

Abstract
The polysaccharide capsule is a major virulence factor of Streptococcus pneumoniae and the target of all currently licensed pneumococcal vaccines. At present, there are 92 serologically distinct pneumococcal serotypes. Structural and antigenic variation of capsular types is the result of genetic variation within the capsular polysaccharide synthesis (CPS) locus; however, genetic variation may not always result in phenotypic differences which produce novel serotypes. With the introduction of high throughput whole genome sequencing, discovery of novel genotypic variants is not unexpected and this study describes a novel variant of the serotype 23B CPS operon. This novel variant was characterized as a novel genotypic subtype (23B1) with ∼70% homology to the published 23B CPS sequence. High sequence variability was determined in eight cps genes involved in sugar biosynthesis. However, there was no distinction between the classic 23B serotype and 23B1 serologically or in terms of polysaccharide structure. Phylogenetic and eBURST analysis revealed a distinct lineage for 23B1 with multiple clones (UK, Thailand, and USA) that arose at different points during pneumococcal evolution. Analysis of the UK S. pneumoniae isolates (n = 121) revealed an upsurge of 23B1 ST2372 in 2011, after which this previously unseen ST increased to reach 50% proportion of the 23B sequenced isolates from 2013 and remained prevalent within our sequenced isolates from later years. Therefore, although the 23B1 variant appears to have no phenotypic impact and cannot be considered as novel serotype, it appears to have led to a genetic restructuring of the UK serotype 23B population.
Keywords: Streptococcus pneumoniae, serotype, genotype, capsular polysaccharide, whole genome sequencing
Introduction
Streptococcus pneumoniae (pneumococcus) is a major cause of morbidity and mortality worldwide. The pneumococcal capsular polysaccharide (CPS) which surrounds the organism is a major virulence factor, enabling the cell to evade phagocytosis (AlonsoDeVelasco etal. 1995). To date, 92 serotypes of pneumococcus have been defined using the Danish classification system, based on the antigenicity of the capsular polysaccharide with rabbit antisera supplied by Statens Serum Institut, Copenhagen, Denmark (Henrichsen 1995; McEllistrem and Nahm 2012). In this system, immunogenically cross-reactive serotypes are assigned to serogroups.
The genes responsible for the synthesis of the polysaccharide capsule are closely linked and are arranged in a capsular polysaccharide biosynthesis (cps) operon (Kolkman etal. 1998). The cps operon sequences for 90 pneumococcal serotypes (serotypes 6C and 6D were not included) were reported by Bentley etal. (2006), thus paving the way for genotypic capsular typing. Since then, several genotypic serotyping methods have been described (microarray [Turner etal. 2011], multiplex PCR [Brito etal. 2003], in silico inference from whole genome sequence [WGS] data [Croucher etal. 2011; Everett etal. 2012; Metcalf etal. 2016]), utilizing the genetic differences between the capsular loci to determine serotype. Although these methods have many advantages over the traditional methods (Jauneikaite etal. 2015) they still do not currently provide full coverage for all 92 serotypes. However, our laboratory has recently released a new automated pipeline (PneumoCaT) for assignment of serotype from WGS data, that can predict to the level of serotype for 87/92 serotypes (+ genotype 6E [Burton etal. 2016]) and to serogroup level for the remaining five (Kapatai etal. 2016). During the early validation of PneumoCaT, it was observed that a large number of serotype 23B isolates (30/62) caused the analysis to terminate prematurely due to low mapping coverage (∼70%) to the reference serotype 23B capsular locus DNA sequence. Following initial investigation, it became apparent that although these isolates were phenotypically serotype 23B, they had a genetically distinct capsular operon and this novel sequence was introduced into PneumoCaT as genotypic subtype 23B1. Although this variant had no apparent phenotypic impact (on the serotype), it appeared frequently in the UK population and appeared to be on the increase. We, therefore, initiated an investigation into the physical structure of the capsular polysaccharide, and differences between the capsular operon DNA sequences, the clonal lineage and global distribution of 23B and 23B1 pneumococcal isolates.
Materials and Methods
Isolate Selection
Public Health England (PHE) routinely seeks submission of all invasive pneumococci (i.e., those isolated from normally sterile sites such as blood and CSF) from hospital laboratories in England and Wales to the pneumococcal national reference laboratory (NRL), Colindale, London. These isolates were serotyped on receipt as part of the PHE enhanced surveillance programme using slide agglutination with Statens Serum Institut typing sera. Isolates with 23B serotype on receipt (n = 121) were randomly selected from the 23B collection (n = 761 isolates collected between 2004 and 2015) hosted in the archives of the PHE NRL (details of all isolates used in this study can be found at the PHE Pathogens BioProject PRJEB14267 at ENA (http://www.ebi.ac.uk/ena/data/view/PRJEB14267; last accessed August 21, 2017, supplementary table S1, Supplementary Material online). Details of the Statens Serum Institut reference strains for all 92 serotypes and representative clinical isolates can also be found at the same BioProject (PRJEB14267; supplementary table S2, Supplementary Material online). In addition, genomic data for nonUK isolates were obtained from the Streptococcus pneumoniae database hosted in BIGSdb website (http://pubmlst.org/software/database/bigsdb/; last accessed August 21, 2017) following searches for isolates belonging to serotype 23B. These WGS data included those derived from isolates originally isolated in Thailand (n = 16), USA (n = 23), and South Korea (n = 1) (supplementary table S1, Supplementary Material online).
DNA Extraction and Sequencing
Pneumococcal isolates were grown overnight on horse blood agar (PHE Media Services) with 5% CO2. DNA was extracted from an entire plate of growth for each isolate using the QIAsymphony SP automated instrument (Qiagen) and QIAsymphony DSP DNA Mini Kit, using the manufacturer’s recommended tissue extraction protocol for Gram negative bacteria (including a 1 h preincubation with proteinase K in ATL buffer and RNAse A treatment). DNA concentrations were measured using the Quant-iT dsDNA Broad-Range Assay Kit (Life Technologies, Paisley, UK) and GloMax® 96 Microplate Luminometer (Promega, Southampton, UK). DNA was sent for WGS by Illumina sequencing using the PHE Genomic Services and Development Unit (Colindale, UK). Illumina Nextera DNA libraries were constructed and sequenced using the Illumina HiSeq 2500.
