

Abstract
Dysregulation of microRNA (miRNA) expression in various cancers and their role in cancer progression is well documented. The purpose of this study was to investigate the biological role of miR‐372 in human pancreatic adenocarcinoma (HPAC). We collected 20 pairs of HPAC tissues and adjacent non‐cancerous tissues to detect miR‐372 expression levels. We transfected BXPC‐3 and PANC‐1 cells with miR‐372 inhibitor/mimics to study their effect on cell proliferation, apoptosis, invasion, migration and autophagy. In addition, miR‐372 mimics and a tumor protein UNC51‐like kinase 1 (ULK1) siRNA were co‐transfected into BXPC‐3 and PANC‐1 cells to explore the mechanism of miR‐372 and ULK1 on HPAC tumorigenesis. We found that the expression of miR‐372 was markedly downregulated in HPAC cells compared to adjacent normal tissues. Furthermore, functional assays showed that miR‐372 inhibited cell proliferation, invasion, migration and autophagy in BXPC‐3 and PANC‐1 cells. An inverse correlation between miR‐372 expression and ULK1 expression was observed in HPAC tissues. Downregulation of ULK1 inhibited the overexpression effects of miR‐372, and upregulation of ULK1 reversed the effects of overexpressed miR‐372. Finally, we found that silencing ULK1 or inhibiting autophagy partly rescued the effects of miR‐372 knockdown in HPAC cells, which may explain the influence of miR‐372/ULK1 in HPAC development. Taken together, these results revealed a significant role of the miR‐372/ULK1 axis in suppressing HPAC cell proliferation, migration, invasion and autophagy.
Keywords: Autophagy, human pancreatic adenocarcinoma, microRNA‐372, migration, proliferation
Human pancreatic adenocarcinoma (HPAC) is an extremely serious malignant neoplasm, with a 5‐year survival rate of <5%.1 Early onset of distant metastasis primarily accounts for the poor prognosis of HPAC.2, 3, 4 Therefore, understanding the mechanisms that regulate HPAC metastasis are critical to improving HPAC treatment.
MicroRNA (miRNA) are small non‐coding RNA approximately 17–24 nucleotides in length that regulate gene expression by inhibiting mRNA translation.5, 6 miRNA play critical roles in cell proliferation, apoptosis and differentiation.7 6‐8nt binding site sequences between miRNA and its target gene is a widespread phenomenon in miRNA study.8, 9, 10 MiRNA are usually downregulated in various kinds of human cancers, suggesting that they act as tumor suppressors in tumor cells.11 Previous work on miR‐371–373 was limited to human embryonic stem cells.12, 13 However, recent work in several kinds of cancer cells has demonstrated that miR‐372 regulates cell cycle, apoptosis, proliferation and invasion. Specifically, misexpression of miR‐372 is observed in human hepatocellular cancer.14 In ovarian carcinoma, miR‐372 plays a role in inhibiting tumor growth through ATPase family‐AAA domain containing 2 (ATAD2), large tumor suppressor kinase 2 (LATS2), sequestosome 1 (P62), dickkopf WNT signaling pathway inhibitor 1 (DKK1) and cyclinA1.15
Autophagy is a highly conserved eukaryotic stress and survival response that degrades cytoplasmic contents and recycles biosynthetic substrates to produce energy. Autophagy involves the lysosomal degradation of cytoplasmic contents for regeneration of anabolic substrates.16 UNC51‐like kinase 1 (ULK1) is a serine/threonine kinase that initiates the autophagy cascade.17 ULK1 is regulated in part by mechanistic target of rapamycin (mTOR) and adenosine 5′‐monophosphate‐activated protein kinase (AMPK), which inhibit and activate ULK1, respectively.18
It is not known whether miR‐372 directly targets ULK1 and negatively regulates cell cycle progression and proliferation in human pancreatic adenocarcinoma cells through the regulation of autophagy. In this study, we investigated the role of miR‐372 in HPAC. We predicted the target genes of miR‐372 and demonstrated a direct relationship between miR‐372 and ULK1.
MATERIALS AND METHODS
Tissues and cell lines
Samples of pancreatic carcinoma and para‐carcinoma normal tissues were collected from Xiangya Hospital. The pancreatic carcinoma tissues and normal tissues were rapidly frozen in liquid nitrogen and stored at −80°C. BXPC‐3 and PANC‐1 cell lines used in the study were purchased from ATCC, and cells were cultured according to ATCC recommendations: RPMI 1640 medium with 10% FBS (Gibco, USA) and supplemented with penicillin/streptomycin (Sigma, USA) at 37°C in an atmosphere of 5% CO2 and 95% air.
Antibodies
Commercially available antibodies were used for all immunoblotting and immunofluorescence studies. Anti‐ULK1, anti‐p‐ULK1 (Ser757), anti‐p‐ULK1 (Ser555), anti‐p‐mTOR, anti‐P62 and anti‐LC3 were obtained from Abcam (UK). Anti‐GAPDH was obtained from Shanghai Kangchen Bio‐tech Company (China). All secondary antibodies were obtained from Boster (China).
Cell transfection
Cells were transfected with anti‐miR‐372 (Ambion, Austin, TX, USA) or miR‐372 mimic using the reverse transfection method using the siPORTTM NeoFXTM transfection reagent (Ambion). The siRNA targeting human ULK1 (si‐ULK1) and the corresponding negative control were purchased from GenePharma (Shanghai, China). ULK1 overexpressing plasmids were constructed using pCDNA3.1 (+) basic vectors in our laboratory. These were transfected into BXPC‐3 and PANC‐1 cells with 80%–90% confluence, using Lipofectamine 2000 (Invitrogen, USA), according to the manufacturer's instructions.
