

Graphical abstract
A nanotoxoid concurrently carrying pathogen-specific antigens is fabricated on-demand by leveraging the interactions between naturally secreted bacterial virulence factors and cell membrane-coated nanoparticles. The nanovaccine is confirmed to carry and neutralize known toxins from a model bacterium, and is capable of significantly controlling bacteria growth when used to vaccinate mice. The reported approach may provide new avenues for controlling the rise of antibiotic-resistant bacteria.
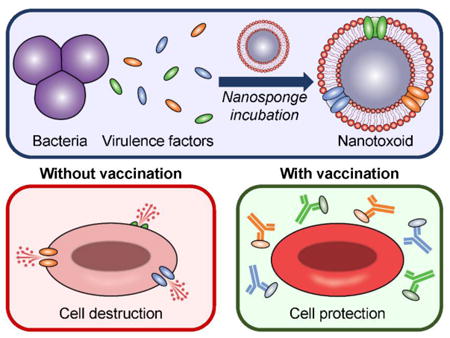
Keywords: nanomedicine, biomimetic nanoparticle, antibiotic-resistant bacteria, vaccination, antivirulence therapy
The continued rise of antibiotic-resistant bacteria poses a significant threat to public health, yet the development of new small-molecule antibiotics remains slow.[1] An increasing number of lives will be at risk as time progresses, and thus finding new and innovative ways to combat these potentially lethal pathogens is of extremely high importance.[2–6] Along these lines, antivirulence therapy is a promising strategy for addressing bacterial infection that focuses on removing the offensive weapons used by bacteria to successfully colonize a host.[7–9] Examples of such factors include protein-based toxins, which can be used to attack host cells via physical disruption, biochemical degradation, or signaling interruption, thereby preventing immune clearance and providing the nutrients necessary for proliferation.[10,11] Neutralization of these bacterial virulence factors can have a marked impact on bacteria survivability.[12] One major advantage of employing antivirulence therapy is that, unlike with antibiotics, the treatment itself doesn't exert direct selective pressure on individual bacterium; by focusing instead on blocking pathogen-to-host interactions, this strategy can ultimately translate into a reduced likelihood of developing resistance.[13] Implementation of this strategy has varied, ranging from traditional antibody neutralization[14,15] to novel nanotechnology-based complexation.[16–19] While antivirulence can be effective in therapeutic settings, arguably the most impactful applications center around prophylactic vaccination. In fact, commonly used vaccines in the clinic against diphtheria and tetanus are targeted against their respective virulence factors,[20] underscoring the usefulness of this strategy.
While the immunity generated through antivirulence vaccination can be used to effectively prevent some bacterial infections, others have proven much more difficult to address.[21] A major challenge for creating vaccines against biological toxins is the balance that must be struck between safety and immunogenicity, which often exhibit an inverse relationship.[22] Toxicity can be attenuated via several different approaches, including heat treatment and chemical modification;[23] however, not all toxins are heat-labile, and denaturation has the potential to compromise vaccine efficacy due to the modified presentation of epitopic targets.[24] Subunit engineering can eliminate virulence, but requires significant upfront investment of resources and is only applicable towards well-characterized targets.[25] Vaccine potency is further challenged by the varying secretion profiles of different bacterial species and strains. As many bacteria produce a wide variety of toxins and other factors,[26,27] it can be difficult to pinpoint which of these are major contributors to pathogenesis. In some cases, vaccination approaches based on multiple known toxins have been shown to carry great utility,[28] but these are hard to pursue given the significant time required for identification and confirmation of new virulence factors. While such approaches will undoubtedly be aided by advances in genomic and proteomic technologies, the history of some well-known toxins, such as streptolysin S secreted by group A streptococcus, underscores the gap that often exists between knowledge and application.[29]
To circumvent the need for identification of individual virulence factors, direct derivation from bacterial protein secretions should represent an attractive method to obtain the material needed for generating antivirulence vaccines. However, this strategy has seldom been studied,[30,31] likely also due to issues in balancing safety and immunogenicity, with the added challenge of having to manage the presence of irrelevant proteins that dilute immune focus. In this work, we report on a facile approach for generating on-demand nanotoxoids from naturally-derived bacterial protein preparations by leveraging the near universal natural affinity of virulence factors for cellular membranes[16,32] (Figure 1). Virulent proteins are biomimetically entrapped using a membrane-coated nanosponge construct,[17] effectively modulating the surface material composition for custom vaccine applications. Following a generalizable workflow that doesn't require prior knowledge of secreted constituents, pathogen-specific formulations that are safe, potentially multi-antigenic, and epitopically faithful can be fabricated. The feasibility of this approach is demonstrated using methicillin-resistant Staphylococcus aureus (MRSA), which employs multiple well-characterized toxins,[26,33] as the model pathogen, along with red blood cell (RBC) membrane-coated nanosponges as the model vector.
Figure 1.
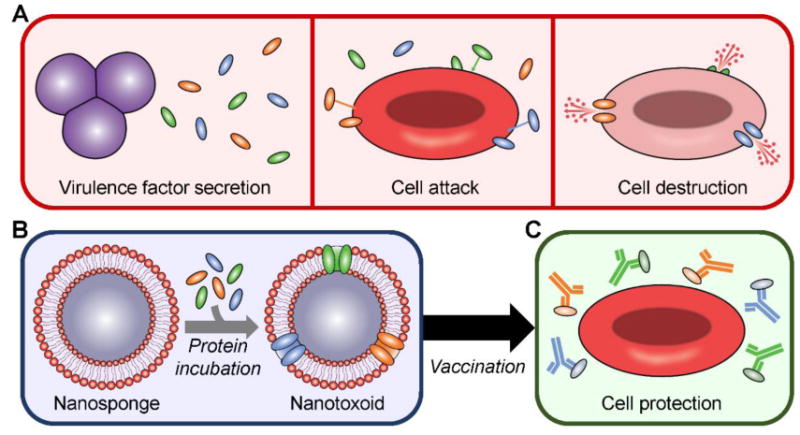
Schematic depicting on-demand fabrication of a pathogen-specific nanotoxoid and its vaccination benefits. A) Pathogens secrete virulence factors, which are capable of inserting into target cells and causing their destruction. B) Using nanosponges prepared with the membrane of target cells and incubating the particles with a bacterial supernatant-derived protein fraction, it is possible to generate a nanotoxoid carrying pathogen-specific virulence factors. C) After vaccination using the nanotoxoid, antibodies against the incorporated virulence factors are elicited and can prevent their toxic effects, leaving the intended targets unharmed.