Demultiplexing and k-mer Analysis
Casava 1.8.2 (Illumina inc. San Diego, CA, USA) was used to demultiplex the samples and FASTQ reads were processed with Trimmomatic (Bolger etal. 2014) to remove bases from the trailing end that fall below a PHRED score of 30. k-mer identification software (https://github.com/phe-bioinformatics/kmerid; last accessed August 21, 2017) was used to compare the sequence reads with a panel of curated NCBI Refseq genomes to identify the species. A sample of k-mers (DNA sequences of length k) in the sequence data were compared against the k-mers of 1,769 reference genomes representing 59 pathogenic genera obtained from RefSeq. The closest percentage match is identified, and provides initial confirmation of the species. This step also identifies samples containing more than one species of bacteria (i.e., mixed cultures) and any bacteria misidentified as Streptococcus pneumoniae by the sending laboratory. Further analysis continued only if S. pneumoniae was identified.
MultiLocus Sequence Typing (MLST) Analysis
Metric-Oriented Sequence Typing (MOST; https://github.com/phe-bioinformatics/MOST; last accessed August 21, 2017) was used to predict MLST type using WGS data (Tewolde etal. 2016). This pipeline is species-specific and uses the MLST scheme available for S. pneumoniae in PubMLST (http://pubmlst.org/spneumoniae/; last accessed August 21, 2017). eBURST analysis (http://eburst.mlst.net) was used to divide the MLST data set into groups of related isolates (clonal complexes) and predict the founding genotype (ST) of each clonal complex (Feil etal. 2004). The allele sequences were concatenated and aligned and this alignment was used to infer evolutionary history using the Minimum Evolution method (Rzhetsky and Nei 1992) in MEGA6 (Tamura etal. 2013).
Capsular Typing Using Whole Genome Sequencing Data
PneumoCaT v.1.0 (https://github.com/phe-bioinformatics/PneumoCaT; last accessed August 21, 2017) was used to predict capsular type from WGS data as described in Kapatai etal. (2016). In brief, processed reads are initially mapped to capsular locus sequences for all 94 serotypes (92 serotypes plus two molecular subtypes) which predicts serotype or genogroup based on mapping coverage (>90%). If genogroup is predicted the analysis uses the capsular type variant (CTV) database and the reads are mapped to genogroup relevant genes allowing for variant analysis. Serotype can be predicted if 100% match with an available variant profile is achieved.
Phylogenetic Tree Generation
The reference strains for the phylogenetic tree generation were selected according to the strains included in each analysis. For the tree comparing only serotype 23B strains, the ATCC 700669 reference strain (23F; Accession NC_011900) was selected from the available S. pneumoniae complete genomes due to the close relationship to the 23B serotype. For the tree comparing the 23B strains with additional serotypes, nonencapsulated reference strain R6 was used instead. Strain NT 100.58 (Accession NZ_CP007593.1) from the NT classical lineage (Hilty etal. 2014) was used as an outgroup to draw a rooted tree. Reads were mapped to the selected reference sequence using bwa (version 0.7.12; Li and Durbin 2009). Variants were called using GATK 2.6.5 (McKenna etal. 2010). Variants were then parsed to retain high quality SNPs based on the following conditions: depth of coverage (DP) ≥5, AD ratio (ratio between variant base and alternative bases) ≥0.8, Mapping Quality (MQ) ≥30, ratio of reads with MQ0 to total number of reads ≤0.05. All positions that fulfilled the filtering criteria in >0.9 of the samples were joined to produce a multiple fasta format file where the sequence for each strain consists of the concatenated variants. This filtered variant alignment file was used as an input to generate a neighbor joining (NJ) tree using MEGA6 (Tamura etal. 2013) and a maximum likelihood (ML) tree using RAxML (Stamatakis 2014) with the following parameters –m (substitutionModel) GTRCAT –b (bootstrapRandomNumberSeed) 12345 -# (numberOfRuns) 1000 (option –o was used to define a sample as an outgroup). The ML tree combined with temporal data was used to detect root to tip divergence in TempEst (formerly known as Path-O-Gen) (Rambaut etal. 2016).
FastTree version 2.1.8 (Price etal. 2010) was used to generate a ML tree using an alignment of the full capsular operon sequences extracted from all isolates in this study (n = 162).
Assembly Based Sequence Analysis
Genomic reads were assembled using SPAdes (version 2.5.1) (Bankevich etal. 2012) de novo assembly software with the following parameters “spades.py –careful -1 strain.1.fastq.gz -2 strain.2.fastq -t 4 -k 33,55,77,85,93.” The resulting contigs.fasta file was converted into a BLAST database using blast+ (version 2.2.27) (Camacho etal. 2009) and queried using selected query sequence (i.e., gene or capsular operon sequences). To extract the capsular operon sequence from 23B1 isolates, the capsular operon sequence for 23B (accession number CR931684.1) was used to query the contigs. fasta file. The capsular operon sequence for 23B1 was previously submitted to NCBI with accession number LT594598.1 (Kapatai etal. 2016). For the full capsular operon sequence alignment, the reference sequence of 23B (CR931684.1) or 23B1 (LT594598.1) were used to extract the cps locus from genomic assemblies of the 23B and 23B1 isolates, respectively.
Capsular Biosynthesis Operon Sequence Analysis
Capsular operon sequence alignment of the 23B and 23B1 sequences was performed using progressiveMauve (Darling etal. 2010), whereas Artemis (Rutherford etal. 2000) was used for annotating the 23B1 capsular locus operon sequence. The NCBI BLAST website (https://blast.ncbi.nlm.nih.gov (https://github.com/phe-bioinformatics/PneumoCaT; last accessed August 21, 2017)) was used to query the BLAST nucleotide collection database (nr/nt) whereas RDP4 was used for recombination detection (Martin etal. 2015). BioEdit version 7.2.5 (Hall 1999) was used to align the capsular operon sequences extracted from all isolates.
Gene Presence/Absence Analysis
The reads from WGS were mapped to the sequences of the capsular genes present in serotype 23B capsular locus and the resulting bam files parsed to find which genes/alleles were present in each isolate. Genes showing ≥80% coverage and nucleotide identity over the full length of their sequences were considered as present; if nucleotide identity is <80%, genes are considered present, but might exhibit difference in function. Genes with <80% coverage over the length of the gene sequence were considered absent.