Quantitative real‐time PCR
Tumor specimens and cells were subjected to total RNA extraction using TRIzol Reagent (Takara, Japan). To detect RNA expression, complementary DNA (cDNA) was synthesized using a Bestar qPCR RT Kit (DBI Bioscience, Germany), according to the instructions. RNA amplification and detection were performed using a Bestar qPCR RT Kit under an ABI 9700 PCR amplifier system (Applied Biosystems, USA). The primer sequences are as follows: for hsa‐miR‐372, forward primer: ACA CTC CAG CTG GGA AAG TGC TGC GAC ATT TG, reverse primer: CTC AAC TGG TGT CGT GGA GTC GGC AAT TCA GTT GAG ACG CTC A; for ULK1 detection, forward primer: CCT GCT GAG CCG AGA ATG, reverse primer: CTG CTT CAC AGT GGA CGA CA. The miRNA and mRNA expression levels were normalized to those of U6 and β‐actin.
Luciferase reporter assay
The target genes of miR‐372 were predicted using miRanda‐mirSVR (http://www.microrna.org/) and TargetScan (http://www.Targetscan.org/). The wild‐type 3′‐UTR segment (508 bp) of the ULK1 mRNA (not the full length ULK1 3′UTR) containing miR‐372 binding sites was cloned into the dual‐luciferase reporter psiCHECK‐2vector (Promega, USA) using Xhol I and NotI sites, and termed WT‐ULK1‐3′UTR. A mutant construct in miR‐372 binding sites of ULK1 3′UTR region was also synthesized, and subcloned into the psiCHECK‐2 vector (Ambion) using Xhol I and NotI sites, and termed Mut‐ULK1‐3′UTR. For the dual‐luciferase reporter assay, BXPC‐3 cells were transfected with miR‐372 or negative control (NC) for 24 h and the cells were then transfected with WT/Mut‐ ULK1‐3′UTR reporter plasmid using Lipofectamine 2000 (Invitrogen). After 48 h, luciferase activity was determined using a Dual‐Luciferase Reporter Assay Kit (Promega, USA), according to the manufacturer's instructions. Renilla‐luciferase was used to normalize the data.
Western blotting
The BXPC‐3 and PANC‐1 cells were grown to approximately 80%–90% confluence, and then washed with ice‐cold PBS. Forty‐eight hours after transfection, the cells were lysed with protein extraction buffer (Beyotime, China) and phenylmethanesulfonyl fluoride (Genebase, China). For western blotting, proteins were mixed with SDS loading buffer (pH = 6.8 250 mM Tris‐Hcl, 10% SDS, 0.5% bromophenol blue, 5% β‐mercaptoethanol, 50% glycerol) and incubated at 98°C for 8 min. Proteins were separated with 5%–15% SDS‐PAGE, then transferred to PVDF membranes (Millipore, USA), and detected with the appropriate primary antibodies. Finally, PVDF membranes were viewed by using chemiluminescent HRP substrate (Millipore, USA). For P62 detection, the anti‐P62 antibody was incubated at a dilution of 1:1000, and the P62 membrane was transferred at a constant current of 300 mA for 55 min. For ULK1 detection, the anti‐ULK1 antibody was incubated at a dilution of 1:3000 dilution, and the ULK1 membrane was transferred at a constant current of 300 mA for 100 min. For P‐ULK1 (ser757) detection, the anti‐P‐ULK1 (ser757) antibody was incubated at a dilution of 1:1500, and the P‐ULK1 (ser757) membrane was transferred at a constant current of 300 mA for 100 min. For P‐ULK1 (ser555) detection, the anti‐P‐ULK1 (ser555) antibody was at diluted at a concentration of 1:2000 dilution, and the P‐ULK1 (ser555) membrane was transferred at a constant current of 300 mA for 100 min. For P‐mTOR detection, the anti‐P‐mTOR antibody was diluted at a concentration of 1:1000, and the P‐mTOR membrane was transferred at a constant current of 300 mA for 240 min. For GAPDH detection, the anti‐GAPDH antibody was incubated at a dilution of 1:10 000, and the GAPDH membrane was transferred at a constant current of 300 mA for 40 min.
5‐ethynyl‐2‐deoxyuridine and Hoechst 33258 staining assay
The BXPC‐3 and PANC‐1 cells were seeded into 96‐well plates. The cell proliferation rate was detected by a 5‐ethynyl‐2′‐deoxyuridine (EdU) kit (Molecular Probes, USA), according to the manufacturer's instructions; 100 μL culture medium and 5 mol/L EdU was added to each well, and incubated for 2 h. The cells were washed three times with PBS and were observed under a fluorescence microscope (Olympus, Japan). In addition, 30 min after fixation with 4% paraformaldehyde, followed by a 10‐min treatment with 0.5% Triton, Hoechst 33258 (10 μg/mL) reaction solution (Sigma, USA) was added to each well, and stained with 4,6‐diamidino‐2‐phenylindole nuclear. After three washes with PBS, the cells were detected at 400 × magnification. Condensed or fragmented nuclei were considered apoptotic.19
Transwell migration and invasion assay
The treated BXPC‐3 and PANC‐1 cells were cultured in a serum‐free medium for 12 h. Then 2 × 104 starved cells were added to the top of a 24‐well Millipore transwell chamber (Millipore, USA) and 600 μL of RPMI 1640 medium containing 10% FBS were added to the lower chamber. After 24 h, the cells on the lower chamber were stained with a crystal violet solution (Sigma‐Aldrich, USA) for microscopic analysis.20 Subsequently, the membranes were mounted on a glass slide, and the cells that had migrated were viewed by light microscopy. Five different fields of cell quantity were compared using a 200 × magnification. Apart from paving the upper well with diluted Matrigel (BD Biosciences, USA), the same procedures were performed to conduct the invasion assay.
