

Abstract
Rationale
The highly conserved NOTCH signaling pathway functions as a key cell-cell interaction mechanism controlling cell-fate and tissue patterning, while its dysregulation is implicated in a variety of developmental disorders and cancers. The pivotal role of endothelial NOTCH in regulation of angiogenesis is widely appreciated; however, little is known about what controls it signal transduction. Our previous study indicated the potential role of post-translational small ubiquitin-like modifier (SUMO) modification (SUMOylation) in vascular disorders.
Objective
To investigate the role of SUMOylation in endothelial NOTCH signaling and angiogenesis.
Methods and Results
Endothelial SENP1 deletion, in newly generated endothelial SENP1 (the major protease of the SUMO system) deficient mice, significantly delayed retinal vascularization by maintaining prolonged NOTCH1 signaling, as confirmed in cultured endothelial cells. An in vitro SUMOylation assay and immunoprecipitation revealed that when SENP1 associated with NOTCH1 intracellular domain (N1ICD) it functions as a deSUMOylase of N1ICD SUMOylation on conserved lysines. Immunoblot and immunoprecipitation analyses and dual luciferase assays of natural and SUMO-conjugated/nonconjugated NOTCH1 forms demonstrated that SUMO conjugation facilitated NOTCH1 cleavage. This released N1ICD from the membrane and stabilized it for translocation to the nucleus where it functions as a co-transcriptional factor. Functionally, SENP1-mediated NOTCH1 deSUMOylation was required for NOTCH signal activation in response to DLL4 stimulation. This in turn suppressed VEGF receptor signaling and angiogenesis, as evidenced by immunoblotted signaling molecules and in vitro angiogenesis assays.
Conclusions
These results establish reversible NOTCH1 SUMOylation as a regulatory mechanism in coordinating endothelial angiogenic signaling; SENP1 acts as a critical intrinsic mediator of this process. These findings may apply to NOTCH-regulated biological events in non-vascular tissues and provide a novel therapeutic strategy for vascular diseases and tumors.
Keywords: SUMOylation, SENP1, NOTCH1, angiogenesis, endothelial cell growth, signaling pathways, receptor, developmental biology
Subject Terms: Angiogenesis, Basic Science Research, Cell Signaling/Signal Transduction, Developmental Biology, Vascular Biology
INTRODUCTION
Vascular network expansion is fundamental to embryonic development, tissue growth, and wound healing. New blood vessels are generated from the preexisting ones by a series of successive steps, including endothelial sprouting and tube formation. Typically, these processes are regulated by angiogenic factors, such as vascular endothelial growth factors,1–3 fibroblast growth factors,4, 5 and platelet derived growth factors.6 In particular, VEGF-A induces tip cell formation leading to vascular ingrowth into avascular areas. Endothelial sprouting is controlled by a feedback signaling loop during vascular network formation.7–9 Tip cells expressing Delta-like 4 (DLL4) activate NOTCH1 in adjoining cells to suppress the tip cell phenotype, thus committing them to stalk cell specification. Once bound to DLL4, activated NOTCH1 is cleaved by proteolytic enzymes within the membrane, thereby releasing its intracellular domain (N1ICD). The latter translocates into the nucleus to combine with co-activators and finally drives specific target genes. Activation of the endothelial NOTCH1 pathway represses expression of VEGF receptors and inhibits VEGF signaling, which leads to reduced endothelial cell (EC) angiogenic capability.10, 11 The critical role of endothelial NOTCH1 in coordinating angiogenesis has been widely appreciated, but the regulatory and adaptive mechanisms of its signaling conduction are largely unclear.
Several studies have highlighted the importance of post-translational modifications on NOTCH functions. Glycosylation of NOTCH receptors, which is modulated by glycosyltransferases–Fringe family, regulates the tip-stalk cell selection by promoting the interaction between Delta ligands and the NOTCH receptor; thus, decreasing Jagged-NOTCH affinity.10, 12, 13 On the other hand, the C-terminal region of N1ICD protein is the target of ubiquitin E3 ligase, FBXW7. It is strongly modified by ubiquitination, which disables NICD protein stability through hydrolysis.14–16 A recent study showed that SIRT1-mediated N1ICD acetylation significantly promotes its stabilization and enhances the activation of NOTCH1 signaling; this results in reduced vascular branching and density.17
Post-translational SUMOylation, like ubiquitination, is covalently attached to substrate proteins via an isopeptide bond between its C-terminal glycine and a lysine residue in the substrate proteins.18 A consensus SUMO acceptor site, consisting of the sequence ψKXE (ψ represents the hydrophobic amino acid and K is the SUMO conjugation site), has been identified.19, 20 SUMOylation is considered to be an essential process that controls gene expression, protein stability, chromatin structure, signal transduction, and maintenance of the genome.17–22 Particularly, SUMOylation is a highly dynamic process attributed to a SUMO-specific proteases family, sentrin-specific protease (SENP), which reversely detaches SUMO molecules from the substrate proteins. Six members of the SENP family (SENP1, 2, 3, 5, 6, and 7) have been identified in mammals. Different members of these SUMO-specific proteases appear to localize in different cellular compartments where they regulate protein functions by altering protein stability, cellular localization, and protein–protein interactions.23–26
To date, most SUMOylation substrates are categorized as nuclear and peri-nuclear proteins. SUMOylation targeting of membrane proteins is indicated; however, a limited number of cell membrane substrates have been identified. On the other hand, recent studies suggest a potential involvement of SUMOylation in the pathogenesis of cardiovascular disease in which vascular cells could be the objects of modulation. Our previous study implies engagement of SENP1-regulated SUMOylation in vasculature formation during embryonic development (Fig. 2A, ref.26) and severe anemia as the major cause of death in SENP1 global knockout mice.26 Therefore, we considered the possibility that SUMO regulates endothelial receptors, the pivotal effectors of vascular extension.
Figure 2. Loss of SENP1 function enhances endothelial NOTCH1 responses.
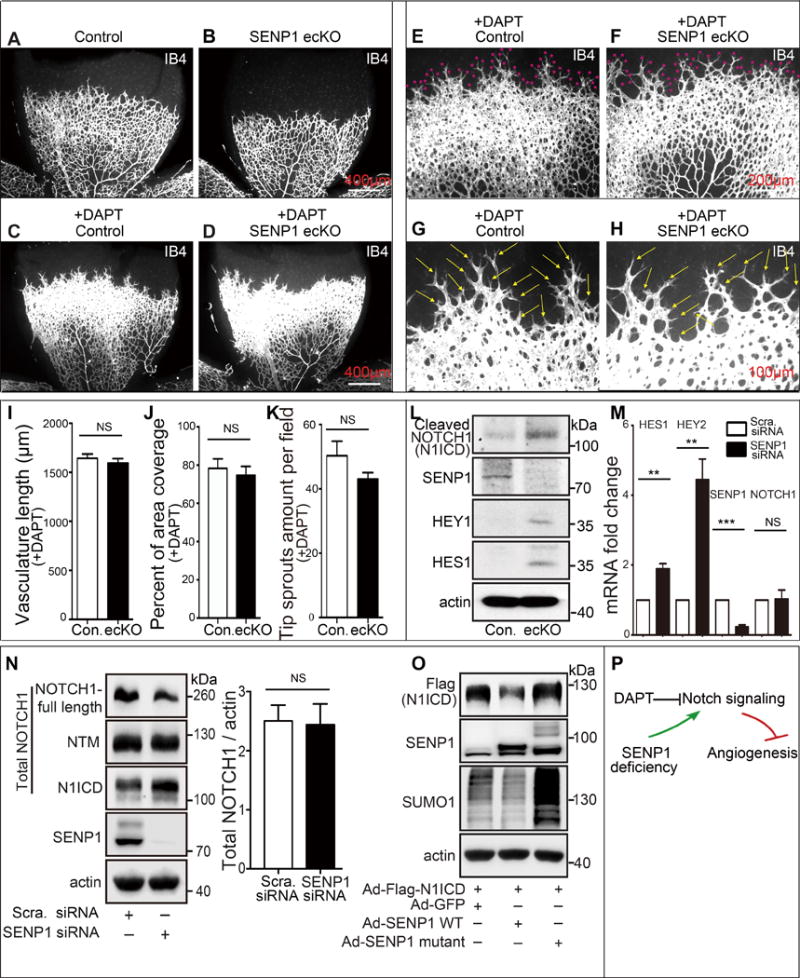
(A–K) NOTCH response inhibitor, DAPT, completely rescues defects in the retinal vasculature of SENP1-ecKO pups. (A, B) Isolectin B4 (IB4) stained vascular plexus in P6 retina whole mounts from control or SENP1-ecKO mice without DAPT treatment (n=7 and n=8 respectively) or (C–H) with daily injection of DAPT (100 mg kg−1) from the P4 stage (n=9 and n=7 respectively). Red asterisks indicate the tip sprouts of vessels and yellow arrows indicate tip cells. (I–K) Statistical summary of vascular parameters in retinas from each genotype. (L–O) SENP1 limits endothelial NOTCH signaling in vitro and in vivo. (L) Protein expression of NOTCH signaling in MLEC isolated from control or SENP1-ecKO mice, and (M) endothelial NOTCH signaling was determined by measuring the mRNA levels of NOTCH1 intracellular domain (N1ICD) and NOTCH1 target genes (HES1 and HEY2), and SENP1 in control (scramble) or SENP1-siRNA-transfected HMVEC cells. (M–N) SENP1 deficiency does not affect endothelial NOTCH1 expression on mRNA (M) and protein levels (N). (O) N1ICD stability was tested in HUVEC bearing SENP1 wild-type or catalytic inactive mutant. (P) A model for a critical role of SENP1 in regulation of endothelial NOTCH1 signaling and NOTCH1-dependent angiogenesis.