NMR Analysis of Capsule Polysaccharide
The extraction and NMR analysis of capsular polysaccharide of types 23B and 23B1 were conducted as described previously (Brugger etal. 2016; Hathaway etal. 2012). In brief, the strains were grown in 40 ml of modified Lacks medium until OD600nm of 0.5 was reached, followed by centrifugation of the culture at 4,000 × g for 10 min and washing of the pellet with PBS (10 ml) and H2O (2 × 5 ml). After ultracentrifugation at 20,000 × g for 30 min at 4 °C, the pellets were resuspended in H2O (5 ml) and incubated at room temperature overnight with the addition of 1% buffer saturated phenol solution. The samples were again ultracentrifuged and the pellet discarded. Polysaccharide was precipitated from the supernatant by addition of NaOAc and EtOH to a final concentration of 7.2% and 60%, respectively, and incubation for 1 h at 4 °C. The polysaccharide was collected by ultracentrifugation and dissolved in 5 ml of H2O. Undesirable protein contaminants were removed by sequential treatment at 37 °C with benzonase nuclease (250 U) for 6 h and with proteinase K (40 µl of a 20 mg/ml solution) overnight. The sample was further purified by centrifugation in an Amicon Ultra 30 kDa cut off membrane centrifugal filter unit (Millipore, Billerica, MA) at 4,000 × g for 10 min and the retained sample was dried at 45°C in a vacuum centrifuge. Capsule extract (4–5 mg) was dissolved in 50 µl of D2O transferred to 1.7 mm NMR tubes. NMR data were collected on a Bruker Avance II (500 MHz; 1H) spectrometer with a 1.7 mm triple-resonance (1H, 13C, 31P) microprobe head at 298 K with the water resonance suppressed using a classical presaturation scheme. All spectra were calibrated to the residual water peak (4.766 ppm).
Results
Capsular Locus Operon Analysis
Capsular locus operon DNA sequences extracted from genomic assemblies of 12 23B1 isolates (ST2372) were used to define a consensus sequence. The 12 sequences were aligned to the reference 23B capsular operon sequence using progressiveMauve and the consensus sequence was defined using the per position majority base (>80%) from this alignment and annotated using Artemis (fig. 1A). The sequence was deposited in EMBL with accession number LT594598.
Fig. 1.

—Genetic analysis of 23B1 capsule. (A) View of annotated 23B1 capsular operon sequence in Artemis. (B) Alignment of 23B and 23B1 capsular operon sequences in progressiveMauve. The capsular gene order is as follows: wzg, wzh, it, wze, wchA, wchF, wzy, wchV, wchW, wzx, wchX, gtp1, gtp2, gtp3, rmlA, rmlC, rmlB, and rmlD.
Subsequent alignment of the 23B1 consensus sequence (length = 21,797 bp) to the 23B cps locus (length = 23,047 bp) revealed high similarity (98%) across the first 14,000 bp, followed by lower homology (82%) for the remaining part of the cps locus, which includes genes wchX, gtp1, gtp2, gtp3, rmlA, rmlC, rmlB, and rmlD (fig. 1B). In addition, the rmlD gene sequence was inverted and found in the reverse DNA strand (green in fig. 1B). All gene sequences were still intact and produced full ORFs with similar functionality as their 23B counterparts based on the functional domains identified during protein BLAST analysis (table 1).
Table 1.
Functions Associated with the Genes Differentiating 23B and 23B1
| Gene | Function |
|---|---|
| wchX | sugar phosphate transferase for Gro-2P |
| gtp1, gtp2, and gtp3 | synthesis of NDP-2-glycerol (Gro-2P) |
| rmlA, rmlB, rmlC, and rmlD | synthesis of the precursor dTDP-rhamnose (L-Rhap) |
The 23B1 reference sequence was used to query the NCBI nucleotide collection database to determine whether the region differing between the two capsules originated from another source as a result of a recombination event. BLAST analysis using the megablast algorithm revealed high similarity with a number of other serotype capsular loci in the conserved region covering the four regulatory and processing genes wzg, wzh, wzd, and wze; the remaining region exhibited high similarity (∼80%) with the 23B and 23F capsular loci but most interestingly, the region with the four rml genes exhibited high similarity (∼90%) with 19A capsular loci suggesting that a recombination event might have occurred between these two capsular loci. To investigate this further, the reference sequences for 23B, 23B1, 23F, and 19A were aligned and analyzed using the recombination detection tool RDP4; however no evidence of recombination was found within this set.
Structural Analysis of Capsular Polysaccharide
Although the 23B1 isolates could not be distinguished from 23B isolates by slide agglutination using rabbit antisera, we investigated whether they possessed capsular polysaccharide (CPS) with any detectable structural differences. CPS was extracted from the 23B isolate (accession number ERS1196211) and the 23B1 isolate (accession number ERS1194164) and analyzed by 31P NMR and 1H NMR (fig. 2). 31P NMR (specific for the phosphate content) analysis of capsular polysaccharide of types 23B and 23B1 showed no significant difference between the two capsules (fig. 2A). 1H NMR analysis of CPS (fig. 2B and C) revealed no significant differences as well, especially in the anomeric region (fig. 2D), from which the most valuable information can be extracted (Abeygunawardana etal. 2000). The NMR spectra quality of this analysis was adequate to identify differences among the two strains, but not to reveal composition of the capsule in detail; further optimization of the spectra baseline and sample purity is required to achieve the latter.
Fig. 2.

—NMR analysis of the 23B and 23B1 capsule polysaccharide. (A) 31P NMR spectrum of type 23B and 23B-1 capsular polysaccharide extract, (B) 1H NMR spectrum of the same samples, (C) details of the region of the large peaks (x axis, region from 3 to 4 ppm; 1H NMR spectrum [B]) but with the height of the peaks reduced so they fit in the frame and (D) details of the anomeric region (x axis, region from 4 to 6 ppm) of the 1H NMR spectrum shown in (B). No significant differences between the two strains have been observed in the spectra.
Population Analyses
WGS data from 162 isolates reported as (phenotypic) serotype 23B were collected from our in-house or public databases; UK, n = 121; USA, n = 23; Thailand, n = 16; South Korea, n = 1 and the SSI-23B reference strain. Genomic data from all isolates were analyzed using MOST and PneumoCaT (version that includes 23B1 cps locus) to assign MLST type and serotype, respectively.