Wound scratch assay
Six‐well plates supplemented with RPMI 1640 and 10% FBS were used to culture cells. A scratch was made in the center of the cell plates with a sterile p200 pipette tip. The medium was then removed and replaced with serum‐free medium. Subsequently, the appropriate transfected cells were incubated in serum‐free medium for 48 h. The ability of the cells to move and close the wound was determined by comparing the 0 h and 48 h phase contrast micrographs of three marked positions.
Immunohistochemistry
Tissue sections were fixed with formalin, embedded with paraffin, and stained immunohistochemically. The tissue sections were deparaffinized in xylene, and then subjected to alcohol dehydration. We incubated the sections in an EDTA buffer (pH 8.0) with microwaves for 20 min to retrieve antigens. To block endogenous peroxidase activity, tissue sections were treated with 100% methanol and 0.3% H2O2 at room temperature for 25 min. These were incubated with anti‐ULK1 monoclonal antibody at 4°C overnight, followed by incubating with biotinylated anti‐rabbit secondary antibody. Finally, the sections were incubated in a streptavidin‐peroxidase solution for 45 min at room temperature. Normal tissue for the primary antibody was the positive control.
Cell proliferation assay
The proliferation rate of cells was determined using a Cell Counting Kit‐8 (CCK‐8, Beyotime, China), according to the specifications. BXPC‐3 and PANC‐1 cells (2 × 103) were seeded in 96‐well plates in RPMI1640 (100 uL) containing 10% FBS, then cultured overnight. Four hours after transfection, the medium was renewed and then continuously cultured for 48 h; 10 uL CCK‐8 solution was added to each well, and incubated at 37°C for 100 min. An automatic microplate reader (BioTeke, China) detected the absorbance at 450 nm. In addition, a standard curve was established to calculate the cell number. Each experiment was repeated three times.
Apoptosis assay
A Coulter Epics XL Flow Cytometer (Beckman Coulter, CA, USA) analyzed apoptotic rates using an annexin V‐Alexa Fluor/PI Apoptosis Detection Kit (Ebioe, China). Annexin V‐Alexa Fluor/PI staining was performed according to the manufacturer's specifications.
Immunofluorescence
Cells were fixed in 4% formaldehyde for 10 min permeabilized with 0.5% Triton X‐100 for 10 min, and blocked with 1% BSA solution for 45 min. The cells were incubated with the appropriate primary antibodies at 4°C overnight and then incubated with Cy3‐labelled or FITC‐labelled secondary antibodies (Beyotime, China) at room temperature for 1 h. Each step was followed by two 10‐min washes. The prepared specimens were counterstained with DAPI (5 g/L) for 5 min, and observed with a fluorescence microscope (Olympus, Japan).
Statistical analyses
SPSS 20.0 software (Chicago, USA) was used to analyze data with independent sample t‐tests. All values were represented as mean ± SD. The statistical significance difference was defined as P < 0.05.
RESULTS
MiR‐372 expression is downregulated in human pancreatic adenocarcinoma tissues
First, we examined differences in miR‐372 expression between pancreatic adenocarcinoma and normal pancreas. To explore the potential biological role of altered miR‐372 expression in human HPAC progression, we evaluated miR‐372 expression in 20 pancreatic adenocarcinoma tissues and 20 normal pancreatic tissues by quantitative RT‐PCR. As shown in Figure 1a, the level of miR‐372 was significantly decreased in pancreatic adenocarcinoma tissues compared with normal tissues. In addition, we determined autophagy associated with LC3 expression and found that the levels of LC3‐II were increased in pancreatic adenocarcinoma tissues compared to normal tissues. The levels of P62, which is an autophagy substrate, were also increased in pancreatic adenocarcinoma tissues (Fig. 1b).
Figure 1.
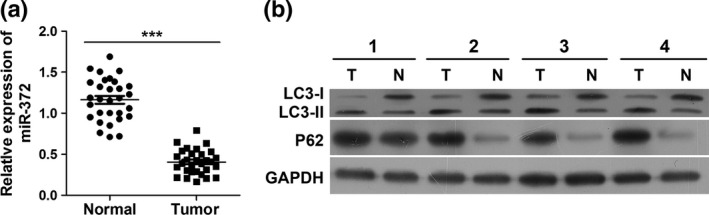
MiR‐372 expression is downregulated in pancreatic carcinoma and may play a positive role on autophagy. (a) Quantitative real‐time polymerase chain reaction (qRT‐PCR) analysis of miR‐372 expression in pancreatic carcinoma tissues and normal pancreatic tissues. ***P < 0.001 versus normal. (b) Both protein levels of LC3‐II and P62 are increased in pancreatic carcinoma tissues. Pancreatic carcinoma tissues (T) and normal pancreas tissues (N) were collected for western blotting analysis.