We first confirmed that RBC nanosponges could be used to effectively neutralize the harmful biological activity of proteins secreted by MRSA strain USA300. Using a hemolytic secreted protein (hSP) fraction collected from bacterial culture supernatant via ammonium sulfate precipitation (see Experimental Section), it was demonstrated that preincubation with a sufficient amount of RBC nanosponges could effectively eliminate the hSP's lytic effects on RBCs (Figure 2A). From the data, approximately 400 μg of nanosponges could be used to neutralize 15 μg of the protein, and this ratio was used to fabricate an hSP-loaded nanosponge vaccine formulation, termed nanotoxoid(hSP), for further study. According to dynamic light scattering measurements, the size of the nanotoxoid(hSP) was slightly larger and the surface zeta potential was less negative when compared to the blank nanosponges without hSP loading, both suggesting the association of the hSP with the membrane-coated nanoparticle substrate (Figure 2B,C). Transmission electron microscopy confirmed that, after protein loading, nanotoxoid(hSP) still exhibited a characteristic core-shell structure,[34,35] with a membrane layer surrounding the polymeric core (Figure 2D).
Figure 2.
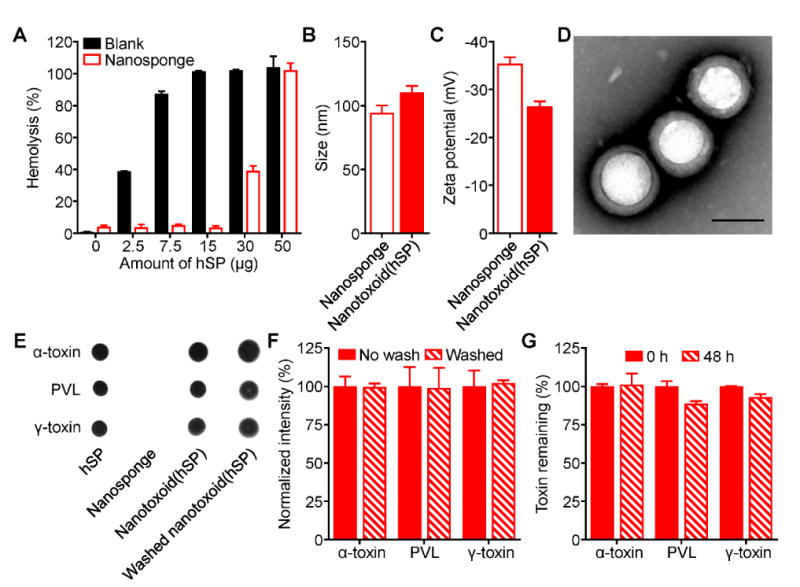
Synthesis and characterization of hemolytic secreted protein (hSP)-loaded nanotoxoid, denoted nanotoxoid(hSP). A) Hemolysis of RBCs when incubated with varying amounts of hSP in the absence or presence of 400 μg of RBC nanosponges (n = 3; mean ± SD). B) Size of RBC nanosponges and nanotoxoid(hSP) as measured by dynamic light scattering (n = 3; mean ± SD). C) Surface zeta potential of nanosponges and nanotoxoid(hSP) (n = 3; mean ± SD). D) Transmission electron microscope image of nanotoxoid(hSP) negatively stained with uranyl acetate (scale bar = 100 nm). E) Dot blots probing for α-toxin, PVL, or γ-toxin in hSP, blank nanosponges, nanotoxoid(hSP), or nanotoxoid(hSP) subject to a wash step. F) Relative band intensities of western blots probing for α-toxin, PVL, or γ-toxin in nanotoxoid(hSP) or nanotoxoid(hSP) subject to a wash step (n = 3; mean ± SD). G) Retention of α-toxin, PVL, or γ-toxin on nanotoxoid(hSP) after dialyzing against 1× PBS for 48 hours (n = 3; mean ± SD).
While previous versions of nanotoxoids have worked with individual, purified toxins,[12,36] the advantage of the present approach is its ability to entrap and neutralize pathogen-specific virulence factors from a protein preparation with unknown composition. To validate this concept, we probed the nanotoxoid(hSP) formulation for the presence of known virulence factors by immunoblotting (Figure 2E). Of the three different antigens that were analyzed, all were easily detectable on the nanotoxoid(hSP). These included α-toxin, a major MRSA virulence factor that has previously been successfully neutralized using RBC nanosponges,[17] as well as Panton-Valentine leukocidin (PVL), a white blood cell attacking toxin expressed in most community-acquired MRSA variants,[37,38] and γ-toxin, a bicomponent toxin formed from combinations of three different monomers.[39] Quantitative western blot analysis demonstrated that α-toxin, PVL, and γ-toxin contributed to 11.0% ± 0.7%, 8.7% ± 0.8%, and 5.6% ± 0.2% of the total bacterial protein, respectively (see Experimental Section). After subjecting the nanotoxoid(hSP) to a wash step, the three toxins remained strongly present (Figure 2F). Additionally, the toxins remained mostly bound to the nanoparticles even after dialyzing against physiological buffer for 48 hours (Figure 2G), which suggested stable and efficient complexation and explained the ability of the nanoparticles to neutralize the toxins' hemolytic activity.
