulfonyl)sulfamoyl)-N-(3,4-difluorophenyl)-2-fluorobenzamide (4q)
Title compound 4q was obtained from 3 using the same procedure as for compound 4o. Yield 59% yield, triethylamine salt. 1H NMR (400 MHz, Acetone-d6) δ 9.86 (s, 1H), 8.93 (s, 1H), 8.26 (dd, J = 6.8, 2.4 Hz, 1H), 8.13 – 8.04 (m, 1H), 8.03 – 7.94 (m, 1H), 7.62 – 7.47 (m, 1H), 7.39 – 7.28 (m, 2H), 3.32 (q, J = 7.3 Hz, 4H), 2.79 – 2.64 (m, 1H), 1.35 (t, J = 7.3 Hz, 6H), 0.97 – 0.76 (m, 4H). 13C NMR (101 MHz, Acetone-d6) δ 161.9, 160.5 (d, J = 253.3 Hz), 149.6 (dd, J = 244.0, 13.2 Hz), 146.5 (dd, J = 242.5, 13.2 Hz), 143.2 (d, J = 3.3 Hz), 135.8 (dd, J = 9.0, 3.0 Hz), 131.9 (d, J = 9.6 Hz), 129.3 (d, J = 3.4 Hz), 123.4 (d, J = 15.3 Hz), 117.2 (d, J = 18.1 Hz), 116.4 – 116.1 (m), 116.0 (d, J = 23.9 Hz), 109.3 (d, J = 22.1 Hz), 46.1, 32.2, 8.1, 4.7. 19F NMR (377 MHz, Acetone-d6) δ −113.7 – −113.8 (m), −139.7 – −139.8 (m), −146.5 – −146.6 (m). HRMS (ESI): m/z [M+H]+ calcd for C16H14F3N2O5S2: 435.0296, found: 435.0289.
4.1.3.18. N-(3,4-Difluorophenyl)-5-(N-(N,N-dimethylsulfamoyl)sulfamoyl)-2-fluorobenzamide (4r)
Title compound 4r was obtained from 3 using the same procedure as for compound 4o. Yield 48%, triethylamine salt. 1H NMR (400 MHz, Acetone-d6) δ 9.90 (s, 1H), 8.27 (dd, J = 6.8, 2.4 Hz, 1H), 8.13 – 8.03 (m, 1H), 8.04 – 7.97 (m, 1H), 7.63 – 7.51 (m, 1H), 7.43 – 7.27 (m, 2H), 3.36 (q, J = 7.3 Hz, 4H), 2.63 (s, 6H), 1.37 (t, J = 7.3 Hz, 6H). 13C NMR (101 MHz, Acetone-d6) δ 162.0, 160.4 (d, J = 253.2 Hz), 149.6 (dd, J = 243.8, 13.2 Hz), 146.5 (dd, J = 242.9, 12.7 Hz), 142.9 (d, J = 3.5 Hz), 135.8 (dd, J = 9.1, 3.1 Hz), 131.8 (d, J = 9.6 Hz), 129.2 (d, J = 3.4 Hz), 123.3 (d, J = 15.2 Hz), 117.2 (d, J = 17.8 Hz), 116.3 (dd, J = 6.1, 3.5 Hz), 116.0 (d, J = 23.9 Hz), 109.3 (d, J = 22.0 Hz), 46.4, 38.1, 8.1. 19F NMR (377 MHz, Acetone-d6) δ −113.8 – −113.9 (m), −139.7 – −139.8 (m), −146.4 – −146.6 (m). HRMS (ESI): m/z [M+H]+ calcd for C15H15F3N3O5S2: 438.0405, found: 438.0399.
4.1.3.19. N-(3,4-Difluorophenyl)-5-(N-((3,4-difluorophenyl)sulfonyl)sulfamoyl)-2-fluorobenzamide (4s)
Title compound 4s was obtained from 3 using the same procedure as for compound 4o. Yield 69%, triethylamine salt. 1H NMR (400 MHz, Acetone-d6) δ 9.81 (s, 1H), 8.13 (dd, J = 6.8, 2.4 Hz, 1H), 8.05 – 7.91 (m, 2H), 7.74 – 7.64 (m, 1H), 7.64 – 7.59 (m, 1H), 7.56 – 7.49 (m, 1H), 7.40 – 7.27 (m, 3H), 3.38 (q, J = 7.3 Hz, 6H), 1.35 (t, J = 7.3 Hz, 9H). 13C NMR (101 MHz, Acetone-d6) δ 161.68 (d, J = 1.7 Hz), 160.58 (d, J = 253.9 Hz), 151.40 (dd, J = 250.6, 12.7 Hz), 149.62 (dd, J = 244.0, 13.3 Hz), 149.12 (dd, J = 249.4, 13.4 Hz), 147.9 – 145.1 (m), 142.8 (d, J = 4.1 Hz), 142.2 (d, J = 3.3 Hz), 135.7 (dd, J = 9.1, 3.1 Hz), 131.8 (d, J = 9.6 Hz), 129.3 (d, J = 3.5 Hz), 124.0 (dd, J = 7.3, 3.8 Hz), 123.3 (d, J = 15.3 Hz), 117.3, 117.1, 117.0, 116.5 – 116.2 (m), 116.1, 109.4 (d, J = 22.0 Hz), 46.6, 8.2. 19F NMR (377 MHz, Acetone-d6) δ −113.0, −137.6 – −137.7 (m), −139.6 – −139.8 (m), −140.1 – −140.2 (m), −146.3 – −146.4 (m). HRMS (ESI): m/z [M+H]+ calcd for C19H12F5N2O5S2: 507.0108, found: 507.0104.
4.1.4. 5-((tert-Butoxycarbonyl)amino)-2-fluorobenzoic acid (6)
To a solution of 5-amino-2-fluorobenzoic acid 5 (2 g, 12.9 mmol) in a mixture of 1,4-dioxane/H2O (14 mL, 1:1) were added Boc2O (4.22 g, 19.3 mmol) and NaHCO3 (2.16 g, 25.8 mmol) at 0 °C. The mixture was stirred for 16 h at room temperature and the volatiles were removed under reduced pressure. The aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with 1M HCl, water, brine and dried over MgSO4. Concentration under reduced pressure afforded 6 (2.45 g, 74%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H), 8.05 (dd, J = 6.6, 2.9 Hz, 1H), 7.74 – 7.54 (m, 1H), 7.21 (dd, J = 10.5, 9.0 Hz, 1H), 1.47 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 165.4 (d, J = 3.3 Hz), 156.7 (d, J = 251.9 Hz), 153.2, 136.1 (d, J = 2.9 Hz), 124.3 (d, J = 8.1 Hz), 121.1, 119.5 (d, J = 11.1 Hz), 117.5 (d, J = 23.5 Hz), 79.9, 28.5. 19F NMR (377 MHz, DMSO-d6) δ −119.1 – −121.9 (m).
