

Abstract
Background
Complex chromosomal rearrangements (CCRs) are constitutional structural rearrangements that involve three or more chromosomes or that have more than two breakpoints.
Case presentation
Here, we describe a four-way CCR involving chromosomes 4, 5, 6 and 8. The patient had mild multisystematic abnormalities during his development, including defects in his eyes and teeth, exomphalos and asthenozoospermia. His wife had two spontaneous abortions during the first trimester. The translocations in 4q27, 5q22, 6q22.3, and 8p11.2 were diagnosed by conventional cytogenetic analysis and confirmed by fluorescence in situ hybridization(FISH). After analysis using a SNP array, we defined three microdeletions, including 0.89 Mb on chromosome 4, 5.39 Mb on chromosome 5 and 0.43 Mb on chromosome 8. His mother had a chimera karyotype of 47, XXX[5]/45, X[4]/46, XX[91]; the other chromosomes were normal. After one cycle of in vitro fertility (IVF) treatment followed by preimplantation genetic diagnosis (PGD), they obtained two embryos, but neither was balanced.
Conclusions
The patient’s phenotype resulted from the CCR and microdeletion of chromosomes 4, 5 and 8. The couple decided to use artificial insemination by donor (AID) technology.
Keywords: CCR, SNP-array, FISH, Microdeletion, Rearrangement, AID
Background
Complex chromosome rearrangements (CCRs) are structural aberrations involving more than two chromosome breaks with exchanges of several chromosomal fragments [1]. The phenotype of individuals with CCRs can be normal; this largely depends on whether or not the CCR is balanced or whether developmentally important gene(s) are disrupted at the breakpoints. Balanced CCRs contain unchanged amounts of genes but unbalanced CCRs do not. According to the literature, approximately 70% of CCRs are detected in people without a phenotype, 20-25% are detected in patients with congenital abnormalities and/or mental retardation and 5-10% are detected during prenatal diagnosis [2]. Occasionally, cases have been detected because of psychiatric trouble [3, 4]. In addition, CCRs are frequently observed in tumor cells, especially in hematological malignancies [5]. Among phenotypically normal CCR carriers, most suffer reproductive failures, including spontaneous abortions, stillbirths, the delivery of children with congenital malformations, and male infertility [6, 7].
There are various classifications of CCRs due to their complex nature. CCRs can be considered familial or de novo, according to the mode of transmission [8]. Based on the number of chromosome breaks, CCRs are divided into two groups: those with four or fewer breaks and those with more than four breaks [9]. CCRs are also divided into three classes according to their structure [10]: 1) three-way rearrangement, which refers to three chromosome breaks and exchanges of chromosomal segments; 2) exceptional CCRs, involving rearrangements in which there is more than one breakpoint per chromosome; and 3) double two-way translocations, which indicates two or three independent, simple reciprocal or Robertsonian translocations that co-exist in the same carrier.
Here, we describe a de novo CCR case that involves four chromosomes and four breakpoints. The patient displayed mild multisystematic abnormalities, which were identified by conventional cytogenetics and molecular genetic technologies. After a failure to obtain normal embryos with PGD, they chose to accept AID with donor spermatozoa.
Case presentation
A 25-year-old man and his 26-year-old wife were referred to our reproductive medical center due to two spontaneous abortions in the past 3 years after their marriage. The abortions occurred at the sixth and seventh week of gestation for the first and second time, respectively.
The physical examination of the husband showed that his eyes had refractive errors; his left eye displayed congenital amblyopia and his vision was 0.2 (Fig. 1a). He also had bilateral primary open angle glaucoma (POAG) and the intraocular pressure of his eyes was more than 40 mmHg. His eyes were subjected to a trabeculotomy and the intraocular pressure was well-controlled. He also had exomphalos (Fig. 1b). His two central incisors were congenitally lost as implant, his lower right primary canine was retained (Fig. 1c) and his lower left permanent canine was congenitally missing (Fig. 1d). He had graduated from high school and is now employed. He can communicate normally. His routine semen analysis demonstrated a sperm deformity rate of 99%, sperm viability rate of 9.56%, DNA fragmentation index (DFI) of 13.58%, and high DNA stainability (HDS) of 15.36%.