In the present study, we identified SENP1-regulated SUMOylation as a novel post-translational modification to NOTCH1, which plays a critical role in coordinating developmental angiogenesis. Mechanistically, NOTCH1 SUMOylation facilitates NOTCH1 cleavage and stabilizes N1ICD to function as a co-transcriptional factor in EC, which consequently promotes NOTCH signaling.
METHODS
A detailed description of methods is provided in the Online Data Supplement and includes information on generating endothelial SENP1 deficient mouse, retina dissection and whole mount staining, endothelial cell culture, transfection and viral infection, immunoprecipitation and immunoblotting, in vitro SUMOylation assay, protein stability assay, quantitative real time PCR, dual luciferase assay, in vitro endothelial cell migration and capillary-like structure formation assays, and statistical analyses.
RESULTS
Endothelial SENP1 deficiency compromises neonatal retina angiogenesis
To investigate the involvement of SENP1 in regulation of angiogenesis, we generated mice with an endothelial cell-specific SENP1 deficiency (SENP1-ecKO) as described in supplementary data. These mice were found to be viable and fertile. Postnatal retina angiogenesis was assessed in P4 and P7 pups. SENP1 expression in control mice was readily evident throughout the retinal endothelium, but was highly expressed in the angiogenic front of the vasculature (Fig. 1A). A similar intracellular expression pattern of SENP1 was also observed in HMVEC (Fig. 1B); this suggested a role for endothelial SENP1 in vascular sprouting. In contrast, SENP1 expression was profoundly reduced in the SENP1-ecKO retina endothelium (Online Figure I A, B). However, SENP1-ecKO mice demonstrated a delayed expansion of the vascular plexus to the periphery, as compared to controls; this was evidenced by a decrease in vascular branching and reduced vessel coverage at P4 (Online Figure I C–L, M, N) and P7 (Fig. 1C–J, M, N). Further analysis revealed that the tip cell number, tip sprouts and vascular length were significantly reduced in the sprouting region in SENP1-ecKO (Fig. 1O, P, Q; Online Figure I O, P). Pulse BrdU labeling did not show any difference in the proliferation rate of retinal endothelial cells between control and SENP1-ecKO mice (Fig. 1K, L, S). The inhibitory effect of SENP1 deficiency on ECs was further confirmed in cultured primary HUVEC or HMVEC; SENP1 knockdown resulted in diminished spheroid sprouting (Online Figure II A left panels, B bar 1 and 3), migration (Online Figure II C–F), and capillary-like structure formation (Online Figure II H, I).
Figure 1. Endothelial SENP1 deficiency significantly delays neonatal retinal angiogenesis.
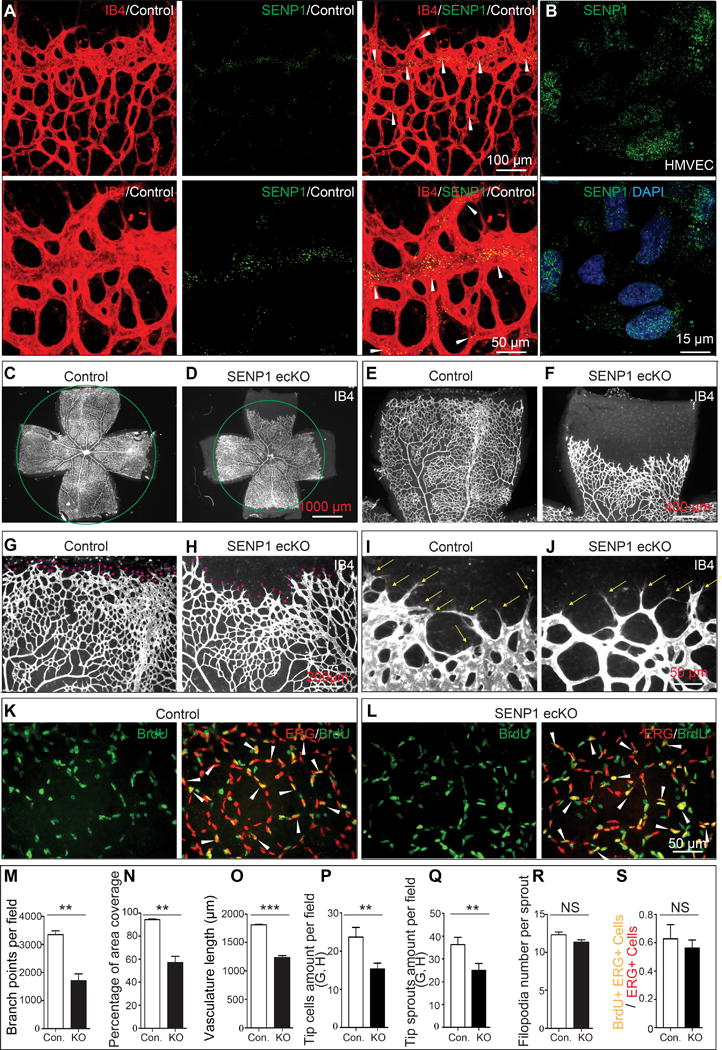
(A) SENP1 expression in neonatal retina endothelium. Whole mount retinas from P5 control or SENP1-ecKO pups were probed for isolectin B4 (IB4, red) and SENP1 (green). (A, upper panel) Confocal images of SENP1 expression in neonatal retina endothelium. Robust SENP1 expression is detected at the angiogenic front in retinal endothelium of control mice. (A, lower panel) Higher magnification of the inset in A, upper panel. The arrowheads indicate abundant SENP1 expression at the angiogenic front. (B) SENP1 expression in cultured primary human microvascular endothelial cell (HMVEC). Confocal images of SENP1 expression (green) is extensively detected in the cultured HMVECs. (C–J) Defects in the P7 SENP1-ecKO retinal vasculature. Representative images of P7 vascular outgrowth in control (n=6) and SENP1-ecKO retinas (n=5) stained with IB4. Red asterisks indicate the tip sprouts of vessels and yellow arrows indicate tip cells. (K, L) ERG (endothelial nuclei marker) stained (red) and BrdU labeled (green) whole mount control (n=5) and SENP1-ecKO (n=4) P7 retinas. Arrows indicate BrdU-ERG double-positive nuclei of the endothelial cells with high proliferation capability. (M–S) Statistical summary of vascular parameters of retinas and the number of BrdU-ERG double positive endothelial nuclei in definitive retinal endothelial cells (ERG positive) in control and SENP1-ecKO mice. Tip cells are defined as leading endothelial cells located at the frontage of the retinal endothelium and are characterized with sprouts that have numerous filopodia at the tip. Statistical significance was determined in an unpaired t test; Error bars represent mean ± s.e.m where *, p≤0.05; **, p≤0.01; ***, p≤0.001; NS, not significant.
Inactivation of SENP1 enhances endothelial NOTCH1 responses
The above findings suggested activation of endothelial Notch1 signaling. Control and SENP1-ecKO pups were treated with DAPT, a γ-secretase inhibitor, to test if increased Notch activity was responsible for the observed phenotype. DAPT treatment resulted in a large increase in vessel density and expansion of the vascular plexus in the postnatal retina of control mice (Fig. 2A, C, E, G). DAPT completely rescued the defect in SENP1-ecKO pups (Fig. 2B, D, F, H), which resulted in vascular length, vascular coverage, and tip sprouts growth comparable to the control group (Fig. 2I–K). The rescue effect of DAPT on SENP1-deficiency-induced angiogenesis defect was further confirmed in an in vitro spheroid-based angiogenesis assay (Online Figure II A bottom panels, B bar 3 and 4).