4.1.5. tert-Butyl (3-((3,4-difluorophenyl)carbamoyl)-4-fluorophenyl)carbamate (7)
To a solution of 6 (3 g, 11.7 mmol) in DMF (20 mL) were added 3,4-difluoroaniline (1.4 mL, 14.1 mmol), DIPEA (6.1 mL, 35.2 mmol) and HATU (6.7 g, 17.6 mmol). The mixture was stirred 1 h at room temperature, diluted with EtOAc and washed with 1N HCl, water and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (8:2) to afford 7 (3.87 g, 90%) as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 9.60 (s, 1H), 7.97 – 7.84 (m, 1H), 7.78 (dd, J = 6.3, 2.8 Hz, 1H), 7.57 (ddd, J = 9.0, 4.5, 2.8 Hz, 1H), 7.51 – 7.36 (m, 2H), 7.28 (t, J = 9.4 Hz, 1H), 1.48 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 163.3, 154.3 (d, J = 244.1 Hz), 153.3, 149.4 (dd, J = 243.5, 13.3 Hz), 146.1 (dd, J = 242.3, 12.7 Hz), 136.9 – 135.7 (m), 136.3 – 136.1 (m), 124.7 (d, J = 16.2 Hz), 122.4, 119.1, 118.0 (d, J = 18.0 Hz), 117.6 – 116.30 (m), 116.6, 109.2 (d, J = 21.6 Hz), 79.9, 28.5. 19F NMR (377 MHz, DMSO-d6) δ −124.1 – −124.2 (m), −138.3 – −138.5 (m), −145.3 – −145.4 (m). HRMS (ESI): m/z [M+H]+ calcd for C18H18F3N2O3: 367.1270, found: 367.1263.
4.1.6. 5-Amino-N-(3,4-difluorophenyl)-2-fluorobenzamide (8)
To a solution of 7 (250 mg, 0.68 mmol) in CH2Cl2 (7 mL) was added TFA (522 μL, 6.8 mmol) at 0 °C. The mixture was stirred 1 h at 0 °C and then 1 h at room temperature. The solution was diluted with CH2Cl2 and neutralized by addition of solid NaHCO3. The mixture was washed with water. The organic layer was separated, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography with hexanes/EtOAc (7:3) to afford 8 (109 mg, 60%). 1H NMR (400 MHz, Chloroform-d) δ 8.51 (d, J = 17.6 Hz, 1H), 7.92 – 7.76 (m, 1H), 7.43 (dd, J = 6.5, 3.1 Hz, 1H), 7.25 – 7.10 (m, 2H), 7.00 (dd, J = 11.9, 8.7 Hz, 1H), 6.86 – 6.75 (m, 1H), 3.77 (s, 2H). 13C NMR (101 MHz, Chloroform-d) δ 161.5 (d, J = 3.8 Hz), 153.8 (d, J = 236.5 Hz), 150.1 (dd, J = 247.2, 13.2 Hz), 147.2 (dd, J = 245.9, 12.8 Hz), 143.5 (d, J = 1.9 Hz), 134.2 (dd, J = 8.7, 3.2 Hz), 120.8 (d, J = 12.1 Hz), 120.0 (d, J = 9.0 Hz), 117.0, 116.7, 116.0 (dd, J = 5.9, 3.6 Hz), 110.3 (d, J = 22.0 Hz). 19F NMR (377 MHz, Chloroform-d) δ −127.9 – −128.0 (m), −136.9 – −137.0 (m), −143.5 – −143.6 (m). HRMS (ESI): m/z [M+H]+ calcd for C13H10F3N2O: 267.0745, found: 267.0738.
4.1.7. 5-(Cyclopropanesulfonamido)-N-(3,4-difluorophenyl)-2-fluorobenzamide (9)
To a solution of 8 (180 mg, 0.68 mmol) in CH2Cl2 (6 mL) were added cyclopropylsulfonyl chloride (69 μL, 0.68 mmol), Et3N (104 μL, 0.75 mmol) and DMAP (4 mg, 0.03 mmol). The mixture was heated at 40 °C during 3 h and cooled down to room temperature. The solution was diluted with CH2Cl2 and washed with 1M HCl and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (7:3) to afford 9 (119 mg, 47%). 1H NMR (400 MHz, Acetone-d6) δ 9.67 (s, 1H), 8.79 (s, 1H), 8.03 – 7.94 (m, 1H), 7.82 (dd, J = 6.2, 2.9 Hz, 1H), 7.63 – 7.56 (m, 1H), 7.56 – 7.48 (m, 1H), 7.41 – 7.27 (m, 2H), 2.72 – 2.56 (m, 1H), 1.07 – 0.96 (m, 4H). 13C NMR (101 MHz, Acetone-d6) δ 161.9 (d, J = 2.2 Hz), 156.6 (d, J = 246.4 Hz), 149.6 (dd, J = 244.0, 13.2 Hz), 146.5 (dd, J = 243.1, 12.8 Hz), 135.7 (dd, J = 9.0, 3.1 Hz), 135.0 (d, J = 2.9 Hz), 126.2 (d, J = 8.8 Hz), 124.1 (d, J = 15.6 Hz), 123.3 (d, J = 2.9 Hz), 117.3, 117.1, 116.9, 116.3 (dd, J = 6.1, 3.6 Hz), 109.4 (d, J = 22.2 Hz), 4.8. 19F NMR (377 MHz, Acetone-d6) δ −121.3 – −121.4 (m), −139.6 – −139.7 (m), −146.3 – −146.4 (m). HRMS (ESI): m/z [M+H]+ calcd for C16H14F3N2O3S: 371.0677, found: 371.0669.
4.1.8. 5-(Cyclopentanesulfonamido)-N-(3,4-difluorophenyl)-2-fluorobenzamide (10)
To a solution of 8 (180 mg, 0.68 mmol) in CH2Cl2 (6 mL) were added cyclopentylsulfonyl chloride (86 μL, 0.68 mmol), Et3N (104 μL, 0.75 mmol) and DMAP (4 mg, 0.03 mmol). The mixture was heated at 40 °C for 3 h and cooled down to room temperature. The solution was diluted with CH2Cl2 and washed with 1M HCl and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (7:3) to afford 10 (62 mg, 23%). 1H NMR (400 MHz, Acetone-d6) δ 9.67 (s, 1H), 8.82 (s, 1H), 8.13 – 7.92 (m, 1H), 7.79 (dd, J = 6.2, 2.9 Hz, 1H), 7.61 – 7.55 (m, 1H), 7.55 – 7.50 (m, 1H), 7.40 – 7.24 (m, 2H), 3.68 – 3.58 (m, 1H), 2.04 – 1.93 (m, 4H), 1.81 – 1.68 (m, 2H), 1.67 – 1.55 (m, 2H). 13C NMR (101 MHz, Acetone-d6) δ 161.9, 156.3 (d, J = 246.1 Hz), 149.6 (dd, J = 244.0, 13.2 Hz), 146.5 (dd, J = 242.9, 12.8 Hz), 135.7 (dd, J = 9.1, 3.1 Hz), 135.3 (d, J = 2.9 Hz), 125.1 (d, J = 8.7 Hz), 124.2 (d, J = 15.6 Hz), 122.1 (d, J = 2.6 Hz), 117.2 (dd, J = 18.2, 0.9 Hz), 117.1 (d, J = 24.7 Hz), 116.2 (dd, J = 6.2, 3.4 Hz), 109.3 (d, J = 22.2 Hz), 60.5, 27.6, 25.5. 19F NMR (377 MHz, Acetone-d6) δ −122.0 – −122.1 (m), −139.7 – −139.8 (m), −146.4 – −146.6 (m). HRMS (ESI): m/z [M+H]+ calcd for C18H18F3N2O3S: 399.0990, found: 399.0985.