Fig. 1.

a Mug shot of the patient. b Exomphalos of the patient. c Two central incisors were congenitally lost as implant; the lower right primary canine was retained. d The lower left permanent canine was congenitally missing
He is the second child of non-consanguineous parents and has two sisters. His parents and both sisters are healthy. After discovering his chromosome abnormalities, his family members underwent genetic testing (except his older sister, who was abroad).
After genetic counseling, the couple insisted on preimplantation genetic diagnosis (PGD). Initially, they obtained two embryos to undergo PGD; both were unbalanced. After counseling, they decided to accept artificial insemination with donor spermatozoa.
Methods and results
Metaphase chromosomes obtained from colchicine-stimulated cultures of peripheral blood lymphocytes and fibroblast cultures were used for GTL-banding and fluorescence in situ hybridization(FISH) analysis. FISH was performed according to the method described by Wieczorek et al. [11]. A SNP-array was performed using Cyto12 genechip (Illumina, USA) according to the manufacturer’s instructions.
The patient, his parents and his younger sister were examined. A classical cytogenetic examination revealed that the patient’s karyotype was 46, XY, t(4; 8; 6; 5) (q27; p11.2; q22.3; q22) (Fig. 2a). The father and younger sister’s G-banded karyotypes were normal. Unexpectedly, his mother’s karyotype was 47, XXX[5]/45, X[4]/46, XX[91] (data not shown). As the patient and his wife had described, his mother was normal during her pregnancy with the patient.
Fig. 2.
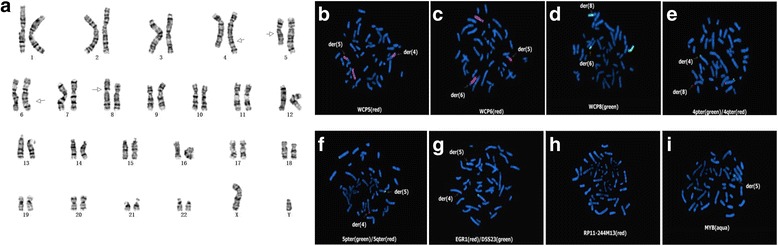
Images of karyotype and FISH. a. Metaphase spread in GTG-banding obtained from the patient’s blood lymphocytes showing t(4;8;6;5) (q27;p11.2;q22.3;q22). Arrows show abnormal chromosomes. b-d. Images of FISH with whole chromosome painting (WCP) probes. Chromosome 5 is red in B, chromosome 6 is red in C, chromosome 8 is green in D. Material from der(4) is present on der(5), whereas material from der(5) is present on der(6). Material from der(6) is present on der(8). e-f. Images of FISH with terminal probes. 4pter is green, 4qter is red in E. 5pter is green, 5qter is red in F. g-i. The metaphase spread image of FISH with the EGR (5q31.2) probe in red, D5S23 (5p15.2) probe in green (G), RP11-244 m13(5q21.1q21.2) probe in red (H) and MYB probe (6q23.2) in green (I). [GRCh38/hg38]
To confirm the presence of a complex translocation involving four chromosomes, FISH was performed with the whole chromosome painting (wcp) probes wcp5, wcp6, wcp8, the chromosome terminal probes 4pter, 4qter, 5pter and 5qter, as well as the specific probes EGR1, D5S23, DMYB and RP11-244 M13. The combined karyotype of the patient was 46, XY,? t(4;8;6;5) (q27;p11.2;q22.3;q22).ish der(4) t(4;8) (q27;p11.2) (4pter+,4qter-,EGR1+,5qter+), der(8) t(8;6) (p11.2;q22.3) (8pter-, 4qter+, WCP8+), der(6) t(6;5) (q22.3;q22) (WCP8+, WCP6+), der(5) del(5) (q21.1q21.3) t(5;4) (q22;q27) (RP11-244 M13-, 5pter+, D5S23+, EGR1-, 5qter-, WCP6+, MYB+) (Fig. 2).
