

Abstract
Background
A better understanding of the molecular mechanisms involved in papillary thyroid cancer (PTC)-associated adverse outcome is needed to effectively manage these patients. Our objectives were to identify molecular pathways associated with unfavorable features and outcome in patients with PTC.
Method
We performed genome-wide expression (GWE) analysis in 64 PTC human tissue samples. Clinical, pathologic and microarray data were analyzed to identify differentially expressed genes/pathways associated with an unfavorable outcome. Gene Set Enrichment Analysis (GSEA) was used to determine which molecular pathways are associated with mortality.
Results
GWE analysis identified 43, 115 and 40 genes that were significantly differentially expressed by gender, tumor differentiation status and mortality, respectively, with a false discovery rate (FDR) of <5%. For mortality, GSEA revealed 7 enriched pathways including transfer RNA synthesis, mitochondria and oxidative phosphorylation, porphyrin and chlorophyll metabolism, and fatty acid synthesis. Leading edge analysis showed 341 genes significantly involved in the enriched pathways. Cluster analysis using 100 differentially expressed genes showed complete separation of patients by mortality.
Conclusions
To our knowledge, this is the first GWE analysis of PTC and adverse outcome. Eleven molecular pathways were significantly associated with PTC mortality. A 100-gene signature completely separates patients with and without PTC-associated mortality.
Keywords: Thyroid cancer, molecular pathways, mortality, gene set enrichment analysis, genome wide expression
Introduction
Thyroid cancer is the most common endocrine malignancy and the fastest growing cancer diagnosis in the United States1. Papillary thyroid cancer (PTC) accounts for over 80% of all thyroid cancer cases. While most patients with PTC have an excellent prognosis, some patients die from loco-regional disease or distant metastasis. The overall mortality due to thyroid cancer is approximately 1%–2%2. Several clinicopathologic prognostic factors such as age (≥ 45 years old at diagnosis), large tumor size (> 4cm), extrathyroidal tumor extension, lymph node and distant metastases, aggressive tumor histologic subtypes, and the presence of BRAF mutation are associated with worse outcome3. A number of prognostic scoring systems have been utilized to accurately provide risk stratification for patients with thyroid cancer. On such widely used prognostic scoring system is the AMES prognostic index. This system categorizes patients into low-risk or high-risk group based on Age, distant Metastases, Extent of primary tumor and tumor Size4. Patients with high-risk tumors have a 54% survival rate at 20 years follow up compared to 98% for patients with low-risk tumors2. Although the prognostic scoring systems accurately predict patient outcome, none can be used preoperatively to risk stratify patients.
Several molecular markers have emerged as candidate diagnostic and/or prognostic markers for thyroid cancer, most notably BRAF mutation in PTC3. These findings not only could provide an improvement in diagnosis and risk stratification but also can be explored for targeted therapy. The success of the human genome project combined with advances in genomic technologies and bioinformatics have provided valuable information to help understand molecular oncogenesis.
The objective of our study was to identify genes and pathways associated with unfavorable outcome in patients with PTC. We used genome wide expression (GWE) analysis in 64 PTC human samples with demographic, clinicopathologic and outcome data to determine the presence of differentially expressed genes and pathways.
Material and Methods
The study was approved by the Office of Human Subject Research at the National Institute of Health.
Patients and tissue samples
Sixty-four primary PTC samples at the time of initial surgery were procured and snap frozen immediately after tumor removal. All the tumor tissue samples were reviewed by an endocrine pathologist to confirm the diagnosis and the presence of > 80% tumor cells. Patient demographics, clinical and pathological information were collected prospectively and follow up of patient was annually. The BRAF mutation status was analyzed in 55 tumor samples (Table 1).
Table 1.