4.1.9. 5-(Cyclohexanesulfonamido)-N-(3,4-difluorophenyl)-2-fluorobenzamide (11)
To a solution of 8 (180 mg, 0.68 mmol) in CH2Cl2 (6 mL) were added cyclohexyllsulfonyl chloride (98 μL, 0.68 mmol), Et3N (104 μL, 0.75 mmol) and DMAP (4 mg, 0.03 mmol). The mixture was heated at 40 °C during 3 h and cooled down to room temperature. The solution was diluted with CH2Cl2 and washed with 1M HCl and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified over silica gel column chromatography using hexanes/EtOAc (7:3) to afford 11 (98 mg, 35%). 1H NMR (400 MHz, Acetone-d6) δ 9.68 (s, 1H), 8.84 (s, 1H), 7.99 (ddd, J = 13.0, 7.4, 2.6 Hz, 1H), 7.79 (dd, J = 6.1, 2.9 Hz, 1H), 7.58 (ddd, J = 8.9, 4.3, 2.9 Hz, 1H), 7.56 – 7.51 (m, 1H), 7.40 – 7.25 (m, 2H), 3.17 – 3.03 (m, 1H), 2.19 – 2.12 (m, 2H), 1.89 – 1.79 (m, 2H), 1.70 – 1.62 (m, 1H), 1.60 – 1.44 (m, 2H), 1.37 – 1.23 (m, 3H). 13C NMR (101 MHz, Acetone-d6) δ 161.9, 156.1 (d, J = 245.6 Hz), 149.6 (dd, J = 243.9, 13.2 Hz), 146.5 (dd, J = 242.9, 12.7 Hz), 135.7 (dd, J = 9.1, 3.2 Hz), 135.4 (d, J = 2.8 Hz), 124.5 (d, J = 8.6 Hz), 124.2 (d, J = 15.7 Hz), 121.6 (d, J = 2.6 Hz), 117.2 (d, J = 18.3 Hz), 117.1 (d, J = 24.6 Hz), 116.2 (dd, J = 5.9, 3.6 Hz), 109.3 (d, J = 22.1 Hz), 60.0, 26.3, 24.9, 24.7. 19F NMR (377 MHz, Acetone-d6) δ −122.4 – −122.5 (m), −139.7 – −139.9 (m), −146.5 – −146.6 (m). HRMS (ESI): m/z [M+H]+ calcd for C19H20F3N2O3S: 413.1147, found: 413.1139.
4.1.10. 3-Acetamido-4-fluorobenzenesulfonyl chloride (13)
To chlorosulfonic acid (20 mL) at 0 °C was added portion wise 2-fluoroacetanilide (5 g, 32.6 mmol). The solution was heated at 80 °C for 5 h, cooled down to room temperature and poured into crushed ice. The white precipitate was filtered, washed with cold water and dried in vacuo. 1H NMR revealed the presence of 2 regioisomers which were separated by silica gel column chromatography using hexanes/EtOAc (8:2) to afford the desired isomer 13 (5.1 g, 62%). 1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.11 (dd, J = 7.7, 2.2 Hz, 1H), 7.35 (ddd, J = 8.5, 4.9, 2.2 Hz, 1H), 7.17 (dd, J = 10.9, 8.5 Hz, 1H), 2.08 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.1, 153.9 (d, J = 246.5 Hz), 144.6, 125.8 (d, J = 12.2 Hz), 122.9 (d, J = 8.4 Hz), 122.2, 115.1 (d, J = 20.4 Hz), 23.9. 19F NMR (377 MHz, DMSO-d6) δ −125.6 – −125.7 (m). HRMS (ESI): m/z [M+H]+ calcd for C8H8ClFNO3S: 251.9897, found: 251.9889.
4.1.11. N-(5-(N-Cyclopentylsulfamoyl)-2-fluorophenyl)acetamide (14)
To a solution of compound 13 (1 g, 3.97 mmol) in CH2Cl2 (10 mL) was added cyclopentylamine (196 μL, 3.97 mmol) and Et3N (610 μL, 4.37 mmol). The mixture was stirred 2 h at room temperature, diluted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (8:2) to afford 14 (1.05 g, 88%). 1H NMR (400 MHz, Chloroform-d) δ 8.81 (dd, J = 7.3, 2.3 Hz, 1H), 7.79 (d, J = 3.1 Hz, 1H), 7.68 – 7.59 (m, 1H), 7.19 (dd, J = 10.3, 8.6 Hz, 1H), 5.17 (d, J = 7.3 Hz, 1H), 3.61 (h, J = 6.8 Hz, 1H), 2.26 (s, 3H), 1.89 – 1.73 (m, 2H), 1.70 – 1.56 (m, 2H), 1.56 – 1.33 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 168.8, 154.3 (d, J = 251.5 Hz), 137.5 (d, J = 3.3 Hz), 127.1 (d, J = 11.3 Hz), 123.6 (d, J = 8.8 Hz), 120.8, 115.4 (d, J = 21.0 Hz), 55.3, 33.4, 24.5, 23.2. 19F NMR (377 MHz, Chloroform-d) δ −124.6. HRMS (ESI): m/z [M+H]+ calcd for C13H18FN2O3S: 301.1022, found: 301.1017.
4.1.12. 3-Amino-N-cyclopentyl-4-fluorobenzenesulfonamide (15)
A solution of compound 14 (1 g, 3.33 mmol) in HCl 6N (5 mL) was stirred at 100 °C for 1 h and neutralized with NaOH 1M. The mixture was extracted with CH2Cl2 (3 × 10 mL). The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography using hexanes/EtOAc (1:1) to afford 15 (639 mg, 74%). 1H NMR (400 MHz, DMSO-d6) δ 7.48 (d, J = 7.0 Hz, 1H), 7.22 (dd, J = 8.4, 2.4 Hz, 1H), 7.16 (dd, J = 11.3, 8.4 Hz, 1H), 6.97 – 6.91 (m, 1H), 5.62 (s, 2H), 3.43 – 3.32 (m, 1H), 1.65 – 1.49 (m, 4H), 1.43 – 1.26 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 152.5 (d, J = 243.2 Hz), 138.2 (d, J = 2.8 Hz), 137.5 (d, J = 13.9 Hz), 115.8 (d, J = 19.9 Hz), 114.6 (d, J = 13.6 Hz), 114.6, 54.9, 32.9, 23.3. 19F NMR (377 MHz, DMSO-d6) δ −131.2 – −131.4 (m). HRMS (ESI): m/z [M+H]+ calcd for C11H16FN2O2S: 259.0917, found: 259.0910.
4.1.13. N-(5-(N-Cyclopentylsulfamoyl)-2-fluorophenyl)-3,4-difluorobenzamide (16)
To a solution of 15 (250 mg, 0.968 mmol) in CH2Cl2 (5 mL) were added 3,4-difluorobenzoyl chloride (122 μL, 0.97 mmol) and Et3N (148 μL, 1.06 mmol) at 0 °C. The mixture was stirred 2 h at room temperature, diluted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (8:2) to afford 16 (138 mg, 36%). 1H NMR (400 MHz, Acetone-d6) δ 9.60 (s, 1H), 8.63 (dd, J = 7.2, 2.4 Hz, 1H), 8.06 – 7.99 (m, 1H), 7.97 – 7.91 (m, 1H), 7.77 – 7.69 (m, 1H), 7.62 – 7.49 (m, 1H), 7.45 (dd, J = 10.4, 8.6 Hz, 2H), 6.67 (d, J = 7.2 Hz, 1H), 3.73 – 3.56 (m, 1H), 1.87 – 1.73 (m, 2H), 1.70 – 1.57 (m, 2H), 1.56 – 1.38 (m, 4H). 13C NMR (101 MHz, Acetone-d6) δ 163.7, 156.2 (d, J = 252.4 Hz), 152.5 (dd, J = 252.4, 12.7 Hz), 149.9 (dd, J = 247.7, 13.1 Hz), 138.4 (d, J = 3.6 Hz), 132.1 – 131.1 (m), 126.8 (d, J = 12.5 Hz), 125.2 (dd, J = 7.5, 3.7 Hz), 124.8 (d, J = 8.9 Hz), 123.5 (d, J = 2.8 Hz), 117.6 (d, J = 18.0 Hz), 117.3 (dd, J = 18.8, 1.3 Hz), 116.1 (d, J = 21.4 Hz), 55.1, 32.8, 23.0. 19F NMR (377 MHz, Acetone-d6) δ −120.3 – −120.4 (m), −134.5 – −134.6 (m), −138.8 – −138.9 (m). HRMS (ESI): m/z [M+H]+ calcd for C18H18F3N2O3S: 399.0990, found: 399.0985.