Given the robust binding of the toxins with the nanosponges, we further sought to evaluate the safety of the nanotoxoid(hSP) formulation in various settings. First, we compared the hemolytic capacity of hSP in its native form, when subjected to rigorous heat denaturation, and when in nanotoxoid(hSP) form (Figure 3A,B). Native hSP demonstrated complete lysis while nanotoxoid(hSP) fabricated with an equivalent amount of hSP had almost no activity; this neutralization effect has previously been shown to be exclusive to membrane-coated nanoparticles.[17] As expected, blank nanosponges were not hemolytic, but it was striking that, even after boiling the hSP for 4 hours, 40% of its hemolytic activity was still preserved. While specific toxins secreted by MRSA are known to be heat-labile,[36] the results demonstrated that the more complex hSP preparation contained elements that were not sensitive to temperature. The data also hints that nanosponge-based neutralization, despite its non-denaturing approach, may also be more universally applicable. The results were mirrored when the same formulations were incubated with bone marrow-derived dendritic cells (Figure 3C). The hSP preparation completely killed the cells in vitro, and the heat-treated proteins also had significant toxicity, leading to only 20% of the cells remaining viable. On the other hand, both the nanotoxoid(hSP) and blank nanosponges showed no signs of cytotoxicity, again demonstrating the ability of nanocomplexation to much more effectively eliminate the harmful biological effects of the toxins.
Figure 3.
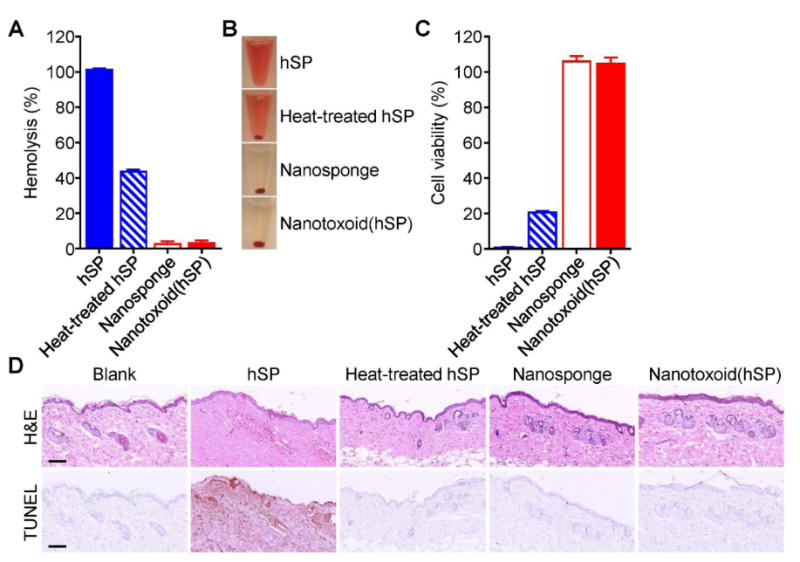
In vitro and in vivo safety studies. A) Comparison of hemolysis induced by hSP, heat-treated hSP, blank nanosponge, and nanotoxoid(hSP) (n = 3; mean ± SD). B) Representative images demonstrating the varying degrees of hemolysis in the samples from (A). C) Comparison of bone marrow-derived dendritic cell viability after 24 hours of incubation with hSP, heat-treated hSP, blank nanosponge, or nanotoxoid(hSP) followed by another 48 hours of culture (n = 4; mean ± SD). D) Hematoxylin and eosin (H&E) and TUNEL staining of skin samples collected from untreated mice or from mice 48 hours after subcutaneous injection of hSP, heat-treated hSP, blank nanosponge, or nanotoxoid(hSP) (scale bars = 100 μm).
In vivo, we assessed potential toxicity by administering the different formulations subcutaneously followed by histological analysis after 48 hours (Figure 3D). Hematoxylin and eosin (H&E) staining showed that native hSP induced significant atrophy in the squamous epithelium and scattered bleeding in the dermal as well as subcutaneous tissues. Disarrangement and degeneration of collagen fibers were also observed. The toxicity of the protein was further demonstrated by TUNEL staining, which revealed widespread apoptosis throughout. In contrast, there was no obvious skin damage in the other three samples; the structure of the skin remained intact and orderly with minimal signs of apoptosis. While the heated hSP displayed considerable toxicity in vitro, the in vivo results suggest that the partial attenuation afforded by the heat treatment was sufficient to prevent it from reaching the threshold required for inducing significant damage in a more complex biological setting. Given the relative safety of the heat-treated hSP demonstrated here, we elected to employ it as a control in subsequent functional studies as a comparison against nanotoxoid(hSP).
Following the safety evaluation, the ability of the nanotoxoid(hSP) formulation to elicit potent humoral immunity was studied. The induction of germinal centers within lymph nodes is one of the critical steps in the immune response against infection, and it is in these regions where affinity-based maturation of B cells occurs.[40] To study the effect of the different formulations on this phenomenon, draining lymph nodes were collected 21 days after immunization and analyzed for the presence of B cells with the corresponding phenotype (Figure 4A,B). Flow cytometric analysis revealed that, of the different formulations, only the nanotoxoid(hSP) could significantly raise the percentage of B cells with the germinal center marker GL-7, with the value increasing to 44% compared with 19% for the blank control. This was also evident by immunofluorescence staining of histological sections, which indicated the presence of several nodules with a high concentration of GL-7+ cells in the lymph nodes of mice from the nanotoxoid(hSP) group. Of note, heat-treated hSP did not result in the formation of germinal centers despite delivering the same antigenic material. Additionally, blank nanosponges had no effect, precluding any adjuvanting contributions from the nanoparticle vector itself and suggesting a favorable biocompatibility profile.[36] From the data, it appears that the particulate delivery of undenatured bacterial hSP facilitates the generation of strong immune responses.
Figure 4.