Demographics, clinical and pathologic characteristics of study cohort
| Variables | Status | Number (%) |
|---|---|---|
| Gender | Male | 24 (37.5%) |
| Median Age (years) Range (years) |
41 17–82 |
|
| Race | White | 38 (59.4%) |
| Asian | 24 (37.5%) | |
| Other | 2 (3.1%) | |
| TNM Stage | I | 42 (65.6%) |
| II | 3 (4.7%) | |
| III | 4 (6.3%) | |
| IV | 15 (23.4%) | |
| Lymph node metastasis | Yes | 34 (53.1%) |
| No | 25 (39.1%) | |
| Unknown | 5 (7.8%) | |
| Extrathyroidal extension | Yes | 22 (34.4%) |
| No | 40 (62.5%) | |
| Unknown | 2 (3.1%) | |
| Synchronous distant metastasis (number of patients) | 1 | |
| Tumor size | > 4cm | 12 (18.8%) |
| Tumor differentiation | Well or moderately differentiated | 37 (57.8%) |
| Poorly differentiated | ^ 4 (6.3%) | |
| Unknown | 23 (35.9%) | |
| AMES classification | High risk | 23 (35.9%) |
| Low risk | 35 (54.7%) | |
| Unknown | 6 (9.4%) | |
| BRAF mutation | V600E Heterozygous | 45.5% |
| Tumor recurrence | Yes | 19 (29.7%) |
| No | 39 (60.9%) | |
| Unknown | 6 (9.4%) | |
| Site of recurrence | Locoregional | 14 (21.9%) |
| Distant | 3 (4.7%) | |
| Unknown | 2 (3.1%) | |
| PTC-associated Mortality | Yes | 10 (15.6%) |
1. RNA extraction and Microarray hybridization
Frozen thyroid tissue was sectioned for RNA extraction and adjacent tissue sections were examined by hemotoxylin and eosin staining to confirm the presence of PTC. Total RNA was extracted from homogenized frozen tissue using the TRIzol® reagent (Invitrogen, Carlsbad, CA) and purified using the RNeasy® Mini Kit (Qiagen, Valencia, CA). Sample purity was confirmed by measuring A260/A280 ratios. The quality of RNA was determined before labeling using the 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA). Two hundred ng of total RNA was amplified and labeled using the MessageAmp™ aRNA Kit (Ambion Inc., Foster City, CA). Twelve micrograms of labeled and fragmented complimentary RNA was hybridized to the Affymetrix Human Genome U133 plus 2.0 GeneChip® (Santa Clara, CA) for 16 hours at 45°C. The GeneChip® arrays were stained and washed, in the Affymetrix Fluidics Station 400, according to the manufacturer’s protocol. The probe intensities were measured using argon laser confocal GeneArray Scanner (Hewlett-Packard).
2. Quantitative Real-Time PCR (qRT-PCR) analysis
A subset of differentially expressed genes, in the GWE analysis, was validated by TaqMan® qRT-PCR using RNA from the same tissue samples. Two hundred ng of total RNA was reverse transcribed using high capacity cDNA reverse transcription kits (Applied Biosystems, Foster City, CA). The thermocycle condition for cDNA reaction was 25°C for 10 minutes, followed by 37°C for 120 minutes and 85°C for 5 minutes. We measured mRNA expression levels relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression. All PCR reactions were performed in a final volume of 10 μL using ABI PRISM® 7900 Sequence Detection System (Applied Biosystems, Foster City, CA). The PCR thermal cycler condition was 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. The following formula was used to determine gene expression levels, ΔCt= Ct of gene of interest - Ct of GAPDH, where Ct is the PCR cycle threshold. The relative fold difference was also calculated.
3. Data analysis and Statistical Methods
To generate intensity values in log2 scale, raw microarray data were analyzed using the affy package in the free statistical environment R/Bioconductor using the robust multiarray average method with default parameters. Gene expression levels were analyzed using R Project for Statistical Computing Program version 2.11.1 (Auckland, NZ) and SAS program version 9.00 (Cary, NC). To identify differentially expressed genes, we used t-statistics to analyze and compare microarray gene expression data by pathology and clinical variables (age, gender, AMES, initial lymph node and distant metastasis, extrathyroidal extension, tumor differentiation, local and distant recurrence, cause specific mortality). Associated p-values were adjusted for multiple testing by controlling for the false discovery rate (FDR) less than 5% using the Benjamini-Hochberg (BH) method5.