The potential link between SENP1 and NOTCH1 signaling in ECs was further explored by examining the protein level of cleaved NOTCH1 and its main downstream effectors, HES1 and HEY1, in MLEC isolated from WT and SENP1-ecKO mice. There was a pronounced increase in the amount of cleaved NOTCH1 N1ICD and expression of HES1 and HEY1 in SENP1 deficient MLEC compared to control cells (Fig. 2L).
The role of SENP1 in NOTCH1 signaling was also examined in HMVEC. siRNA interference of SENP1 in HMVEC resulted in up-regulation of NOTCH1 target genes, while the level of NOTCH1 itself was not affected (Fig. 2M). This was confirmed by immunoblot analysis of protein levels of all the NOTCH1 forms (full-length, NTM and N1ICD) and quantification of total NOTCH1 (Fig. 2N). Furthermore, upregulation of N1ICD in HUVEC was observed in the presence of a SENP1 catalytic inactive mutant (SENP1-mutant) (Fig. 2O). Taken together, these results pointed to a strong link between SENP1 and NOTCH1 during angiogenesis (Fig. 2P).
SENP1 regulates NOTCH1 SUMOylation
The hyperactivation of NOTCH signaling was induced by either deleting endothelial SENP1 or altering its catalytic function in vivo and in vitro. SENP1 is an endopeptidase that deconjugates SUMOs from substrate proteins. Therefore, we reasoned that SUMOylation may directly regulate NOTCH1 and its signaling activation in ECs. This was tested by analyzing the amino acid sequence of NOTCH1 protein using computational-system based software. Several classic SUMO binding motifs were predicted in the C-terminal N1ICD region (Online Figure III A). A truncated N1ICD fragment, containing putative SUMO binding motifs, was used as a potential substrate in an in vitro SUMOylation assay in the presence of SUMO E1 (AOS1/UBA2), SUMO E2 (UBC9), and ATP. SUMOylated bands were observed, which indicated a SUMO modification of NOTCH1 (Online Figure III C–E).
Next, we examined whether NOTCH1 is indeed SUMOylated in ECs. N1ICD was the predominant SUMOylated form of NOTCH1 in HMVEC (Fig. 3A), although some amount of SUMOylation was also detected on full length NOTCH1 (Online Figure IV A). The occurrence of the endogenous modification was further confirmed by overexpressing SUMO1 and N1ICD constructs in 293 cells (Fig. 3B). These results corresponded to the computational prediction, which indicated the N1ICD region as the acceptor of SUMO modification. The predicted bioinformatical analysis (Online Figure III B) prompted our further investigation of putative SUMO binding sites in N1ICD, which are evolutionarily conserved among vertebrates. Accordingly, we generated single mutants bearing lysine (K)-to-arginine (R) substitutions at three putative N1ICD SUMOylation sites K2049, K2150, K2252, which we refer to as N1ICD K2049R, K2150R, and K2252R. Additionally, we generated a construct containing the triple mutation N1ICD 3KR. The K2049R, K2150R, and K2252R substitutions significantly diminished SUMOylation of N1ICD when co-expressed with SUMO1. This was further reduced in cells expressing N1ICD 3KR, thus, indicating that K2049, K2150, and K2252 are the major SUMO biding sites on NOTCH1 (Fig. 3C).
Figure 3. SENP1 regulates the SUMOylation of NOTCH1.
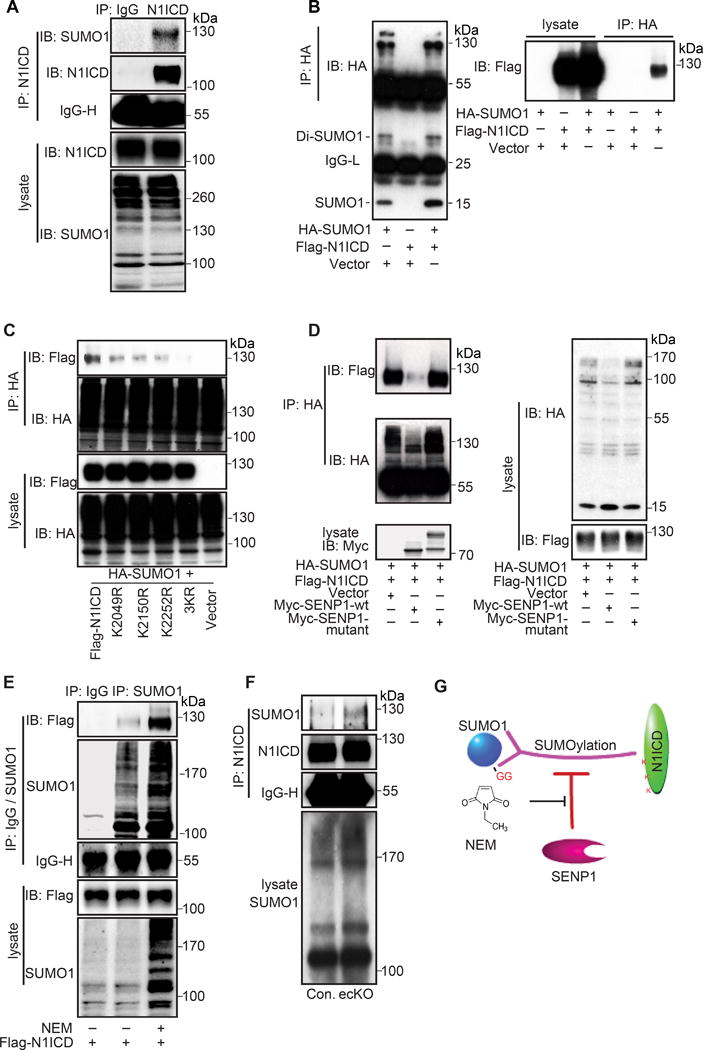
(A) Endogenous SUMOylation of cleaved NOTCH1 (N1ICD) in HMVEC. Immunoprecipitated (IP) cleaved NOTCH1 was immunoblotted (IB) for SUMO1. (B) Exogenous SUMOylation of N1ICD in 293T cells co-transfected with HA-SUMO1 and Flag-N1ICD. SUMO1 (HA) was IP followed by IB for SUMO1 (HA) and N1ICD (Flag). (C) Identification of SUMOylation sites in N1ICD in 293T cells transfected with mutants bearing single lysine (K) to arginine (R) substitutions at 3 putative SUMOylation sites or triple mutations (N1ICD 3KR). SUMO1 (HA) was IP followed by IB for N1ICD (Flag). (D–F) SENP1 deSUMOylates NOTCH1 SUMOylation. (D) N1ICD SUMOylation in 293T cells co-transfected with Flag-N1ICD plus SUMO1, SUMO1 and SENP1-WT or SUMO1 and SENP1-mutant (a catalytic inactive form). SUMO1 (HA) was IP followed by IB for SUMO1 (HA) and N1ICD (Flag). (E) Exogenous N1ICD SUMOylation in 293T cells treated with SENP activity inhibitor NEM (20mM). SUMO1 was IP followed by IB for N1ICD (Flag). (F) Endogenous N1ICD SUMOylation in MLEC isolated from control or SENP1-ecKO mice. Cleaved NOTCH1 (N1ICD) was IP followed by IB for SUMO1. (G) Model for SENP1-mediated endothelial NOTCH1 SUMOylation.
N1ICD SUMOylation in MLEC from control and SENP1-ecKO mice was examined to determine if SENP1 functions as a NOTCH1 deSUMOylase in ECs. Intriguingly, the amount of SUMOylated N1ICD was significantly increased in SENP1-deficient ECs (Fig. 3F). Also, N1ICD was co-expressed with SENP1 WT or a catalytic inactive mutant (SENP1-mutant) in an exogenous system; N1ICD SUMOylation was clearly detected in the presence of SUMO1. Importantly, SUMOylated N1ICD was robustly attenuated by SENP1 WT, while SENP1-mutant increased the amount of SUMOylated N1ICD (Fig. 3D). Moreover, we observed that N-ethylmaleimide, a SENP inhibitor that blocks SENP activity by modifying the active-site cysteine, intensely promoted N1ICD SUMOylation (Fig. 3E). SENP1, SENP2, and SENP5 were reported as major functional deSUMOylases in mammalian cells.24,25 Co-immunoprecipitation experiments revealed a complex formed between N1ICD and SENP1; furthermore, N1ICD predominantly associated with SENP1, but only weakly with other SENPs (Online Figure IV B, C). These results suggested that SENP1 specifically mediates NOTCH1 deSUMOylation in ECs by deconjugating SUMO modification of N1ICD (Fig. 3G).
