

Abstract
Immune modulation of the tumor microenvironment has been reported to participate in the therapeutic efficacy of many chemotherapeutic agents. Recently, we reported that liposomal encapsulation of oxaliplatin (l‐OHP) within PEGylated liposomes conferred a superior antitumor efficacy to free l‐OHP in murine colorectal carcinoma‐bearing mice through permitting preferential accumulation of the encapsulated drug within tumor tissue. However, the contribution of the immune‐modulatory properties of liposomal l‐OHP and/or free l‐OHP to the overall antitumor efficacy was not elucidated. In the present study, therefore, we investigated the effect of liposomal encapsulation of l‐OHP within PEGylated liposomes on the antitumor immunity in both immunocompetent and immunodeficient mice. Liposomal l‐OHP significantly suppressed the growth of tumors implanted in immunocompetent mice, but not in immunodeficient mice. In immunocompetent mice, liposomal l‐OHP increased the tumor MHC‐1 level and preserved antitumor immunity through decreasing the number of immune suppressor cells, including regulatory T cells, myeloid‐derived suppressor cells, and tumor‐associated macrophages, which collectively suppress CD8+ T cell‐mediated tumor cells killing. In contrast, free l‐OHP ruined antitumor immunity. These results suggest that the antitumor efficacy of liposomal l‐OHP is attributed, on the one hand, to its immunomodulatory effect on tumor immune microenvironment that is superior to that of free l‐OHP, and on the other hand, to its direct cytotoxic effect on tumor cells.
Keywords: Antitumor immunity, drug delivery system, immunomodulation, liposome, oxaliplatin
Oxaliplatin (l‐OHP) is a third‐generation platinum agent that exhibits distinct pharmacological properties compared with earlier‐generation agents such as cisplatin and carboplatin.1 Clinically, l‐OHP is frequently used as a first‐line antitumor agent for the treatment of advanced colorectal cancer in conjunction with other agents. The cytotoxic effect of l‐OHP is exerted through the formation of platinum‐DNA adducts. The intrastrand cross‐links formed by l‐OHP inhibit the replication and transcription of DNA, which has a direct cytotoxic effect against tumor cells.2 However, its clinical efficacy is limited, at least in part, by its dose‐limiting side effects, including neurotoxicity.3 In addition, l‐OHP alone has shown limited antitumor efficacy in vivo because of low distribution in tumor tissues.4 Accordingly, overcoming these limitations requires the use of a nanocarrier system to ensure the selective and/or adequate delivery of l‐OHP to tumor tissue.
Liposomes, a bilayer liquid‐filled vesicle made from phospholipids, have been reported to improve the pharmacokinetics and tumor accumulation of encapsulated drugs. Although conventional liposomes are rapidly taken up by cells of the mononuclear phagocyte system (MPS), surface modification with polyethylene glycol (PEGylation) has been proven to prevent recognition by the cells of the MPS, and consequently prolongs the circulating time of liposomes.5 Such long circulation characteristics confer passive tumor‐targeting to PEGylated liposomes through the so‐called enhanced permeability and retention (EPR) effect.6
In an earlier study, we reported that encapsulation of l‐OHP within PEGylated liposomes permitted the preferential accumulation of l‐OHP within the tumor tissue through the EPR effect, resulting in antitumor effects that were greater than those of free l‐OHP in murine colorectal carcinoma‐bearing mice.7, 8, 9 Similarly, Yang et al.10 also report that intravenous injection of neutral PEGylated liposome encapsulating l‐OHP induced a significant apoptotic response against a human colorectal carcinoma xenograft model. These reports suggest that the selective delivery of l‐OHP by encapsulation into PEGylated liposome resulted in enhanced antitumor activity.
Many reports have emphasized the contribution of immune modulation of the tumor microenvironment to the therapeutic efficacy of many chemotherapeutic agents. Previously, antitumor agents have been considered immunosuppressive and have been credited with ruining antitumor immunity. However, recent studies have indicated that some of these antitumor agents have demonstrated a positive effect on antitumor immunity, and their clinical outcomes partially depend on their immunomodulation properties.11 Anthracyclines, particularly doxorubicin, are known to cause tumor cells to undergo immunogenic death and to induce tumor‐specific immune responses. In fact, the depletion of CD8+ T cells, which can kill tumor cells following the recognition of a tumor antigen, has led to a loss of the antitumor effect of doxorubicin.12 This suggests that part of the therapeutic efficacy of doxorubicin depends on CD8+ T cells. In addition, antitumor agents are known to suppress protumor immunity, which includes regulatory T cells (Treg),13 myeloid‐derived suppressor cells (MDSC) 14 and tumor‐associated macrophage (TAM).15 However, it is unclear whether liposomal antitumor agents such as l‐OHP can modulate antitumor and protumor immunity, which would increase therapeutic efficacy in combination with its direct tumor‐cell killing activity.