Salford Systems’ data mining software, CART® 6.0 (San Diego, CA) was used to calculate Relative Importance Score of differentially expressed genes identified by BH method using FDR<5% to pinpoint the most significant genes associated with mortality for validation. The closer the relative Importance Score is to 100 the stronger the association with the variable. We also performed Gene Set Enrichment Analysis (GSEA), as described by Subramanian et al.6 to help understand biological pathways involved in PTC in patients with mortality after initial surgery. The GSEA-P software for desktop (Cambridge, MA), freely available online at http://www.broad.mit.edu/GSEA, was used. The primary result of GSEA is the enrichment score (ES). For categorical phenotypes, such as mortality in this study, a positive enrichment score indicates correlation with no-mortality phenotype and a negative enrichment score indicates correlation with mortality phenotype. The proportion of false positives was controlled for using FDR. For the GSEA analysis, we used the built-in C2 curated gene sets version 2.5. The statistical significance of GSEA was analyzed using 1000 permutations. Since not all of the members in a gene set may have a significant role in a particular biological process, the members of genes that mainly contribute to ES, leading-edge subset, of differentially expressed gene sets were then further analyzed. A comparison of patients’ characteristics by mortality was performed using Fisher’s exact test, independent t-test and ANOVA. The normalized gene expression levels from qRT-PCR were analyzed using Mann-Whitney U test to identify significantly differentially expressed genes in patients stratified by mortality.
Results
The demographic and clinical data of the study cohort are summarized in Table 1. We found a number of significantly differentially expressed genes by gender, tumor differentiation status and mortality (Table 2).
Table 2.
Genome wide expression analysis in papillary thyroid cancer samples
| Group Stratification | Number of Differentially Expressed Genes | FDR adjusted p-value Range (BH* method) | |
|---|---|---|---|
| Mortality | Presence vs. Absence | 40 | <0.05 |
| Gender | Male vs. Female | 43 | <0.05 |
| Tumor differentiation | Poorly vs. Well differentiated | 115 | <0.05 |
| Age | ≥ 45 vs. <45 Years | 0 | NS** |
| AMES risk classification | Low-risk vs. high-risk | 0 | NS |
| Tumor size | ≥ 4 vs. <4 cm. | 0 | NS |
| Lymph node metastasis | Presence vs. Absence | 0 | NS |
| Extrathyroidal extension | Presence vs. Absence | 0 | NS |
| Recurrent tumor | Presence vs. Absence | 0 | NS |
BH: Benjamini-Hochberg method
NS: not statistically significant
We focused further analysis on genes and pathways associated with mortality in PTC. As expected, there were several unfavorable variables that were significantly different between patients with and without PTC-related mortality (Table 3). There was, however, no significant difference in mortality by BRAF mutation (V600E heterozygous) status, gender and tumor differentiation.
Table 3.
Demographic, clinical and pathologic variables by mortality status
| No mortality (n=54) | Mortality (n=10) | p-value | |
|---|---|---|---|
| Age | 41 +/− 16 years | 76 +/− 13 years | <0.01 |
| Male | 20/54 (37%) | 4/6 (66.7%) | 1.0 |
| Tumor size | 2.6 +/− 1.8 cm | 4.5 +/− 2.8 cm | <0.01 |
| Extrathyroidal extension | 15/52 (28.9%) | 7/10 (70%) | 0.03 |
| Lymph node metastasis | 25/49 (51%) | 9/10 (90%) | 0.03 |
| High-risk AMES | 16/48 (33.3%) | 7/8 (87.5%) | <0.01 |
| Poorly differentiated tumor | 2/36 (5.6%) | 2/5 (40%) | 0.07 |
| Tumor recurrence | 12/49 (24.5%) | 7/9 (77.8%) | <0.01 |
| BRAF V600E mutation | 20/25 (80%) | 5/25 (20%) | 0.45 |
| Synchronous metastasis | 0/48 | 1/10 (10%) | 0.14 |
| Follow up time | 74 +/− 31 months | 42 +/− 22 months | <0.01 |
We found 40 differentially expressed genes by mortality with FDR <0.05. The fold difference ranged from 1.1 to 3.3 fold (Table 2). All except one gene (Ras-related associated with diabetes) were independent to differentially expressed genes found in gender and tumor differentiation groups. Of the 40 genes differentially expressed, 6 genes had a relative important score above 20 and were validated to be differentially expressed by mortality (Table 4).
Table 4.