Furthermore, we investigated the status of NOTCH1 SUMOylation under hypoxia, a circumstance that is typically a critical inductive factor for both physiological and pathological angiogenesis. To this end, HUVEC cells were cultured in regular normoxia or hypoxia condition for 24 hours. Cell lysates were then subjected to immunoprecipitation with anti-N1ICD followed by immunoblotting with anti-N1ICD or anti-SUMO1. Hypoxia treatment significantly increased HIF1α expression level and nuclear accumulation, but decreased the SUMO conjugation on N1ICD (Online Figure V A), as well as the expression of HES1 and HEY1 (Online Figure V A). These data indicated a suppressive effect of hypoxia on NOTCH1 SUMOylation and signaling.
SENP1-regulated NOTCH1 SUMOylation facilitates its cleavage
The interaction between SENP1 and NOTCH1 suggested that manipulation of SENP1 expression level or activity can affect NOTCH1 activity. Notably, SENP1 directly interacted with full length NOTCH1, the cell membrane receptor form, in EC (Online Figure IV D). Proteolytic cleavage of NOTCH1 is the key event involved in activation of its signaling cascade that leads to release of N1ICD. The Dll4-NOTCH1 binding leads to proteolytic cleavage of full-length NOTCH1 into the NOTCH1 transmembrane subunit form (NTM, the intermediate cleaved form composed of transmembrane and intracellular domains) and NOTCH1 extracellular domain form (N1ECD); NTM is further cleaved into N1ICD, while N1ECD is removed by bound Dll4 (Fig. 4A). Therefore, we tested whether SENP1-regulated NOTCH1 SUMOylation affects its cleavage. To this end, NOTCH1 and its cleaved forms were examined in HMVEC following SENP1 knockdown. In control ECs, NOTCH1 was mostly detected as full length and the NTM subunit; N1ICD was detected to a lesser extent (Fig. 4B, lane 1). The amounts of full length and NTM were somewhat reduced in the absence of SENP1, while N1ICD was increased (Fig. 4B, lane 2). This was accompanied by elevated HES1 expression, which demonstrated increased NOTCH signaling output (Fig. 4B, lane 2); these data were consistent with the results in mouse retinal endothelium and MLEC. A similar role for SENP1 was observed in Dll4-mediated NOTCH1 cleavage, which resulted in an even higher level of N1ICD (Fig. 4B, lanes 3 and 4). Moreover, SENP1 WT, but not high levels of SENP1 mutant (a catalytic inactive form that smothers the deSUMOylase activity), reversed the extensive N1ICD cleavage induced by SENP1 deficiency (Fig. 4C). This suggested that SENP1-regulated SUMOylation may facilitate NOTCH1 cleavage. Indeed, SUMO1 knockdown significantly inhibited NOTCH1 cleavage and activation, which was completely opposite of the inactive SENP1 effect (Fig. 4D). These data strongly support the hypothesis that SENP1-regulated SUMOylation facilitates NOTCH1 cleavage (Fig. 4G). Furthermore, we examined the regulatory role of Dll4 in NOTCH1 SUMOylation in Dll4-treated HMVECs. Immunoprecipitation assays revealed that Dll4 induced increased NTM SUMOylation, which consequently promoted cleavage into N1ICD (Fig. 4E). A similar enhancement of N1ICD SUMOylation was also detected in the assay using a comparable level of N1ICD input adjusted for better discernment (Fig. 4F). Conversely, decreased N1ICD SUMOylation was exhibited by immunoprecipitation in the presence of soluble Dll4 (sDll4), thus blocking the binding of NOTCH receptor with Dll4 (Online Figure V B). Taken together, we conclude that Dll4 positively regulates N1ICD SUMOylation for NOTCH1 cleavage and signaling in endothelial cells.
Figure 4. SENP1 mediated NOTCH1 SUMOylation facilitates cleavage of NOTCH1 receptor.
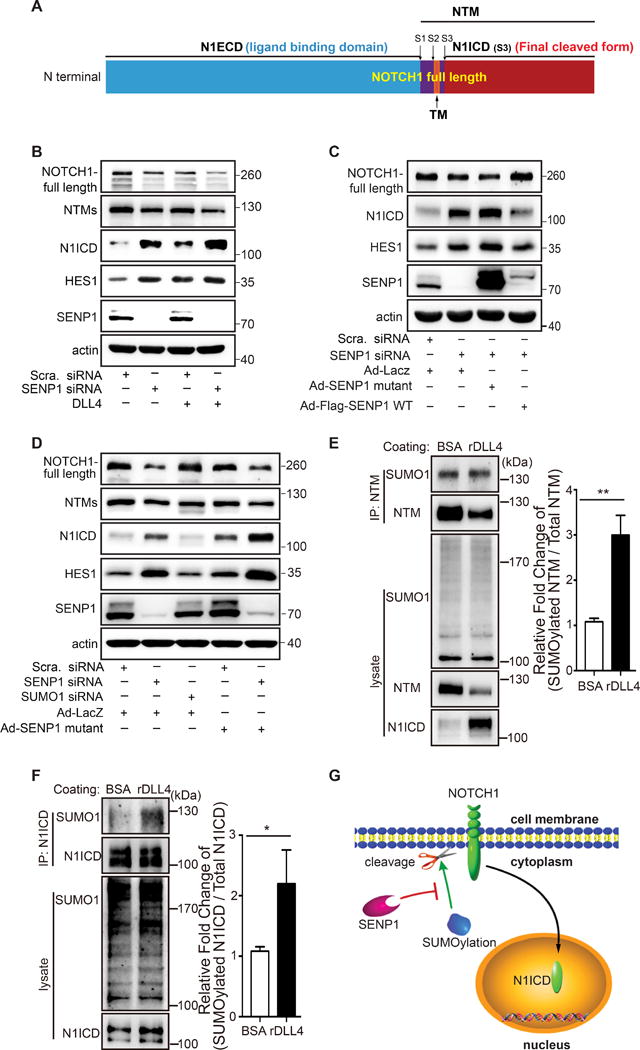
(A) Mapped forms of NOTCH1 protein: N1ECD, the extracellular domain of NOTCH1 (ligand binding domain), NTM, NOTCH1 protein containing the transmembrane region (the intermediate form composed of transmembrane and intracellular domains), N1ICD, intracellular domain of NOTCH1 protein (final cleaved form), S1-3, endoproteolytic cleavage sites. (B) Immunoblot of full length NOTCH1, NOTCH1 transmembrane subunit (NTM, the intermediate cleaved form composed of transmembrane and intracellular domains), cleaved N1ICD, and HES1 in control (scrambled) or SENP1 siRNA transfected HMVEC cells. Note that siRNA knockdown of SENP1 enhanced Dll4 stimulated NOTCH1 cleavage. (C) Immunoblot of full length NOTCH1, N1ICD, and HES1 in control (scrambled) or SENP1-siRNA-transfected HMVEC cells with SENP1-WT or SENP1-mutant overexpression. Note that SENP1-knockdown enhanced NOTCH1 cleavage was rescued by SENP1-WT, but not SENP1 mutant (a catalytic inactive form). (D) Immunoblots containing cell lysate proteins from HMVEC, HMVEC transfected with SUMO1 siRNA or SENP1 siRNA with or without SENP1-mutant overexpression were probed for full length NOTCH1, NTM, cleaved N1ICD, and HES1. Note that NOTCH1 cleavage was inhibited by SUMO1 siRNA, but enhanced by SENP1 siRNA and further enhanced by SENP1-mutant. (E–F) SUMOylation of NTM and N1ICD in response to DLL4 in HMVEC. (E) Immunoprecipitated (IP) NTM was immunoblotted (IB) for SUMO1 and NTM with or without recombinant DLL4 (rDLL4) treatment in HMVEC. Fold change of SUMOylated NTM in total NTM was quantified. (F) Immunoprecipitated (IP) N1ICD was immunoblotted (IB) for SUMO1 and N1ICD with or without rDLL4 treatment in HMVEC. The fold change of SUMOylated N1ICD in total N1ICD was quantified. Note that both SUMOylation of NTM and N1ICD were significantly enhanced by Dll4 treatment. (G) Model depicting a critical role of SENP1-mediated SUMOylation in regulating endothelial NOTCH1 cleavage.