MiR‐372 plays a negative role in human pancreatic adenocarcinoma cell proliferation, migration and invasion
To further explore the effect of miR‐372 on HPAC, we transfected miR‐372 mimics and anti‐miR‐372 into BXPC‐3 and PANC‐1 cells, respectively, and measured their proliferation, migration and invasion. First, as shown in Figure 2b, the transfected miR‐372 mimics increased the expressions of miR‐372, while anti‐miR‐372 decreased the expressions of miR‐372. BXPC‐3 cells transfected with miR‐372 mimics and PANC‐1 cells transfected with anti‐miR‐372 cells were seeded into 96‐well plates. After 24, 48 and 72 h, the CCK‐8 assay was used to evaluate cell proliferation. We found that overexpression of miR‐372 reduced BXPC‐3 and PANC‐1 cell proliferation rates, whereas inhibition of miR‐372 promoted BXPC‐3 and PANC‐1 cell proliferation rates compared with control cells (Fig. 2b). These results were consistent with results obtained by the EdU staining assay (Fig. 2d), suggesting that miR‐372 is involved in inhibiting cell proliferation. We next examined the effect of miR‐372 on cell apoptosis. As shown in Figure 2c, we found that miR‐372 increased the apoptosis of cultured BXPC‐3 and PANC‐1 cells. We then performed the transwell migration/invasion assay to investigate the in vitro biological functions of miR‐372 on HPAC cells. We found that the expression of miR‐372 in BXPC‐3 and PANC‐1 cells inhibited cell migration and invasion (Fig. 2e,f).
Figure 2.
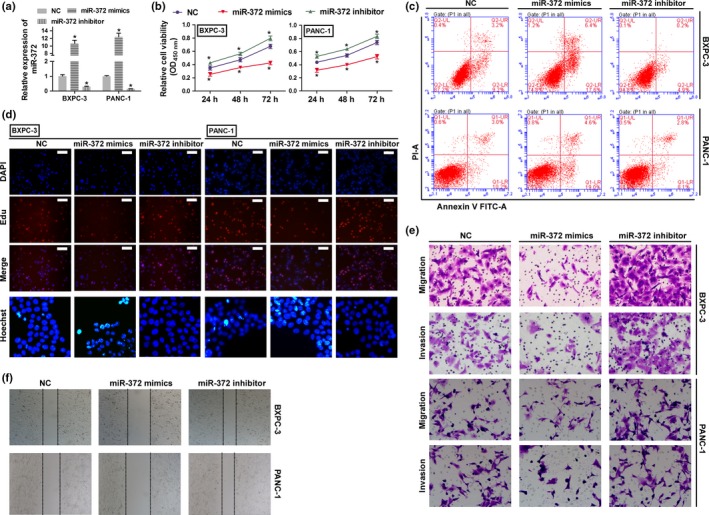
MiR‐372 plays a negative role in cell proliferation, migration and invasion in human HPAC cells. BXPC‐3 and PANC‐1 cells were transfected with miR‐372 mimics or anti‐miR‐372 and used in following assay. (a) The expression levels of miR‐327 were detected by qRT‐PCR. (b) Cell proliferation rates were determined by the CCK8 assay. *P < 0.01 versus negative control (NC). (c) Cell apoptosis was determined by flow cytometry. (d) Cell proliferation and apoptosis were detected by EdU (Scale bar: 100 μm) and Hoechst 33 258 (400 × magnification) staining assay. (e,f) Migration and invasion were determined by transwell migration (200 × magnification) assay, transwell invasion (200 × magnification) assay and wound scratch assay (40 × magnification).
ULK1 is a target of miR‐372 in human pancreatic adenocarcinoma cells
We used bioinformatics prediction (miRanda‐mirSVR and Target‐Scan) software to determine the targets of miR‐372 in HPAC cells. We found that ULK1 3′UTR has a sequence that binds to miR‐372 at position 474–480 (Fig. 3a). To further confirm that miR‐372 directly targets ULK1, the luciferase activity assay was performed. Our results showed that miR‐372 significantly inhibited the luciferase activity of 3′‐UTR of ULK1 in BXPC‐3 and PANC‐1 cells (Fig. 3a). We also measured the expression levels of miR‐372 in cells transfected with miR‐372 mimics or anti‐miR‐372 by qRT‐PCR. We found that expression of miR‐372 inhibited the mRNA and protein levels of ULK1, and inhibition of miR‐372 promoted the expression of ULK1 (Fig. 3b,c). Furthermore, we found that the phosphorylation levels of ULK1 at Ser555 were decreased when miR‐372 was overexpressed, while the phosphorylation levels were increased when expression of miR‐372 was inhibited (Fig. 3c). Furthermore, the levels of LC3‐II and P62 were negatively regulated by miR‐372 in HPAC cells (Fig. 3d). These results suggest that miR‐372 regulates autophagy of HPAC cells by targeting ULK1.
Figure 3.
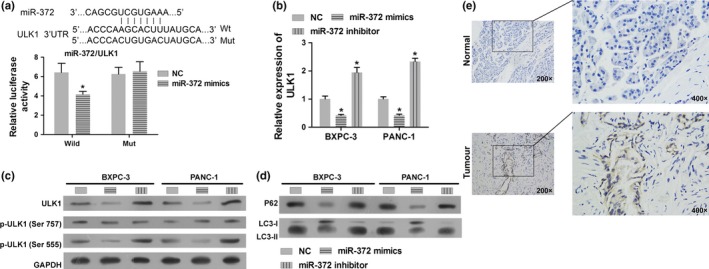
MiR‐372 directly targets ULK1 and regulates autophagy. (a) Detection of dual luciferase reporter gene activity: BXPC‐3 cells were co‐transfected with miR‐372 mimics and ULK1 3′UTR WT/Mut recombinant plasmids. The luciferase activity assay suggested that miR‐372 inhibits the luciferase activity of WT plasmid but had no effect on the activity of Mut plasmid. (b) ULK1 expression on mRNA level as determined by qRT‐PCR in BXPC‐3 and PANC‐1 cells transfected with miR‐372 mimics and anti‐miR‐372 or control. (c) Total protein and phosphorylation levels of ULK1 were determined by miR‐372 mimics and anti‐miR‐372 transfected cells by western blotting in BXPC‐3 and PANC‐1 cells. (d) P62 and LC3 levels were determined in miR‐372 mimics and anti‐miR‐372 transfected cells by western blotting. GAPDH was used as an internal control. (e) Immunohistochemical analysis of ULK1 expression in pancreatic adenocarcinoma tissues and normal pancreas tissues. *P < 0.01 versus negative control (NC).