4.1.14. 1-(3,4-Difluorophenyl)cyclopropan-1-amine (18)
To a solution of 3,4-difluorobenzonitrile (278 mg, 2 mmol) in dry Et2O (10 mL) at −78 °C was added dropwise Ti(OiPr)4 (0.64 mL, 2.2 mmol) followed by EtMgBr (1.5 mL, 2.2 mmol, 3M in Et2O). After 10 minutes, BF3.Et2O (0.5 mL, 4 mmol) was added and the solution was stirred for 1 h at −78 °C. The reaction was quenched by addition of 1N HCl (2 mL) and diluted with Et2O (10 mL). The organic layer was separated, washed with water, brine and dried over MgSO4. The volatiles were evaporated under reduced pressure and the residue was purified by silica gel column chromatogrpahy using hexanes/EtOAc (1:1) to afford 18 (189 mg, 56%). 1H NMR (400 MHz, Chloroform-d) δ 7.16 – 7.04 (m, 2H), 7.03 – 6.98 (m, 1H), 1.91 (s, 2H), 1.16 – 1.05 (m, 2H), 1.01 – 0.90 (m, 2H). 13C NMR (101 MHz, Chloroform-d) δ 150.1 (dd, J = 247.5, 12.8 Hz), 148.6 (dd, J = 246.2, 12.8 Hz), 144.2 (dd, J = 5.0, 3.5 Hz), 121.2 (dd, J = 6.2, 3.4 Hz), 116.9 (d, J = 17.0 Hz), 114.7 (d, J = 17.7 Hz), 36.2, 18.2. 19F NMR (377 MHz, Chloroform-d) δ −139.3 – −139.4 (m), −143.3 – −143.4 (m). HRMS (ESI): m/z [M+H]+ calcd for C9H10F2N: 170.0781, found: 170.0776.
4.1.15. 5-(N-Cyclopentylsulfamoyl)-N-(1-(3,4-difluorophenyl)cyclopropyl)-2-fluorobenzamide (19)
A solution of 2 (240 mg, 1 mmol) in SOCl2 (2.5 mL) was heated at 80 °C for 16 h. The mixture was concentrated under reduced pressure and co-evaporated with toluene. The crude mixture was dissolved in toluene (2.5 mL) and a solution of 18 (170 mg, 1 mmol) in toluene (2 mL) was added via cannula. The mixture was heated at 110 °C for 2 h and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (8:2) to afford the desired sulfonyl chloride derivative. To a solution of this sulfonyl chloride (314 mg) in CH2Cl2 (5 mL) were added cyclopentylamine (83 μL, 0.844 mmol) and Et3N (129 μL, 0.929 mmol). The mixture was stirred 2 h at room temperature, diluted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (8:2) to afford 19 (149 mg, 34% over two steps). 1H NMR (400 MHz, Acetone-d6) δ 8.49 (s, 1H), 8.25 (dd, J = 6.6, 2.5 Hz, 1H), 8.07 – 7.99 (m, 1H), 7.47 (dd, J = 10.4, 8.7 Hz, 1H), 7.41 – 7.32 (m, 1H), 7.31 – 7.12 (m, 2H), 6.68 (d, J = 7.2 Hz, 1H), 3.68 – 3.48 (m, 1H), 1.80 – 1.69 (m, 2H), 1.67 – 1.56 (m, 2H), 1.53 – 1.31 (m, 8H). 13C NMR (101 MHz, Acetone-d6) δ 162.7 (d, J = 2.3 Hz), 161.7 (d, J = 254.9 Hz), 149.8 (dd, J = 244.8, 12.9 Hz), 148.5 (dd, J = 244.4, 12.6 Hz), 140.9 (dd, J = 5.5, 3.6 Hz), 138.8 (d, J = 3.5 Hz), 131.5 (d, J = 10.2 Hz), 130.0 (d, J = 4.3 Hz), 124.2 (d, J = 15.9 Hz), 122.2 (dd, J = 6.3, 3.4 Hz), 117.1 (d, J = 25.0 Hz), 116.8 (d, J = 17.2 Hz), 115.0 (d, J = 18.3 Hz), 55.1, 34.7, 32.8, 22.9, 17.6. 19F NMR (377 MHz, Acetone-d6) δ −110.5 – −110.6 (m), −141.9 – −142.0 (m), −145.2 – −145.3 (m). HRMS (ESI): m/z [M+H]+ calcd for C21H22F3N2O3S: 439.1303, found: 439.1300.
4.1.16. 2-(3,4-Difluorophenyl)propan-2-amine (20)
A solution of anhydrous CeCl3 (2.84g, 11.5 mmol) in THF (18 mL) was stirred at 45 °C for 3 h and cooled down to room temperature. Then, 3,4-difluorobenzonitrile (800 mg, 5.75 mmol) was added and the mixture was cooled down to −25 °C before addition of MeLi (9.6 mL, 14.4 mmol, 1.5 M in Et2O). The solution was stirred 1 h at −25 °C and quenched with NaOH 30% (4 mL). The mixture was stirred for 16 h at room temperature and the cerium salts were filtered and washed with THF. The filtrate was dried over MgSO4 and concentrated under reduced pressure. The residue was dissolved in THF and HCl (4N in dioxane) was added and the solution was concentrated in vacuo. The resulting salts were filtered, washed with hexanes and then treated with aqueous ammonium hydroxide (5 mL). The solution was then extracted with CH2Cl2, dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography using CH2Cl2/MeOH (98:2) to afford 20 (502 mg, 51%). 1H NMR (400 MHz, Chloroform-d) δ 7.41 – 7.32 (m, 1H), 7.27 – 7.19 (m, 1H), 7.11 (dt, J = 10.2, 8.4 Hz, 1H), 1.49 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 150.51 (dd, J = 121.6, 12.6 Hz), 148.83 – 147.43 (m), 147.48 (d, J = 16.9 Hz), 120.69 (dd, J = 6.1, 3.5 Hz), 116.62 (d, J = 16.8 Hz), 114.26 (d, J = 17.8 Hz), 52.12 (d, J = 1.4 Hz), 33.01. 19F NMR (377 MHz, Chloroform-d) δ −139.4 – −139.5 (m), −143.4 – −143.5 (m). HRMS (ESI): m/z [M+H]+ calcd for C9H12F2N: 172.0938, found: 172.0932.