Germinal center formation. A) Flow cytometric analysis of cells at the draining lymph node 21 days after administration with blank solution, heat-treated hSP, blank nanosponge, or nanotoxoid(hSP) (n = 4; mean ± SD). Cells were first gated on the B220+IgDlow population and values are expressed as percentage GL-7+. B) Fluorescent images of draining lymph node histological sections stained with antibodies against B220 (green), IgD (blue), and GL-7 (red) at different magnifications (top: 4× objective, scale bar = 500 μm; bottom: 20× objective, scale bar = 100 μm). *** p < 0.001, **** p < 0.0001, one-way ANOVA.
To test how the increased response to the nanoformulation would translate into antigen-specific immunity, we quantified the titers generated against known constituents present on the nanotoxoid(hSP), including α-toxin, PVL, and γ-toxin (Figure 5A-C). To compare the different antigen-containing formulations, mice were vaccinated with a prime injection plus two boosts on days 7 and 14. On day 21, around the peak of IgG responses, the serum was sampled and titers analyzed by indirect enzyme-linked immunosorbent assays (ELISAs). For α-toxin, which is one of the most highly secreted by MRSA, there was an easily detectable difference in antibody production. This is consistent with previous reports on a nanotoxoid formulated with purified α-toxin.[12,36] Heat-treated hSP was approximately two orders of magnitude less effective. For PVL, 57% of the mice exhibited highly elevated titers when vaccinated with the nanotoxoid(hSP), while the other portion were non-responders. This represented a large improvement compared with the heat-treated hSP group, which had titer values near baseline. While the results for γ-toxin were less pronounced, the effect of nanotoxoid(hSP) vaccination still bordered on significance. It appeared that the trend in titer production reflected the relative amounts of each toxin in the hSP preparation. In total, the nanotoxoid(hSP) formulation was more adept at eliciting anti-toxin immune responses compared with the heat-treated protein formulation, despite both delivering the same antigenic material.
Figure 5.
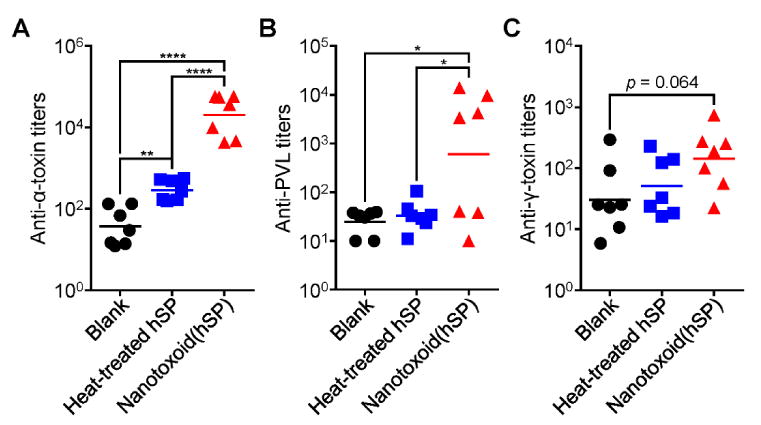
Multivalent antibody responses in vivo. Mice were vaccinated with blank solution, heat-treated hSP, or nanotoxoid(hSP) on day 0 with boosts on days 7 and 14. On day 21, the serum was sampled and analyzed for the presence of IgG antibody titers against α-toxin (A), PVL (B), and γ-toxin (C) (n = 7; geometric mean). * p < 0.05, ** p < 0.01, **** p < 0.0001, one-way ANOVA.
Finally, we evaluated the effectiveness of nanotoxoid(hSP) vaccination in preventing live bacterial infection by employing MRSA strain USA300 in two separate in vivo models reflective of how the disease presents in the clinic.[41,42] For both studies, mice were vaccinated with a prime injection plus two boosts on days 7 and 14. On day 35 after the first administration, mice were challenged with bacteria, and the impact of antivirulence immunity on bacterial survival was assessed. In the subcutaneous model, which mimics the skin infections common to MRSA,[43] the nanotoxoid(hSP) had a striking effect on skin lesion formation (Figure 6A). On the final day of the study, those receiving the nanotoxoid(hSP) had, on average, a 3-fold smaller affected area compared to mice vaccinated with heat-treated hSP. Similarly, the nanotoxoid(hSP) performed well in controlling bacterial growth upon intravenous injection, which was used to model potentially life-threatening systemic MRSA infections[44] (Figure 6B,C). Looking at the total bacterial load 3 days after challenge, mice vaccinated with the nanotoxoid(hSP) were able to much more effectively clear out the MRSA bacteria compared to those receiving heat-treated hSP. At the organ level, the effect was most apparent in the heart, lungs, and especially the kidneys. Overall, the results are a reflection of the differences seen in titer production among the formulations and highlight the stronger immunity generated by the nanotoxoid(hSP) formulation, which inhibits the ability of the bacteria to survive over time.
Figure 6.
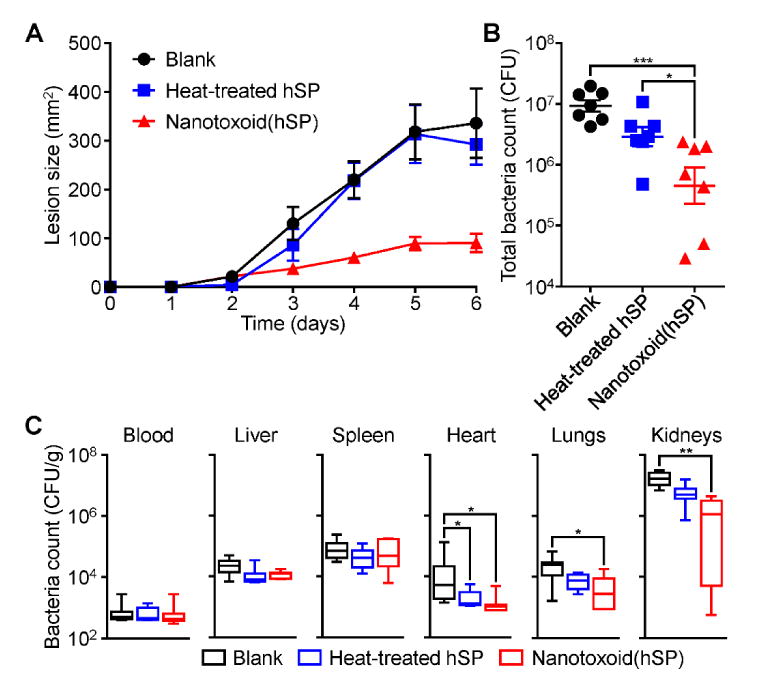
Protection against challenge with live bacteria. Mice were vaccinated with blank solution, heat-treated hSP, or nanotoxoid(hSP) on day 0 with boosts on days 7 and 14. A) Lesion size over time after subcutaneous challenge with MRSA USA300 on day 35 (n = 7; mean ± SEM). B) Total bacterial load summed from major organs 3 days after intravenous challenge with MRSA USA300 on day 35 (n = 7; geometric mean ± SEM). C) Individual, weight-normalized bacteria counts in major organs from (B) (n = 7; min to max). * p < 0.05, ** p < 0.01, *** p < 0.001, one-way ANOVA.