In the present study, therefore, we compared the efficacy and toxicity of free l‐OHP and liposomal l‐OHP in tumor‐bearing immunocompetent mice and in immunodeficient mice. We here showed that liposomal l‐OHP significantly suppressed protumor immunity and preserved antitumor immunity, whereas free l‐OHP disrupted both forms of immunity. It appears that the encapsulation of antitumor drugs into liposomes may modulate the immunological effect of these drugs.
Materials and Methods
Materials
Hydrogenated soy phosphatidylcholine (HSPC) and 1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐n‐(methoxy [polyethylene glycol]‐2000) (mPEG2000‐DSPE) were generously donated by NOF (Tokyo, Japan). Cholesterol (Chol) was purchased from Wako Pure Chemical (Osaka, Japan). Oxaliplatin (l‐OHP) was generously donated by Taiho Pharmaceutical (Tokyo, Japan). All lipids were used without further purification. All other reagents were of analytical grade.
Animals and tumor cells
Male immunocompetent BALB/c mice (5 weeks old) and male immuno‐deficient BALB/c nu/nu mice (nude mice, 5 weeks old) were purchased from Japan SLC (Shizuoka, Japan). All animal experiments were evaluated and approved by the Animal and Ethics Review Committee of Tokushima University. Colon 26 murine colorectal carcinoma (C26) was purchased from the Cell Resource Center for Biomedical Research (Institute of Development, Aging and Cancer, Tohoku University). The C26 cell line was maintained in RPMI‐1640 medium supplemented with 10% heat‐inactivated FBS (Mediatech, VA, USA), 100 units/mL penicillin and 100 μg/mL streptomycin (MP Biomedicals, CA, USA) under a humidified atmosphere with 5% CO2/95% air at 37°C.
Preparation of l‐OHP‐containing PEGylated liposomes
l‐OHP‐containing PEGylated liposomes (liposomal l‐OHP), composed of HSPC/Chol/mPEG2000‐DSPE (2/1/0.2 molar ratio), were prepared using a reverse‐phase evaporation method that was described earlier.7 Unencapsulated l‐OHP was removed using a dialysis cassette (Slide‐A‐Lyzer, 10000MWCO; Thermo Fisher Scientific, MA, USA) against 5% dextrose. The concentration of l‐OHP in the liposomes was quantified by an atomic absorption photometer (Z‐5700, Hitachi, Tokyo, Japan) after destroying the liposomes with 1% Triton‐X solution. The phospholipid concentration of the liposomes was quantified by phosphorus assay.16 The mean diameter of the liposomes was approximately 100 nm, as determined using a NICOMP 380 ZLS (Particle Sizing System, CA, USA).
Treatment of tumor‐bearing mice with l‐OHP formulations
To develop tumor‐bearing mice, C26 cells (2 × 106 cells) were inoculated subcutaneously into the left flank of either BALB/c or BALB/c nude mice. On day 0 when the tumor volume reached 50–100 mm3, the mice were divided into three groups: a control group (non‐treated), a free l‐OHP treatment group and a liposomal l‐OHP treatment group. In the previous study, we observed that a low dose (4.2 mg/kg) of l‐OHP had little therapeutic effect in a similar experimental animal model.7 To obtain the optimal therapeutic and immunomodulatory effect of l‐OHP, in the current study, we selected 8.3 mg/kg of l‐OHP as an experimental dose. On days 0, 7 and 14, free l‐OHP or liposomal l‐OHP (8.3 mg l‐OHP/kg body) was intravenously injected into the mice. Tumor volume was measured every 3 days using a caliper. The tumor volume was calculated using the following formula: 0.5 × (length) × (width)2. Body weight was measured simultaneously and was taken as a parameter of systemic toxicity.
Treatment of tumor cells with l‐OHP formulations in vitro
C26 cells (105 cells) were seeded onto 12‐well plates 24 h prior to drug exposure. The culture medium was replaced with fresh medium containing free l‐OHP or liposomal l‐OHP (15, 300 μM). After culture for 6, 24 and 48 h, the medium was removed, and the cells were collected. The cell suspension was used for flow cytometry to examine the expression levels of MHC class 1 (MHC‐1) molecules. [Correction added on 31 July 2017, after first online publication: The incubation times in the sentence “After culture for 6, 12 and 48 h, the medium was removed, and the cells were collected” has been changed to “6, 24 and 48h”.]