After using wcp, we observed material from der(5) present on der(4), material from der(6) present on der(5), and material from der(8) present on der(6) (Fig. 2b-d). The rearrangement of these chromosomes was confirmed by the terminal and specific probes (Fig. 2e-i). When the probes were hybridized with 4pter and 4qter, we observed that 4qter was present on der(8), whereas 5qter was present on der(4) (Fig. 2e-f). When we used the probes EGR and D5S23, which are specific for 5q31.2 and 5p15.2, respectively, one EGR signal was observed on der(4) (Fig. 2g). When we used the probe RP11-244 M13, which is specific for 5q21.1q21.2, only one copy was observed, indicating a deletion on chromosome 5 (Fig. 2h). A signal from the MYB probe, which is specific for 6q23.2, was observed on der(5) (Fig. 2i). The FISH results were in accordance with the karyotype.
To confirm the FISH results and to determine the presence of microdeletions during chromosome rearrangement, we examined DNA from the patient’s peripheral blood using a SNP-array assay according to the manufacturer’s instructions (Illumina). The results were as follows: arr[hg19] 4q25(110499958-111,393,691)×1, arr[hg19] 5q21.1(100718992-106,104,465)×1 and arr[hg19] 8p22(14547284-14,972,402)×1, which revealed three microdeletions on three different chromosomes (Fig. 3). The genes involved in these regions are shown in Table 1. These genes included five OMIM genes (which belong to the phospholipase A2 group XIIA (PLA2G12A)), ELOVL fatty acid elongase 6 (ELOVL6), solute carrier organic anion transporter family member 4C1 (SLCO4C1), diphosphoinositol pentakisphosphate kinase 2 (PPIP5K2, also known as HISPPD1) and nudix hydrolase 12 (Nudt12). These genes are not associated with known disorders. Among them, SLCO4C1 is an organic anion transporter, HISPPD1 is a kinase (which acts as a cell signaling molecule), and the remaining genes are enzyme-encoding genes that are involved in several metabolic processes, including phospholipid, fatty acid and nucleotide metabolism. Interestingly, two genes, RRH and LRIT3, were related to his ocular disorder. RRH (retinal pigment epithelium-derived rhodopsin homolog) belongs to the seven-exon subfamily of mammalian opsin genes [12]; mutation of this gene has been linked to retinitis pigmentosa and allied diseases [13].
Fig. 3.
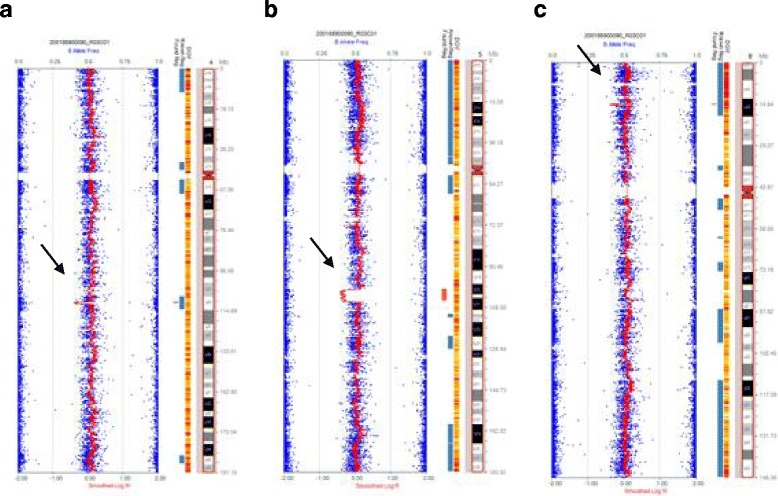
The SNP-array results of the patient. Microdeletions on chromosome 4 (a), 5 (b) and 8 (c) were shown. The arrows indicate the sites of microdeletions
Table 1.