In conclusion, we have reported on a method of fabricating on-demand nanotoxoids for use as vaccines against pathogenic bacteria. The nanoformulation was able to entrap virulence factors from protein preparations of unknown composition, was safe both in vitro and in vivo, and could elicit functional immunity capable of combating live bacterial infections. Despite containing the same bacterial antigens, the nanotoxoid(hSP) formulation consistently outperformed a denatured protein preparation in all of the metrics studied, which underscores the utility of biomimetic nanoparticle-based neutralization and delivery. Overall this strategy helps to address major hurdles in the design of antivirulence vaccines, enabling increased antigenic breadth while maintaining safety. Looking forward, the workflow presented here can easily be modified for application towards a variety of different pathogens. It may be possible to employ personalized culture isolates or to change culture conditions such that virulence factor production is modulated. Alternatively, nanotoxoid formulations can be screened to identify a broadly neutralizing option that is effective across multiple bacterial strains. Other purification or fractionation strategies can be tested to emphasize non-hemolytic virulence factors, and different membrane substrates derived from other cell types can be leveraged.[45–47] The inclusion of immunological adjuvants can also be considered to further boost efficacy.[48–50] Ultimately, the success of antivirulence vaccines may help to control the spread of many deadly diseases and abate the rising threat of antibiotic-resistant bacteria.
Experimental Section
Preparation of Hemolytic Secreted Protein (hSP) Fraction
The MRSA strain USA300 (BAA-1717; American Type Culture Collection) was first plated onto a tryptic soy agar (Sigma Aldrich) plate overnight at 37 °C. A single colony was cultured in 5 mL of tryptic soy broth (TSB; Sigma Aldrich) for 24 hours at 37 °C, and 1 mL was then transferred to another 100 mL of TSB and cultured for 24 hours. The media was collected after spinning down the bacteria at 3,000× g for 20 minutes. Saturated ammonium sulfate (Sigma Aldrich) solution was added slowly to the media in a glass beaker while stirring at 4 °C up to a 25% volume ratio. After stirring for 1 hour, the solution was centrifuged at 3,000× g for 20 minutes to pellet the first fraction. Fractions at 50% and 75% volume ratios were collected in the same manner. Finally, solid ammonium sulfate was added to obtain the equivalent of a 95% saturated solution volume ratio and stirred overnight before collection of the last fraction. All precipitated protein pellets were dissolved in water and desalted using columns packed with fine G-25 Sephadex (GE Healthcare). Only the first protein fraction to pass through each column was collected, ultimately yielding concentrated samples free from most other non-protein contaminants. Hemolytic activity was assessed by adjusting protein solutions to 1× phosphate buffered saline (PBS) and incubating at 1 mg/mL with an equal volume of 2.5% purified RBCs collected from male ICR mice (Envigo). All animal experiments were performed in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Diego. After 30 minutes of incubation, the samples were spun down at 2,000× g for 5 minutes. Hemolysis was determined by measuring the absorbance of the supernatant at 540 nm using a Tecan Infinite M200 plate reader. Fractions demonstrating considerable signal were combined together for further use as the hSP fraction.
Preparation and Physicochemical Characterization of Nanosponges and Nanotoxoid(hSP)
RBC membrane-coated nanosponges were prepared by a previously reported method.[51] Membrane vesicles collected from male ICR mice were coated by a sonication process onto preformed polymeric cores prepared with carboxyl-terminated poly(lactic-co-glycolic acid) (0.67 dL/g, 50:50 monomer ratio; LACTEL Absorbable Polymers). To assess the ability of nanosponge preincubation to prevent hemolysis by the hSP fraction, 400 μg of the nanosponges was incubated with varying amounts of protein ranging from 1 to 50 μg in 10 wt% sucrose at 37 °C for 30 minutes. The mixtures in a volume of 100 μL were added to an equal volume of 2.5% mouse RBCs in PBS. Equivalent amounts of free hSP in the absence of nanosponges were used for comparison. After another 30 minutes of incubation at 37 °C, samples were spun down at 2,000× g for 5 minutes. Hemolysis was determined by measuring the absorbance of the supernatant at 540 nm using a Tecan Infinite M200 plate reader. A 100% lysis control was prepared by treating the RBCs with Triton X-100 (Sigma Aldrich). Subsequent studies were carried out using a ratio of 400 μg nanosponges incubated directly with 15 μg of hSP, the product of which was referred to as the nanotoxoid(hSP) formulation. The size and the surface zeta potential of the nanoformulations was measured by dynamic light scattering using a Malvern ZEN 3600 Zetasizer. The structure of the nanotoxoid(hSP) was examined after negative staining with 1 wt% uranyl acetate (Electron Microscopy Sciences) on a carbon-coated 400-mesh copper grid (Electron Microscopy Sciences) using a Zeiss Libra 120 PLUS EF-TEM transmission electron microscope.