Flow cytometry analysis
Tumor cell suspensions were prepared using a gentleMACS Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer's instructions. Briefly, tumors were dissected, chopped into small pieces and homogenized in RPMI‐1640 medium using the gentleMACS Dissociator. After the addition of a mixture of enzymes (collagenase type I [Wako Pure Chemical] and Dispase II [Roche Diagnostic, Mannheim, Germany]), the suspensions were incubated for 40 min at 37°C. Next, the suspensions were homogenized again after the addition of DNase I (Roche Diagnostic). After digestion, the cells were filtered through a cell strainer (100 μm, Becton Dickinson, NJ, USA).
Spleen cell suspensions were prepared as described previously.17 Briefly, single‐cell suspensions were prepared using a gentleMACS Dissociator. The cells were suspended in PBS containing 0.5 mM EDTA (EDTA‐PBS). Red blood cells in the suspensions were lysed with ammonium chloride solution (0.83% NH4Cl) for 3 min. Cells were washed with EDTA‐PBS and filtered with a cell strainer to remove clumps. For in vitro re‐stimulation with an antigen, spleen cells (107 cells) were cultured in vitro with mitomycin C‐inactivated C26 cells (2 × 105 cells) in a 24‐well plate for 24 h. During the last 4 h, brefeldin A (Life Technologies, NY, USA) was added (5 μg/mL) to the culture.
For extracellular staining, the prepared cells were incubated with the combinations of antibodies (CD8+ T cell; FITC‐labeled anti‐mouse CD8a, Treg; PE‐labeled anti‐mouse CD4 and Alexa488‐labeled anti‐mouse CD25, MDSC; PE‐labeled anti‐mouse Ly‐6G and FITC‐labeled anti‐mouse CD11b (eBioscience, CA, USA), TAM; Alexa488‐labeled anti‐mouse CD206 (BioLegend, CA, USA) and PE‐labeled anti‐F4/80 [GmbH, CA, USA]). To examine the expression level of MHC‐1, cells were stained with mouse anti‐mouse MHC‐1 (H‐2Dd) antibody (AbD serotec, Oxford, UK) and Alexa647‐labeled anti‐mouse IgG (Life Technologies). For intracellular staining, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% saponin for 20 min, and stained with FITC‐labeled anti‐mouse IFN‐γ (eBioscience). The cells were analyzed using a flow cytometer, Guava EasyCyte Mini (Millipore, MA, USA) or Gallios (Beckman Coulter, CA, USA). The data were analyzed using WinMDI version 2.9 (The Scripps Research Institute, CA, USA).
Statistics
Data are expressed as the mean ± SD. Statistical analysis was performed using a two‐tailed unpaired t‐test and one‐way anova followed by the Tukey post hoc test using graphpad instat software (GraphPad Software, CA, USA). The level of significance was set at P < 0.05.
Results
Liposomal l‐OHP and free l‐OHP had greater therapeutic effect in C26 tumor‐bearing immunocompetent mice than in C26 tumor‐bearing immunodeficient mice
C26 tumor‐bearing immunocompetent BALB/c mice received three intravenous injections (once a week) of either free l‐OHP or liposomal l‐OHP. Non‐treated mice served as the control. Free l‐OHP modestly reduced the tumor growth compared with the control (P < 0.05, Fig. 1a). Compared with free l‐OHP, liposomal l‐OHP significantly reduced the tumor growth (P < 0.05). In addition, the treatment of free l‐OHP led to a small amount of body weight loss and a significant reduction in the number of splenocytes, whereas that of liposomal l‐OHP led to even less body weight loss (Fig. 1b), and the number of splenocytes (Fig. 1c) was maintained similar to that of an untreated state.
Figure 1.
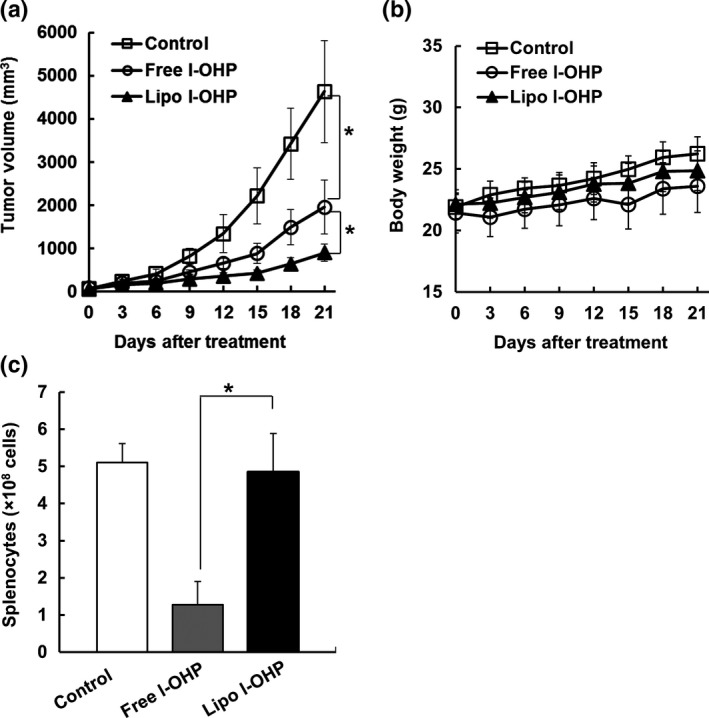
l‐OHP formulations exhibited antitumor effect without systemic toxicity in C26 tumor‐bearing immunocompetent BALB/c mice. C26 cells (2 × 106 cells) were inoculated subcutaneously into BALB/c mice. On days 0, 7 and 14, C26 tumor‐bearing mice received three intravenous injections of either l‐OHP (Free l‐OHP) or l‐OHP‐containing PEGylated liposome (Lipo l‐OHP) at a dose of 8.3 mg l‐OHP/kg. Non‐treated mice served as the control. (a) Tumor volume. (b) Body weight. (c) Number of splenocytes. Each value represents the mean ± SD (n = 5). *P < 0.05.