SENP1-regulated SUMOylation enhances transcriptional activity and stability of N1ICD
Next, we considered that enhanced NOTCH1 signaling in SENP1 deficient EC may also be due to SUMO modification on N1ICD in the cytoplasm. SUMOylation has multiple effects on its substrates, including intracellular localization, protein stability, and protein activity; this is unlike ubiquitination, which usually induces degradation of target proteins. The biological effect of SUMOylation on N1ICD was examined by generating a SUMO1-N1ICD fusion protein. We used a SUMO fusion strategy described in our previous study,26 which mimics constant N1ICD SUMOylation (Online Figure VI A, B). N1ICD, with or without SUMO fusion, and SUMOylation defective mutant N1ICD-3KR showed a similar pattern of specific localization to the nucleus, as detected by immunofluorescence (Online Figure VI C). On the other hand, N1ICD entered the nucleus and activated downstream signaling by forming a complex with the transcriptional factor RBP-J. SUMO conjugation had little effect on intracellular localization of N1ICD, but it significantly promoted the binding of N1ICD to RBP-J, as demonstrated by co-immunoprecipitation (Fig. 5A, B). In contrast, N1ICD-3KR had a weaker involvement in the RBP-J complex (Fig. 5C, D).
Figure 5. SENP1 mediated N1ICD SUMOylation reinforces N1ICD co-transcriptional activity and protein stability.
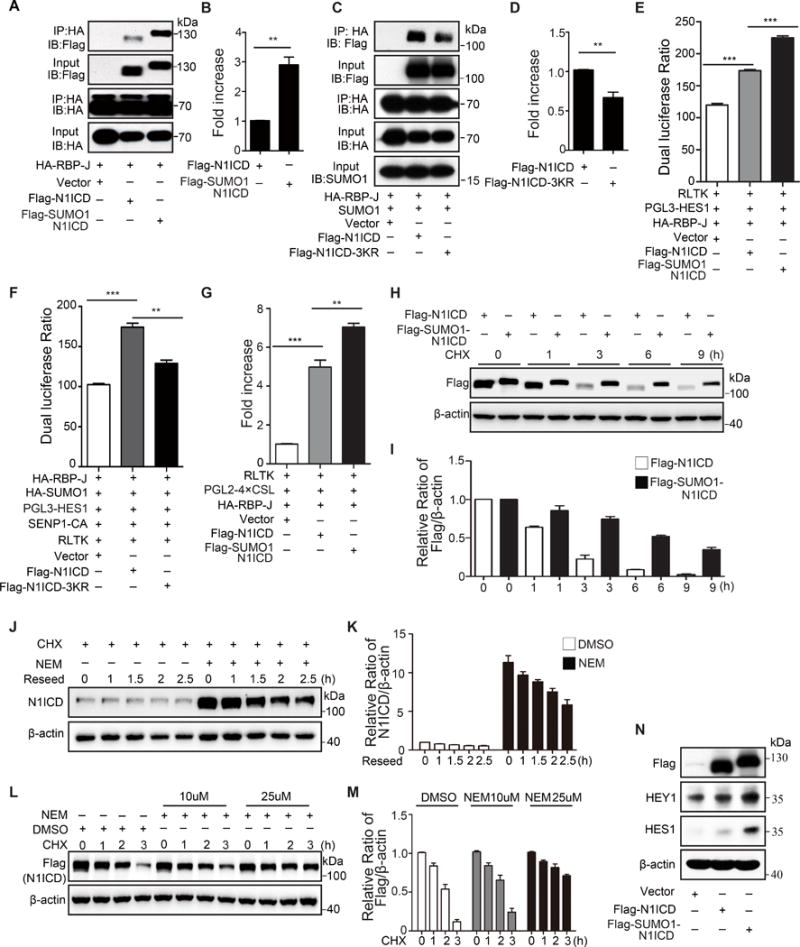
(A, B) RBP-J binding association with N1ICD or SUMO1-N1ICD (fusion protein mimics constant N1ICD SUMOylation) in transfected 293T cells. RBP-J (HA) was immunoprecipitated (IP) from the cell lysate followed by immunoblotting (IB) for N1ICD (Flag), SUMO-N1ICD (Flag), and RBP-J (HA). (A) Representative image of co-immunoprecipitated proteins in the complex and (B) quantification of the IP protein (n=3, biologically independent experiments; unpaired t test; bars are mean ± s.e.m.). (C, D) RBP-J association with N1ICD or N1ICD-3KR mutant in transfected 293T cells with SUMO1 overexpression. RBP-J (HA) was IP then IB for N1ICD (Flag), N1ICD-3KR (Flag), and RBP-J (HA). (C) Representative image of co-immunoprecipitation. (D) Quantification of the IP proteins. (E, F) HES1 and (G) CSL luciferase activity in 293T cells transfected with Flag-N1ICD, Flag-SUMO1-N1ICD or Ad-N1ICD-3KR. (H-M) SENP1-mediated SUMOylation preserves the N1ICD stability. (H, I) Measurement of N1ICD or SUMO1-N1ICD protein stability in transfected 293T cells using a cycloheximide (CHX) half-life assay; (J, K) endogenous N1ICD stability in HUVEC with or without NEM treatment; and (L, M) exogenous N1ICD stability in 293T cells with or without NEM treatment. (N) IB of NOTCH target genes, HES1 and HEY1, in HUVEC infected with Ad-N1ICD or Ad-SUMO1-N1ICD. The data represent 3 biologically independent experiments. Statistical significance was calculated in an unpaired t test; Bars represent mean ± s.e.m. where *, p≤0.05; **, p≤0.01; ***, p≤0.001.
The effect of SUMOylation on NOTCH1 co-transcriptional activity was further examined in a dual-luciferase assay. As expected, SUMO-N1ICD strengthened while N1ICD-3KR diminished RBP-J binding to the HES1 promoter, the classic downstream gene driven by NOTCH1 signaling in EC (Fig. 5E, F). A trend was confirmed by a similar finding, which showed that SUMOylated-N1ICD promoted transcriptional binding of RBP-J to its general binding motif (Fig. 5G). Furthermore, SUMO conjugation to N1ICD substantially enhanced the amplitude and duration of the N1ICD half-life in the presence of protein synthesis inhibitor cycloheximide (CHX) (Fig. 5H, I). Importantly, treatment with the SENP inhibitor, N-ethylmaleimide, significantly improved the stability of endogenous N1ICD in HMVEC (Fig. 5J, K) and overexpressed exogenous N1ICD (Fig. 5L, M).
Concomitantly, expression of NOTCH1 target genes was extensively elevated in Ad-SUMO1-N1ICD infected HUVECs compared to the moderate increase induced by N1ICD overexpression (Fig. 5N). Together, these data revealed that SENP1-regulated N1ICD SUMOylation positively modulates its co-transcriptional activity and protein stability, but not intracellular localization.
NOTCH1 SUMOylation negatively regulates angiogenic signaling in endothelial cells
VEGFR signaling is the dominant driver of endothelial sprouting. Its impact on tip/stalk cell balance is mediated via activation of NOTCH signaling. We examined angiogenic signaling in ECs to define the biological function of SENP1-regulated NOTCH1 SUMOylation in angiogenesis. SENP1 knockdown in HUVEC resulted in a significant decrease in VEGFR2 phosphorylation and an increase in VEGFR1 expression level; both were further promoted by Dll4 treatment (Fig. 6A). The SENP1 deficiency-induced VEGFR2 phosphorylation blockage was confirmed in SENP1-ecKO retinal endothelium compared with control mice (Fig. 6B). On the contrary, SUMO1 knockdown reversed the effect of SENP1 deficiency on VEGFR2 activation and VEGFR1 expression (Fig. 6C). We further observed that SUMO1 augmented N1ICD regulated VEGFR1 expression (Fig. 6D, E), which was attributed to N1ICD SUMO conjugation (Fig. 6F, G).
Figure 6. NOTCH1 SUMOylation suppresses VEGFR signaling in endothelial cells.