All isolates, except the South Korean, were assigned a 23B or 23B1 serotype (table 2). The South Korean isolate caused the analysis to terminate prematurely due to low coverage (<90%) and with highest coverage observed for 19F (78.7%). In addition MLST analysis yielded ST81, a type observed in 19A, 19F, and 23F isolates. This isolate was removed from further analyses.
Table 2.
Geographical and Temporal Distribution of the 23B and 23B1 Types
| Thailand | UK | USA | Total | Grand Total (per year) | ||
|---|---|---|---|---|---|---|
| 2001 | 23B | 1 | 1 | 1 | ||
| 23B1 | ||||||
| 2004 | 23B | 2 | 6 | 8 | 8 | |
| 23B1 | ||||||
| 2005 | 23B | 3 | 3 | 4 | ||
| 23B1 | 1 | 1 | ||||
| 2006 | 23B | 3 | 3 | 3 | ||
| 23B1 | ||||||
| 2007 | 23B | 10 | 9 | 19 | 26 | |
| 23B1 | 7 | 7 | ||||
| 2008 | 23B | 8 | 8 | 14 | ||
| 23B1 | 6 | 6 | ||||
| 2009 | 23B | 12 | 12 | 16 | ||
| 23B1 | 4 | 4 | ||||
| 2010 | 23B | 10 | 10 | 16 | ||
| 23B1 | 6 | 6 | ||||
| 2011 | 23B | 9 | 9 | 10 | ||
| 23B1 | 1 | 1 | ||||
| 2012 | 23B | 5 | 5 | 10 | ||
| 23B1 | 5 | 5 | ||||
| 2013 | 23B | 11 | 11 | 22 | ||
| 23B1 | 11 | 11 | ||||
| 2014 | 23B | 3 | 3 | 15 | ||
| 23B1 | 12 | 12 | ||||
| 2015 | 23B | 8 | 8 | 15 | ||
| 23B1 | 7 | 7 | ||||
| Grant Total (per country) | 16 | 121 | 23 | 160 | 160 |
Note.—Distribution of the 23B and 23B1 molecular capsular types per country and per year of collection. The South Korean isolate was excluded from this analysis as it failed typing.
The UK isolates were selected randomly to cover a period from 2004 to 2015 to allow for genotypic composition analysis of the serotype 23B population based on the distribution of 23B and 23B1 genotypes. Unfortunately, the nonUK isolates represented isolates from specific research projects (Thailand: 2008–2010; USA: 2001, 2004, 2007, and 2013) therefore they could only offer a snapshot of the 23B/23B1 distribution during the period covered. PneumoCaT analysis revealed that all Thailand isolates (n = 16) carried the 23B1 cps locus whereas the USA isolates carried either 23B or 23B1 cps locus with the 23B1 present only in the 2007 and 2013 isolates.
As part of the enhanced surveillance scheme currently in place for pneumococcal isolates, 761 23B isolates were sent to the PHE reference laboratory between 2004 and 2015. A graph analysis of these isolates revealed an upsurge of 23B following the introduction of PCV7 vaccine in April 2006 (fig. 3A). The isolates used in this study, were sampled randomly from the 23B population so that the period 2004–2015 was, as far as possible, evenly represented (n = 121) (fig. 3A). Graph analysis of the study isolates was able to show the genetic composition of the 23B population for the years sampled. This analysis revealed an emergence and subsequent increase in the numbers of 23B1 isolates from 2011, suggesting a possible introduction of this subtype from abroad (fig. 3B). Only one UK 23B1 isolate was found before 2010, a 2005 isolate with a unique MLST profile (ST2448).
Fig. 3.

—(A) Temporal analysis of the UK serotype 23B population collected between 2004 and 2015 (n = 761). Line plot of the total number (n = 761; orange line) and sampled number (n = 121; green line) of the serotype UK 23B isolates received between 2004 and 2015. (B) Temporal analysis of the 23B and 23B1 genotype distribution in the UK panel of serotype 23B isolates sampled from the serotype 23B population collected between 2004 and 2015 (n = 121). Line plot of the serotype 23B UK isolates based on molecular subtype showing temporal distribution between 2004 and 2015. (C) Temporal analysis of UK isolates within clonal complexes CC439 and CC1349 (n = 112) in relation to isolates within the most common STs, ST439, and ST2372 (n = 97). Line plot of UK isolates within the two main clonal complexes CC439 for 23B and CC1349 for 23B1 (solid line; blue and red, respectively) and within the two most common STs, ST439 for 23B and ST2372 for 23B1 (dashed line; blue and red, respectively).
MLST Analyses
MLST analysis of all 161 serotype 23B isolates revealed distinct MLST profiles for the 23B and 23B1 isolates with no MLST profiles shared between the two subtypes (fig. 4A). The majority of the genotype 23B isolates (84%) belonged to ST439 a sequence type generally associated with 23B isolates, whereas the 23B1 isolates belonged to ST2372 (35.9%), ST6707 (21.9%) and ST1373 (10%). ST6707 was only present in Thailand isolates (14/16) and ST2372 was only present in the UK isolates, whereas ST1373 was found in both USA and UK isolates (supplementary table S1, Supplementary Material online).
Fig. 4.

—(A) Multilocus sequence type (ST) distribution of the 23B and 23B1 clinical isolates from UK, USA, and Thailand during 2001–2016 (n = 161). (B) Population snapshot of the UK and nonUK 23B and 23B1 clinical isolates (n = 161). Visualization of the number and sizes of clusters of linked STs in a single eBURST diagram. Founder genotypes are colored in yellow and edges correspond to SLVs. Clonal complex (CC) 439 is the main cluster amongst the 23B isolates whereas CC1349 with subgroup founder ST2372 is the main cluster amongst the 23B1 isolates. Main CCs based on SLVs are circled using solid line whereas CCs based on DLVs are circled using dashed lines.
