

Abstract
The Retinoblastoma (RB) tumor suppressor regulates G1/S transition during cell cycle progression by modulating the activity of E2F transcription factors. The RB pathway plays a central role in the suppression of most cancers, and RB mutation was initially discovered by virtue of its role in tumor initiation. However, as cancer genome sequencing has evolved to profile more advanced and treatment‐resistant cancers, it has become increasingly clear that, in the majority of cancers, somatic RB inactivation occurs during tumor progression. Furthermore, despite the presence of deregulation of cell cycle control due to an INK4A deletion, additional CCND amplification and/or other mutations in the RB pathway, mutation or deletion of the RB gene is often observed during cancer progression. Of note, RB inactivation during cancer progression not only facilitates G1/S transition but also enhances some characteristics of malignancy, including altered drug sensitivity and a return to the undifferentiated state. Recently, we reported that RB inactivation enhances pro‐inflammatory signaling through stimulation of the interleukin‐6/STAT3 pathway, which directly promotes various malignant features of cancer cells. In this review, we highlight the consequences of RB inactivation during cancer progression, and discuss the biological and pathological significance of the interaction between RB and pro‐inflammatory signaling.
Keywords: IL‐6, inflammation, metabolism, RB, STAT3, stem cell
The RB tumor suppressor protein forms a transcriptional repression complex with the E2F family of transcription factors and various chromatin modifiers, and thereby negatively regulates G1/S transition during the cell cycle through the suppression of E2F target genes such as CCNA1 and CCNE1. In response to growth stimuli, the cyclin D–CDK4/6 complex phosphorylates RB, releasing E2F transcription factors and facilitating the G1/S transition. As almost all human cancer cells carry aberrations in components of the RB pathway, including INK4A, CCND, CDK4/6, RB, and E2Fs, genetic or functional inactivation of the RB pathway seems to be indispensable for dysregulated proliferation in cancer cells.1, 2
Retinoblastoma mutations are found in retinoblastoma, osteosarcoma, and SCLC at a high frequency, but less frequently in more common types of cancer.2, 3, 4 Mutations in INK4A are frequently detected in pancreatic cancer and NSCLC. CCND amplification is often seen in breast cancer cells.2, 3, 4 Although, in part, this may be related to different mutational mechanisms in distinct tissue types, these observations also suggest that the RB pathway is not strictly linear, and that loss of function due to genetic ablation of the RB gene and loss of E2F binding activity due to hyperphosphorylation of the RB protein are not completely synonymous.
The RB protein shows E2F‐independent functions through binding to other nuclear or extra‐nuclear partners. For example, RB cooperates with transcription factors such as MYOD or RUNX2 to regulate cell differentiation in an E2F‐independent manner.5 The RB protein suppresses the degradation of p27 by SKP2 through direct binding to SKP2; this allows RB to attenuate cell cycle progression in an E2F‐independent manner.6, 7 The RB protein is located in the mitochondrial fraction, where it promotes BAX‐dependent apoptosis.8, 9 Hyperphosphorylated RB is not simply inactivated, but rather contributes to suppression of the mTORC2‐AKT pathway, leading to enhanced sensitivity to chemotherapy.10 In addition to genes involved in cell cycle control, the RB–E2F complex suppresses a number of genes involved in pluripotency, cellular metabolism, innate immunity, and cytokine signaling (Fig. 1).11, 12, 13, 14, 15 The RB–E2F complex colocalizes with EZH2 at intronic and intergenic regions in the genome, and mediates silencing of repetitive DNA sequences.16 Conversely, in particular contexts, the RB–E2F complex positively regulates gene transcription by forming a complex with transcriptional activators.3 To date, more than 300 proteins have been identified as possible binding partners of RB. The variability in these binding partners could explain the multifunctional aspects of the RB protein.
Figure 1.
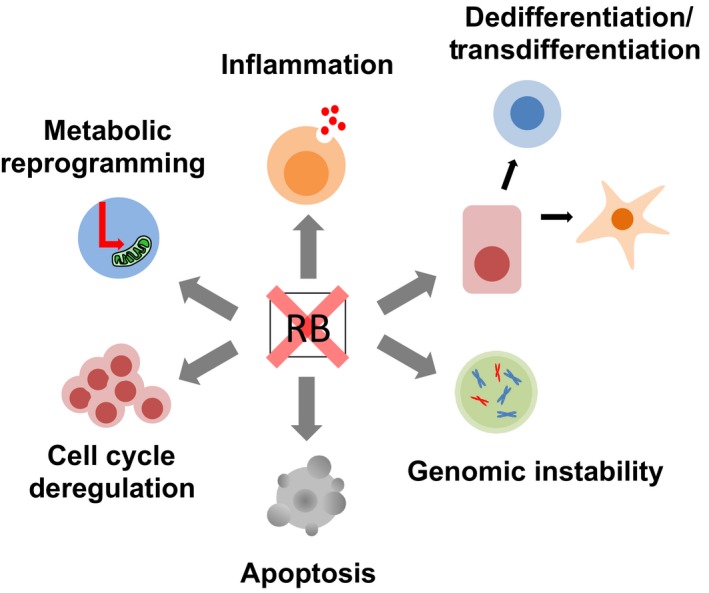
Inactivation of RB during cancer progression results in multiple malignant phenotypes.