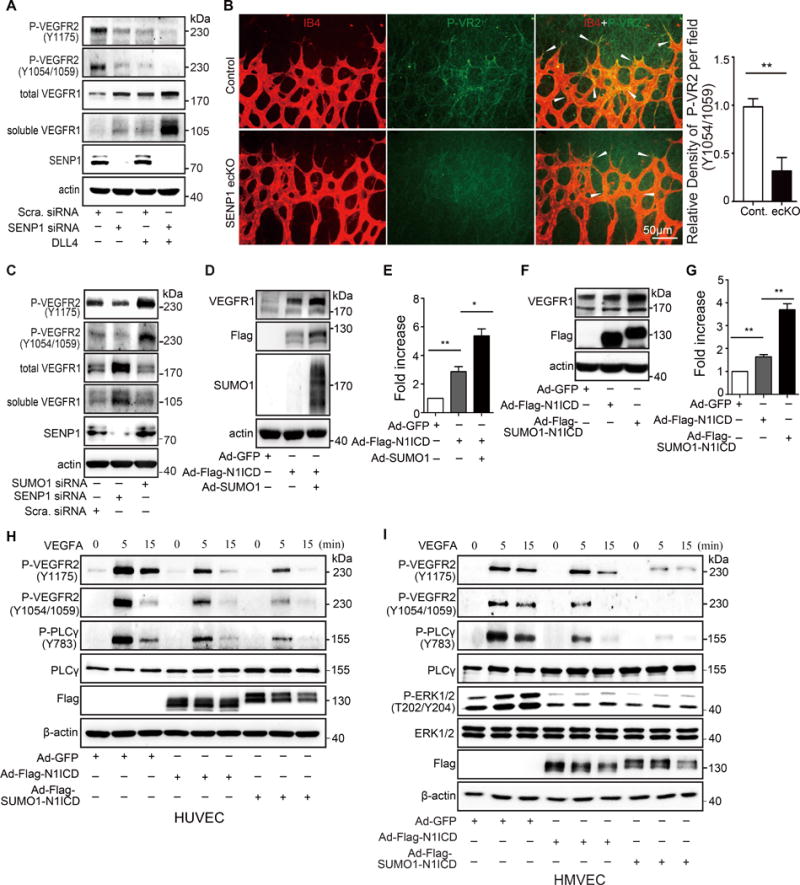
(A) Immunoblot (IB) of phosphorylated VEGFR2 (Y1175, Y1054/1059), total VEGFR1, soluble VEGFR1 in HUVEC with or without SENP1 siRNA transfection and with or without Dll4 treatment. Note that SENP1 knockdown in HUVEC decreases VEGFR2 phosphorylation and increases total or soluble VEGFR1, which reinforces negative regulation of Dll4-NOTCH activation. (B) Phosphorylated VEGFR2 (P-VR2, Y1054/1059, green) expression and isolectin B4 (IB4, red) were visualized by immunofluorescence in the retinal endothelium in whole-mount retinas from P5 control (n=6) or SENP1-ecKO (n=6) mice. Arrow heads indicate endothelial P-VR2. Representative images are shown with quantification. (C) IB of phosphorylated VEGFR2 (Y1175, Y1054/1059), total VEGFR1, soluble VEGFR1 in HUVEC with or without SENP1 or SUMO1 siRNA transfection. Note that the effect of siRNA-induced SUMO1 knockdown had the opposite effect on VEGFR2 phosphorylation and total or soluble VEGFR1 level in HUVEC cells compared to siRNA-induced SENP1 knockdown. (D–G) N1ICD SUMOylation promotes VEGFR1 expression level in HUVEC. (D) Representative image of IB of total VEGFR1 in HUVEC co-expressing N1ICD with or without SUMO1; (E) Quantification of D. (F) Representative IB of total VEGFR1 in HUVEC infected with Ad-N1ICD or Ad-SUMO1-N1ICD. (G) Quantification of F. (H, I) N1ICD SUMOylation blocks VEGF-A/VEGFR2 signaling in human endothelial cells. IB of phosphorylated VEGFR2 (Y1175, Y1054/1059), PLCγ, and ERK in Ad-GFP, Ad-N1ICD or Ad-SUMO1-N1ICD infected (H) HUVEC and (I) HMVEC. Statistical significance was analyzed in an unpaired t test of data from 3 biologically independent experiments. Bars represent mean ± s.e.m. where *, p≤0.05; **, p≤0.01.
We further examined the role of SUMOylated N1ICD in VEGFR signaling by transducing HUVECs and HMVEC with adenoviral vectors containing N1ICD (Ad-Flag-N1ICD) or Flag-SUMO1-N1ICD (Ad-Flag-SUMO1-N1ICD). VEGF-A treatment resulted in significant inhibition of VEGFR2 phosphorylation and its downstream signaling molecules in ECs overexpressing N1ICD. However, inhibition of VEGFR2 signaling was augmented in the SUMO1-N1ICD transgene group (Fig. 6H, I). A similar trend was observed in VEGF-C-induced VEGFR3 activation (Online Figure VII A), with a slight decrease of VEGFR3 expression in the presence of SUMO1-N1ICD in EC (Online Figure VII A) or SENP1 deficiency in retina endothelium (Online Figure VII B, C). Thus, SENP1-regulated NOTCH1 SUMOylation negatively modulates angiogenesis by upregulating VEGFR1 expression, thereby, suppressing VEGFR2 and VEGFR3 angiogenic signaling in ECs.
N1ICD and SUMO1-N1ICD were each expressed in HUVEC or HMVEC to verify the role of NOTCH1 SUMOylation in EC angiogenesis. Vessel sprouting (Online Figure VI D, E), migration (Online Figure VI F, G) and capillary-like structure formation capabilities (Online Figure VI H, I) were significantly diminished in EC expressing SUMO1-N1ICD compared to the N1ICD infected ECs. This further proved the negative regulation of NOTCH1 SUMOylation in endothelial angiogenic activity.
DISCUSSION
Endothelial tip cell-derived Dll4 binds to NOTCH receptor on adjacent stalk EC during sprouting angiogenesis, which tightly coordinates the growth and patterning of the vessel network.10, 11, 27 However, the complete regulatory mechanism of endothelial NOTCH activation remains unknown. This study revealed that SENP1, the major protease for post-translational SUMO modification, acts as an intrinsic positive modulator of endothelial NOTCH1 signaling in developmental angiogenesis. The diminished NOTCH1 activity and cellular growth of EC deficient in SENP1 or bearing SENP1 deSUMOylase inactive mutant confirmed the in vivo retina phenotype; thus, indicating the regulatory effect of SUMOylation on the NOTCH response in EC. Indeed, constitutive SUMOylation of endothelial NOTCH1 was identified both endogenously and exogenously, which was reversibly regulated by SENP1. Functionally, SENP1-regulated endothelial NOTCH1 SUMOyaltion is required for NOTCH1 cleavage at the cell membrane, maintenance of N1ICD protein stability, and co-transcriptional activity in the nucleus. Augmented NOTCH1 signaling, in turn, counteracts VEGF/VEGFR signaling in EC and consequent angiogenic growth (Fig. 7).
Figure 7. Model for a critical role of SENP1-mediated deSUMOylation in the regulation of endothelial NOTCH1 and angiogenesis.
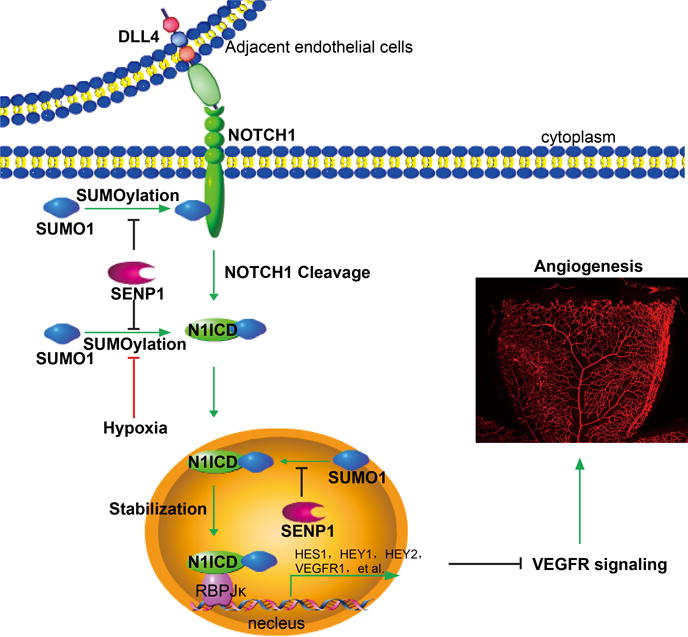
The endothelial cell Dll4-activated NOTCH signal pathway requires SUMOylation of N1ICD for NOTCH1 cleavage to occur, which releases N1ICD from the cell membrane, stabilizes N1ICD and initiates substantial co-transcriptional activity in the nucleus. SENP1 associates with N1ICD of NOTCH1 and deconjugates the SUMO modification as an intrinsic controller, whereas NOTCH1 SUMOylation facilitates dynamic NOTCH responses and subsequent angiogenic homeostasis regulated by hypoxia. In the case of endothelial SENP1 deficiency or functional mutation, augmented N1ICD SUMOylation leads to exaggerated NOTCH signaling, which in turn suppresses VEGFR signaling and endothelial angiogenesis.