Differentially expressed genes identified by relative Important Score.
| Important Score | Gene Symbol | Gene Title | Normalized expression level in mortality vs. no mortality (fold) | Standard deviation | p-value |
|---|---|---|---|---|---|
| 100 | CPSF2 | cleavage and polyadenylation specific factor 2, 100kDa | 6.04 vs. 9.29 | 3.09 vs. 3.76 | 0.01 |
| 81 | LARS | leucyl-tRNA synthetase | 7.04 vs. 6.29 | 0.67 vs. 0.52 | 0.003 |
| 81 | AURKC | aurora kinase C | 0.90 vs. 1.86 | 1.12 vs. 2.05 | 0.002 |
| 81 | TFDP1 | Transcription factor Dp-1 | 1.38 vs. 0.94 | 1.6 vs. 1.35 | 0.77 |
| 81 | TRNT1 | tRNA nucleotidyl transferase, CCA-adding, 1 | 0.39 vs. 0.82 | 0.27 vs. 0.81 | 0.02 |
| 22 | BCL11A | B-cell CLL/lymphoma 11A (zinc finger protein) | 0.97 vs. 4.9 | 1.04 vs. 10.3 | 0.01 |
By GSEA, we identified 100 top ranked genes by mortality. Supervised cluster analysis of the 100 top ranked genes, showed complete clustering by mortality status (Figure 1). Further analysis showed 11 differentially expressed gene sets (Table 5), representing 7 pathways (transfer ribonucleic acid (tRNA) biosynthesis, mitochondrial oxygen transport and ATP metabolism, heme, porphyllin and chlorophyll metabolism, fatty acid synthesis, genes up-regulated by MYC, genes up-regulated in hepatocellular cancer (HCC), and down-regulated genes by anticancer drugs in pro-B cells). Leading edge analysis identified 341 genes from 11 differentially enriched gene sets that contributed to ES. The 6 most common leading edge genes present in 4 of 11 gene sets are summarized in Table 6.
Figure 1.
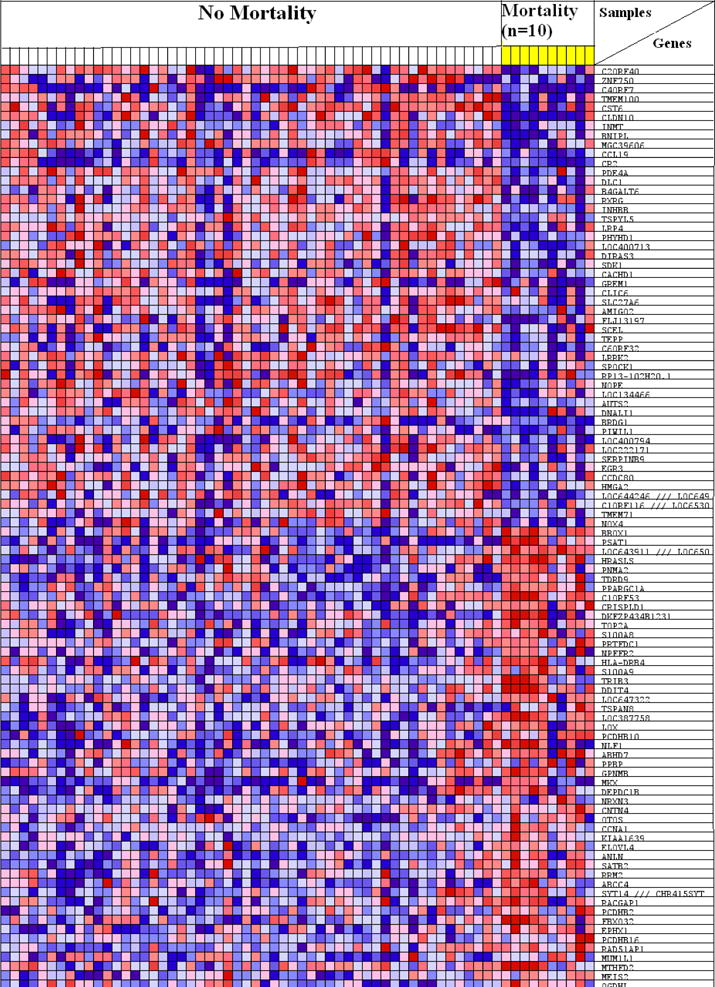
Heat map of top 100 ranked genes separates patients with and without mortality. Yellow columns represent samples from patients with mortality. In the heat map, relative gene expression levels are represented as colors (range: high (in red) to low (blue).