As shown in Figure 1a, liposomal l‐OHP exhibited therapeutic effects that were superior to those of free l‐OHP in tumor bearing‐immunocompetent mice, which was consistent with the results shown in our previous study.7, 8 To investigate the contribution of the immune system to the increased therapeutic efficacy, either free l‐OHP or liposomal l‐OHP was injected into C26 tumor‐bearing immunodeficient BALB/c nude mice (Fig. 2). Surprisingly, the antitumor effect of both liposomal l‐OHP and free l‐OHP was diminished in immunodeficient mice; free l‐OHP exhibited no tumor growth inhibition effect in C26 tumor‐bearing BALB/c nude mice, while liposomal l‐OHP exhibited a slight level of inhibition (Fig. 2a). In addition, the treatment with l‐OHP formulations caused a slight level of weight loss (Fig. 2b). To find further direct evidence indicating that CD8+ T cells are responsible for the liposomal l‐OHP‐mediated antitumor effect, the antitumor effect of liposomal l‐OHP was investigated in CD8+ T cell‐depleted immunocompetent mice which had been treated with anti‐CD8 antibody. As we expected, the therapeutic effect of liposomal l‐OHP was decreased by depletion of CD8+ T cells (Fig. S1). These results indicate that host immunity, particularly CD8+ T cells, contributes to the tumor growth suppression effect of l‐OHP formulations.
Figure 2.
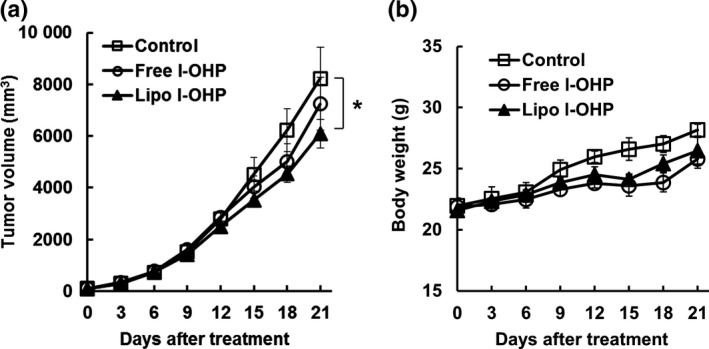
l‐OHP formulations exhibited no antitumor effect in C26 tumor‐bearing immunodeficient BALB/c nude mice. C26 cells (2 × 106 cells) were inoculated subcutaneously into BALB/c nude mice. On days 0, 7 and 14, C26 tumor‐bearing mice received three intravenous injections of either Free l‐OHP or Liposomal l‐OHP at a dose of 8.3 mg l‐OHP/kg. Non‐treated mice served as the control. (a) Tumor volume. (b) Body weight. Each value represents the mean ± SD (n = 5). *P < 0.05.
Liposomal l‐OHP and free l‐OHP suppressed protumor host immunity
We next investigated the impact of l‐OHP formulations on immunosuppressive cell components in tumor tissue. After three sequential intravenous injections with l‐OHP formulations into C26‐bearing immunocompetent BALB/c mice, the frequency of Treg, MDSC and TAM in the tumors was determined through flow cytometry. Liposomal l‐OHP significantly reduced the frequency of Treg (Fig. 3a), MDSC (Fig. 3b) and TAM (Fig. 3c). In contrast, free l‐OHP significantly reduced the frequency of Treg (Fig. 3a) and MDSC (Fig. 3b), but not that of TAM (Fig. 3c). However, there was no significant difference in terms of the number of Treg, MDSC and TAM between the free l‐OHP‐treated group and the liposomal l‐OHP‐treated group. These results indicate that both l‐OHP formulations eliminated immunosuppressive cells in the tumor tissue; it was particularly clear that liposomal l‐OHP had eliminated TAM.