Downregulation of ULK1 exhibited a similar effect with miR‐372 overexpression in human pancreatic adenocarcinoma cells
To investigate the biological functions of ULK1 in HPAC cells, endogenous ULK1 was knocked down in BXPC‐3 and PANC‐1 cells with a specific siRNA against ULK1 (si‐ULK1). We found that ULK1 was significantly inhibited in BXPC‐3 and PANC‐1 cells by si‐ULK1 (Fig. 4a,g). Knockdown of ULK1 in BXPC‐3 and PANC‐1 cells significantly inhibited cell proliferation (Fig. 4b,d), invasion, migration (Fig. 4e,f) and promoted apoptosis (Fig. 4c,d). These results suggest that the inhibition of ULK1 plays a negative role in HPAC growth, and has a similar effect with overexpressed miR‐372 in HPAC cells. In addition, the phosphorylation levels of ULK1 at Ser555 and LC3‐II were decreased by si‐ULK1, while the levels of P62 and LC3‐I were increased (Fig. 4g,h). This indicates that si‐ULK1 inhibits the autophagy processes, resulting in p62 accumulation in the cell.
Figure 4.
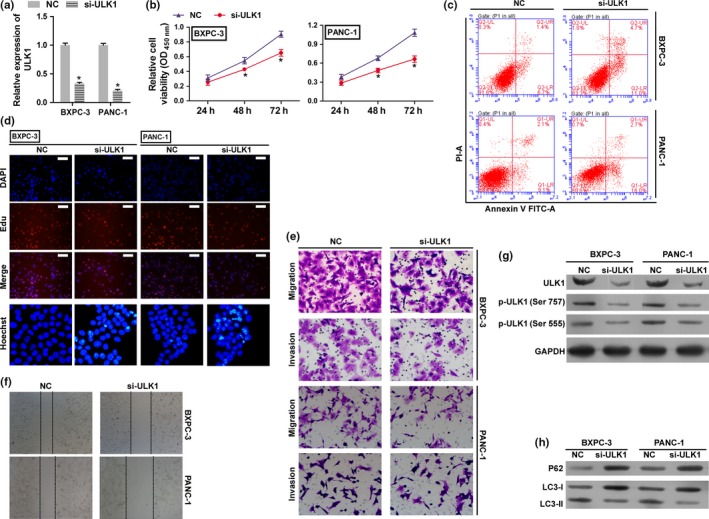
Loss of expression of ULK1 inhibits tumor cell growth in HPAC cells. BXPC‐3 and PANC‐1 cells were transfected with either the negative control (NC) siRNA or si‐ULK1 and used in following assay. (a) The expression levels of ULK1 were detected by qRT‐PCR. (b) The cell proliferation rates were determined by the CCK8 assay. *P < 0.01 versus negative control (NC). (c) Cell apoptosis was determined by flow cytometry. (d) Cell proliferation and apoptosis were detected by EdU (Scale bar: 100 μm) and Hoechst 33 258 (400 × magnification) staining assay. (e,f) Migration and invasion were determined by transwell migration (200 × magnification) assay, transwell invasion (200 × magnification) assay and wound scratch assay (40 × magnification). (g) Total protein and phosphorylation levels of ULK1 were determined by western blotting. (h) P62 and LC3 levels were determined in si‐ULK1 transfected cells by western blotting. GAPDH was used as an internal control. *P < 0.01 versus negative control (NC).
Overexpression of ULK1 rescues the effects of miR‐372 in human pancreatic adenocarcinoma cells
To investigate the functional relevance of ULK1 targeting by miR‐372, we assessed whether ULK1 overexpression could rescue the inhibitory effects of miR‐372 on BXPC‐3 and PANC‐1 cell proliferation, migration, invasion and apoptosis. BXPC‐3 and PANC‐1 cells were co‐transfected with miR‐372 mimics and ULK1 overexpression plasmids. QRT‐PCR and western blot analysis were used to validate the ULK1 mRNA and protein levels in the rescue experiment (Fig. 5a,g). Results showed that the exogenous expression of ULK1 rescued the inhibitory effect of miR‐372 on cell proliferation, migration and invasion (Fig. 5b,d–f). Apoptosis levels were reduced in the ULK1 overexpressed group (Fig. 5c). Moreover, protein levels of LC3‐II were increased and protein levels of P62 were reduced by exogenous ULK1 (Fig. 5h).
Figure 5.