Analysis using eBURST revealed two main clonal complexes representing the majority of 23B and 23B1 isolates (fig. 4B and supplementary table S3, Supplementary Material online). Clonal complex 439 (Group 1; n = 96) comprised 95.1% of the 23B isolates (UK, n = 80; USA, n = 16) with ST439 as the predicted founder genotype. Clonal complex CC1349 (Group 2; n = 32) comprised 86.5% of the UK 23B1 isolates (32/37) and 53.3% of the total 23B1 data set (32/60). Although ST1349 was predicted as the founder genotype, ST2372 was the most common genotype and the predicted subgroup founder. Population analysis using only the UK isolates within the two clonal complexes and the two most common STs (ST439 and ST2372) demonstrated that the increase of 23B1 isolates after 2010 was mainly due to increase of ST2372 (fig. 3C) suggesting a clonal expansion might have taken place. All Thailand isolates form a smaller clonal complex (Group 4) with ST6707 and its SLV 10567. The remaining UK (23B n = 4, 23B1 n = 5) and USA (23B1 n = 9) isolates fall within mostly singleton groups (no SLV) with the majority falling within ST1373 (23B1; UK = 4, USA = 2). The eBURST analysis was rerun with a less stringent analysis of relatedness (5/7 matches) to investigate any relations within the singletons and the smaller groups. Following this analysis group 5 and singletons 36 and its DLV 1448 were incorporated within group 1 (CC439) (fig. 4B). This suggests that the 23B1 sequence types ST36 and ST1448 are more closely related to the 23B clonal complex than the 23B1. The presence of the 23B1 capsule in these diverse clonal complexes suggests that horizontal gene transfer might have been involved. MultiLocus Sequence Analysis (MLSA) analysis using the Minimum Evolution method on an alignment of concatenated MLST sequences confirms the eBURST results (supplementary fig. S1, Supplementary Material online). As seen with the eBURST analysis, the two groups are separated into two main branches (23B leaf nodes: CC439 and 23B1 leaf nodes: ST2372) whereas the Thailand isolates (CC6707) located into a smaller branch between the main 23B leaf node cluster and SSI-23B isolate. The 23B1 sequence types ST36 and ST1448 are located on a branch close to the main 23B leaf node cluster whereas the two “odd” 23B are found on a branch close to SSI-23B and the remaining 23B1 singletons are spread between SSI-23B and the main 23B1 leaf node cluster (supplementary fig. S1, Supplementary Material online).
Phylogenetic Analysis
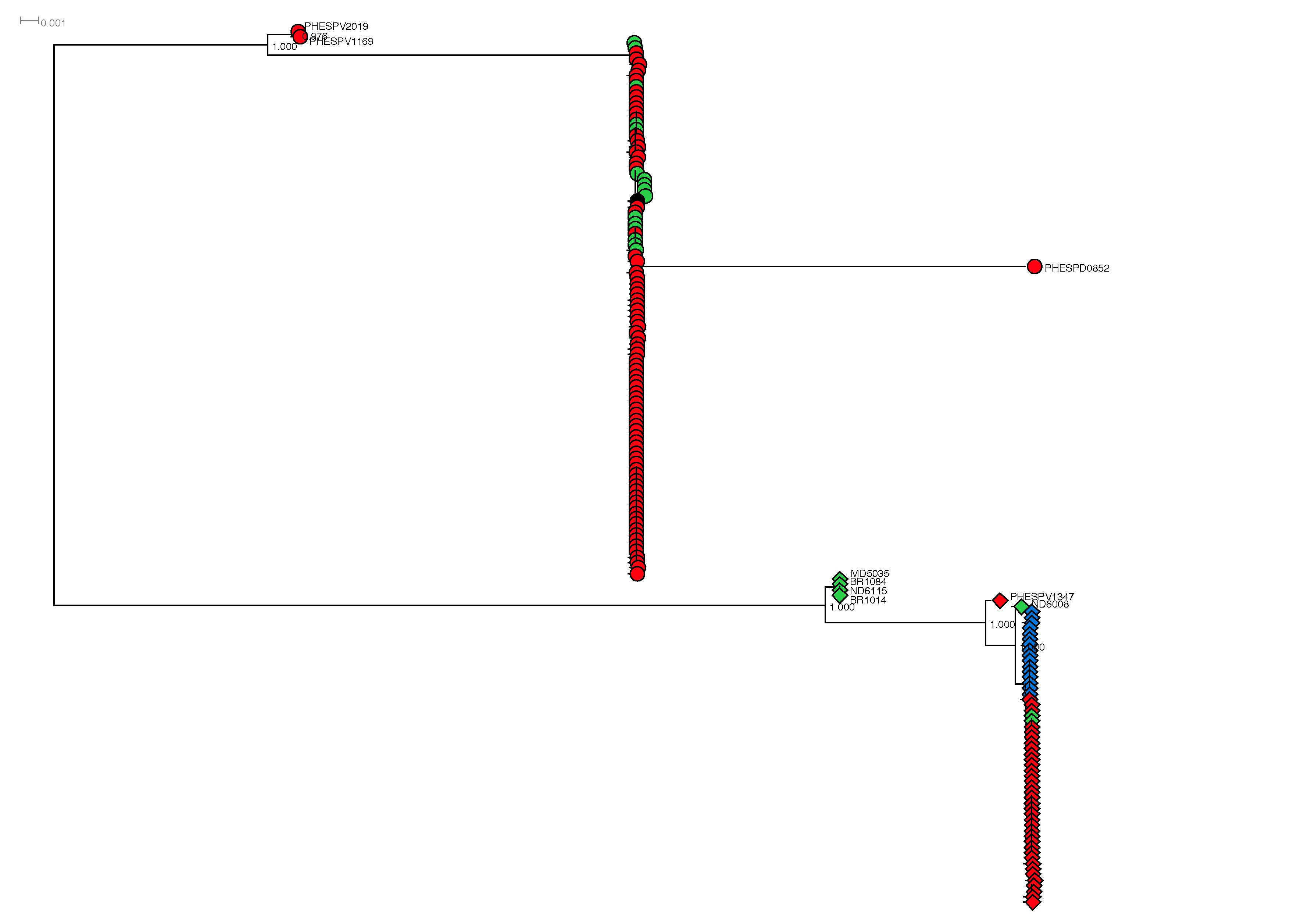
This part of the study utilized three approaches to define the evolutionary history of the 23B1 genotype and its relation to 23B. The first approach utilized the maximum likelihood (ML) method to further investigate the relationship between the 23B and 23B1 isolates based on the capsular operon sequence alone; the full capsular operon sequence alignment of the 161 isolates was used for this analysis (supplementary fig. S2, Supplementary Material online). The two genotypes are separated into two main branches with minimal sequence variability within the majority of the isolates of each group. Within the genotype 23B main branch 0–10 SNP variance is observed (23B CPS length: 19,652 bps); only three isolates diverge from the main cluster. Isolate PHESPD0852 extends from the main branch into a subbranch with branch length of ∼380 SNPs. Two additional isolates, PHESPV2019 and PHESPV1169, comprising eBURST group3 (supplementary table S3, Supplementary Material online; ST4423 and its SLV 11172, respectively), are located ∼370 SNPs apart from the main 23B branch and closer to the 23B1 branch. Similar variance (0–12 SNPs) is observed in the main 23B1 (23B1 CPS length: 21,797 bps) branch, however within the Thailand isolates 0–2 SNPs variance is observed and 0–8 within the remaining 23B1 isolates in this main branch. Six isolates diverge from this main branch; the four ST36 isolates are located ∼183 SNPs apart from the main 23B1 branch closer to the 23B branch whereas their DLV ST1448 is the closest of the six outliers to the main branch with only 18 SNPs difference from the main branch capsular locus sequence, further supporting the possibility of horizontal gene transfer for this clonal group. Isolate PHESPV1347 (ST11178 SLV of ST2372) is the sixth outlier from the main 23B1 group and differs by ∼43 SNPs from the main 23B1 capsular locus sequence.