Figure 3.
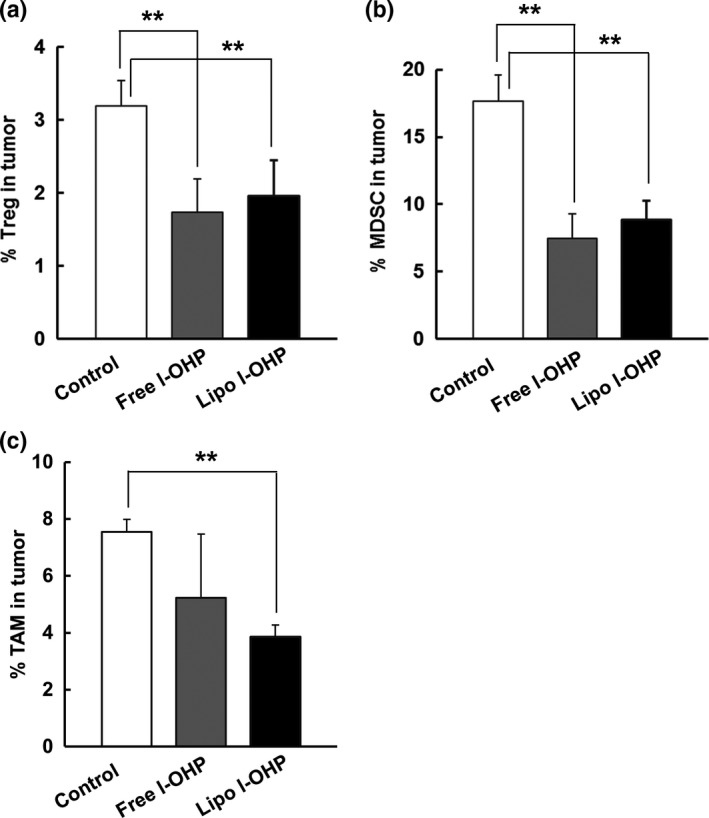
l‐OHP formulations eliminated immunosuppressive cell components from tumors. On days 0, 7 and 14, C26 tumor‐bearing immunocompetent BALB/c mice received three intravenous injections of either Free l‐OHP or Liposomal l‐OHP. Non‐treated mice served as the control. On day 21, tumors were collected and tumor tissue suspensions were prepared. Those cells were stained with various antibodies before flow cytometry analysis. Cells were gated on size and expression of each marker. (a) The frequency of Treg (CD4+ CD25+), (b) MDSC (CD11b+ Gr‐1+) and (c) TAM (F4/80+ CD206+) in tumors. The percentage of each population was calculated by dividing each cell population number by the total cell number in tumor tissue. Each value represents the mean ± SD (n = 3). **P < 0.01.
Liposomal l‐OHP preserved CD8+ T cell‐mediated antitumor immunity
We further investigated the effect of free l‐OHP or liposomal l‐OHP treatment on CD8+ T cell populations in the spleen and tumor tissues. Compared with non‐treated mice, the mice treated with free l‐OHP showed smaller numbers of CD8+ T cells in the spleen (Fig. 4a) and a lower frequency of tumor‐infiltrating CD8+ T cells (Fig. 4b). However, as expected, liposomal l‐OHP preserved the number of splenic CD8+ T cells (Fig. 4a) as well as the frequency of tumor‐infiltrating CD8+ T cells (Fig. 4b). To confirm the presence of activated tumor‐specific CD8+ T cells, the numbers of IFN‐γ+ T cells in CD8+ T cell populations were determined following the incubation of splenocytes with C26 cells. Liposomal l‐OHP did not increase, but did maintain, the number of activated tumor‐specific CD8+ T cells, while the number of activated tumor‐specific CD8+ T cells was decreased with free l‐OHP (Fig. 4c). These results indicate that liposomal l‐OHP treatment preserved CD8+ T cell‐mediated antitumor immunity against C26 tumors in the immunocompetent BALB/c mice.
Figure 4.