Stem cell‐like features induced by RB inactivation
In the past decade, it has been proposed that tissue stem/progenitor cells with multipotency and self‐renewing activity could be the cells of origin for various cancers.13 To maintain a normal balance between differentiation and self‐renewal in tissue stem cells, RB family proteins, including p107 and p130, strictly regulate the G1/S transition. In general, depletion of all RB family proteins in tissue stem/progenitor cells causes defects in differentiation potency and promotes self‐renewing activity, leading to stem cell expansion and tumor initiation.13 However, post‐mitotic cells, that is, those that have completed terminal differentiation, could also be cells of origin for cancer. Depletion of RB family proteins in post‐mitotic cells induces cell cycle re‐entry and dedifferentiation, and even leads to tumor initiation in some contexts. For example, MEFs in which all RB family proteins are inactivated become resistant to G1 arrest, and acquire cell characteristics similar to those of stem cells, such as increased sphere‐forming activity and expression of pluripotent genes.17, 18, 19 In addition, RB depletion on an ARF null background induces cell cycle re‐entry and dedifferentiation in post‐mitotic muscle cells.20 Moreover, an RB family triple KO in post‐mitotic horizontal interneurons of the retina induces metastatic retinoblastoma.21 As expected from these findings, RB inactivation contributes to the generation of inducible pluripotent stem cells from human fibroblasts.22 The RB–E2F1 complex directly suppresses the expression of pluripotent factors such as POU5F1 and SOX2 by binding directly to their regulatory regions with recruiting repressive chromatin modifiers, which consequently antagonizes inducible pluripotent stem cell induction.23 Thus, in addition to deregulation of the cell cycle, RB inactivation in tissue stem/progenitor cells and post‐mitotic cells contributes to tumor formation by promoting self‐renewal activity and dedifferentiation.
It should be noted that studies using genetically engineered mouse models have shown that a single RB gene deletion in stem/progenitor cells and post‐mitotic cells is not sufficient to initiate tumor formation. One of the underlying mechanisms is the induction of apoptosis and/or cell cycle arrest through feedback activation of the p53 pathway, which could be an E2F‐dependent or ‐independent function. For example, suppression of RB function by an SV40 T antigen mutant (T121) induces p53‐dependent cell death and cell cycle arrest in human embryonic stem cells, even though RB is strongly phosphorylated in these cells.24 Another possible mechanism is functional compensation by p107 and p130. In Rb heterozygous mice, p130‐dependent cellular senescence is induced in C cells following Rb loss of heterozygosity.25, 26
Simultaneous mutation in the p53 and RB genes is frequently observed in human cancer cells. A number of studies using genetically engineered mouse models have reported that RB inactivation in a p53‐null background results in tumors that are less differentiated compared to a WT p53 background.27, 28 Rb heterozygous mice develop low‐grade thyroid medullary cancer derived from C cells as a consequence of biallelic loss of Rb. The p53‐null background allowed this C cell tumor to develop dedifferentiated phenotypes depicted by lower expression of calcitonin. This was not seen in backgrounds lacking other p53 pathway genes such as Arf and Cdkn1a, even though thyroid tumors from all of these mice showed similar levels of Ki67 positivity.29 Similarly, RB depletion in p53 KO MEFs, but not in Arf and Cdkn1a KO MEFs, induced sphere‐forming activity in 3‐D culture supplemented with limited growth factors, an indicator of stem‐like activity, despite the fact that these cells showed nearly identical proliferation rates in 2‐D culture (Fig. 2).29 Depletion of RB in p53 KO MEFs, however, did not induce tumorigenicity in immunodeficient mice, suggesting that the acquisition of stem‐like activity caused by Rb/p53 double inactivation is independent of the transformation from normal to cancer cells. Taken together, these findings show that the genetic interaction between Rb and p53 is a critical determinant of the undifferentiated state in both normal and tumor cells.
Figure 2.