Protein Characterization
To visually confirm the presence of bacterial virulence factors on the resulting nanotoxoid(hSP), dot blots were performed to probe for three known toxins secreted by MRSA (α-toxin, PVL, and γ-toxin). In addition to hSP, nanosponge, and nanotoxoid(hSP), a washed nanotoxoid(hSP) sample was obtained by centrifugation at 21,100× g to separate out unbound proteins. Nanoparticle samples were run at equivalent nanosponge concentrations, and hSP was run at the same concentration as inputted into the nanotoxoid(hSP) formulation. The samples were prepared using lithium dodecyl sulfate sample loading buffer (Invitrogen), heated at 70 °C for 15 minutes, and 5 μL of each was dropped onto a nitrocellulose membrane (Thermo Scientific) followed by drying under vacuum. Membranes were probed using either a polyclonal rabbit anti-staphylococcal α-toxin (Sigma Aldrich), polyclonal rabbit anti-PVL LukS subunit (IBT Bioservices), or polyclonal rabbit anti-staphylococcal γ-toxin B (IBT Bioservices) as the primary antibody along with an HRP-conjugated anti-rabbit IgG (Biolegend) as the secondary antibody. Blots were developed with ECL western blotting substrate (Pierce) using an ImageWorks Mini-Medical/90 Developer.
Western blotting was carried out to quantitatively determine the amount of toxins that remained bound to the nanoparticles. Nanotoxoid(hSP) and washed nanotoxoid(hSP) were prepared in the same manner as above and run on NuPAGE Novex 4%–12% Bis-Tris minigels (Invitrogen) in MOPS running buffer (Invitrogen). After transferring onto nitrocellulose membranes, the blots were probed for α-toxin, PVL LukS subunit, or γ-toxin B. Band intensities were measured using Adobe Photoshop and normalized to the average values of the no wash nanotoxoid(hSP) sample for each toxin. To determine the composition of the final hSP preparation, different dilutions of the hSP protein, alongside purified α-toxin (Sigma Aldrich), PVL LukS subunit (IBT Bioservices), and γ-toxin B (IBT Bioservices), were subjected to western blot analysis. Linear standard curves were generated using the hSP dilutions upon probing for each toxin. Composition percentages were determined as the concentration of each purified toxin divided by the interpolated hSP concentration based on the band intensities measured for that specific toxin (n = 3; mean ± SD). To perform the release study, nanotoxoid(hSP) at a concentration of 2 mg/mL was placed into a 300 kDa MWCO Float-A-Lyzer G2 (Spectrum Laboratories) and dialyzed against 2 L of 1× PBS. Samples were collected at 0 and 48 hours and probed for α-toxin, PVL LukS subunit, or γ-toxin B by western blotting. Values were normalized to the average band intensities of the 0 hour samples for each toxin.
In Vitro Safety
To assess hemolytic activity, hSP (15 μg), heat-treated hSP (15 μg heated for 4 hours at 100 °C), nanosponge (400 μg), and nanotoxoid(hSP) (400 μg of nanosponge incubated with 15 μg of hSP for 30 minutes) were added in 150 μL of solution to an equal volume of 2.5% mouse RBCs in PBS. Note that the nanoparticle concentration employed was near the maximum feasible value allowed by the nanotoxoid fabrication process. After 30 minutes of incubation at 37 °C, each sample was spun down and the absorbance of hemoglobin in the supernatant was measured at 540 nm using a Tecan Infinite M200 plate reader. Bone marrow-derived dendritic cells were isolated from ICR mice and cultured as reported before.[52] To assess cytotoxicity, the cells were plated into 96-well plates and incubated with hSP (7.5 μg), heat-treated hSP (7.5 μg), nanosponge (200 μg), or nanotoxoid(hSP) (200 μg nanosponge with 7.5 μg hSP). After 24 hours of incubation with the different samples, the cells were cultured for another 48 hours in fresh media. Cell viability was assayed using an MTT reagent (Invitrogen) following the manufacturer's instructions. Untreated cells were used as the 100% viability control.
In Vivo Safety
Male ICR mice were first shaved to remove the hair on their back. Subsequently, 150 μL of blank solution, hSP (22.5 μg), heat-treated hSP (22.5 μg), nanosponge (600 μg), or nanotoxoid(hSP) (600 μg nanosponge with 22.5 μg hSP) was injected subcutaneously. After 48 hours, the mice were euthanized and skin samples at the site of injection, where most of the nanoparticles were expected to remain, were collected for histological processing. Sections were stained either by hematoxylin and eosin (H&E) using SelecTech reagents (Leica Biosystems) or by TUNEL using an ApopTag peroxidase in situ apoptosis detection kit (EMD Millipore). Brightfield images were acquired using a Hamamatsu Nanozoomer slide scanning system.
Germinal Center Formation
Six week-old male ICR mice were administered with blank solution, hSP (7.5 μg), nanosponge (200 μg), or nanotoxoid(hSP) (200 μg nanosponge with 7.5 μg hSP) by hock injection. On day 21 after immunization, the draining popliteal lymph nodes were collected for analysis. For immunohistochemistry, the lymph nodes were cryosectioned and stained with Pacific Blue-labeled anti-mouse/human B220 (Clone: RA3-6B2; Biolegend), Alexa488-labeled anti-mouse IgD (Clone: 11-26c.2a; Biolegend), and Alexa647-labeled anti-mouse/human GL-7 (Clone GL7; Biolegend). Fluorescence imaging was conducted on a Keyence BZ-9000 microscope. For flow cytometric analysis, the popliteal lymph nodes were dissociated into single cell suspensions using 1 mg/mL collagenase D (Roche) and 1 mg/mL DNAse I (Roche). The cells were then stained with the above antibodies followed by data collection on a BD FACSCanto-II flow cytometer. Analysis was performed using Flowjo software.