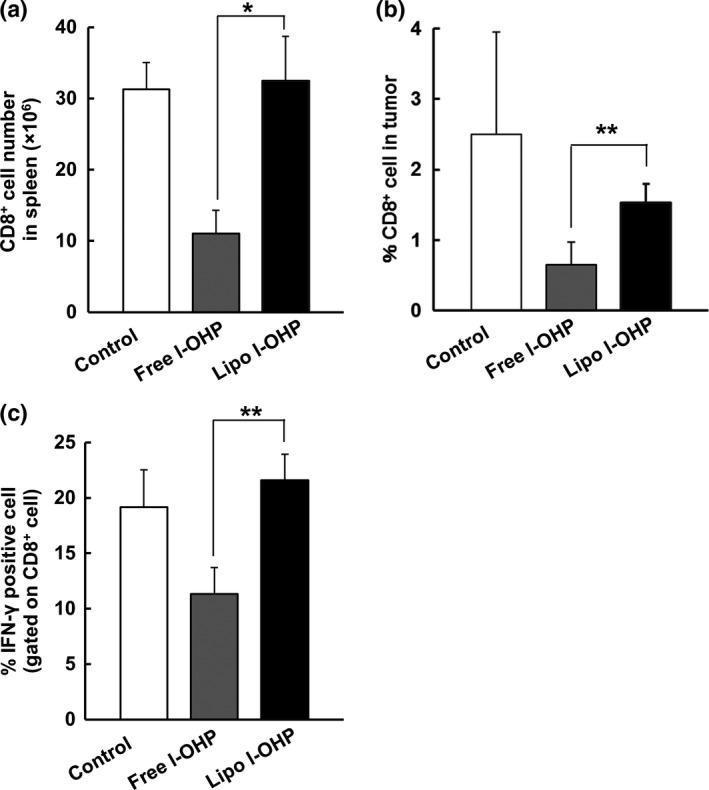
Liposomal l‐OHP CD8+ T cell‐mediated antitumor immunity. On days 0, 7 and 14, C26 tumor‐bearing BALB/c mice received three intravenous injections with either Free l‐OHP or Liposomal l‐OHP. Non‐treated mice served as the control. On day 21, the tumors and spleens were collected, and cell suspensions were prepared. The suspensions were stained with anti‐CD8 antibody and then analyzed using flow cytometry. (a) The number of CD8+ T cells in the spleens. (b) The frequency of CD8+ T cells in tumors. The percentage of CD8 + T cells was calculated by dividing CD8 + T cell number by total cell number in tumor tissue. (c) On days 0 and 7, C26‐bearing BALB/c mice received two intravenous injections of either Free l‐OHP or Liposomal l‐OHP. Non‐treated mice served as the control. On day 10, the spleens were collected and cell suspensions were prepared. The cells were pulsed with mitomycin C‐treated C26 tumor cells in vitro. IFNγ+ CD8+ T cells were analyzed through flow cytometry. Each value represents the mean ± SD (n = 3). *P < 0.05, **P < 0.01.
Liposomal l‐OHP and free l‐OHP increased the MHC‐1 level of tumor cells
Tumor cells can escape immune surveillance through the downregulation of MHC‐1 on their surface, which causes a reduction in their antigenicity.18 We investigated the effect of both free l‐OHP and liposomal l‐OHP treatment on the level of MHC‐1 in C26 tumor cells in vitro and in vivo. Short exposure did not change the level of MHC‐1 molecules in vitro (Fig. 5a). With more time, however, free l‐OHP increased the expression of MHC‐1 in an exposure‐dependent manner. Liposomal l‐OHP also increased the expression level of MHC‐1, but to a smaller extent than that of free l‐OHP. Under in vivo conditions, both l‐OHP formulations increased the expression of MHC‐1 to a similar extent (Fig. 5b). These results indicated that both l‐OHP formulations increased the MHC‐1 level in vitro and in vivo, which may correspond to tumor cell antigenicity.
Figure 5.
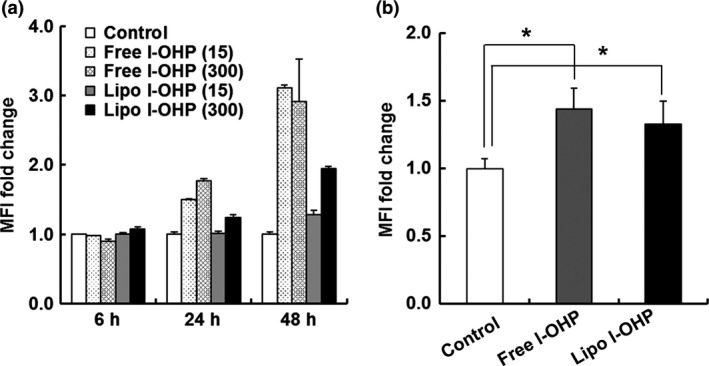
l‐OHP formulations increased the MHC‐1 level of tumor cells. (a) C26 cells were cultured for 6, 24 and 48 h in the presence of Free l‐OHP or Liposomal l‐OHP (15, 300 μM). (b) On days 0 and 7, C26 tumor‐bearing BALB/c mice received two intravenous injections of either Free l‐OHP or Liposomal l‐OHP. Non‐treated mice served as the control. On day 10, tumors were collected. The treated cells in vitro and in vivo were stained with anti‐mouse MHC‐1 (H‐2Dd) antibody and then analyzed using flow cytometry. Each value represents the mean ± SD (n = 3). *P < 0.05. [Correction added on 31 July 2017, after first online publication: In the figure legend of figure 5, the incubation times in the sentence “C26 cells were cultured for 6, 12 and 48 h in the presence of Free l‐OHP or Liposomal l‐OHP (15, 300 μM)” has been changed to “6, 24 and 48h”.]