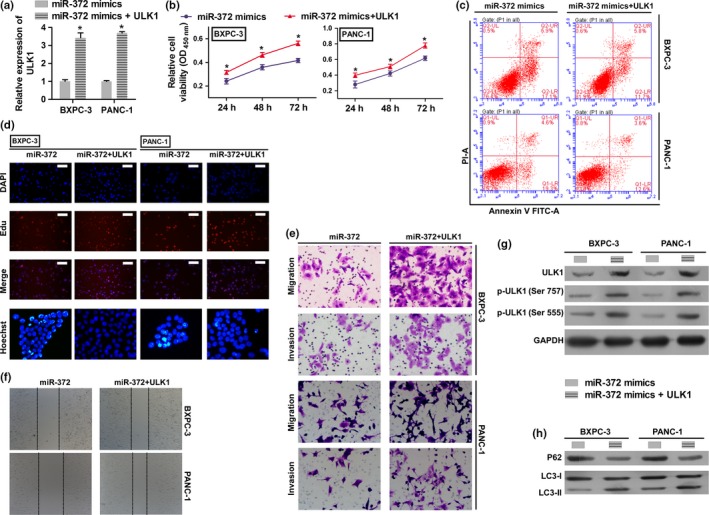
Overexpression of ULK1 rescues the effects of miR‐372 in HPAC cells. BXPC‐3 and PANC‐1 cells were transfected with miR‐372 mimic, or co‐transfected with ULK1 overexpression plasmid and miR‐372 mimic. (a) ULK1 expression on mRNA levels by qRT‐PCR. (b) Cell proliferation rates were determined by the CCK8 assay. (c) Cell apoptosis was determined by flow cytometry. (d) Cell proliferation and apoptosis were detected by EdU (Scale bar: 100 μm) and Hoechst 33 258 (400 × magnification) staining assay. (e,f) Migration and invasion were determined by transwell migration (200 × magnification) assay, transwell invasion (200 × magnification) assay and wound scratch assay (40 × magnification). (g) Total protein and phosphorylation levels of ULK1 were determined by western blotting. (h) P62 and LC3‐II levels were determined by western blotting. GAPDH was used as an internal control. *P < 0.01 versus miR‐372 mimic group.
Inhibitor of autophagy and knockdown of ULK1 reversed the effects of miR‐372 inhibition in human pancreatic adenocarcinoma cells
Because ULK1 is a serine/threonine kinase that initiates the autophagy cascade, we next explored whether ULK1 knockdown and an inhibitor of autophagy could reverse the effects of miR‐372 loss on BXPC‐3 and PANC‐1 cell proliferation, migration and invasion. BXPC‐3 and PANC‐1 cells were co‐transfected with miR‐372 inhibitor and/or ULK1 siRNA with/without chloroquine treatment. The results showed that knockdown of ULK1 and inhibitor of autophagy with chloroquine treatment reversed all the effects of miR‐372 loss on cell proliferation, migration and invasion (Fig. 6b,d–f), while apoptosis levels were increased (Fig. 6c). Levels of LC3‐II were decreased by knockdown of ULK1 and increased by chloroquine treatment; however, levels of P62 were increased in this group when compared with the miR‐372 inhibition group (Fig. 6h). These results suggest that miR‐372 influences the survival of BXPC‐3 and PANC‐1 cells by targeting P62, and by regulating autophagy by targeting ULK1.
Figure 6.
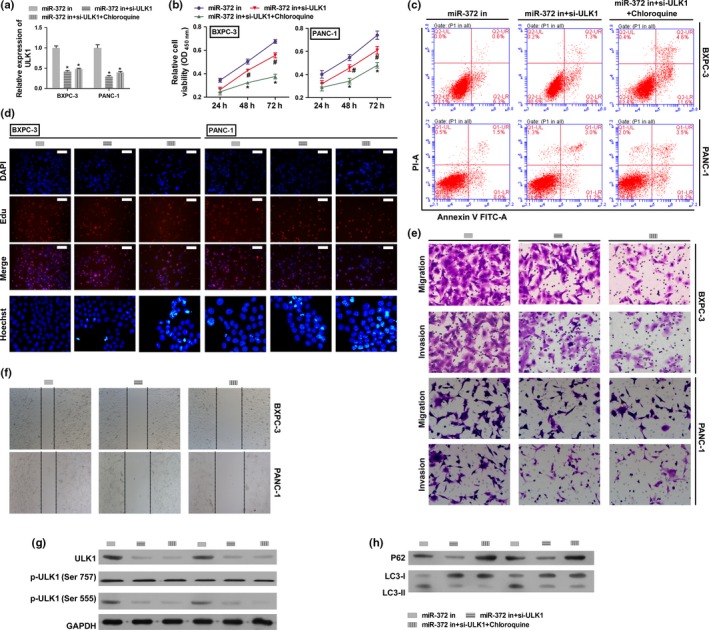
Autophagy inhibitor and ULK1 knockdown reversed the effects of miR‐372 inhibition in HPAC cells. BXPC‐3 and PANC‐1 cells were transfected with miR‐372 inhibitor, co‐transfected with ULK1 siRNA and miR‐372 inhibitor, or co‐transfected with ULK1 siRNA and miR‐372 inhibitor and additionally stimulated with chloroquine. (a) ULK1 expression on mRNA levels by qRT‐PCR. (b) Cell proliferation rates were determined by the CCK8 assay. (c) Cell apoptosis was determined flow cytometry. (d) Cell proliferation and apoptosis were detected by EdU (Scale bar: 100 μm) and Hoechst 33 258 (400 × magnification) staining assay. (e,f) Migration and invasion were determined by transwell migration (200 × magnification) assay, transwell invasion assay (200 × magnification) and wound scratch assay (40 × magnification). (g) Total protein and phosphorylation levels of ULK1 were determined by western blotting. (h) P62 and LC3‐II level were determined by western blotting. GAPDH was used as an internal control. *P < 0.01 versus miR‐372 inhibitor group.