A second ML tree was generated using RAxML and an alignment of high quality variants, derived following mapping of all 23B/23B1 isolates (n = 161) to the 23F ATCC 700699 genome as described in material and methods (fig. 5A). 23B and 23B1 isolates fall within two main branches; 99/101 23B isolates cluster in one main branch (fig. 5A; red circle), whereas the 59/60 23B1 isolates are separated into 4 lineages. The UK CC1349 isolates (n = 32) cluster in the second main branch (fig. 5A; green circle), whereas the Thailand CC6707 isolates (n = 16; fig. 5A blue circle) and the ST1373 isolates (UK and USA; n = 6; fig. 5A pink circle) are found in subbranches off the main 23B1 branch. However, the USA 23B1 isolates from 2007 (ST36 and double locus variant (DLV) ST1448; n = 5) previously shown to be closely related to CC439 (supplementary fig. S1, Supplementary Material online) fall within a subbranch of the main 23B branch (fig. 5A; orange circle). The remaining three isolates (23B: PHESPV2019 and PHESPV1169; 23B1: PHESPV2011) are located within a distinct subbranch equidistant to the two main branches.
Fig. 5.

—Phylogenetic analysis of 23B and 23B1 isolates. Maximum likelihood (ML) trees generated following SNP analyses using RAxML with the complete genome of Streptococcus pneumoniae 23F ATCC700699 strain as reference for (A) and the nonencapsulated S. pneumoniae R6 strain for (B). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The scale bar corresponds to the number of nucleotide substitutions per site. All positions with <90% site coverage were eliminated. That is, <10% alignment gaps, missing data, and ambiguous bases were allowed at any position. (A) Radial phylogram representation of ML tree for the full 23B and 23B1 data set (n = 161). A total of 18,730 variant positions were included in the analysis. Lineages 23B and 23B1 are represented with shapes, circle, and diamond, respectively, whereas the shape color is used to differentiate the country of origin for each isolates (UK: red, Thailand: blue, USA: green). The various clusters are circled and colored based on lineage, country and/or MLST clonal complex (CC); 23B CC439: red, 23B1 CC1349: green, CC6707: blue, ST1373: pink, CC36: orange. Zoomed in views of the two main branches can be seen in supplementary figure S3, Supplementary Material online. (B) Radial phylogram representation of ML tree for the full 23B and 23B1 data set (n = 161) plus reference strains for all serotypes (n = 92) and representative clinical isolates (n = 76). A total of 87,965 variant positions were included in the analysis. The lineages and country of origin representation remains the same as (A). The various clusters are circled and colored based on lineage, country and/or MLST CC as described in (A). This figure is available in Microreact (https://microreact.org/project/rJpKzoXTe; last accessed August 21, 2017).
In order to investigate the evolution of the two 23B lineages in relation to other S. pneumoniae serotypes, the SNP analysis was repeated, but this time including WGS data from the Statens Serum Institut reference strains for all 92 serotypes and representative contemporaneous clinical isolates for each serotype where possible (fig. 5B, Supplementary Material online). For this analysis, the nonencapsulated S. pneumoniae R6 strain was used as reference. Strain NT 100.58 from the NT classical lineage (Hilty etal. 2014) was used as an outgroup to draw a rooted tree. The results are shown in figure 5B and a zoomed-in view of the 23B and 23B1 main branches is shown in supplementary figure S3, Supplementary Material online. Based on the origin of the two main serotype 23B branches in the center of the radial phylogram it is apparent that the 23B1 genotype did not diverge from the 23B genotype and indeed it is as distant to the 23B genotype as to any other serotype. A closer view on the 23B branch (supplementary fig. S3A, Supplementary Material online) shows once again the close relationship of the ST36 and DLV ST1448 group to the main genotype 23B branch and reinforces the hypothesis of this 23B1 lineage emerging following introduction of the 23B1 capsular operon into a CC439 strain.
Gene-Based Analysis
Gene presence/absence analysis of the 23B capsular genes for all isolates in this study (n = 161) presented next to a neighbor joining tree of the isolates (based on SNP analysis from WGS data) (fig. 6) revealed a clear differentiation between the two main branches of 23B and 23B1 lineages consistent with the sequence variability observed in figure 1 within the region covering the eight affected genes (wchX, gtp1, gtp 2, gtp3, rmlA, rmlB, rmlC, and rmlD). Furthermore, some distinction was evident amongst the gene profiles of the different lineages of 23B1 with larger variation observed between the gene profiles of the UK, Thailand and USA 23B1 isolates (fig. 6). This supports the hypothesis that the emergence of the 23B1 capsule constitutes a deep historical event followed by introduction of this capsular operon into various lineages at different points in time resulting to multiple lineages exhibiting the same phenotype even though some variance has been introduced in the capsule over time. Interestingly, the two classic 23B isolates that cluster with the 2005 23B1 isolate in a distinct branch (branch indicated with * in fig. 6) show variability in the wchX gene, suggesting that these two isolates do not have a classic ST439 23B type capsular operon.