Antibody Titer Responses
Six week-old male ICR mice were vaccinated by subcutaneous injections at the neck region with blank solution, hSP (75 μg), or nanotoxoid(hSP) (2 mg of nanosponge with 75 μg hSP) on days 0, 7, and 14. On day 21, the blood of each mouse was collected, and the serum was subsequently derived by centrifugation at 700× g. Antibody titers were assessed by an indirect ELISA using plates coated with purified α-toxin, PVL LukS subunit, or γ-toxin A (IBT Bioservices) following a previously reported protocol.[12]
Protective Efficacy against MRSA Infection
Six week-old male ICR mice were immunized using the same formulations and schedule as above. For the subcutaneous model, 1×109 CFU of MRSA USA300 was inoculated into the shaved back region away from the site of vaccination on day 35. The lesion on the skin of each mouse was monitored daily and reported as the width multiplied by the length of the visibly affected area. For the systemic model, 2×106 CFU of MRSA USA300 was injected via the tail vein. On day 3 after challenge, the blood was first collected prior to euthanasia. The mice were then perfused with PBS, and the liver, spleen, heart, lungs, and kidneys of each mouse were collected and processed for bacterial enumeration following a previously published protocol.[12] To obtain the total bacteria count, the values from all collected organs for each individual mouse were summed.
Acknowledgments
X.W. and J.G. contributed equally to this work. This work is supported by the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense under Grant Number HDTRA1-14-1-0064 and by the National Institutes of Health under Award Number R01EY025947. The authors acknowledge the Waitt Advanced Biophotonics Core Facility of the Salk Institute for TEM imaging, which is supported with funding from NIH-NCI CCSG: P30 014195, NINDS Neuroscience Core Grant, and the Waitt Foundation.
Contributor Information
Dr. Xiaoli Wei, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A.; Department of Pharmaceutics, School of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery (Fudan University), Ministry of Education, Shanghai 201203, PR China.
Jie Gao, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A.; Department of Pharmaceutics, School of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery (Fudan University), Ministry of Education, Shanghai 201203, PR China.
Fei Wang, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A.; Department of Pharmaceutics, School of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery (Fudan University), Ministry of Education, Shanghai 201203, PR China.
Man Ying, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A.; Department of Pharmaceutics, School of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery (Fudan University), Ministry of Education, Shanghai 201203, PR China.
Pavimol Angsantikul, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A.
Ashley V. Kroll, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A
Jiarong Zhou, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A.
Dr. Weiwei Gao, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A
Prof. Weiyue Lu, Department of Pharmaceutics, School of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery (Fudan University), Ministry of Education, Shanghai 201203, PR China
Dr. Ronnie H. Fang, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A
Prof. Liangfang Zhang, Department of NanoEngineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, U.S.A.
References
- 1.Lewis K. Nat Rev Drug Discov. 2013;12:371. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 2.Arias CA, Murray BE. N Engl J Med. 2009;360:439. doi: 10.1056/NEJMp0804651. [DOI] [PubMed] [Google Scholar]
- 3.Wright GD, Sutherland AD. Trends Mol Med. 2007;13:260. doi: 10.1016/j.molmed.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 4.Spellberg B, Bartlett JG, Gilbert DN. N Engl J Med. 2013;368:299. doi: 10.1056/NEJMp1215093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng VWL, Ke X, Lee ALZ, Hedrick JL, Yang YY. Adv Mater. 2013;25:6730. doi: 10.1002/adma.201302952. [DOI] [PubMed] [Google Scholar]
- 6.Xiong MH, Li YJ, Bao Y, Yang XZ, Hu B, Wang J. Adv Mater. 2012;24:6175. doi: 10.1002/adma.201202847. [DOI] [PubMed] [Google Scholar]
- 7.Rasko DA, Sperandio V. Nat Rev Drug Discov. 2010;9:117. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 8.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. Nat Rev Microbiol. 2008;6:17. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clatworthy AE, Pierson E, Hung DT. Nat Chem Biol. 2007;3:541. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 10.Henkel JS, Baldwin MR, Barbieri JT. EXS. 2010;100:1. doi: 10.1007/978-3-7643-8338-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lubran MM. Ann Clin Lab Sci. 1988;18:58. [PubMed] [Google Scholar]
- 12.Wang F, Fang RH, Luk BT, Hu CMJ, Thamphiwatana S, Dehaini D, Angsantikul P, Kroll AV, Pang Z, Gao W, Lu W, Zhang L. Adv Funct Mater. 2016;26:1628. doi: 10.1002/adfm.201505231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mellbye B, Schuster M. MBio. 2011;2:e00131. doi: 10.1128/mBio.00131-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hua L, Cohen TS, Shi Y, Datta V, Hilliard JJ, Tkaczyk C, Suzich J, Stover CK, Sellman BR. Antimicrob Agents Chemother. 2015;59:4526. doi: 10.1128/AAC.00510-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiGiandomenico A, Keller AE, Gao C, Rainey GJ, Warrener P, Camara MM, Bonnell J, Fleming R, Bezabeh B, Dimasi N, Sellman BR, Hilliard J, Guenther CM, Datta V, Zhao W, Gao C, Yu XQ, Suzich JA, Stover CK. Sci Transl Med. 2014;6:262ra155. doi: 10.1126/scitranslmed.3009655. [DOI] [PubMed] [Google Scholar]