Table 5.
Differentially enriched gene sets in papillary thyroid cancer associated with mortality.
| Gene sets | DETAILS | No. Members in Gene Set | No. Members in Leading Edge(%) | NES* | FDR** q-val | FWER# p-val |
|---|---|---|---|---|---|---|
| AMINOACYL_TRNA _BIOSYNTHESIS | Gene set involved in Aminoacyl-tRNA biosynthesis | 23 | 14 (61%) 1 | −2.03 | 0.003 | 0.003 |
| HSA00970_AMINOACYL_TRNA_BIOSYNTHESIS | Gene set involved in Aminoacyl-tRNA biosynthesis | 31 | 18 (58%) | −2.00 | 0.007 | 0.01 |
| MITOCHONDRIA | Large gene set involved in oxygen transport, ATP metabolism, glycolysis | 391 | 214 (55%) | −1.94 | 0.016 | 0.04 |
| TRNA_SYNTHETASES | Gene set involved in Aminoacyl-tRNA biosynthesis | 19 | 12 (63%) | −1.92 | 0.020 | 0.06 |
| PORPHYRIN_AND_CHLOROPHYLL_MET ABOLISM | Gene set involved in heme, porphyrin and chlorophyll metabolism | 20 | 12 (60%) | −1.91 | 0.017 | 0.06 |
| HUMAN_MITODB_6_2002 | A set of genes involved in oxidative phosphorylation decreased in human diabetic muscle. Expression of these genes is high at sites of insulin-mediated glucose disposal, activated by PGC-1alpha and correlated with total-body aerobic capacity. | 382 | 229 (60%) | −1.88 | 0.026 | 0.09 |
| ZELLER_MY C_UP | genes that are up-regulated, or responsive, to Myc. | 23 | 10 (43%) | −1.86 | 0.030 | 0.11 |
| FATTY_ACID_SYNT HESIS | Genes involved in fatty acid synthesis | 16 | 11 (69%) | −1.84 | 0.037 | 0.16 |
| HSA00190_OXIDATIVE_PHO SPHORYLATION | Gene set involved in oxygen transport, ATP metabolism, glycolysis | 113 | 83 (73%) | −1.83 | 0.038 | 0.18 |
| SMITH_HCV_INDUC ED_HCC_UP | Potential marker genes specifically up-regulated in the majority of hepatocellular carcinoma (HCC) tumors. | 33 | 20 (61%) | −1.80 | 0.046 | 0.25 |
| CANCERDRUGS_PROBCELL_DN | Down-regulated by at least two of four cancer drugs (cisplatin, camptothecin, methotrexate and/or paclitaxel) in pro-B cells | 15 | 11 (73%) | −1.81 | 0.048 | 0.25 |
NES: normalized enriched score,
FDR: false discovery rate,
FWER: family wise error rate
Table 6.
Six most common genes from differentially enriched gene sets in papillary thyroid cancer associated with mortality.