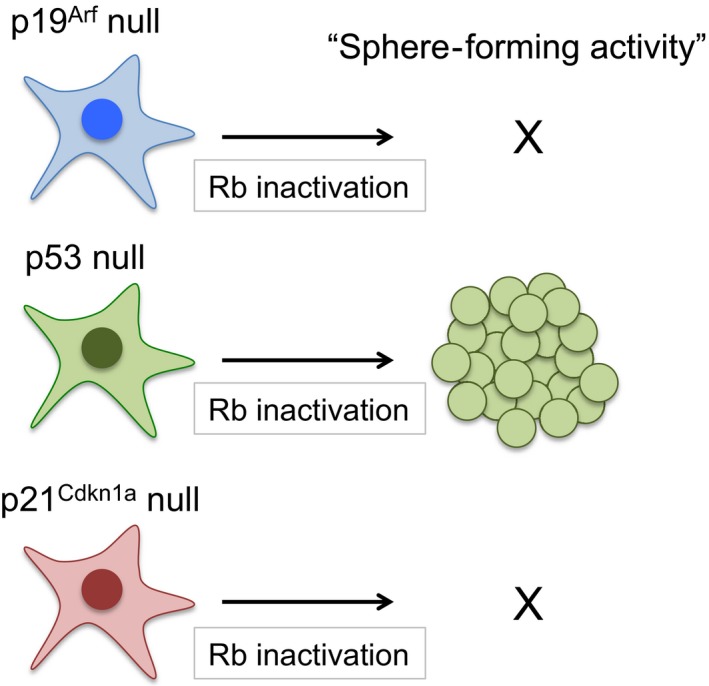
Inactivation of Rb increases the frequency of sphere‐forming activity in p53‐null background but not in Arf or Cdkn1a‐null backgrounds.
Inactivation of RB promotes pro‐inflammatory signaling
It is becoming increasingly clear that in lung, prostate, and breast cancer the acquisition of therapy resistance associated with dedifferentiation, transdifferentiation, and hormone‐independent growth is tightly correlated with aberration of the RB gene during cancer progression. For example, acquisition of drug resistance to EGFR‐targeted therapy in EGFR mutant NSCLC is achieved by transdifferentiation to SCLC, which occurred due to loss of the RB gene.30 In prostate cancer, simultaneous inactivation of RB and p53 in luminal‐type cancer cells leads to increased lineage plasticity and induces the formation of AR‐independent basal‐like cells.31, 32 Another study indicated that E2F1 activation following RB inactivation directly enhances AR expression, thereby promoting hormone‐independent growth.33 In breast cancer, RB inactivation induces epithelial–mesenchymal transition and hormone‐independent growth in ER‐positive luminal‐type cancer cells.34, 35 We have reported that RB inactivation in luminal‐type cancer cells induces stem cell‐like features associated with an enhanced secretion of pro‐inflammatory cytokines, including IL‐6, resulting in hormone‐independent growth.36
Estrogen receptor‐negative breast cancers generally carry RB mutations at a higher frequency, contain a stem cell‐like fraction, and show a higher secretion of pro‐inflammatory cytokines compared to other subtypes of breast cancer.37, 38, 39 Previous studies have shown that pro‐inflammatory cytokines, and the major downstream STAT3 and nuclear factor‐κB signaling pathways in breast cancer, are essential for emergence and maintenance of the stem cell‐like fraction, and for acquisition of drug resistance. For example, single supplementation of IL‐6 or IL‐8 in luminal‐type breast cancer cells induces mammosphere‐forming activity, which is often used as a marker activity of breast cancer stem cells.40, 41, 42, 43 Similarly, another study indicated that activation of the IL‐6/STAT3 pathway in luminal‐type breast cancer cells induces hormone‐independent growth.44 Activation of the IL‐6/STAT3 pathway contributes to the acquisition of drug resistance in various other types of cancer, such as lung and liver, in addition to breast cancer.45, 46, 47
It has been proposed that RB directly regulates pro‐inflammatory signaling. Activation of E2F following RB inactivation directly induces prostaglandin‐endoperoxide synthase 2/COX2 expression in basal‐like breast cancer cells.48 In addition, RB depletion induces IL‐6 secretion in the Cdkn1a‐deficient epidermis.49 Indeed, E2F binding sites have been reported to be localized in the gene regulatory region of some cytokines and their receptors, and the RB–E2F complex directly suppresses cytokine expression, including chemokine (C‐X‐C motif) ligand 1 and 2 and IL‐8.50 In contrast, we recently reported an inverse correlation between RB and IL‐6 expression in breast cancer, as mentioned above,36 but this is not due to direct E2F binding to the IL‐6 promoter (discussed below).
Based on these findings, it has been assumed that activation of pro‐inflammatory signaling by RB inactivation directly contributes to the promotion of self‐renewal and the undifferentiated state. Indeed, we showed that hormone‐independent growth, sphere‐forming activity, and tumor initiation activity induced by RB depletion in MCF‐7 cells, a luminal‐type breast cancer cell line, is antagonized by suppression of IL‐6 or STAT3.36 These results indicate that RB inactivation contributes to the acquisition of these malignant features, not only through “cell intrinsic mechanisms” such as defects in cell cycle control, but also through “cell extrinsic mechanisms” such as hypersecretion of pro‐inflammatory cytokines. As the humanized anti‐IL‐6 receptor antibody, tocilizumab, shows a specific therapeutic effect on RB‐depleted MCF‐7 cells, RB inactivation‐induced IL‐6 hypersecretion may be a potential therapeutic target.