Post-translational SUMOylation tightly regulates molecular properties of NOTCH1 in endothelial cell
NOTCH1 undergoes a number of post-translational modifications, including glycosylation,12, 13, 28 ubiquitination,16, 29 and acetylation.17 In the present study, we discovered that SUMOylation is a novel post-translational modification of the NOTCH receptor. NOTCH1 is normally modified in a SUMO-1-conjugation preferred manner throughout its cellular functions. Dll4 binding promotes SUMOylation upon which SENP1 selectively acts as the key deSUMOylation regulator. SUMOyaltion is not a typical post-translational modification. The biochemical effect of SUMO modification is still controversial, because SUMOyaltion of specific targets correlates with a plethora of altered protein properties. Our investigation reveals that SUMOylation does not disturb N1ICD localization in the nucleus. However, intranuclear SUMOylation directly participates in the binding of N1ICD to RBP-J, the dominant transcriptional factor for driving downstream molecules in the NOTCH signaling cascade and formation of a NOTCH active transcriptional complex. The SUMO modification-induced exposure or creation of a relevant interaction surface may provide an elegant explanation for the biochemical change. Accordingly, the SUMOylation-promoted N1ICD interaction may result in a complex choreography by facilitating the recruitment of other components in the same transcriptional complex in a context-dependent pattern.
SUMOylated N1ICD was found to have greater stability. On the other hand, the SUMOylation-deficient mutant displayed attenuated protein stability. SUMO-1 shares about 18% homology and a similar three-dimensional structure with ubiquitin. However, SUMOylation usually does not trigger proteolysis of a conjugated protein, but mediates an abundance of substrate via interplay with the ubiquitin-proteasome system.30 It is well established that ubiquitin-dependent proteasomal degradation mediates NOTCH processing and NICD accumulation,28 but evidence for an exact antagonism between SUMOylation and ubiquitination on NOTCH levels remains under investigation. Additionally, SUMOylation shares at least one common lysine residue (K2150) with acetylated NICD, which is based on the 3 major SUMOylation sites identified in our study.17 NICD acetylation and SUMOylation play similar roles in the maintenance of N1ICD stability and promotion of NOTCH signature signaling in spite of the fact that they are within different molecular frameworks. Therefore, it is worthwhile to determine the crosstalk between SUMOylation and acetylation of NOTCH in future studies.
Biological significance of SUMOylation on NOTCH response in angiogenesis
Transmembrane receptor proteins in ECs are a predominant part of the communication system for endothelial growth and homeostasis. Their molecular flexibility in response to angiogenic stimuli permits fast adaptation to the cellular microenvironment to promote angiogenesis. Protein SUMOylation predominantly occurs in nuclei. However, a number of cell membrane proteins were identified as SUMO modification substrates in a highly dynamic but specific pattern, which suggests that the SUMOylation system could be well suited to managing and fine-tuning the angiogenic ligand-receptor network.31, 32 Thus far, little is known about SUMOylation of endothelial receptor proteins and how it impacts their function. Our current study reveals the significant involvement of SUMOylation in endothelial NOTCH functions during angiogenesis. The underlying biological consequences apply to both NOTCH cleavage and activation of NOTCH signaling.
The ligand-bound NOTCH receptors require successive proteolytic cleavage across the cell membrane to trigger NOTCH signaling in ECs during angiogenesis. Importantly, cleavage of S2 by ADAM metalloproteinases and γ-secretase cleavage of S3 result in NICD release from the cell membrane, which activates intracellular NOTCH signaling.28, 33 The receptor processing delineates the framework for initiation of the NOTCH cascade, but the regulatory machinery for converting the molecular information into proper biological output is still unclear. Our study identified SENP1-regulated SUMOylation as an essential requirement for the NOTCH cleavage process in angiogenic ECs. The positive regulation of Dll4 in NOTCH SUMOylation indicates an intrinsic modulatory mechanism of Dll4-activated NOTCH cleavage and signaling. Ubiquitination, which targets N1ICD for degradation within recycling endosomes, contrasts with SUMO conjugation to the NOTCH C-terminal that promotes N1ICD turnover and ensures its stability. We further demonstrate that intracellular N1ICD SUMOylation enhances the intensity and duration of NOTCH target gene expression, which is a result of augmented assembly of the co-transcriptional complex. Thus, SUMOylation of NOTCH functions positively on the spatiotemporal regulation of the NOTCH responses for EC behavior. Moreover, the suppressive effect of hypoxia on NOTCH signaling and its required N1ICD SUMOylation might be new contribution to the regulatory mechanism for hypoxia-induced endothelial sprouting and angiogenesis. Consequently, our study provides an appealing approach for tailoring the linear transmission of NOTCH signaling to adjust ongoing EC growth and communication during angiogenesis. In view of the current finding that SENP1 associates with full or cleaved forms of NOTCH, we propose that SENP1 functions as an autonomous rheostat to reconcile NOTCH signaling scenarios in different spatial and temporal contexts in ECs. To our knowledge, our finding is the first demonstration of the critical role of the SUMO system in angiogenesis.
An understanding of the details of SUMOylation regulation of the dynamic fluctuations of endothelial NOTCH responses should be of great importance given the highly dynamic characteristic of the SUMOylation procedure. The initial focus could be on the effect of NOTCH SUMOylation on the γ-secretase complex composition and its subcellular localization. This event influences the rapid-turnover of NICD in response to NOTCH activation. On the other hand, we also observed that SENP1 mediates HES-1 deSUMOylation in endothelial cells (Online Figure VIII), based on a previous report.34 N1ICD SUMOylation occurs in the cytosol or on the membrane, while HES-1 SUMOylation may occur in the nucleus; therefore, the synergistic modulation of SENP1 in NOTCH signaling-controlled endothelial growth is worthy of investigation. Moreover, there is little knowledge about the cooperation of SUMOylation with other post-translational modifications, such as ubiquitination and phosphorylation, to manipulate the efficiency of NOTCH signaling.
Novel modulatory machinery of NOTCH-VEGFR signaling interaction
The NOTCH pathway has been identified as a prominent negative modulator of angiogenesis, which counteracts VEGFR signaling in ECs. However, the precise interacting mechanism is not completely understood. VEGF-stimulated VEGFR2 and VEGFR3 signaling potently promote angiogenesis and vascular development, while VEGFR1 negatively regulates signaling by trapping VEGF with high ligand affinity.2, 8, 10, 35, 36 Our study shows that over-SUMOylation of NOTCH leads to direct suppression of VEGFR2 and 3 activation in EC, which accounts for the dysfunction of EC growth and new vessel formation. A potential explanation is that it forms another link between endothelial VEGFR and NOTCH pathways, which would indicate the importance of crosstalk at a post-translational level. Comprehensively, the effect of NOTCH SUMOylation spans endothelial VEGFR1-3 by targeting VEGFR expression and its corresponding signal transduction. On the other hand, the positive regulation of Dll4 and negative regulation of hypoxia in NOTCH SUMOylation in ECs may indicate another interaction mode between Dll4-NOTCH signaling and hypoxia-VEGF/VEGFR signaling in coordination of angiogenesis. Therefore, we speculate a novel fine-tuned mechanism for the VEGFR-NOTCH balance in angiogenesis via SUMOylation or deSUMOylation of NOTCH. The direct effect of NOTCH signaling on VEGFR2 expression is via HEY1/HEY2/HEYL binding to the VEGFR2/VEGFR3 promoter.37 It will be interesting to investigate whether SENP1 synergistically regulates other NOTCH pathway components during different stages of angiogenesis.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The NOTCH pathway is the prominent negative regulator of endothelial sprouting and vascular growth.
Activation of NOTCH leads to its proteolytic cleavage, which releases its intracellular domain and activates transcription of downstream genes.
Post-translational modification of proteins by SUMOylation, regulatesgene expression, protein stability, chromatin structure, and signal transduction. It is reversed by the sentrin-specific protease (SENP).
What New Information Does This Article Contribute?
Endothelial SENP1 deficiency delays developmental angiogenesis in mice and elevates NOTCH signaling.
NOTCH1 SUMOyaltion is required for NOTCH1 cleavage at the cell membrane, maintenance of N1ICD protein stability, and co-transcriptional activity in the nucleus.
SENP1 functions as a critical intrinsic mediator of NOTCH1 SUMOylation to reconcile NOTCH signaling scenarios in endothelial cells.
NOTCH1 SUMOylation coordinates hypoxia-induced angiogenesis by facilitating NOTCH signaling which in turn suppresses VEGF receptor (VEGFR) signaling.
Angiogenesis is essential for embryonic development and tissue growth. NOTCH signaling plays a pivotal role in angiogenesis; however, the mechanisms that regulate NOTCH signaling are still obscure. Herein, we show that deletion of endothelial SENP1 (the major protease of the SUMO system) delays retinal vascularization by prolonging NOTCH1 signaling. Importantly, NOTCH1 SUMOylation is required for NOTCH signal activation to fine-tune endothelial growth and angiogenesis, in which SENP1 selectively functions as the key regulator of deSUMOylation. The current results reveal a novel mechanism for the VEGFR-NOTCH balance in angiogenesis via SUMOylation/deSUMOylation of NOTCH. They also indicate an additional mode of interaction between Dll4-NOTCH signaling and hypoxia-VEGF/VEGFR signaling in coordination of angiogenesis. These findings may be of significance in understanding other NOTCH-regulated biological events and could lead to the development of novel diagnostic and therapeutic strategies for vascular diseases and tumors.