DISCUSSION
Accumulating evidence shows that miRNA are involved in tumorigenesis and metastasis of various human cancers, including HPAC.21, 22, 23 Furthermore, miRNA have been implicated to function as both tumorigenic or tumor‐suppressing genes.11 The overexpression of miR‐372 in some cancers24, 25 has been reported to play an oncogenic role.26 For instance, miR‐372 suppresses human prostate cancer cell migration and invasion by targeting p65,27 miR‐372 inhibits endometrial carcinoma development by targeting the expression of the Ras homolog gene family member C (RhoC),28 and miR‐372 suppresses renal cell carcinoma proliferation and invasion by targeting IGF2BP1.29 This is interesting, as our study showed that miR‐372 was downregulated in human pancreatic adenocarcinoma tissues. Overexpression of miR‐372 in human pancreatic adenocarcinoma cell lines suppresses cell growth. Depending on the specific cellular milieu, the same miRNA can act as a tumorigenic agent in some cancers and a tumor suppressor in others. Therefore, we speculate that cell‐specific environments can account for the differences observed between the biological functions of miR‐372 in pancreatic adenocarcinomas compared with other cancers.
The ULK1 kinase complex initiates the autophagy process.30 The activity of the ULK1 complex is inhibited by mTOR signaling and is positively regulated by the adenosine monophosphate‐activated protein kinase (PRKAA2/AMPK). The phosphorylation of ULK1 on Ser 555 (stimulates the activity) or on ULK1 Ser 757 (inhibits the activity) allows specific antibodies to recognize the activity of the ULK1 complex.30, 31, 32 Luke R. G. reported that the autophagy‐initiating kinase ULK1 contributes to cancer cell survival, and that ULK1 is a direct target of ATF4 (activating transcription factor 4), which promotes the expression of ULK1 mRNA and protein.33 Similarly, in our study, we found that the loss of miR‐372 promotes the expression of ULK1 mRNA and protein, and enhances the pancreatic adenocarcinoma cell proliferation, migration and invasion. Finally, our results showed that knockdown of ULK1 and the autophagy inhibitor with chloroquine treatment can partly reverse the effect of miR‐372 loss on cell proliferation, migration and invasion. These indicate that miR‐372 induces the loss of phosphorylation in ULK1 on Ser 555, and has a regulatory role in HPAC cell autophagy. A previous study also reported that miR‐372 inhibits P62 in head and neck squamous cell carcinoma in vitro and in vivo.34 Jin et al. report that YY1 was stimulated to promote P62 expression and subsequent autophagy activation by suppressing miR‐372 expression,35 Increasing P62 promotes tumorigenesis while inhibition of autophagy prevents tumor growth in nude mice.36 However, the mechanism of how autophagy is activated under different conditions remains largely unknown. In our study, we detected protein levels of P62 and found that they changed with autophagy levels and miR‐372 expression levels. Impaired autophagy promotes accumulation of P62,37 while P62 is directly suppressed by miR‐372. P62, a multifunctional protein that directs ubiquitinated protein aggregates to autophagosomes for degradation,36 may also play a dual‐role in this signaling pathway. Our results suggest that miR‐372 inhibits survival of BXPC‐3 and PANC‐1 cells through regulating autophagy by targeting ULK1, rather than directly targeting P62 (Fig. 7).
Figure 7.
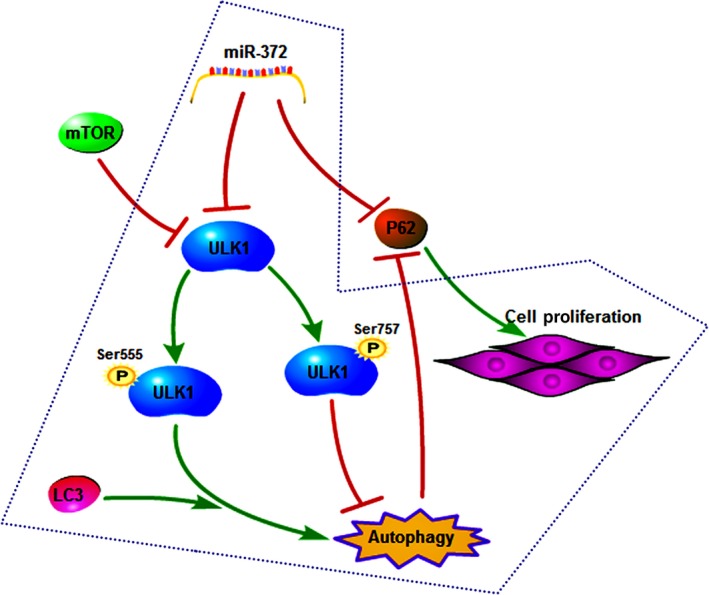
Regulation pathway: miR‐372 inhibits survival of human pancreatic adenocarcinoma (HPAC) cells through regulating autophagy by targeting ULK1.
Altogether, our results demonstrated that miR‐372 plays a tumor‐suppressor role in HPAC. In addition, miR‐372 targets ULK1 (which is associated with autophagy) to suppress the growth of BXPC‐3 and PANC‐1 cells.
DISCLOSURE STATEMENT
The authors have no conflicts of interest to declare.
ACKNOWLEDGMENTS
This work was supported by grants from the Key Projects of Hunan Province Science and Technology Plan (No. 2016JC2040).
Cancer Sci 108 (2017) 1811–1819
Funding information
This work was supported by grants from the Key Projects of Hunan Province Science and Technology Plan (No. 2016JC2040).