Detailed information on the microdeletions resulting from chromosomal rearrangement
| Chromosome | Genomic position (GRCh37/hg19) | Cytobands | Size (M kb) | Copy number | Genes |
|---|---|---|---|---|---|
| 4 | 110,499,958-111,393,691 | q25 | 0.89 | 1 | CCDC109B; CASP6; PLA2G12A; CFI; GAR1; RRH; LRIT3; EGF; ELOVL6; |
| 5 | 100,718,992-106,104,465 | q21.1 q21.2 q21.3 | 5.39 | 1 | SLCO4C1; SLCO6A1; PAM; GIN1; HISPPD1; C5orf30; NUDT12; RAB9P1; |
| 8 | 14,547,284-14,972,402 | p22 | 0.43 | 1 | SGCZ; MIR383; |
The LRIT3 (leucine rich repeat, Ig-like and transmembrane domains 3) encoded protein may regulate fibroblast growth factor receptors and affect the modification of these receptors, which are glycosylated differently in the Golgi and endoplasmic reticulum. Mutations in this gene are associated with congenital stationary night blindness, type 1F [14]. Our results demonstrate that although these genes are not associated with known disorders, they show haploinsufficiency.
To determine whether the patient’s microdeletions were inherited from his parents or whether they appeared de novo, the parents were subjected to a SNP-array analysis. The results demonstrated that the parents do not have microdeletions in the three chromosomes mentioned above (Fig. 4), indicating that the loss of chromosome fragments was derived from rearrangement.
Fig. 4.

The CCR patient’s pedigree. I represents the patient’s parents; II represents the patient and his sisters; II-2 is the patient
Discussion and conclusion
Here, we describe a four-way CCR involving several microdeletions on chromosomes 4, 5, 6 and 8. The patient had mild multisystematic abnormalities during development, including defects in his eyes and teeth, exomphalos and asthenozoospermia. After completing a cycle of PGD, he did not obtain normal embryos and decided to use AID.
CCRs are rare events with an estimated frequency of 0.1% [15]. Most CCR cases are unknown to the carriers or their families. Some chromosomes, including 2, 3, 4, 7, and 11, are more frequently implicated in CCR than would be expected. This is the first CCR case to involve chromosomes 4, 5, 6, and 8 [16, 17].
CCRs can involve up to 15 breakpoints. According to a 2011 summary, cases that included four breakpoints accounted for 29.1% of all 251 CCR cases [2].
However, this CCR occurred de novo; the patient’s mother’s karyotype was 47, XXX[5]/45, X[4]/46,XX[91], and she had a low level of mosaic 47, XXX and 45, X, which was less than 10%. She had three children at 19, 21 and 35 years of age and had no fertility issues during her childbearing age. Her mosaic karyotype is possibly due to a gain or a loss of X chromosomes as she aged [18, 19] or chromosomal nondisjunction during the culture of peripheral blood lymphocytes.
Breakpoint analysis of a growing number of complex rearrangements has revealed that translocations involving three or more chromosomes are likely formed via chromothripsis [20–23]. Most constitutional chromothripsis events occur de novo and those investigated thus far have been verified as paternal in origin [20–23]. Alternatively, mitotic errors in the early embryo [24] or the pulverization of micronuclei [25] could be responsible for numerous DNA breaks. We speculate that this de novo CCR is due to chromothripsis.
According to the literature, a three-way CCR would theoretically form 64 different gametes: one normal, one balanced, and the rest unbalanced [2]. A four-way CCR, as in this case study, has a probability of producing normal and balanced gametes of less than 1/32. We disclosed this possible risk and as a result of genetic counseling, the couple opted for PGD. After a failure to obtain normal embryos with PGD, they chose to accept AID with donor spermatozoa.
In conclusion, we systematically investigated this CCR and the accompanying microdeletions and were able to characterize the genetic defects that resulted in the patient’s multisystematic abnormalities, which had bothered him for many years. After receiving genetic counseling, the couple understood that they could not conceive a chromosomally balanced child because the husband had microdeletions in three chromosomes. They chose to undergo AID.