| Genes | Name | Chromosome Location | Function |
|---|---|---|---|
| WARS2 | Tryptophanyl -tRNA synthetase, mitochondrial | Chr 1p12 | Aminoacyl-tRNA synthetases catalyze the aminoacylation of tRNA by their cognate amino acid. This gene encodes the mitochondrial tryptophanyl-tRNA synthetase. |
| EPRS | Glutamyl-prolyl-tRNA synthetase | Chr 1 | This gene is a multifunctional aminoacyl-tRNA synthetase that catalyzes the aminoacylation of glutamic acid and proline tRNA species. |
| BCL2 | B-cell CLL/lymphoma 2 | Chr 18q21.33 | This gene encodes an integral outer mitochondrial membrane protein that blocks the apoptotic death of some cells such as lymphocytes. |
| SDHC | Succinate dehydrogenase complex, subunit C, integral membrane protein | Chr 1q23.3 | One of four nuclear-encoded subunits that comprise succinate dehydrogenase, also known as mitochondrial complex II, a key enzyme complex of the tricarboxylic acid cycle and aerobic respiratory chains of mitochondria. |
| LARS2 | leucyl-tRNA synthetase 2, mitochondrial | Chr 3p21.3 | Aminoacyl-tRNA synthetases catalyze the aminoacylation of tRNA by their cognate amino acid. This gene encodes mitochondrial leucyl-tRNA synthetase. |
| SLC25A1 | solute carrier family 25 (mitochondrial carrier; citrate transporter), member 1 | Chr 22q11.21 | The mitochondrial tricarboxylate transporter (also called citrate transport protein, or CTP) is responsible for the movement of citrate across the mitochondrial inner membrane |
Discussion
Recent advances in high throughput, genomic technology have made personalized oncologic treatment available for some cancers. For example, the 21-gene recurrent score in breast cancer is being used to tailor adjuvant treatment recommendation based on individual patient sample analysis7. The application of genomic approaches to thyroid cancer could greatly improve our ability to identify the group of patients, who are at risk of mortality from their thyroid cancer and to tailor therapy as most cases of thyroid cancer are associated with a good prognosis. The goal of our study was to identify genes and pathways associated with mortality in PTC. The clinical application of molecular markers could not only confirm the prognosis preoperatively in patients, but also could guide therapeutic decision towards more aggressive approach for those without obvious clinical risk factors. We validated 5 genes that were most significantly associated with mortality from PTC. Patients with PTC-associated mortality usually have several features of aggressive tumor behavior (Table 3) but we found no differentially expressed genes among these known risk factors. This finding emphasizes the independent association of the differentially expressed genes identified and mortality. Analysis of microarray data demonstrated 11 enriched gene sets from various pathways associated with mortality in PTC (Table 5).
Three of eleven enriched gene sets, three of six most common genes from leading edge analysis and two of five significantly differentially expressed genes associated with mortality in PTC are involved in tRNA biosynthesis. This finding suggests abnormality in tRNA biosynthesis may have an important role in the aggressiveness of PTC. Our study showed overexpression of LARS and downregulation of TRNT1 which may contribute to aggressive behavior of PTC. Shin et al. demonstrated a reduction in LARS expression leads to decreased lung cancer cell migration and decreased colony formation8. On the other hand, inactivation of leucyl-tRNA synthetase in mitochondria (LARS2) was described in nasopharyngeal cancer cells9. Dysregulation of aminoacyl-tRNA synthesis has also been associated with many types of cancers including nasopharyngeal, prostate, breast, colon, and lung cancer10.
The role of tryptophanyl-tRNA synthetase (WARS) and EPRS genes, identified by leading edge analysis (Table 6), in thyroid cancer is not yet understood. Low expression of WARS in tumor tissue of colorectal cancer has been associated with increased risk of recurrence and worse survival11. WARS is involved in inflammatory and angiogenic signaling pathways11. After proteolysis or alternative splicing, WARS is an active inhibitor of angiogenesis12. EPRS gene has multifunctional tandem repeats that can bind with various tRNA synthetases, which can result in diverse cellular activities. Phosphorylation of EPRS is reported to be essential for the formation of GAIT (Gamma-interferon Activated Inhibitor of Translation) complex that regulates the translation of multiple genes in monocytes and macrophages13.
Our study showed downregulated Aurora kinase-C gene in patients with mortality associated with PTC. Aurora kinases (AURK) are the members of serine/threonine kinases which are important to cell cycle control, chromosomal segregation and cytokenesis. There are 3 members in the family AURK-A, -B and –C. Aurora-A is associated with centrosomes positioning, forming mitotic spindle and microtubules stability14. AURKB forms a complex with other chromosome passenger proteins such as inner centromere protein (INCENP). AURKB is associated with chromatin at the beginning of mitosis14. Disruption of AURKB causes chromosome misalignment and cytokinesis failure15. The function of AURKC is not as well understood. AURKC can phosphorylate endogenous histone H3 and evidence suggests that AURKB and AURKC are functionally overlapping16. Baldini and colleagues reported deregulated expression of all three aurora kinases in testicular germ cell tumor17. The downregulation of AURKC found in our study may result in dysregulated mitosis and more aggressive PTC. However, the increased expression of AURKA and AURKB protein levels have been reported in PTC and anaplastic thyroid cancer (ATC) cells. In-vivo treatment of ATC cell lines using AURK inhibitors (MLN8054, VX 680) resulted in marked tumor volume and vascular reduction by inducing apoptosis, causing cell cycle arrest18.