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66: 7–30. [DOI] [PubMed] [Google Scholar]
- 2. Xu Y, Chang R, Peng Z et al Loss of polarity protein AF6 promotes pancreatic cancer metastasis by inducing Snail expression. Nat Commun 2015; 6: 7184. [DOI] [PubMed] [Google Scholar]
- 3. Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet 2004; 363: 1049–57. [DOI] [PubMed] [Google Scholar]
- 4. Feldmann G, Dhara S, Fendrich V et al Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res 2007; 67: 2187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat Genet 2011; 43: 854–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pichinuk E, Broday L, Wreschner DH. Endogenous RNA cleavages at the ribosomal SRL site likely reflect miRNA (miR) mediated translational suppression. Biochem Biophys Res Commun 2011; 414: 706–11. [DOI] [PubMed] [Google Scholar]
- 7. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6: 857–66. [DOI] [PubMed] [Google Scholar]
- 8. Song J, Li Y. miR‐25‐3p reverses epithelial–mesenchymal transition via targeting Sema4C in cisplatin‐resistance cervical cancer cells. Cancer Sci 2017; 108: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuan B, Liang Y, Wang D, Luo F. MiR‐940 inhibits hepatocellular carcinoma growth and correlates with prognosis of hepatocellular carcinoma patients. Cancer Sci 2015; 106: 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huan L, Bao C, Chen D et al MicroRNA‐127‐5p targets the biliverdin reductase B/nuclear factor‐κB pathway to suppress cell growth in hepatocellular carcinoma cells. Cancer Sci 2016; 107: 258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol 2007; 302: 1–12. [DOI] [PubMed] [Google Scholar]
- 12. Stadler B, Ivanovska I, Mehta K et al Characterization of microRNAs involved in embryonic stem cell states. Stem Cells Dev 2010; 19: 935–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lavon I, Zrihan D, Granit A et al Gliomas display a microRNA expression profile reminiscent of neural precursor cells. Neuro Oncol 2010; 12: 422–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang J, Liu X, Wu H et al CREB up‐regulates long non‐coding RNA, HULC expression through interaction with microRNA‐372 in liver cancer. Nucleic Acids Res 2010; 38: 5366–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guan X, Zong ZH, Chen S et al The role of miR‐372 in ovarian carcinoma cell proliferation. Gene 2017; 624: 14–20. [DOI] [PubMed] [Google Scholar]
- 16. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147: 728–41. [DOI] [PubMed] [Google Scholar]
- 17. Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6: 764–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hollville E, Martin SJ. Measuring apoptosis by microscopy and flow cytometry. Curr Protoc Immunol 2016; 112: 14.38.1–38.24 [DOI] [PubMed] [Google Scholar]
- 20. Queiroz KC, Shi K, Duitman J et al Protease‐activated receptor‐1 drives pancreatic cancer progression and chemoresistance. Int J Cancer 2014; 135: 2294–304. [DOI] [PubMed] [Google Scholar]
- 21. Guo J, Feng Z, Huang Z, Wang H, Lu W. MicroRNA‐217 functions as a tumour suppressor gene and correlates with cell resistance to cisplatin in lung cancer. Mol Cells 2014; 37: 664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang W, Liu M, Xu J et al MiR‐377 inhibits the proliferation of pancreatic cancer by targeting Pim‐3. Tumour Biol 2016; 37: 14813–24. [DOI] [PubMed] [Google Scholar]
- 23. Yonemori K, Kurahara H, Maemura K, Natsugoe S. MicroRNA in pancreatic cancer. J Hum Genet 2017; 62: 33–40. [DOI] [PubMed] [Google Scholar]
- 24. Voorhoeve PM, le Sage C, Schrier M et al A genetic screen implicates miRNA‐372 and miRNA‐373 as oncogenes in testicular germ cell tumors. Cell 2006; 124: 1169–81. [DOI] [PubMed] [Google Scholar]
- 25. Ason B, Darnell DK, Wittbrodt B et al Differences in vertebrate microRNA expression. Proc Natl Acad Sci USA 2006; 103: 14385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cho WJ, Shin JM, Kim JS et al miR‐372 regulates cell cycle and apoptosis of ags human gastric cancer cell line through direct regulation of LATS2. Mol Cells 2009; 28: 521–7. [DOI] [PubMed] [Google Scholar]
- 27. Kong X, Qian X, Duan L, Liu H, Zhu Y, Qi J. MicroRNA‐372 suppresses migration and invasion by targeting p65 in human prostate cancer cells. DNA Cell Biol 2016; 35: 828–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu BL, Sun KX, Zong ZH, Chen S, Zhao Y. MicroRNA‐372 inhibits endometrial carcinoma development by targeting the expression of the Ras homolog gene family member C (RhoC). Oncotarget 2016; 7: 6649–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang X, Huang M, Kong L, Li Y. miR‐372 suppresses tumour proliferation and invasion by targeting IGF2BP1 in renal cell carcinoma. Cell Prolif 2015; 48: 593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR‐mediated autophagy regulation. Mol Biol Cell 2009; 20: 2004–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roach PJ. AMPK → ULK1 → autophagy. Mol Cell Biol 2011; 31: 3082–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Egan DF, Shackelford DB, Mihaylova MM et al Phosphorylation of ULK1 (hATG1) by AMP‐activated protein kinase connects energy sensing to mitophagy. Science 2011; 331: 456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pike LR, Singleton DC, Buffa F et al Transcriptional up‐regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem J 2013; 449: 389–400. [DOI] [PubMed] [Google Scholar]
- 34. Yeh LY, Liu CJ, Wong YK, Chang C, Lin SC, Chang KW. miR‐372 inhibits p62 in head and neck squamous cell carcinoma in vitro and in vivo . Oncotarget 2015; 6: 6062–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feng L, Ma Y, Sun J et al YY1‐MIR372‐SQSTM1 regulatory axis in autophagy. Autophagy 2014; 10: 1442–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Komatsu M, Waguri S, Koike M et al Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy‐deficient mice. Cell 2007; 131: 1149–63. [DOI] [PubMed] [Google Scholar]
- 37. Mathew R, Karp C, Beaudoin B et al Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137: 1062–75. [DOI] [PMC free article] [PubMed] [Google Scholar]