Discussion
Immune modulation of the tumor microenvironment has been reported to contribute to the antitumor activity of many anticancer drugs.11 In the current study, we showed that liposomal l‐OHP significantly inhibited C26 tumor growth in immunocompetent mice (Fig. 1a), which is consistent with our previous results.7, 8 However, the therapeutic effect was substantially diminished in immunodeficient nude mice, which is suggestive of the contribution of the immune system to the therapeutic effect that was obtained by liposomal l‐OHP. Nude mice lack a thymus, so they cannot generate mature T lymphocytes relating to T cell‐dependent antitumor immunity. As shown in Figure 4c, in the immunocompetent mice, liposomal l‐OHP preserved the C26 tumor‐specific activated CD8+ T cells in tumor tissue, thereby indicating the generation of a CD8+ T cell‐mediated antitumor immune response. To the best of our knowledge, ours is the first study to suggest that liposomal l‐OHP elicits a strong antitumor effect on tumor cells through not only the direct cytotoxic effect of l‐OHP against tumor cells but modulating the antitumor immunity as well.
Conventional anticancer chemotherapy is generally thought to reduce tumor progression through direct cytotoxic effects on tumor cells. We also showed that free l‐OHP and liposomal l‐OHP had a direct cytotoxic effect on tumor cells in vitro and in vivo using immunocompetent mice.8 Several recent studies have reported that antitumor activities induced by antitumor agents such as doxorubicin,12 cyclophosphamide,13 bortezomib19 and gemcitabine20 are severely alleviated under immunocompromised conditions, indicating that their antitumor effects are partially and/or mainly related to the host antitumor immunity. In the same context, in the current study, free l‐OHP failed to exhibit tumor growth suppression in immunodeficient nude mice (Fig. 2a). In addition, in immunocompetent mice, free l‐OHP also tended to ruin protumor immunity involving regulatory T cells (Treg) and myeloid‐derived suppressor cells (MDSC) (Fig. 3), which combine to suppress CD8+ T cell‐mediated antitumor immune responses. Furthermore, l‐OHP increased the expression of MHC‐I on tumor cells (Fig. 5) and made them sensitive to CD8+ T cells, as with gemcitabine.21 Therefore, free l‐OHP has the ability to enhance CD8+ T cell‐mediated antitumor immunity. However, due to the lack of T cells in the immunodeficient nude mice, l‐OHP failed to induce antitumor immunity (Fig. 2a). These data suggest that free l‐OHP also elicits its antitumor effect through T cell‐mediated antitumor immunity along with its inherent cytotoxicity on tumor cells. Accordingly, the liposomal l‐OHP‐induced antitumor immune response observed in the present study was due mainly to the antitumor immune response induced by l‐OHP encapsulated in the PEGylated liposome, in combination with the direct cytotoxicity effect against tumor cells.
The tumor growth suppression effect of l‐OHP in the current study was much higher in liposomal l‐OHP treatment than in free l‐OHP treatment in immunocompetent mice (Fig. 1a). It is well known that antitumor agents encapsulated in PEGylated liposomes passively accumulate in tumor tissue through the EPR effect.6 Such preferential tumor accumulation increases the intratumor concentration of antitumor agents. Therefore, liposomal antitumor agents are believed to achieve a suppression of tumor growth that is superior to that of free antitumor agents.7, 8 In the present study, we demonstrated that liposomal l‐OHP significantly reduced protumor immunity (Fig. 3) and preserved antitumor immunity (Fig. 4), while free l‐OHP reduced not only protumor immunity but also antitumor immunity. Free l‐OHP is distributed throughout the body following intravenous injection and thereby causes not only tumor suppression but also adverse reactions such as sensory neuropathy, nausea, vomiting, diarrhea and hematologic dyscrasias.3 Such non‐selective l‐OHP distribution consequently appeared to deplete CD8+ T cells in both tumors (Fig. 4b) and the spleen (Fig. 4a), and it also depleted total splenocytes (Fig. 1c). Conservation of T cells through the liposomalization of l‐OHP might be one of the reasons that liposomal l‐OHP exhibited a tumor growth suppressive effect that was higher than that of free l‐OHP in immunocompetent mice (Fig. 1a).