4.1.17. 3-(N-Cyclopentylsulfamoyl)-N-(2-(3,4-difluorophenyl)propan-2-yl)benzamide (21)
A solution of 2 (507 mg, 2.12 mmol) in SOCl2 (5 mL) was heated at 80 °C for 16 h. The mixture was concentrated under reduced pressure and co-evaporated with toluene. The crude product was dissolved in toluene (3 mL) and a solution of 20 (360 mg, 2.12 mmol) in toluene (2 mL) was added via cannula. The mixture was heated at 110 °C again for 2 h and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using hexanes/EtOAc (8:2) to afford the sulfonyl chloride intermediate. To a solution of this sulfonyl chloride (338 mg) in CH2Cl2 (5 mL) were added cyclopentylamine (89 μL, 0.904 mmol) and Et3N (139 μL, 0.995 mmol). The mixture was stirred 2 h at room temperature, diluted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (8:2) to afford 21 (230 mg, 25% over two steps). 1H NMR (400 MHz, Acetone-d6) δ 8.15 (dd, J = 6.7, 2.5 Hz, 1H), 8.03 – 7.98 (m, 1H), 7.97 (s, 1H), 7.54 – 7.42 (m, 2H), 7.40 – 7.33 (m, 1H), 7.33 – 7.21 (m, 1H), 6.66 (d, J = 7.2 Hz, 1H), 3.73 – 3.34 (m, 1H), 1.80 (s, 6H), 1.78 – 1.69 (m, 2H), 1.68 – 1.57 (m, 2H), 1.54 – 1.33 (m, 4H). 13C NMR (101 MHz, Acetone-d6) δ 161.6 (d, J = 253.8 Hz), 161.4 (d, J = 2.0 Hz), 149.7 (dd, J = 244.4, 12.7 Hz), 148.5 (dd, J = 244.4, 12.7 Hz), 146.0 – 144.6 (m), 138.7 (d, J = 3.3 Hz), 131.2 (d, J = 10.0 Hz), 129.7 (d, J = 4.3 Hz), 125.0 (d, J = 16.3 Hz), 121.6 (dd, J = 6.4, 3.4 Hz), 117.1, 116.9, 116.7 (d, J = 17.0 Hz), 114.4 (d, J = 18.3 Hz), 55.8 (d, J = 1.3 Hz), 55.1, 32.8, 22.9. 19F NMR (377 MHz, Acetone-d6) δ −111.0 – −111.1 (m), −141.9 – −142.0 (m), −145.4 – −145.5 (m). HRMS (ESI): m/z [M+H]+ calcd for C21H23F3N2O3S: 441.1460, found: 441.1453.
4.1.18. 3,4-Dihydroisoquinolin-1(2H)-one (24)
To a solution of phenethylamine (3 mL, 24 mmol) and Et3N (3.6 mL, 26.4 mmol) in DMF (50 mL) was added methyl chloroformate (2.0 mL, 26.4 mmol) at 0 °C. The reaction mixture was stirred 1 h at room temperature and diluted with EtOAc (100 mL). The solution was washed three times with water (3 × 30 mL). The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography using hexanes/EtOAc (1:1 to 0:1) to afford the carbamate intermediate (3.72 g, 86%) as a colorless liquid. CAS: 26011-68-7. The obtained carbamate (3 g, 16.7 mmol) was dissolved in trifluoromethanesulfonic acid (30 mL) at 0 °C and the mixture was stirred for 24 h at 70 °C. The mixture was then poured into crushed ice and extracted with CH2Cl2. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography using hexanes/EtOAc (1:1) to afford 24 (2.34 g, 95%) as a yellow oil. CAS: 1196-38-9.
4.1.19. 2,3,4,5-Tetrahydro-1H-benzo[c]azepin-1-one (25)
To a solution of phenylpropylamine (3 mL, 21.1 mmol) and Et3N (3.2 mL, 23.2 mmol) in DMF (50 mL) was added methyl chloroformate (1.8 mL, 23.2 mmol) at 0 °C. The reaction mixture was stirred 1 h at room temperature and diluted with EtOAc (100 mL). The solution was washed three times with water (3 × 30 mL). The organic layer was dried over MgSO4 and concentrated in vacuo. The resulting liquid was purified by silica gel column chromatography using hexanes/EtOAc (1:1 to 0:1) to afford the carbamate intermediate (3.80 g, 93%) as a colorless liquid. CAS: 111944-09-3. This carbamate (3.65 g, 18.9 mmol) was dissolved in trifluoromethanesulfonic acid (30 mL) at 0 °C and the mixture was stirred for 24 h at 70 °C. The mixture was poured into crushed ice and extracted with CH2Cl2. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography using hexanes/EtOAc (1:1) to afford 25 (2.74 g, 90%) as a yellow oil. CAS: 6729-50-6.
4.1.20. N-Cyclopentyl-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-sulfonamide (28)
To chlorosulfonic acid (25 mL) cooled to 0 °C was added portion wise compound 24 (2 g, 13.6 mmol). After complete addition, the yellow solution was allowed to warm up to room temperature, then heated at 60 °C for 16 h. The reaction mixture was then cooled down to room temperature and poured dropwise into crushed ice. The light yellow precipitate was filtered, washed with water and cold Et2O and dried in vacuo to afford the desired sulfonyl chloride intermediate 26. To a solution of compound 26 (1 g, 4.07 mmol) in CH2Cl2 (10 mL) were added cyclopentylamine (400 μL, 4.07 mmol) and Et3N (624 μL, 4.48 mmol). The mixture was stirred 2 h at room temperature and quenched with 1M HCl. The precipitate was filtered and washed with water and cold Et2O. The solid was dried in vacuo to afford 28 (971 mg, 81%) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.27 (d, J = 2.1 Hz, 1H), 8.19 (s, 1H), 7.87 (dd, J = 7.9, 2.1 Hz, 1H), 7.75 (d, J = 6.9 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 3.46 – 3.39 (m, 2H), 3.06 – 2.96 (m, 2H), 1.64 – 1.49 (m, 4H), 1.44 – 1.23 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.7, 144.0, 140.7, 130.4, 129.7, 129.1, 125.7, 54.9, 39.2, 32.9, 28.0, 23.2. HRMS (ESI): m/z [M+H]+ calcd for C14H19N2O3S: 295.1116, found: 295.1110.
4.1.21. N-Cyclopentyl-1-oxo-2,3,4,5-tetrahydro-1H-benzo[c]azepine-8-sulfonamide (29)
To chlorosulfonic acid (10 mL) cooled to 0 °C was added portion wise compound 25 (955 mg, 5.92 mmol). After complete addition, the yellow solution was allowed to warm up to room temperature, then heated at 60 °C for 16 h. The reaction mixture was cooled down to room temperature and poured dropwise into crushed ice. The light yellow precipitate was filtered, washed with water and cold Et2O and dried in vacuo to afford the desired sulfonyl chloride intermediate 27. To a solution of compound 27 (250 mg g, 0.963 mmol) in CH2Cl2 (5 mL) were added cyclopentylamine (144 μL, 0.963 mmol) and Et3N (148 μL, 1.06 mmol). The mixture was stirred 2 h at room temperature, diluted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (7:3 to 8:2) to afford the desired sulfonamide derivative 29 (183 mg, 97%) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.27 (t, J = 5.9 Hz, 1H), 7.91 (d, J = 2.1 Hz, 1H), 7.82 (dd, J = 7.9, 2.1 Hz, 1H), 7.70 (d, J = 7.1 Hz, 1H), 7.49 (d, J = 7.9 Hz, 1H), 3.44 – 3.37 (m, 1H), 2.91 (q, J = 6.3 Hz, 2H), 2.82 (t, J = 7.1 Hz, 2H), 1.93 (p, J = 6.7 Hz, 2H), 1.65 – 1.22 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 170.9, 142.7, 140.7, 137.1, 130.1, 128.9, 126.7, 54.9, 38.8, 32.9, 30.11, 30.04, 23.3. HRMS (ESI): m/z [M+H]+ calcd for C15H21N2O3S: 309.1273, found: 309.1266.