Acknowledgments
Not applicable.
Funding
This work was supported by the Chinese National Natural Science Foundation Grant (81472004), the National Key Research and Development Program, Ministry of Science and Technology, P.R. China (Grant No. 2016YFC1000500, 2016YFC1000501). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
Data sharing is not applicable for this article as no datasets were generated or analyzed during the current study.
Abbreviations
- AID
Artificial insemination by donor
- CCR
Complex chromosome rearrangements
- FISH
Fluorescence in situ hybridization
- PGD
Preimplantation diagnosis
Authors’ contributions
CT and DL performed the genetic analysis, collected data from the patient, and wrote the manuscript. PL is the patient’s doctor. LJ and XG performed the genetic analysis. JQ designed the study and revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Peking University Third Hospital. The committee’s reference number is 2016S2-021. The patient gave his consent for participation in this study.
Consent for publication
Informed written consent for publication of the clinical details was obtained from the patient. A copy of the consent form is available for inspection by the editors.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chan Tian, Email: tianchan_cdc@126.com.
Dan Li, Email: lidan040901@126.com.
Ping Liu, Email: pingliu7703@sina.com.
Liping Jiao, Email: jlp9100@163.com.
Xuefeng Gao, Email: xuefenggao4034@126.com.
Jie Qiao, Email: jie.qiao@263.net.
References
- 1.Pai GS, Thomas GH, Mahoney W, Migeon BR. Complex chromosome rearrangements. Report of a new case and literature review. Clin Genet. 1980;18(6):436–444. doi: 10.1111/j.1399-0004.1980.tb01790.x. [DOI] [PubMed] [Google Scholar]
- 2.Pellestor F, Anahory T, Lefort G, et al. Complex chromosomal rearrangements: origin and meiotic behavior. Hum Reprod Update. 2011;17(4):476–494. doi: 10.1093/humupd/dmr010. [DOI] [PubMed] [Google Scholar]
- 3.Lespinasse J, Bugge M, Rethore MO, North MO, Lundsteen C, Kirchhoff M. De novo complex chromosomal rearrangements (CCR) involving chromosome 1, 5, and 6 resulting in microdeletion for 6q14 in a female carrier with psychotic disorder. Am J Med Genet A. 2004;128A(2):199–203. doi: 10.1002/ajmg.a.30064. [DOI] [PubMed] [Google Scholar]
- 4.Goumy C, Mihaescu M, Tchirkov A, et al. De novo balanced complex chromosome rearrangement (CCR) involving chromosome 8, 11 and 16 in a boy with mild developmental delay and psychotic disorder. Genet Couns. 2006;17(3):371–379. [PubMed] [Google Scholar]
- 5.De Braekeleer E, Meyer C, Douet-Guilbert N, et al. Complex and cryptic chromosomal rearrangements involving the MLL gene in acute leukemia: a study of 7 patients and review of the literature. Blood Cells Mol Dis. 2010;44(4):268–274. doi: 10.1016/j.bcmd.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Bartels I, Starke H, Argyriou L, Sauter SM, Zoll B, Liehr T. An exceptional complex chromosomal rearrangement (CCR) with eight breakpoints involving four chromosomes (1;3;9;14) in an azoospermic male with normal phenotype. Eur J Med Genet. 2007;50(2):133–138. doi: 10.1016/j.ejmg.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Kim JW, Chang EM, Song SH, Park SH, Yoon TK, Shim SH. Complex chromosomal rearrangements in infertile males: complexity of rearrangement affects spermatogenesis. Fertil Steril. 2011;95(1):349–352. doi: 10.1016/j.fertnstert.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 8.Kleczkowska A, Fryns JP, Van den Berghe H. Complex chromosomal rearrangements (CCR) and their genetic consequences. J Genet Hum. 1982;30(3):199–214. [PubMed] [Google Scholar]