Acknowledgments
We thank Dr. Adolfo Ferrando (Columbia University) for providing Notch1 full-length construct, Dr. R. Adams (Max Planck Institute, Munster) for Cdh5-CreERT2 mouse, Dr. Xuri Li (Sun Yat-sen University, China) for helpful discussion of the manuscript.
SOURCES OF FUNDING
The present study was supported by the National Natural Science foundation of China (grant Nos. 81422005, 31470057 and 81270357), the Zhejiang Provincial Natural Science foundation of China (grant no. LR14H020002), and the Fundamental Research Funds for the Central Universities to Luyang Yu. China Postdoctoral Science Foundation (grant No. 508000-X91402) to Xiaolong Zhu. National Natural Science foundation of China (grant No. 81600354) to Cong Qiu. This work was partly supported by National Key Research and Development Program of China (2016YFC1300600), National Natural Science Foundation of China (No.91539110) to WM, R01 HL109420 and R01 HL115148, and CT Stem Cell Innovation Award (Established Investigator Grant) 14-SC B-YALE-17 to WM.
Nonstandard Abbreviations and Acronyms
- Cre
Cre recombinase
- DLL4
Delta-like 4
- EC
endothelial cell
- HMVEC
human microvascular endothelial cell
- HUVEC
human umbilical vein endothelial cell
- MLEC
mouse lung endothelial cell
- N1ICD
NOTCH1 intracellular domain
- SENP
sentrin-specific protease
- SUMO
small ubiquitin-like modifier
- VEGF
vascular endothelial growth factor
- WT
wild type
Footnotes
DISCLOSURES
None.
References
- 1.Shubbar E, Vegfors J, Carlstrom M, Petersson S, Enerback C. Psoriasin (S100A7) increases the expression of ROS and VEGF and acts through RAGE to promote endothelial cell proliferation. Breast Cancer Res Treat. 2012;134:71–80. doi: 10.1007/s10549-011-1920-5. [DOI] [PubMed] [Google Scholar]
- 2.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nature reviews Molecular cell biology. 2006;7:359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, Simons M. Receptor tyrosine kinases endocytosis in endothelium: biology and signaling. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:1831–7. doi: 10.1161/ATVBAHA.114.303217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends in pharmacological sciences. 2001;22:201–7. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- 6.Cao R, Brakenhielm E, Pawliuk R, Wariaro D, Post MJ, Wahlberg E, Leboulch P, Cao Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nature medicine. 2003;9:604–13. doi: 10.1038/nm848. [DOI] [PubMed] [Google Scholar]
- 7.Siekmann AF, Covassin L, Lawson ND. Modulation of VEGF signalling output by the Notch pathway. BioEssays: news and reviews in molecular, cellular and developmental biology. 2008;30:303–13. doi: 10.1002/bies.20736. [DOI] [PubMed] [Google Scholar]
- 8.Holderfield MT, Hughes CC. Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circulation research. 2008;102:637–52. doi: 10.1161/CIRCRESAHA.107.167171. [DOI] [PubMed] [Google Scholar]
- 9.Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012;484:110–4. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- 10.Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–35. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 11.Thurston G, Noguera-Troise I, Yancopoulos GD. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nature reviews Cancer. 2007;7:327–31. doi: 10.1038/nrc2130. [DOI] [PubMed] [Google Scholar]
- 12.Bruckner K, Perez L, Clausen H, Cohen S. Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature. 2000;406:411–5. doi: 10.1038/35019075. [DOI] [PubMed] [Google Scholar]
- 13.Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, Vogt TF. Fringe is a glycosyltransferase that modifies Notch. Nature. 2000;406:369–75. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- 14.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–33. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, Look AT. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. The Journal of experimental medicine. 2007;204:1813–24. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsunematsu R, Nakayama K, Oike Y, Nishiyama M, Ishida N, Hatakeyama S, Bessho Y, Kageyama R, Suda T, Nakayama KI. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. The Journal of biological chemistry. 2004;279:9417–23. doi: 10.1074/jbc.M312337200. [DOI] [PubMed] [Google Scholar]
- 17.Guarani V, Deflorian G, Franco CA, Kruger M, Phng LK, Bentley K, Toussaint L, Dequiedt F, Mostoslavsky R, Schmidt MH, Zimmermann B, Brandes RP, Mione M, Westphal CH, Braun T, Zeiher AM, Gerhardt H, Dimmeler S, Potente M. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011;473:234–8. doi: 10.1038/nature09917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hay RT. SUMO: a history of modification. Molecular cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 19.Xue Y, Zhou F, Fu C, Xu Y, Yao X. SUMOsp: a web server for sumoylation site prediction. Nucleic acids research. 2006;34:W254–7. doi: 10.1093/nar/gkl207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Q, Xie Y, Zheng Y, Jiang S, Liu W, Mu W, Liu Z, Zhao Y, Xue Y, Ren J. GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic acids research. 2014;42:W325–30. doi: 10.1093/nar/gku383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li T, Evdokimov E, Shen RF, Chao CC, Tekle E, Wang T, Stadtman ER, Yang DC, Chock PB. Sumoylation of heterogeneous nuclear ribonucleoproteins, zinc finger proteins, and nuclear pore complex proteins: a proteomic analysis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8551–6. doi: 10.1073/pnas.0402889101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vassileva MT, Matunis MJ. SUMO modification of heterogeneous nuclear ribonucleoproteins. Molecular and cellular biology. 2004;24:3623–32. doi: 10.1128/MCB.24.9.3623-3632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–95. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hay RT. SUMO-specific proteases: a twist in the tail. Trends in cell biology. 2007;17:370–6. doi: 10.1016/j.tcb.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 25.Mikolajczyk J, Drag M, Bekes M, Cao JT, Ronai Z, Salvesen GS. Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. The Journal of biological chemistry. 2007;282:26217–24. doi: 10.1074/jbc.M702444200. [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Ji W, Zhang H, Renda MJ, He Y, Lin S, Cheng EC, Chen H, Krause DS, Min W. SENP1-mediated GATA1 deSUMOylation is critical for definitive erythropoiesis. The Journal of experimental medicine. 2010;207:1183–95. doi: 10.1084/jem.20092215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalen M, Gerhardt H, Betsholtz C. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–80. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 28.Haines N, Irvine KD. Glycosylation regulates Notch signalling. Nature reviews Molecular cell biology. 2003;4:786–97. doi: 10.1038/nrm1228. [DOI] [PubMed] [Google Scholar]
- 29.Yeh ET, Gong L, Kamitani T. Ubiquitin-like proteins: new wines in new bottles. Gene. 2000;248:1–14. doi: 10.1016/s0378-1119(00)00139-6. [DOI] [PubMed] [Google Scholar]
- 30.Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nature reviews Molecular cell biology. 2001;2:202–10. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- 31.Kang JS, Saunier EF, Akhurst RJ, Derynck R. The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nature cell biology. 2008;10:654–64. doi: 10.1038/ncb1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sehat B, Tofigh A, Lin Y, Trocme E, Liljedahl U, Lagergren J, Larsson O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Science signaling. 2010;3:ra10. doi: 10.1126/scisignal.2000628. [DOI] [PubMed] [Google Scholar]
- 33.Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes & development. 2007;21:2511–24. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- 34.Chiou HY, Liu SY, Lin CH, Lee EH. Hes-1 SUMOylation by protein inhibitor of activated STAT1 enhances the suppressing effect of Hes-1 on GADD45alpha expression to increase cell survival. Journal of biomedical science. 2014;21:53. doi: 10.1186/1423-0127-21-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nature reviews Molecular cell biology. 2011;12:551–64. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R, Freitas C, Duarte A, Isoniemi H, Laakkonen P, Christofori G, Yla-Herttuala S, Shibuya M, Pytowski B, Eichmann A, Betsholtz C, Alitalo K. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454:656–60. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 37.Heisig J, Weber D, Englberger E, Winkler A, Kneitz S, Sung WK, Wolf E, Eilers M, Wei CL, Gessler M. Target gene analysis by microarrays and chromatin immunoprecipitation identifies HEY proteins as highly redundant bHLH repressors. PLoS genetics. 2012;8:e1002728. doi: 10.1371/journal.pgen.1002728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.