4.1.22. N-Cyclopentyl-N-methyl-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-sulfonamide (30)
To a solution of compound 26 (1 g, 4.07 mmol) in CH2Cl2 (10 mL) were added N-methylcyclopentylamine (480 μL, 4.07 mmol) and Et3N (624 μL, 4.48 mmol). The mixture was stirred for 2 h at room temperature, diluted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (7:3 to 8:2) to afford the desired sulfonamide derivative 30 (1.13 g, 90%) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 8.17 (d, J = 2.1 Hz, 1H), 7.87 (dd, J = 8.0, 2.1 Hz, 1H), 7.56 (d, J = 8.0 Hz, 1H), 4.25 (p, J = 8.1 Hz, 1H), 3.48 – 3.38 (m, 2H), 3.02 (t, J = 6.6 Hz, 2H), 2.64 (s, 3H), 1.55 – 1.45 (m, 4H), 1.45 – 1.35 (m, 2H), 1.35 – 1.25 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 163.6, 144.6, 137.7, 130.5, 130.2, 129.4, 126.0, 58.3, 39.2, 28.9, 28.0, 27.8, 24.0. HRMS (ESI): m/z [M+H]+ calcd for C15H21N2O3S: 309.1273, found: 309.1267.
4.1.23. N-Cyclopentyl-N-methyl-1-oxo-2,3,4,5-tetrahydro-1H-benzo[c]azepine-8-sulfonamide (31)
To a solution of compound 27 (250 mg, 0.963 mmol) in CH2Cl2 (5 mL) were added N-methylcyclopentylamine (144 μL, 0.963 mmol) and Et3N (148 μL, 1.06 mmol). The mixture was stirred for 2 h at room temperature, diluted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (7:3 to 8:2) to afford the desired sulfonamide derivative 31 (292 mg, 94%) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (t, J = 5.9 Hz, 1H), 7.82 (s, 2H), 7.52 (d, J = 8.7 Hz, 1H), 4.24 (p, J = 8.1 Hz, 1H), 2.92 (q, J = 6.3 Hz, 2H), 2.83 (t, J = 7.1 Hz, 2H), 2.64 (s, 3H), 1.93 (t, J = 6.8 Hz, 2H), 1.56 – 1.45 (m, 4H), 1.44 – 1.36 (m, 2H), 1.35 – 1.23 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 170.7, 143.3, 137.6, 137.4, 130.4, 129.4, 127.0, 58.3, 38.8, 30.09, 30.04, 28.9, 27.7, 24.1. HRMS (ESI): m/z [M+H]+ calcd for C16H23N2O3S: 323.1429, found: 323.1423.
4.1.24. N-Cyclopentyl-2-(3,4-difluorophenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-sulfonamide (32)
To a solution of compound 28 (50 mg, 0.170 mmol) in DMF (0.5 mL) were added 3,4-difluorobromobenzene (23 μL, 0.170 mmol), CuI (32 mg, 0.170 mmol) and K2CO3 (47 mg, 0.340 mmol). The mixture was heated at 150 °C for 16 h and then diluted with EtOAc and water and filtered over a pad of Celite. The filtrate was washed with a saturated solution of Na2CO3, water, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (9:1 to 7:3) to afford 32 (39 mg, 56%). 1H NMR (400 MHz, DMSO-d6) δ 8.36 (d, J = 2.1 Hz, 1H), 7.95 (dd, J = 7.9, 2.1 Hz, 1H), 7.80 (d, J = 6.9 Hz, 1H), 7.67 – 7.58 (m, 2H), 7.57 – 7.47 (m, 1H), 7.38 – 7.28 (m, 1H), 5.76 (s, 1H), 4.04 – 3.95 (m, 2H), 3.24 (t, J = 6.4 Hz, 2H), 1.68 – 1.50 (m, 4H), 1.42 – 1.27 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.6, 149.9 (dd, J = 137.6, 13.0 Hz), 147.4 (dd, J = 137.0, 12.9 Hz), 143.8, 141.0, 139.9 (dd, J = 8.3, 3.4 Hz), 130.2 (d, J = 21.4 Hz), 129.1, 126.4, 123.0 (dd, J = 6.5, 3.3 Hz), 117.7 (d, J = 17.8 Hz), 115.8 (d, J = 18.8 Hz), 55.4, 54.9, 49.0, 32.9, 28.0, 23.3. 19F NMR (377 MHz, DMSO-d6) δ −139.0, −142.5. HRMS (ESI): m/z [M+H]+ calcd for C20H21F2N2O3S: 407.1241, found: 407.1235.
4.1.25. N-Cyclopentyl-2-(3,4-difluorophenyl)-1-oxo-2,3,4,5-tetrahydro-1H-benzo[c]azepine-8-sulfonamide (33)
To a solution of compound 29 (50 mg, 0.170 mmol) in DMF (0.5 mL) were added 3,4-difluorobromobenzene (23 μL, 0.170 mmol), CuI (32 mg, 0.170 mmol) and K2CO3 (47 mg, 0.340 mmol). The mixture was heated at 150 °C for 16 h and then diluted with EtOAc and water and filtered over a pad of Celite. The filtrate was washed with a saturated solution of Na2CO3, water, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (9:1 to 7:3) to afford 33 (63 mg, 58%). 1H NMR (400 MHz, Chloroform-d) δ 8.25 (d, J = 2.0 Hz, 1H), 7.95 (dd, J = 7.9, 2.1 Hz, 1H), 7.37 (d, J = 7.9 Hz, 1H), 7.32 – 7.19 (m, 2H), 7.17 – 7.08 (m, 1H), 5.21 (d, J = 7.3 Hz, 1H), 3.67 – 3.54 (m, 3H), 3.05 (t, J = 7.1 Hz, 2H), 2.19 (p, J = 6.8 Hz, 2H), 1.88 – 1.71 (m, 2H), 1.66 – 1.53 (m, 2H), 1.51 – 1.41 (m, 2H), 1.40 – 1.27 (m, 2H). 13C NMR (101 MHz, Chloroform-d) δ 169.5, 150.1 (dd, J = 249.8, 13.4 Hz), 149.0 (dd, J = 248.9, 12.5 Hz), 141.8, 140.5, 138.6 (dd, J = 7.7, 3.7 Hz), 136.6, 129.8, 129.3, 127.8, 122.3 (dd, J = 6.4, 3.6 Hz), 117.6 (d, J = 18.2 Hz), 116.0 (d, J = 18.5 Hz), 55.3, 49.6, 33.3, 30.0, 29.2, 23.2. 19F NMR (377 MHz, Chloroform-d) δ −136.6 – −136.7 (m), −140.2 – −140.3 (m). HRMS (ESI): m/z [M+H]+ calcd for C21H23F2N2O3S: 421.1397, found: 421.1392.