- 16.Fang RH, Luk BT, Hu CMJ, Zhang L. Adv Drug Deliv Rev. 2015;90:69. doi: 10.1016/j.addr.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu CMJ, Fang RH, Copp J, Luk BT, Zhang L. Nat Nanotechnol. 2013;8:336. doi: 10.1038/nnano.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoshino Y, Koide H, Urakami T, Kanazawa H, Kodama T, Oku N, Shea KJ. J Am Chem Soc. 2010;132:6644. doi: 10.1021/ja102148f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang F, Gao W, Thamphiwatana S, Luk BT, Angsantikul P, Zhang Q, Hu CMJ, Fang RH, Copp JA, Pornpattananangkul D, Lu W, Zhang L. Adv Mater. 2015;27:3437. doi: 10.1002/adma.201501071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitchin NRE. Expert Rev Vaccines. 2011;10:605. doi: 10.1586/erv.11.60. [DOI] [PubMed] [Google Scholar]
- 21.Watkins RR, David MZ, Salata RA. J Med Microbiol. 2012;61:1179. doi: 10.1099/jmm.0.043513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cryz SJ, Jr, Fürer E, Germanier R. Infect Immun. 1982;38:21. doi: 10.1128/iai.38.1.21-26.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fulthorpe AJ, Thomson RO. Immunology. 1960;3:126. [PMC free article] [PubMed] [Google Scholar]
- 24.Metz B, Kersten GFA, Hoogerhout P, Brugghe HF, Timmermans HAM, de Jong A, Meiring H, ten Hove J, Hennink WE, Crommelin DJA, Jiskoot W. J Biol Chem. 2004;279:6235. doi: 10.1074/jbc.M310752200. [DOI] [PubMed] [Google Scholar]
- 25.Zhang W, Sack DA. Clin Vaccine Immunol. 2015;22:983. doi: 10.1128/CVI.00224-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon RJ, Lowy FD. Clin Infect Dis. 2008;46(Suppl 5):S350. doi: 10.1086/533591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wannamaker LW. Rev Infect Dis. 1983;5(Suppl 4):S723. doi: 10.1093/clinids/5.supplement_4.s723. [DOI] [PubMed] [Google Scholar]
- 28.Fujita Y, Taguchi H. Chem Cent J. 2011;5:48. doi: 10.1186/1752-153X-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molloy EM, Cotter PD, Hill C, Mitchell DA, Ross RP. Nat Rev Microbiol. 2011;9:670. doi: 10.1038/nrmicro2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shewen PE, Wilkie BN. Can J Vet Res. 1988;52:30. [PMC free article] [PubMed] [Google Scholar]
- 31.Ferreira RBR, Valdez Y, Coombes BK, Sad S, Gouw JW, Brown EM, Li Y, Grassl GA, Antunes LCM, Gill N, Truong M, Scholz R, Reynolds LA, Krishnan L, Zafer AA, Sal-Man N, Lowden MJ, Auweter SD, Foster LJ, Finlay BB. MBio. 2015;6:e01421. doi: 10.1128/mBio.01421-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geny B, Popoff MR. Biol Cell. 2006;98:667. doi: 10.1042/BC20050082. [DOI] [PubMed] [Google Scholar]
- 33.Otto M. Curr Opin Microbiol. 2014;17:32. doi: 10.1016/j.mib.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao W, Hu CMJ, Fang RH, Luk BT, Su J, Zhang L. Adv Mater. 2013;25:3549. doi: 10.1002/adma.201300638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Proc Natl Acad Sci U S A. 2011;108:10980. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu CMJ, Fang RH, Luk BT, Zhang L. Nat Nanotechnol. 2013;8:933. doi: 10.1038/nnano.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shallcross LJ, Fragaszy E, Johnson AM, Hayward AC. Lancet Infect Dis. 2013;13:43. doi: 10.1016/S1473-3099(12)70238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, Long RD, Dorward DW, Gardner DJ, Lina G, Kreiswirth BN, DeLeo FR. J Infect Dis. 2006;194:1761. doi: 10.1086/509506. [DOI] [PubMed] [Google Scholar]
- 39.Cooney J, Kienle Z, Foster TJ, O'Toole PW. Infect Immun. 1993;61:768. doi: 10.1128/iai.61.2.768-771.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Victora GD, Nussenzweig MC. Annu Rev Immunol. 2012;30:429. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 41.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. Active Bacterial Core surveillance (ABCs) MRSA Investigators. JAMA. 2007;298:1763. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 42.Gorwitz RJ. Pediatr Infect Dis J. 2008;27:1. doi: 10.1097/INF.0b013e31815819bb. [DOI] [PubMed] [Google Scholar]
- 43.Boucher H, Miller LG, Razonable RR. Clin Infect Dis. 2010;51(Suppl 2):S183. doi: 10.1086/653519. [DOI] [PubMed] [Google Scholar]
- 44.Gonzalez BE, Martinez-Aguilar G, Hulten KG, Hammerman WA, Coss-Bu J, Avalos-Mishaan A, Mason EO, Jr, Kaplan SL. Pediatrics. 2005;115:642. doi: 10.1542/peds.2004-2300. [DOI] [PubMed] [Google Scholar]
- 45.Fang RH, Jiang Y, Fang JC, Zhang L. Biomaterials. 2017;128:69. doi: 10.1016/j.biomaterials.2017.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dehaini D, Wei X, Fang RH, Masson S, Angsantikul P, Luk BT, Zhang Y, Ying M, Jiang Y, Kroll AV, Gao W, Zhang L. Adv Mater. 2017;29:1606209. doi: 10.1002/adma.201606209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu CMJ, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L. Nature. 2015;526:118. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Wang F, Ju E, Liu Z, Chen Z, Ren J, Qu X. Adv Funct Mater. 2016;26:6454. [Google Scholar]
- 49.Tao Y, Ju E, Li Z, Ren J, Qu X. Adv Funct Mater. 2014;24:1004. [Google Scholar]
- 50.Li Z, Liu Z, Yin M, Yang X, Ren J, Qu X. Adv Healthc Mater. 2013;2:1309. doi: 10.1002/adhm.201200364. [DOI] [PubMed] [Google Scholar]
- 51.Copp JA, Fang RH, Luk BT, Hu CMJ, Gao W, Zhang K, Zhang L. Proc Natl Acad Sci U S A. 2014;111:13481. doi: 10.1073/pnas.1412420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang RH, Hu CMJ, Luk BT, Gao W, Copp JA, Tai Y, O'Connor DE, Zhang L. Nano Lett. 2014;14:2181. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]