Fig. 6.

—Gene profiling analysis of the 23B and 23B1 isolates (n = 161). Heatmap representing the presence/absence of the 23B capsular locus genes for 23B and 23B1 isolates in the order they appear in the Neighbor-Joining tree. The NJ tree was generated from SNP data analysis using the full genome of ATCC 700669 23F reference strain (gene profiling analysis not shown). The capsular biosynthesis genes are ordered as in the capsular operon (fig. 1) with the affected genes (wchX, gtp1, gtp2, gtp3, rmA, rmlC, rmlB, and rmlD) highlighted in yellow. The 23B1 lineages are marked and colored as defined in figure 5A.
Discussion
In Streptococcus pneumoniae and other vaccine-preventable organisms, serological agglutinating properties have traditionally been used to perform type differentiation. However, since the introduction of genotypic predictive “serotyping” methods a number of genetic variants have been identified leading to an increase in the number of serotypes described in the literature (e.g., 6C and 6D [McEllistrem and Nahm 2012]), but also some novel genotypes (i.e., 6E [Burton etal. 2016]) which have impacted the genetic makeup of the serogroup 6 population (van Tonder etal. 2015). In this study, we present a novel 23B genotypic variant which, like 6E, shares little similarity with the serologically equivalent clones, and investigate the genetic and structural differences and impact on the serotype 23B population.
Variant 23B1 is a novel genotypic subtype of serotype 23B with <70% similarity to the traditional 23B capsular operon sequence. The sequence variability is located towards the 3′-end of the cps operon and covers eight capsular polysaccharide biosynthetic genes (wchX, gtp1, gtp2, gtp3, rmlA, rmlB, rmlC, and rmlD). Despite this variability, the serological type remains unchanged. Furthermore, even though the DNA sequence is distinct, the functionality of the resulting proteins is unchanged and structural analysis by NMR methods confirmed that the 23B1 capsular polysaccharide structure is identical to the 23B structure. Although the rml genes share high homology with the respective genes found in serotype 19A CPS operon, recombination analysis failed to detect any recombination within the capsular locus operon.
Phylogenetic analysis on data derived by mapping to the complete genome of 23F strain ATCC700699 indicates that 23B1 is a distinct lineage that emerged early on, possibly more than once during pneumococcal evolution and has evolved independently from the classic 23B lineage. Tempest analysis was used on the ML tree for initial root to tip divergence analysis to determine whether the data are suitable for more in-depth analysis with BEAST and concluded that there was not sufficient temporal signal in the data to proceed with phylogenetic molecular clock analysis. However, a second ML analysis, using the nonencapsulated R6 strain as reference for SNP analysis, classical NT lineage 100.58 strain as an outgroup to root the tree and including reference and contemporaneous strains of all other serotypes to provide context (fig. 5B), illustrates that the main groups of the two genotypes are as distant to each other as to any other serotype. This is further supported by MLST, MLSA and eBURST data; the two lineages share no MLST types and belong to distinct clonal complexes. Furthermore, whereas 23B is quite clonal with most isolates belonging to the same clonal complex (CC439), the 23B1 lineage has multiple clonal complexes (CC1349, CC6707, CC4423, ST36 (+DLV ST1448), and singletons ST1373 and 2448) which support the phylogenetic data suggesting a multiple emergence pattern. It appears that there is a distinct dominant 23B1 ST in the isolates of each of the countries we tested (UK: ST2372 in 32/36 23B1 isolates, Thailand: ST6707 in 14/16 isolates plus two SLV ST 10567 isolates and USA: ST36 in 4/7 isolates plus a single DLV ST1448 isolate). Although, both in the SNP analysis and MLST analysis, the UK (ST2372) and Thailand (ST6707) lineages are quite distinct, their capsular locus sequence differs by only 8–10 SNPs (supplementary fig. S2, Supplementary Material online). This suggests that the 23B1 operon was likely introduced by recombination into endemic strains for the two countries. A similar mechanism is suspected for the 23B1 USA isolates from 2007 (ST36 and DLV ST1448). These isolates seem to be genetically closer to the main 23B lineage, making this the suspected recipient of the 23B1 CPS operon, but is also close to the SSI strains for 23F and 23A, which is unsurprising since ST36 is also found in 23F isolates. Based on the capsular locus sequence of these strains, the recombination event that resulted to this lineage was an earlier event that the ones responsible for the UK and Thailand isolates (∼183 SNPs variance from the UK and Thailand CPS operon sequence). The only lineage for which there is some evidence for importation is the 23B1 clone ST1373, which is present in both USA from 2007 and UK isolates from 2012 to 2014, although the direction of this importation cannot be ascertained. The limited scope of this study does not allow any comments on the possibility of importation for the other lineages or exportation of the 23B1 ST2372 to other countries, although there is no evidence for importation of the Thailand ST6707 lineage into the UK.
Temporal analysis of the frequency of these two subtypes in the UK population (n = 121; 2004–2015) revealed an increasing dominance of ST2372 after 2010 (fig. 3C, Supplementary Material online); that is the year the PCV13 vaccine was introduced in the UK (Waight etal. 2015) and replaced the PCV7 vaccine (PCV7; serotypes 4, 6B, 9V, 14, 18C, 19F, 23F). PCV13 targets the PCV7 serotypes plus serotypes 1, 3, 5, 6A, 7F, and 19A. The timing of this emergence warrants a consideration of a possible link between the two events. However, since the vaccine evasion mechanism depends on the emergence of new polysaccharide structures and the polysaccharides of the 23B and 23B1 lineages are indistinguishable, it seems unlikely that vaccine evasion is involved in the emergence of this lineage. Enhanced surveillance data shows an overall increase in serotype 23B after introduction of the PCV7 vaccine, along with other nonvaccine serotypes, which continues to increase to the present day. Based on the random sampling of the 2004–2015 UK 23B enhanced surveillance isolates, the 23B1 lineage has been contributing in increasing proportion to the overall increase of the 23B population since 2010. Further sampling and sequencing would be able to confirm this contribution but is outside of the scope of this study.
Gene profile analysis further demonstrates the clonal nature of 23B1 between the different countries. The presence of the 23B1 capsular locus in multiple long-lived lineages is indicative of horizontal transfer of this locus from an unknown donor into multiple lineages.