4.1.26. N-Cyclopentyl-2-(3,4-difluorophenyl)-N-methyl-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-sulfonamide (34)
To a solution of compound 30 (300 mg, 0.972 mmol) in DMF (2 mL) were added 3,4-difluorobromobenzene (220 μL, 1.95 mmol), CuI (371 mg, 1.95 mmol) and K2CO3 (269 mg, 1.95 mmol). The mixture was heated at 150 °C for 90 minutes under microwave irradiation. The mixture was then diluted with EtOAc and filtered over a pad of Celite. The filtrate was washed with a saturated solution of Na2CO3, water, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (9:1) to afford 34 (116 mg, 28%). 1H NMR (400 MHz, Acetone-d6) δ 8.43 (d, J = 2.0 Hz, 1H), 7.97 (dd, J = 8.0, 2.1 Hz, 1H), 7.63 (dd, J = 8.0, 0.7 Hz, 1H), 7.59 – 7.51 (m, 1H), 7.46 – 7.29 (m, 2H), 4.39 (p, J = 8.1 Hz, 1H), 4.14 (dd, J = 6.9, 6.0 Hz, 2H), 3.35 (t, J = 6.4 Hz, 2H), 2.76 (s, 3H), 1.73 – 1.34 (m, 8H). 13C NMR (101 MHz, Acetone-d6) δ 162.2, 149.5 (dd, J = 245.7, 13.5 Hz), 148.0 (dd, J = 245.1, 12.7 Hz), 143.6, 139.9 (dd, J = 8.2, 3.5 Hz), 138.7, 130.4, 128.6, 126.8, 122.0 (dd, J = 6.4, 3.5 Hz), 116.9 (d, J = 18.4 Hz), 115.2 (d, J = 19.2 Hz), 58.3, 48.8, 28.1, 28.0, 27.6, 23.7. 19F NMR (377 MHz, Acetone-d6) δ −140.4 – −140.5 (m), −144.2 – −144.3 (m). HRMS (ESI): m/z [M+H]+ calcd for C21H23F2N2O3S: 421.1397, found: 421.1391.
4.1.27. N-Cyclopentyl-2-(3,4-difluorophenyl)-N-methyl-1-oxo-2,3,4,5-tetrahydro-1H-benzo[c]azepine-8-sulfonamide (35)
To a solution of compound 31 (300 mg, 0.930 mmol) in DMF (2 mL) were added 3,4-difluorobromobenzene (210 μL, 1.86 mmol), CuI (354 mg, 1.86 mmol) and K2CO3 (258 mg, 1.86 mmol). The mixture was heated at 150 °C for 90 minutes under microwave irradiation. The mixture was then diluted with EtOAc and filtered over a pad of Celite. The filtrate was washed with a saturated solution of Na2CO3, water, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes/EtOAc (9:1) to afford 35 (141 mg, 35%). 1H NMR (400 MHz, Acetone-d6) δ 8.03 (d, J = 2.1 Hz, 1H), 7.90 (dd, J = 7.9, 2.0 Hz, 1H), 7.64 – 7.50 (m, 2H), 7.47 – 7.28 (m, 2H), 4.37 (p, J = 8.2 Hz, 1H), 3.71 (t, J = 6.4 Hz, 2H), 3.11 (t, J = 7.1 Hz, 2H), 2.74 (s, 3H), 2.23 (p, J = 6.8 Hz, 2H), 1.72 – 1.31 (m, 8H). 13C NMR (101 MHz, Acetone-d6) δ 168.8, 149.6 (dd, J = 246.1, 13.3 Hz), 148.3 (dd, J = 245.3, 12.7 Hz), 142.3, 139.8 (dd, J = 8.2, 3.5 Hz), 138.4, 137.2, 127.4, 122.8 (dd, J = 6.4, 3.5 Hz), 117.2 (d, J = 18.0 Hz), 116.0 (d, J = 18.9 Hz), 58.3, 49.2, 28.1, 27.6, 23.8. 19F NMR (377 MHz, Acetone-d6) δ −138.7 – −138.8 (m), −142.4 – −142.6 (m). HRMS (ESI): m/z [M+H]+ calcd for C22H25F2N2O3S: 435.1554, found: 435.1546.
4.2. Biological evaluation
Cell culture – HepAD38 cells were seeded at 50,000 cells/well in collagen-coated 96-well plates with DMEM/F12 medium (Thermo Scientific) supplemented with 10% heat-inactivated fetal bovine serum. Cells were treated with 0.3 μg/ml tetracycline as needed. Test compounds and controls were added to cells to a final concentration of 10 μM or in a dose-dependent manner ranging from 0.001 to 10 μM. Cells were cultured in the absence of tetracycline for 7 days to induce DNA synthesis and cccDNA formation, at day 7, test compounds plus tetracycline were added back to the cultures to inhibit transcription of viral RNA from integrated viral genome. Medium and test compounds were replenished every 5 days in culture. Supernatants were harvested at day 14, clarified by centrifugation at 5,000 rpm for 5 min, and stored at −70 °C until use. ELISA – The levels of HBeAg secreted in the culture medium were measured by using HBeAg ELISA kit (BioChain Institute Inc. Hayward, CA) according to the manufacturer’s protocol. The effective concentration of compound that reduced levels of secreted HBeAg by 50% (EC50) was determined by linear regression.
4.3. Electron microscopy
Expression and isolation of HBV Cp149 dimeric protein was carried out following previous literature [23]. The truncated HBV protein (residues 1–149) was cloned into a pET-29b vector at the BAMHI and XhoI sites with a c-terminal stop codon (no tag). This vector was transformed into BL21 e. coli grown on LB media with AMP100 restriction. The e. coli were grown in LB broth at 37 °C to an OD600 = 0.8, and expression of HBV Cp149 was induced with the addition of 1mM IPTG at 16 °C overnight. The cell pellet was solubilized 3 g/10 ml lysis buffer [50 mM Tris, 5 mM DTT, 1 mM EDTA, 0.1 mg/ml RNase/DNase, pH = 7.4] and lysed by sonication. Cell debris was pelleted by centrifugation at 26 k × g for 1 hr. Sucrose was added to the supernatant at a final concentration of 0.15M, and the debris was pelleted by centrifugation at 100 k × g for 1 hr. The remaining solubilized protein, including HBV Cp149, was pelleted by ammonium sulfate (40% saturation, 26 k × g for 1 hr). The ammonium sulfate precipitant was resolubilized in Capsid Buffer (50 mM Tris, 500 mM NaCl, 2 mM DTT, pH = 7.4). High molecular weight assemblies (include HBV Cp149 capsids) were separated from low molecular weight protein using size exclusion chromatography. The capsid fractions were dialized in Dimer Buffer (100 mM carbonate, 2 mM DTT, pH = 9.5), and capsids were completely dissociated into the composite HBV Cp149 dimers by addition of 4 M urea. The HBV Cp149 dimers were isolated from remaining high molecular weight proteins using size exclusion chromatography. The yield of HBV Dimers was ~8–10 mg/L at > 95% purity as determined by SDS-PAGE.
HBV Capsids were prepared for electron microscopy (EM) imaging by adding capsid buffer at a 3:2 ratio (final concentration of NaCl was 300 mM) to a sample of HBV Cp149 dimers (in dimer buffer) yielding a final concentration of HBV Cp149 monomer of 10 μM. The sample was maintained at 4 °C overnight. To evaluate the effects of compounds on capsid assembly, HBV Cp149 dimers were incubated with the agents for 1 hr prior to the addition of Capsid buffer. HBV Cp149 capsid assemblies were foxed onto a charged carbon grid and stained by uranyl acetate contrast agent for 15 minutes. EM images were collected using a JEOL JEM-1400 electron microscope operating at 120 kV at 25,000 – 35,000 × magnification (Emory University Robert P. Apkarian electron microscopy core facility).