The role of cleavage and polyadenylation specificity factor (CPSF) in cancer biology is not fully understood. Cleavage and polyadenylation of precursor mRNA (pre-mRNA) is an essential process during mRNA biogenesis, including transcription initiation, elongation and termination19, Rozenblatt-Rosen et al. demonstrated an association of CDC73 gene, a tumor suppressor gene found inactivated in hereditary and sporadic parathyroid tumors, and a complex of CPSF and cleavage stimulation factor20. A downregulation of this gene in PTC associated with mortality may result in decrease of tumor suppressor protein.
The role of BCL11A, a transcription factor involved lymphoid proliferation21 and hemoglobin F synthesis22, in non-hematologic malignancy is unknown. Our study demonstrated a downregulation of this gene in PTC associated mortality. BCL2 translocation [t(14;18)(q32.3;q21.3)] are common in follicular lymphoma (80%)23. Overexpression of BCL2 results in a decrease in apoptosis which has been reported in a variety of solid tumors24. The presence of BCL2 in autoimmune thyroiditis was significant lower in nonthyroiditis tissue samples25. Both BCL11A and BCL2 involve in lymphoid proliferation and they may be associated with lymphocytic thyroiditis in PTC associated mortality specimen.
The results from this study provided an important background for future research to identify genetic markers associated with mortality in PTC. Ultimately, more aggressive treatment and surveillance in these patients may be beneficial. In addition, pathways and genes identified in our study, particularly tRNA biosynthesis should be further explored for a potential use as a targeted therapy.
To our knowledge, this is the first study to use GWE analysis and GSEA to gain a better insight of the genes and pathways associated with adverse outcome in PTC. It will be important to validate these results in independent PTC samples, as well as, to perform functional study in cell lines to elucidate their roles in thyroid cancer biology.
Summary
Our study demonstrates several genes and pathways associated with mortality from PTC. The function of many of the genes and pathways is not fully understood. Further research to gain better understanding of their mechanisms is warranted.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. Jama. 2006;295(18):2164–7. doi: 10.1001/jama.295.18.2164. [DOI] [PubMed] [Google Scholar]
- 2.Cady B, Rossi R. An expanded view of risk-group definition in differentiated thyroid carcinoma. Surgery. 1988;104(6):947–53. [PubMed] [Google Scholar]
- 3.Sipos JA, Mazzaferri EL. Thyroid cancer epidemiology and prognostic variables. Clin Oncol (R Coll Radiol) 22(6):395–404. doi: 10.1016/j.clon.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Haigh PI, Urbach DR, Rotstein LE. AMES prognostic index and extent of thyroidectomy for well-differentiated thyroid cancer in the United States. Surgery. 2004;136(3):609–16. doi: 10.1016/j.surg.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Hochberg YBY. Controlling of the False Discovery Rate: A practical and powerful approach to multiple testing. Journal of the Royal statistical Society, Series B. 1995;57(1):289–300. [Google Scholar]
- 6.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mamounas EP, Tang G, Fisher B, et al. Association between the 21-gene recurrence score assay and risk of locoregional recurrence in node-negative, estrogen receptor-positive breast cancer: results from NSABP B-14 and NSABP B-20. J Clin Oncol. 28(10):1677–83. doi: 10.1200/JCO.2009.23.7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin SH, Kim HS, Jung SH, Xu HD, Jeong YB, Chung YJ. Implication of leucyl-tRNA synthetase 1 (LARS1) over-expression in growth and migration of lung cancer cells detected by siRNA targeted knock-down analysis. Experimental & molecular medicine. 2008;40(2):229–36. doi: 10.3858/emm.2008.40.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou W, Feng X, Li H, et al. Inactivation of LARS2, located at the commonly deleted region 3p21.3, by both epigenetic and genetic mechanisms in nasopharyngeal carcinoma. Acta biochimica et biophysica Sinica. 2009;41(1):54–62. doi: 10.1093/abbs/gmn006. [DOI] [PubMed] [Google Scholar]