Regulatory feedback mechanisms for fine‐tuning intracellular ROS in RB‐inactivated cells
Many groups have reported that RB and E2Fs control DNA replication and mitosis as well as the G1/S transition during the cell cycle. Inactivation of RB therefore induces genomic instability. For example, RB–E2Fs form a complex with condensin II, which is essential for normal chromosome condensation and subsequent DNA replication.51, 52 Inactivation of RB therefore leads to abnormal chromosome segregation. In addition, excessive expression of Mad2, a spindle‐assembly checkpoint protein, which is directly induced by E2F, causes chromosome instability.53 Furthermore, accelerated cell cycle progression by RB inactivation results in over‐consumption of nucleotides, leading to DNA damage.54 In fact, according to a comprehensive genomic analysis of human tumors, genomic instability is thought to be higher in tumors with mutations in the RB pathway.51, 55
Inactivation of RB is also known to cause oxidative stress by increasing ROS production. This could be related to aberrant autophagy in RB‐inactivated cells,56 because proper degradation of mitochondria by autophagy (i.e. mitophagy) prevents the accumulation of dysfunctional mitochondria, one of the main sources of intracellular ROS production. In myoblast cells, defects in autophagy induced by RB inactivation lead to an accumulation of dysfunctional mitochondria, which in turn inhibits the differentiation of myoblasts to myotubes.57 In contrast, mitochondrial abnormalities in myoblast cells due to RB inactivation have been shown to depend on deregulation of KDM5A, a binding partner of RB.58 Comprehensive proteomic analysis of RB‐inactivated cells identified functional abnormalities in the mitochondria.59 In senescent cells, RB directly promotes mitochondrial oxidative phosphorylation through activation of metabolic genes.60 From these findings, it is assumed that RB‐inactivated cells produce high levels of mitochondrial superoxide due to abnormal electron transfer to oxygen as a result of mitochondrial dysfunction. Conversely, enhanced mitochondrial activity also increases ROS production associated with elevated oxygen consumption. The RB–E2F complex is known to directly suppress the expression of oxidative metabolism‐related enzymes and mitochondrial protein translation genes.61, 62 Disruption of this complex therefore seems to provoke oxidative metabolism, such as the tricarboxylic acid cycle and FAO. Indeed, we reported that RB depletion in MCF‐7 cells promotes mitochondrial superoxide production with elevated oxygen consumption and FAO.36 Taken together, these results suggest that RB‐inactivated cancer cells are exposed to more DNA damage and oxidative stress than RB‐intact cancer cells, even though cancer cells in general already show higher levels of DNA damage and ROS production compared to normal cells.
Accumulation of DNA damage and oxidative stress induces apoptosis and cellular senescence, although lower levels of ROS production act as a growth signal. Thus, RB‐inactivated cancer cells tend to undergo cell death in response to DNA‐damaging agents and excessive ROS production.35, 63 In order for RB‐inactivated cancer cells to display a high proliferation rate in the presence of high ROS production, activation of antioxidant pathways seems to be essential to maintain redox balance in cells at a non‐toxic level. A number of reports have indicated that activation of antioxidative pathways during cancer progression contributes to the acquisition of stem‐like activity, including drug resistance and tumor‐initiating activity. For example, antioxidative pathways are highly activated in the stem cell‐like fraction of cancer cells compared to other non‐tumorigenic fractions, leading to lower levels of ROS and resistance to various cellular stresses.64 Moreover, CD44 and NRF2 directly induce cancer stem‐like activity through activation of antioxidative and detoxification signaling pathways.65, 66
Interleukin‐6 hypersecretion induced by RB inactivation provides cells with an antioxidative function that neutralizes mitochondrial superoxide (Fig. 3). It has been reported that the promoter region of the IL‐6 gene contains an antioxidant responsive element, which is a binding sequence for NRF2, a master regulator of the transcriptional response to oxidative stress, which itself can upregulate IL‐6 secretion.67, 68 We showed that treatment of RB‐depleted MCF‐7 cells with a neutralizing antibody to IL‐6 induced further mitochondrial superoxide production and subsequent cell death.36 In contrast, treatment with recombinant IL‐6 antagonized mitochondrial superoxide production, and promoted sphere‐forming activity and hormone‐independent growth.36 These findings suggest that the induction of stem cell‐like activity induced by RB inactivation as described above depends, at least in part, on the antioxidative function of IL‐6.
Figure 3.