In conclusion, this study presents a detailed analysis of the 23B1 genetic variant. The use of serotype-specific genotypic profiles in PneumoCaT led to the discovery of a number of variants that diverge from the consensus CPS “fingerprint” defined for a serotype. Furthermore, recently, Van Tonder etal. (2016) presented a comprehensive study of available pneumococcal genomes demonstrating the capsular locus diversity among certain serotypes, including 23B. The Thailand 23B1 lineage (CC6707) was amongst the 23B variants presented by Van Tonder et al., as well as the seven USA 23B1 isolates (Van Tonder: ST1373 is represented as CC338 and ST36 and its DLV ST1448 is part of CC439; [van Tonder etal. 2015]). The UK ST2372 lineage has not been previously described. Limited research resources may not allow all genetic variants to be investigated but those, as in the case of UK 23B1 lineage, that form a large part of the serotype population warrant detailed analysis, to determine whether the variant could indeed exhibit phenotypic changes that affect vaccination efficacy and immunity.
This study (based on isolates collected between 2004 and 2015) demonstrated the expansion of a 23B1 clone in the UK and current data suggest that ST2372 has become successful in recent years (2011–2015) and has contributed to the observed increase of 23B isolates (along with other nonvaccine serotypes) following the introduction of PCV13. Possible reasons behind this clonal expansion (e.g., increased fitness, virulence or antibiotic resistance compared with ST439) remain to be investigated. Continued molecular surveillance is required to monitor its possible impact on the bacterial population.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank McDonald Prest, Doris Omoigui, and Tim Chambers for retrieving isolates from archives and preparing DNA for sequencing. Cath Arnold and the team in Genome Services and Development Unit, PHE Colindale for sequencing all isolates. Ella Campion, Gurkiran Mankoo, and John Duncan for performing routine S. pneumoniae serotyping and repeat testing some isolates and Anthony Underwood for providing helpful comments on the manuscript.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Literature Cited
- Abeygunawardana C, Williams TC, Sumner JS, Hennessey JP.. 2000. Development and validation of an NMR-based identity assay for bacterial polysaccharides. Anal Biochem. 279:226–240. [DOI] [PubMed] [Google Scholar]
- AlonsoDeVelasco E, Verheul AF, Verhoef J, Snippe H.. 1995. Streptococcus pneumoniae: virulence factors, pathogenesis, and vaccines. Microbiol Rev. 59:591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley SD, et al. 2006. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito DA, Ramirez M, Lencastre HD.. 2003. Serotyping Streptococcus pneumoniae by multiplex PCR serotyping Streptococcus pneumoniae by multiplex PCR. J Clin Microbiol. 41:2378–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger SD, et al. 2016. Polysaccharide capsule composition of pneumococcal serotype 19A subtypes: unaltered among subtypes and independent of the nutritional environment. Infect Immun. 84:3152–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton RL, Geno KA, Saad JS, Nahm MH.. 2016. Pneumococcus with the ‘6E’ cps locus produces serotype 6B capsular polysaccharide. J Clin Microbiol. 54:967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croucher NJ, et al. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT.. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett DB, et al. 2012. Genetic characterisation of Malawian pneumococci prior to the roll-out of the pCV13 vaccine using a high-throughput whole genome sequencing approach. PLoS One 9:e44250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG.. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 186:1518–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. 41:95–98. [Google Scholar]
- Hathaway LJ, et al. 2012. Capsule type of Streptococcus pneumoniae determines growth phenotype. PLoS Pathog. 8:e1002574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrichsen J. 1995. Six newly recognized types of Streptococcus pneumonia. J Clin Microbiol. 33:2759–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilty M, et al. 2014. Global phylogenomic analysis of nonencapsulated Streptococcus pneumoniae reveals a deep-branching classic lineage that is distinct from multiple sporadic lineages. Genome Biol Evol. 6:3281–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauneikaite E, et al. 2015. Current methods for capsular typing of Streptococcus pneumoniae. J Microbiol Methods 113:41–49. [DOI] [PubMed] [Google Scholar]
- Kapatai G, et al. 2016. Whole genome sequencing of Streptococcus pneumoniae : development, evaluation and verification of targets for serogroup and serotype prediction using an automated pipeline. PeerJ 4:e2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolkman MA, van der Zeijst BA, Nuijten PJ.. 1998. Diversity of capsular polysaccharide synthesis gene clusters in Streptococcus pneumoniae. J Biochem. 123:937–945. Available from: http://jb.oxfordjournals.org/content/123/5/937.abstract (Accessed 2016 February 17). [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Murrell B, Golden M, Khoosal A, Muhire B.. 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:vev003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEllistrem MC, Nahm MH.. 2012. Novel pneumococcal serotypes 6C and 6D: anomaly or harbinger. Clin Infect Dis. 55:1379–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf BJ, et al. 2016. Strain features and distributions in pneumococci from children with invasive disease before and after 13-valent conjugate vaccine implementation in the USA. Clin Microbiol Infect. 22:60.e9–60.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP, Rojas M, Brodie E.. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Lam TT, Max Carvalho L, Pybus OG.. 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2:vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, et al. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. [DOI] [PubMed] [Google Scholar]
- Rzhetsky A, Nei M.. 1992. A simple method for estimating and testing minimum-evolution trees. Mol Biol Evol. 9:945–967. [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S.. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewolde R, et al. 2016. MOST: a modified MLST typing tool based on short read sequencing. PeerJ 4:e2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tonder AJ, et al. 2015. Genomics reveals the worldwide distribution of multidrug-resistant serotype 6E Pneumococci. J Clin Microbiol. 53:2271–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tonder AL, et al. 2016. Putatively novel serotypes and the potential for reduced vaccine effectiveness: capsular locus diversity revealed among 5,405 pneumococcal genomes. Microb Genomics 2:e000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner P, et al. 2011. Improved detection of nasopharyngeal cocolonization by multiple pneumococcal serotypes by use of latex agglutination or molecular serotyping by microarray. J Clin Microbiol. 49:1784–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waight PA, et al. 2015. Effect of the 13-valent pneumococcal conjugate vaccine on invasive pneumococcal disease in England and Wales 4 years after its introduction: an observational cohort study. Lancet Infect Dis. 15:535–543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.