- 10.Park SG, Schimmel P, Kim S. Aminoacyl tRNA synthetases and their connections to disease. Proc Natl Acad Sci U S A. 2008;105(32):11043–9. doi: 10.1073/pnas.0802862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghanipour A, Jirstrom K, Ponten F, Glimelius B, Pahlman L, Birgisson H. The prognostic significance of tryptophanyl-tRNA synthetase in colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2009;18(11):2949–56. doi: 10.1158/1055-9965.EPI-09-0456. [DOI] [PubMed] [Google Scholar]
- 12.Otani A, Kinder K, Ewalt K, Otero FJ, Schimmel P, Friedlander M. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nature medicine. 2002;8(9):1004–10. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- 13.Arif A, Jia J, Mukhopadhyay R, Willard B, Kinter M, Fox PL. Two-site phosphorylation of EPRS coordinates multimodal regulation of noncanonical translational control activity. Mol Cell. 2009;35(2):164–80. doi: 10.1016/j.molcel.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carmena M, Ruchaud S, Earnshaw WC. Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol. 2009;21(6):796–805. doi: 10.1016/j.ceb.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams RR, Maiato H, Earnshaw WC, Carmena M. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J Cell Biol. 2001;153(4):865–80. doi: 10.1083/jcb.153.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasai K, Katayama H, Stenoien DL, et al. Aurora-C kinase is a novel chromosomal passenger protein that can complement Aurora-B kinase function in mitotic cells. Cell Motil Cytoskeleton. 2004;59(4):249–63. doi: 10.1002/cm.20039. [DOI] [PubMed] [Google Scholar]
- 17.Baldini E, Arlot-Bonnemains Y, Mottolese M, et al. Deregulation of Aurora kinase gene expression in human testicular germ cell tumours. Andrologia. 42(4):260–7. doi: 10.1111/j.1439-0272.2009.00987.x. [DOI] [PubMed] [Google Scholar]
- 18.Wunderlich A, Fischer M, Schlosshauer T, et al. Evaluation of Aurora kinase inhibition as a new therapeutic strategy in anaplastic and poorly differentiated follicular thyroid cancer. Cancer Sci. doi: 10.1111/j.1349-7006.2011.01853.x. [DOI] [PubMed] [Google Scholar]
- 19.Proudfoot N. New perspectives on connecting messenger RNA 3′ end formation to transcription. Curr Opin Cell Biol. 2004;16(3):272–8. doi: 10.1016/j.ceb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Rozenblatt-Rosen O, Nagaike T, Francis JM, et al. The tumor suppressor Cdc73 functionally associates with CPSF and CstF 3′ mRNA processing factors. Proc Natl Acad Sci U S A. 2009;106(3):755–60. doi: 10.1073/pnas.0812023106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao L, Kang SH, Li J, et al. Array comparative genomic hybridization detects chromosomal abnormalities in hematological cancers that are not detected by conventional cytogenetics. J Mol Diagn. 12(5):670–9. doi: 10.2353/jmoldx.2010.090192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sankaran VG, Xu J, Orkin SH. Transcriptional silencing of fetal hemoglobin by BCL11A. Ann N Y Acad Sci. 1202:64–8. doi: 10.1111/j.1749-6632.2010.05574.x. [DOI] [PubMed] [Google Scholar]
- 23.Willis TG, Dyer MJ. The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood. 2000;96(3):808–22. [PubMed] [Google Scholar]
- 24.Almubarak H, Jones A, Chaisuparat R, Zhang M, Meiller TF, Scheper MA. Zoledronic acid directly suppresses cell proliferation and induces apoptosis in highly tumorigenic prostate and breast cancers. J Carcinog. 10:2. doi: 10.4103/1477-3163.75723. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Chen S, Fazle Akbar SM, Zhen Z, et al. Analysis of the expression of Fas, FasL and Bcl-2 in the pathogenesis of autoimmune thyroid disorders. Cell Mol Immunol. 2004;1(3):224–8. [PubMed] [Google Scholar]