4.4. Molecular modeling
Molecular modeling was carried out using Schrödinger Suite version 2016-3 using an EXXACT MD GPU server [24]. Unless otherwise stated, the default parameters for all modules were used. The HBV Cp149 Y132A crystal structure (PDBID 5T2P) was prepared using Protein Prep in Maestro that assigned bond orders, added hydrogens and disulfide bonds. Waters within 5 Å of ligands were retained in the model, and hydrogen bonding was optimized using Epik for a pH = 7 [25]. Hydrogen positions were minimized using OPLS3 forcefield in implicit solvation model [26]. A docking grid was created using Glide centered on the cocrystallized ligand (K89) located at the interface of chains B and C [27, 28]. The grid box was 10 × 10 × 10 Å and no groups were permitted to rotate. Re-docking of the co-crystallized ligand K89 using flexible Glide docking at the standard precision yielded a pose similar to that observed in the crystal structure (RMSD = 0.37 Å) supporting the accuracy of the methods. Compound 4f was prepared and docked into a grid using Glide dock with standard precision and permitted flexibility. Desmond molecular dynamics was used to simulation the capsid complexed to compound 4f [29–31]. To avoid artifacts from misplaced amino acids not present in the structure, the proteins were pruned to 1 – 141/142/143aa. All co-crystallized small molecules were removed leaving a system of only six HBV capsid proteins with compound 4f between chains B and C. This system was placed in a periodic SPC water box of buffered by solvent at 10 Å in all directions. Salt was added to a final concentration of 150 mM with an additional 30 Na+ ions to neutralize the system. The final number of atoms for the simulation was 115,834. The molecular dynamics simulation was carried out in the default eight steps of progressive relaxation by Desmond molecular dynamics. The final simulation stage lasted 10 ns at 300K and 1.01325 bar pressure under NPT conditions with frames recorded every 10ps. The simulation analysis was performed using the Simulation Interactions Diagram tool.
Supplementary Material
The synthesis and characterization of new sulfamoyl derivatives is described
All 27 compounds were evaluated for their anti-HBV activity and cytotoxicity
Several compounds reduced cccDNA level in an HBeAg reporter cell-based assay
Disruption of HBV capsid formation was investigated using electron microscopy
Molecular modelling into the capsid protein was used to rationalize the SAR
Acknowledgments
This work was supported in part by NIH Grant 1-R01-AI-132833, and 5P30-AI-50409 (CFAR). The authors declare no competing financial interest. We gratefully acknowledge Elizabeth Wright, Ph.D. and Hong Yi (Emory University Robert P. Apkarian electron microscopy core) for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization. website, available at: http://www.who.int/mediacentre/factsheets/fs204/en/Accessed June 26th, 2017.
- 2.Kang L, Pan J, Wu J, Hu J, Sun Q, Tang J. Viruses. 2015;7:4960–4977. doi: 10.3390/v7092854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei W, Wu Q, Zhou J, Kong Y, You H. Int J Environ Res Public Health. 2015;12:10039–10055. doi: 10.3390/ijerph120810039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang J, Guo F, Zhao X, Guo J-T. Acta Pharm Sin B. 2014;4:248–257. doi: 10.1016/j.apsb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alaluf MB, Shlomai A. Liver Int. 2016;36:775–82. doi: 10.1111/liv.13086. [DOI] [PubMed] [Google Scholar]
- 6.Bourne CR, Finn M, Zlotnick A. J Virol. 2006;80:11055–11061. doi: 10.1128/JVI.00933-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourne C, Lee S, Venkataiah B, Lee A, Korba B, Finn MG, Zlotnick A. J Virol. 2008;82:10262–10270. doi: 10.1128/JVI.01360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boucle S, Bassit L, Ehteshami M, Schinazi RF. Clin Liver Dis. 2016;20:747–749. doi: 10.1016/j.cld.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu G, Liu B, Zhang Y, Li J, Arzumanyan A, Clayton MM, Schinazi RF, Wang Z, Goldmann S, Ren Q, Zhang F, Feitelson MA. Antimicrob Agents Chemother. 2013;57:5344–54. doi: 10.1128/AAC.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katen SP, Chirapu SR, Finn MG, Zlotnick A. ACS Chem Biol. 2010;5:1125–1136. doi: 10.1021/cb100275b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo J. Virol. 2013;87:6931–6942. doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gane EJ, Schwabe C, Walker K, Flores L, Hartman GD, Klumpp K, Liaw S, Brown NA. Hepatology. 2014;60:LB19. [Google Scholar]
- 13.Hartman GD, Flores OA. 2013096744. Int Patent. 2013 Jun 27;
- 14.Kaiser F, Gross S, Narine A, Gunjima K. 2013030319. Int Patent. 2013 Mar 7;
- 15.Lind KE, Cao K, Lin EY, Nguyen T, Tangonan BT, Erlanson DA, Guckian K, Simmons RL, Lee W, Sun L, Hansen S, Pathan N, Zhang L. 2008005457. Int Patent. 2008 Jan 10;
- 16.Clements MJ, Debenham JS, Hale JJ, Madsen–Duggan CB, Walsh TF. 2009117269. Int Patent. 2009 Sep 24;
- 17.Stuyver LJ, Lostia S, Adams M, Mathew JS, Pai BS, Grier J, Tharnish PM, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2002;46:3854–3860. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou T, Guo H, Guo J-T, Cuconati A, Mehta A, Block TM. Antiviral Res. 2006;72:116–124. doi: 10.1016/j.antiviral.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Cai D, Mills C, Yu W, Yan R, Aldrich CE, Saputelli JR, Mason WS, Xu X, Guo J-T, Block TM, Cuconati A, Guo H. Antimicrob Agents Chemother. 2012;56:4277–4288. doi: 10.1128/AAC.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sung JJY, Wong M-L, Bowden S, Liew C-T, Hui AY, Wong VWS, Leung NWY, Locarnini S, Chan HLY. Gastroenterology. 2005;128:1890–1897. doi: 10.1053/j.gastro.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 21.Zoulim F. Antiviral Chem Chemother. 2004;15:299–305. doi: 10.1177/095632020401500602. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Z, Hu T, Zhou X, Wildum S, Garcia-Alcalde F, Xu Z, Wu D, Mao Y, Tian X, Zhou Y, Shen F, Zhang Z, Tang G, Najera I, Yang G, Shen HC, Young JAT, Qin N. Sci Rep-Uk. 2017;7:42374. doi: 10.1038/srep42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zlotnick A, Cheng N, Conway JF, Booy FP, Steven AC, Stahl SJ, Wingfield PT. Biochemistry. 1996;35:7412–7421. doi: 10.1021/bi9604800. [DOI] [PubMed] [Google Scholar]
- 24.Small-Molecule Drug Discovery Suite 2016-3. Schrödinger, LLC; New York, NY: 2016. [Google Scholar]
- 25.Schrödinger Release 2016-3. Epik, Schrödinger, LLC; New York, NY: 2016. [Google Scholar]
- 26.Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. JCTC. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 27.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 28.Schrödinger Release 2016-3. Glide, Schrödinger, LLC; New York, NY: 2017. [Google Scholar]
- 29.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Scalable algorithms for molecular dynamics simulations on commodity clusters; Proceedings of the 2006 ACM/IEEE conference on Supercomputing, ACM; Tampa, Florida. 2006. p. 84. [Google Scholar]
- 30.Schrödinger Release 2017-2: Desmond Molecular Dynamics System. D. E. Shaw Research; New York, NY: 2017. [Google Scholar]
- 31.Maestro-Desmond Interoperability Tools. Schrödinger; New York, NY: 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.