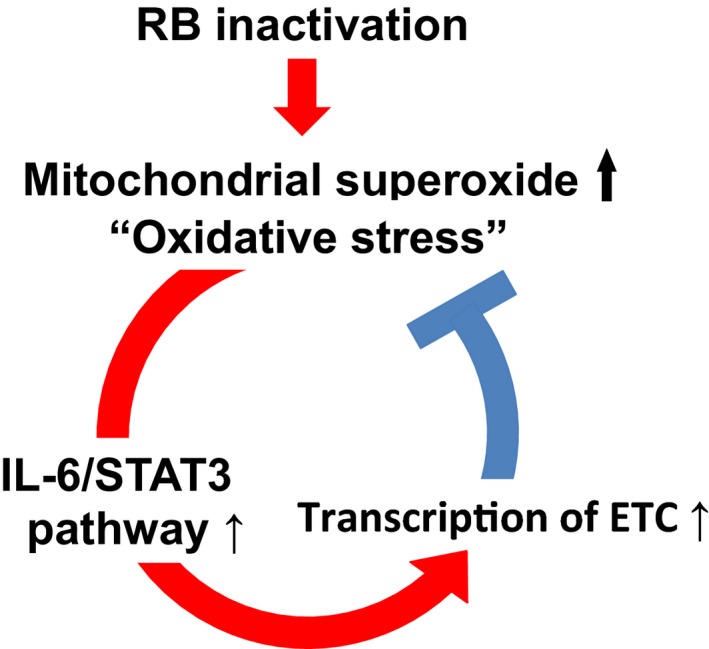
Regulatory feedback loop between interleukin‐6 (IL‐6) secretion and mitochondrial superoxide production for fine‐tuning intracellular reactive oxygen species (ROS) to non‐toxic levels in RB‐inactivated cells. Inactivation of RB induces oxidative stress through enhanced mitochondrial ROS production, leading to hypersecretion of IL‐6. In contrast, IL‐6/signal transducer and activator of transcription 3 (STAT3) activation enhances the function of respiratory chain complex in mitochondria and prevents deregulated ROS production.
Metabolic reprogramming associated with RB inactivation is thought to promote intracellular antioxidant activity. For example, RB‐inactivated cells show an increased dependency on glutamine consumption, which is an amino acid essential for glutathione synthesis, a representative intracellular antioxidant.69, 70 Fatty acid oxidation also provides an antioxidative metabolic pathway by promoting NADPH production, which is another intracellular antioxidant.71 In fact, it has been shown that FAO is essential for maintaining self‐renewal activity in both hematopoietic stem cells and cancer stem‐like cells, and FAO inhibition results in a profound decrease in NADPH levels, an accumulation of ROS, and subsequent cell death.72, 73 Similarly, we reported that RB inactivation in MCF‐7 cells activates FAO, and treatment with an FAO inhibitor increases ROS accumulation.36 Collectively, IL‐6 hypersecretion and metabolic reprogramming, including FAO activation, associated with RB inactivation play key roles in the regulation of redox balance, which leads to cancer progression.
Transcriptional and post‐transcriptional regulation of pro‐inflammatory cytokines by RB
While RB cooperates with various binding partners to directly regulate the expression of various targets as mentioned above, the expression of some pro‐inflammatory cytokines, including IL‐6, is induced by cellular stresses such as oxidative stress and DNA damage associated with RB inactivation. We showed that increased mitochondrial superoxide production due to RB inactivation promotes IL‐6 secretion through activation of the JNK pathway in MCF‐7 cells (Fig. 4).36 Given that IL‐6 possesses an antioxidative function, hypersecretion of IL‐6 induced by RB inactivation can be considered part of a feedback mechanism to maintain the intracellular redox balance within a non‐toxic range.
Figure 4.
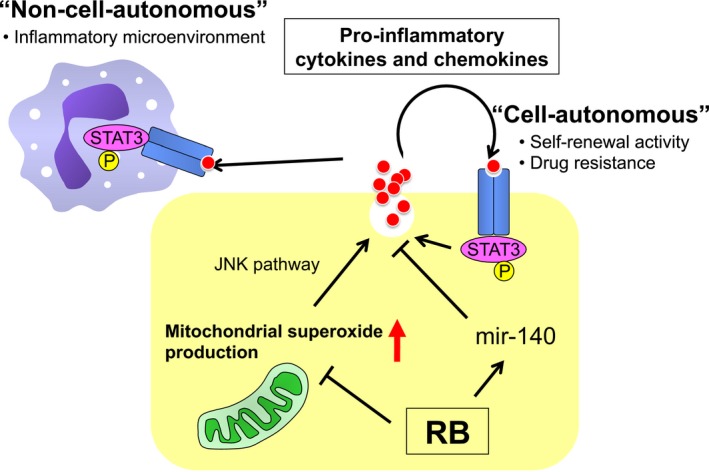
Schematic presentation of the summary in this review. Inactivation of RB induces mitochondrial reactive oxygen species production and subsequent activation of pro‐inflammatory and the signal transducer and activator of transcription 3 (STAT3) loop in a JNK pathway‐dependent manner. Hypersecretion of interleukin‐6 is also post‐transcriptionally promoted by downregulation of microRNA‐140 (mir‐140) following RB inactivation. Hypersecretion of pro‐inflammatory cytokines might contribute to establishment of the inflammatory tumor microenvironment.