- 9.Kousseff BG, Papenhausen P, Essig YP, Torres MP. Complex chromosome rearrangement with ankyloblepharon filiforme adnatum. J Med Genet. 1993;30(2):167–170. doi: 10.1136/jmg.30.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kausch K, Haaf T, Kohler J, Schmid M. Complex chromosomal rearrangement in a woman with multiple miscarriages. Am J Med Genet. 1988;31(2):415–420. doi: 10.1002/ajmg.1320310221. [DOI] [PubMed] [Google Scholar]
- 11.Wieczorek D, Engels H, Viersbach R, Henke B, Schwanitz G, Passarge E. Analysis of a familial three way translocation involving chromosomes 3q, 6q, and 15q by high resolution banding and fluorescent in situ hybridisation (FISH) shows two different unbalanced karyotypes in sibs. J Med Genet. 1998;35(7):545–553. doi: 10.1136/jmg.35.7.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellingham J, Wells DJ, Foster RG. In silico characterisation and chromosomal localisation of human RRH (peropsin)--implications for opsin evolution. BMC Genomics. 2003;4(1):3. doi: 10.1186/1471-2164-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rivolta C, Berson EL, Dryja TP. Mutation screening of the peropsin gene, a retinal pigment epithelium specific rhodopsin homolog, in patients with retinitis pigmentosa and allied diseases. Mol Vis. 2006;12:1511–1515. [PubMed] [Google Scholar]
- 14.Zeitz C, Jacobson SG, Hamel CP, et al. Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2013;92(1):67–75. doi: 10.1016/j.ajhg.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nonaka T, Ooki I, Enomoto T, Takakuwa K. Complex chromosomal rearrangements in couples affected by recurrent spontaneous abortion. Int J Gynaecol Obstet. 2015;128(1):36–39. doi: 10.1016/j.ijgo.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 16.Zhang F, Carvalho CM, Lupski JR. Complex human chromosomal and genomic rearrangements. Trends Genet. 2009;25(7):298–307. doi: 10.1016/j.tig.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Dai Y, Tu Z, Li Q, Zhang L, Wang L. Array-CGH detection of three cryptic submicroscopic imbalances in a complex chromosome rearrangement. J Genet. 2009;88(3):369–372. doi: 10.1007/s12041-009-0056-4. [DOI] [PubMed] [Google Scholar]
- 18.Guttenbach M, Koschorz B, Bernthaler U, Grimm T, Schmid M. Sex chromosome loss and aging: in situ hybridization studies on human interphase nuclei. Am J Hum Genet. 1995;57(5):1143–1150. [PMC free article] [PubMed] [Google Scholar]
- 19.Nowinski GP, Van Dyke DL, Tilley BC, et al. The frequency of aneuploidy in cultured lymphocytes is correlated with age and gender but not with reproductive history. Am J Hum Genet. 1990;46(6):1101–1111. [PMC free article] [PubMed] [Google Scholar]
- 20.Weckselblatt B, Hermetz KE, Rudd MK. Unbalanced translocations arise from diverse mutational mechanisms including chromothripsis. Genome Res. 2015;25(7):937–947. doi: 10.1101/gr.191247.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kloosterman WP, Tavakoli-Yaraki M, van Roosmalen MJ, et al. Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell Rep. 2012;1(6):648–655. doi: 10.1016/j.celrep.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Kloosterman WP, Guryev V, van Roosmalen M, et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum Mol Genet. 2011;20(10):1916–1924. doi: 10.1093/hmg/ddr073. [DOI] [PubMed] [Google Scholar]
- 23.Weckselblatt B, Rudd MK. Human structural variation: mechanisms of chromosome rearrangements. Trends Genet. 2015;31(10):587–599. doi: 10.1016/j.tig.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanneste E, Voet T, Le Caignec C, et al. Chromosome instability is common in human cleavage-stage embryos. Nat Med. 2009;15(5):577–583. doi: 10.1038/nm.1924. [DOI] [PubMed] [Google Scholar]
- 25.Crasta K, Ganem NJ, Dagher R, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482(7383):53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable for this article as no datasets were generated or analyzed during the current study.