In addition, we have shown that RB regulates the expression of many genes, including pro‐inflammatory cytokines, in a miRNA‐dependent manner. MicroRNAs are non‐coding RNAs, consisting of 20–25 bases, that bind to the 3′‐untranslated region of mRNA and regulate gene expression through degradation and/or translational repression of mRNA.74 From a comprehensive analysis of miRNA expression, we found that RB inactivation significantly reduced mir‐140 expression in mouse sarcoma cells and MCF‐7 cells.75 MicroRNA‐140 has been implicated in the suppression of hepatocellular carcinoma, NSCLC, colon cancer, breast cancer, and ovarian cancer through inhibition of growth factor signaling.76, 77, 78, 79, 80 Although the molecular mechanism that underlies the regulation of mir‐140 by RB is still unclear, mir‐140 downregulation following RB inactivation upregulates the expression of multiple pro‐inflammatory cytokines, including IL‐6, and other growth and angiogenic factors that have a mir‐140 binding sequence in the 3′‐untranslated region (Fig. 4).75
Conclusion
Inactivation of RB is frequently observed during cancer progression. Cell cycle deregulation is one of the most important features associated with RB inactivation, however, RB is a multifunction protein interacting with wide varieties of binding partners and has cell cycle‐independent functions. Therefore, RB inactivation during cancer progression induces various malignant phenotypes, including undifferentiated state, drug resistance, and cytokine hypersecretion, in addition to cell cycle deregulation. In this review, we especially focused on stem‐like features induced by RB inactivation associated with hypersecretion of pro‐inflammatory cytokines such as IL‐6. This phenomenon is context‐dependent to some extent, and the molecular mechanisms underlying the control of their expression and its impact on the tumor microenvironment remains incompletely characterized. By clarifying these unsolved questions, we could propose not only novel aspects of RB function but also a new window for development of targeted therapy to RB‐inactivated cancer cells.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- AR
androgen receptor
- C cell
thyroid calcitonin‐producing cell
- E2F
E2 factor
- ER
estrogen receptor
- FAO
fatty acid oxidation
- IL
interleukin
- MEF
mouse embryonic fibroblast
- miRNA
microRNA
- NRF2
nuclear factor erythroid‐related factor 2
- NSCLC
non‐small‐cell lung cancer
- RB
retinoblastoma
- ROS
reactive oxygen species
- SCLC
small‐cell lung cancer
- SKP2
S‐phase kinase‐associated protein 2
- STAT
signal transducer and activator of transcription
Acknowledgment
We thank Drs. D. Barbie and S. Kitajima for critical reading of this manuscript and Mrs. N. Nagatani for secretarial assistance.
Cancer Sci 108 (2017) 1726–1731
Funding Information
Ministry of Education, Culture, Sports, Science and Technology of Japan; Cabinet Office of Japan; Japan Society for the Promotion of Science; Japan Agency for Medical Research and Development; Kanazawa University; Harvard Medical School.
Contributor Information
Shunsuke Kitajima, Email: shunsuke_kitajima@dfci.harvard.edu.
Chiaki Takahashi, Email: chtakaha@staff.kanazawa-u.ac.jp.
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–74. [DOI] [PubMed] [Google Scholar]
- 2. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 2008; 8: 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev 2016; 30: 1492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 2008; 8: 714–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chinnam M, Goodrich DW. RB1, development, and cancer. Curr Top Dev Biol 2011; 94: 129–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ji P, Jiang H, Rekhtman K et al An Rb‐Skp2‐p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial‐penetrance Rb mutant. Mol Cell 2004; 16: 47–58. [DOI] [PubMed] [Google Scholar]
- 7. Binne UK, Classon MK, Dick FA et al Retinoblastoma protein and anaphase‐promoting complex physically interact and functionally cooperate during cell‐cycle exit. Nat Cell Biol 2007; 9: 225–32. [DOI] [PubMed] [Google Scholar]
- 8. Hilgendorf KI, Leshchiner ES, Nedelcu S et al The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev 2013; 27: 1003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferecatu I, Le Floch N, Bergeaud M et al Evidence for a mitochondrial localization of the retinoblastoma protein. BMC Cell Biol 2009; 10: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J, Xu K, Liu P et al Inhibition of Rb phosphorylation leads to mTORC2‐mediated activation of Akt. Mol Cell 2016; 62: 929–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kohno S, Kitajima S, Sasaki N, Takahashi C. Retinoblastoma tumor suppressor functions shared by stem cell and cancer cell strategies. World J Stem Cells 2016; 8: 170–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nicolay BN, Dyson NJ. The multiple connections between pRB and cell metabolism. Curr Opin Cell Biol 2013; 25: 735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sage J. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev 2012; 26: 1409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hutcheson J, Witkiewicz AK, Knudsen ES. The RB tumor suppressor at the intersection of proliferation and immunity: relevance to disease immune evasion and immunotherapy. Cell Cycle 2015; 14: 3812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Benevolenskaya EV, Frolov MV. Emerging links between E2F control and mitochondrial function. Can Res 2015; 75: 619–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ishak CA, Marshall AE, Passos DT et al An RB‐EZH2 complex mediates silencing of repetitive DNA sequences. Mol Cell 2016; 64: 1074–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y, Clem B, Zuba‐Surma EK et al Mouse fibroblasts lacking RB1 function form spheres and undergo reprogramming to a cancer stem cell phenotype. Cell Stem Cell 2009; 4: 336–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dannenberg JH, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth‐restricting conditions. Genes Dev 2000; 14: 3051–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sage J, Mulligan GJ, Attardi LD et al Targeted disruption of the three Rb‐related genes leads to loss of G(1) control and immortalization. Genes Dev 2000; 14: 3037–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pajcini KV, Corbel SY, Sage J, Pomerantz JH, Blau HM. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell Stem Cell 2010; 7: 198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ajioka I, Martins RA, Bayazitov IT et al Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell 2007; 131: 378–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li F, He Z, Shen J et al Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell 2010; 7: 508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kareta MS, Gorges LL, Hafeez S et al Inhibition of pluripotency networks by the rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 2015; 16: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Conklin JF, Baker J, Sage J. The RB family is required for the self‐renewal and survival of human embryonic stem cells. Nat Commun 2012; 3: 1244. [DOI] [PubMed] [Google Scholar]
- 25. Takahashi C, Contreras B, Iwanaga T et al Nras loss induces metastatic conversion of Rb1‐deficient neuroendocrine thyroid tumor. Nat Genet 2006; 38: 118–23. [DOI] [PubMed] [Google Scholar]
- 26. Shamma A, Takegami Y, Miki T et al Rb Regulates DNA damage response and cellular senescence through E2F‐dependent suppression of N‐ras isoprenylation. Cancer Cell 2009; 15: 255–69. [DOI] [PubMed] [Google Scholar]
- 27. Jiang Z, Deng T, Jones R et al Rb deletion in mouse mammary progenitors induces luminal‐B or basal‐like/EMT tumor subtypes depending on p53 status. J Clin Investig 2010; 120: 3296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choi J, Curtis SJ, Roy DM, Flesken‐Nikitin A, Nikitin AY. Local mesenchymal stem/progenitor cells are a preferential target for initiation of adult soft tissue sarcomas associated with p53 and Rb deficiency. Am J Pathol 2010; 177: 2645–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kitajima S, Kohno S, Kondoh A et al Undifferentiated state induced by Rb‐p53 double inactivation in mouse thyroid neuroendocrine cells and embryonic fibroblasts. Stem Cells 2015; 33: 1657–69. [DOI] [PubMed] [Google Scholar]
- 30. Niederst MJ, Sequist LV, Poirier JT et al RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small‐cell lung cancer. Nat Commun 2015; 6: 6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mu P, Zhang Z, Benelli M et al SOX2 promotes lineage plasticity and antiandrogen resistance in TP53‐ and RB1‐deficient prostate cancer. Science 2017; 355: 84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ku SY, Rosario S, Wang Y et al Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017; 355: 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharma A, Yeow WS, Ertel A et al The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Investig 2010; 120: 4478–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arima Y, Inoue Y, Shibata T et al Rb depletion results in deregulation of E‐cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial‐to‐mesenchymal transition. Can Res 2008; 68: 5104–12. [DOI] [PubMed] [Google Scholar]
- 35. Bosco EE, Wang Y, Xu H et al The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Investig 2007; 117: 218–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kitajima S, Yoshida A, Kohno S et al The RB‐IL‐6 axis controls self‐renewal and endocrine therapy resistance by fine‐tuning mitochondrial activity. Oncogene 2017; https://doi.org/10.1038/onc.2017.124. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 37. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Investig 2011; 121: 3804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang Z, Jones R, Liu JC et al RB1 and p53 at the crossroad of EMT and triple‐negative breast cancer. Cell Cycle 2014; 10: 1563–70. [DOI] [PubMed] [Google Scholar]
- 39. Barbie TU, Alexe G, Aref AR et al Targeting an IKBKE cytokine network impairs triple‐negative breast cancer growth. J Clin Investig 2014; 124: 5411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sansone P, Storci G, Tavolari S et al IL‐6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Investig 2007; 117: 3988–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hinohara K, Gotoh N. Inflammatory signaling pathways in self‐renewing breast cancer stem cells. Curr Opin Pharmacol 2010; 10: 650–4. [DOI] [PubMed] [Google Scholar]
- 42. Marotta LL, Almendro V, Marusyk A et al The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(‐) stem cell‐like breast cancer cells in human tumors. J Clin Investig 2011; 121: 2723–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non‐stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA 2011; 108: 1397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sansone P, Ceccarelli C, Berishaj M et al Self‐renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat Commun 2016; 7: 10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Korkaya H, Kim GI, Davis A et al Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell 2012; 47: 570–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. He G, Dhar D, Nakagawa H et al Identification of liver cancer progenitors whose malignant progression depends on autocrine IL‐6 signaling. Cell 2013; 155: 384–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu Z, Aref AR, Cohoon TJ et al Inhibition of KRAS‐driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov 2014; 4: 452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gauthier ML, Berman HK, Miller C et al Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal‐like breast tumors. Cancer Cell 2007; 12: 479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saiz‐Ladera C, Lara MF, Garin M et al p21 suppresses inflammation and tumorigenesis on pRB‐deficient stratified epithelia. Oncogene 2014; 33: 4599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ferrari R, Gou D, Jawdekar G et al Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 2014; 16: 663–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Coschi CH, Ishak CA, Gallo D et al Haploinsufficiency of an RB‐E2F1‐Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov 2014; 4: 840–53. [DOI] [PubMed] [Google Scholar]
- 52. Manning AL, Dyson NJ. RB: mitotic implications of a tumour suppressor. Nat Rev Cancer 2012; 12: 220–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011; 19: 701–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bester AC, Roniger M, Oren YS et al Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011; 145: 435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 2013; 45: 1127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jiang H, Martin V, Gomez‐Manzano C et al The RB‐E2F1 pathway regulates autophagy. Can Res 2010; 70: 7882–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ciavarra G, Zacksenhaus E. Rescue of myogenic defects in Rb‐deficient cells by inhibition of autophagy or by hypoxia‐induced glycolytic shift. J Cell Biol 2010; 191: 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Varaljai R, Islam AB, Beshiri ML, Rehman J, Lopez‐Bigas N, Benevolenskaya EV. Increased mitochondrial function downstream from KDM5A histone demethylase rescues differentiation in pRB‐deficient cells. Genes Dev 2015; 29: 1817–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nicolay BN, Danielian PS, Kottakis F et al Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev 2015; 29: 1875–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takebayashi S, Tanaka H, Hino S et al Retinoblastoma protein promotes oxidative phosphorylation through upregulation of glycolytic genes in oncogene‐induced senescent cells. Aging Cell 2015; 14: 689–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Blanchet E, Annicotte JS, Lagarrigue S et al E2F transcription factor‐1 regulates oxidative metabolism. Nat Cell Biol 2011; 13: 1146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jones RA, Robinson TJ, Liu JC et al RB1 deficiency in triple‐negative breast cancer induces mitochondrial protein translation. J Clin Investig 2016; 126: 3739–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li B, Gordon GM, Du CH, Xu J, Du W. Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer Cell 2010; 17: 469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou D, Shao L, Spitz DR. Reactive oxygen species in normal and tumor stem cells. Adv Cancer Res 2014; 122: 1–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mitsuishi Y, Taguchi K, Kawatani Y et al Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012; 22: 66–79. [DOI] [PubMed] [Google Scholar]
- 66. Ishimoto T, Nagano O, Yae T et al CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(‐) and thereby promotes tumor growth. Cancer Cell 2011; 19: 387–400. [DOI] [PubMed] [Google Scholar]
- 67. Wruck CJ, Streetz K, Pavic G et al Nrf2 induces interleukin‐6 (IL‐6) expression via an antioxidant response element within the IL‐6 promoter. J Biol Chem 2011; 286: 4493–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bulua AC, Simon A, Maddipati R et al Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1‐associated periodic syndrome (TRAPS). J Exp Med 2011; 208: 519–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Reynolds MR, Lane AN, Robertson B et al Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014; 33: 556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nicolay BN, Gameiro PA, Tschop K et al Loss of RBF1 changes glutamine catabolism. Genes Dev 2013; 27: 182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 2013; 13: 227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Viale A, Pettazzoni P, Lyssiotis CA et al Oncogene ablation‐resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014; 514: 628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ito K, Carracedo A, Weiss D et al A PML‐PPAR‐delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med 2012; 18: 1350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15: 509–24. [DOI] [PubMed] [Google Scholar]
- 75. Yoshida A, Kitajima S, Li F et al MicroRNA‐140 mediates RB tumor suppressor function to control stem cell‐like activity through interleukin‐6. Oncotarget 2017; 8: 13872–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang H, Fang F, Chang R, Yang L. MicroRNA‐140‐5p suppresses tumor growth and metastasis by targeting transforming growth factor beta receptor 1 and fibroblast growth factor 9 in hepatocellular carcinoma. Hepatology 2013; 58: 205–17. [DOI] [PubMed] [Google Scholar]
- 77. Yuan Y, Shen Y, Xue L, Fan H. miR‐140 suppresses tumor growth and metastasis of non‐small cell lung cancer by targeting insulin‐like growth factor 1 receptor. PLoS One 2013; 8: e73604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang W, Zou C, Pan L et al MicroRNA‐140‐5p inhibits the progression of colorectal cancer by targeting VEGFA. Cell Physiol Biochem 2015; 37: 1123–33. [DOI] [PubMed] [Google Scholar]
- 79. Lan H, Chen W, He G, Yang S. miR‐140‐5p inhibits ovarian cancer growth partially by repression of PDGFRA. Biomed Pharmacother 2015; 75: 117–22. [DOI] [PubMed] [Google Scholar]
- 80. Li Q, Yao Y, Eades G, Liu Z, Zhang Y, Zhou Q. Downregulation of miR‐140 promotes cancer stem cell formation in basal‐like early stage breast cancer. Oncogene 2014; 33: 2589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]