Liposomalization of l‐OHP may have other advantages in l‐OHP‐mediated antitumor immunity; an increased reduction of protumor immune cells which have phagocytic activity and increase of the MHC‐1 level of tumor cells possibly corresponding to tumor cell antigenicity. Nano‐sized liposomes are preferentially taken up by phagocytic cells. Protumor immune cells such as MDSC22 and TAM15 have phagocytic activity, but CD8+ T cells do not. Liposomal l‐OHP might lead to a remarkable reduction in the numbers of MDSC (Fig. 3b) and TAM (Fig. 3c) through phagocytosis of the liposomes accumulated in a tumor. Thus, the ratio of CD8+ T cell/MDSC and CD8+ T cell/TAM becomes higher in liposomal l‐OHP‐treated mice than in free l‐OHP‐treated mice. This might lead to a preservation of antitumor immunity following liposomal l‐OHP treatment. In our previous study, liposomal l‐OHP achieved a much higher tissue concentration of l‐OHP in tumors compared with free l‐OHP7, 23 and exposed its payload to the tumor cells for a longer period of time compared with free l‐OHP due to their sustained release characteristics. Accordingly, the released l‐OHP might promote immunogenic death in tumor cells24 and could increase the MHC‐1 level of tumor cells, possibly corresponding to tumor cell antigenicity.
Immunotherapy is a new class in cancer therapy that exploits the innate powers of the immune system to fight cancer. However, single treatments using immunotherapeutic agents has had limited efficacy in many cases due to the unfavorable immune environment in tumors. Recently, immunotherapy has been combined with both chemotherapy25, 26 and radiotherapy,27 with the ever‐expanding knowledge of the immune‐modulating ability of these therapies. Hazama et al.28 investigated the efficacy of peptide cancer vaccine combined with free l‐OHP‐based chemotherapy for the treatment of colorectal cancer in a phase II study. Unfortunately, the efficacy of their combined therapy was relatively low, suggesting that the vaccine should be combined with other agents to modulate antitumor immunity. Instead of free l‐OHP, the use of liposomal l‐OHP, which can modulate protumor and antitumor immunities, in combination with peptide vaccine may provide a much stronger therapeutic effect in the treatment of tumors. Liposomal l‐OHP therapy has shown promise when used in combination with immunotherapy. The study of such combined treatment is in progress in our laboratory.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. Depletion of CD8+ T cells attenuated the antitumor effect of liposomal l‐OHP in C26 tumor‐bearing immunocompetent BALB/c mice.
Acknowledgments
The authors thank Mr James L. McDonald for his helpful advice in developing the English manuscript. This work was partially supported by The Japan Society for the Promotion of Science, a Grant‐in‐Aid for Young Scientists (B) (15K18921) and a research program for the development of an intelligent Tokushima artificial exosome (iTEX) from Tokushima University.
Cancer Sci 108 (2017) 1864–1869
Funding Information
The Japan Society for the Promotion of Science, a Grant‐in‐Aid for Young Scientists (B) (15K18921) and Tokushima University.
[Correction added on 31 July 2017, after first online publication: The affiliation of the author, Tatsuhiro Ishida has been changed from “Department of Pharmaceutics, Faculty of Pharmacy, Zagazig University, Zagazig 44519, Egypt” to “Department of Pharmacokinetics and Biopharmaceutics, Graduate School of Pharmaceutical Science, Tokushima University, Tokushima 770‐8505, Japan”.]
References
- 1. Stordal B, Pavlakis N, Davey R. Oxaliplatin for the treatment of cisplatin‐resistant cancer: a systematic review. Cancer Treat Rev 2007; 33: 347–57. [DOI] [PubMed] [Google Scholar]
- 2. Woynarowski JM, Faivre S, Herzig MC et al Oxaliplatin‐induced damage of cellular DNA. Mol Pharmacol 2000; 58: 920–7. [DOI] [PubMed] [Google Scholar]
- 3. Extra JM, Espie M, Calvo F, Ferme C, Mignot L, Marty M. Phase I study of oxaliplatin in patients with advanced cancer. Cancer Chemother Pharmacol 1990; 25: 299–303. [DOI] [PubMed] [Google Scholar]
- 4. Suzuki R, Takizawa T, Kuwata Y et al Effective anti‐tumor activity of oxaliplatin encapsulated in transferrin‐PEG‐liposome. Int J Pharm 2008; 346: 143–50. [DOI] [PubMed] [Google Scholar]
- 5. Allen TM, Hansen C, Martin F, Redemann C, Yau‐Young A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half‐lives in vivo . Biochim Biophys Acta 1991; 1066: 29–36. [DOI] [PubMed] [Google Scholar]
- 6. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387–92. [PubMed] [Google Scholar]
- 7. Doi Y, Okada T, Matsumoto H, Ichihara M, Ishida T, Kiwada H. Combination therapy of metronomic S‐1 dosing with oxaliplatin‐containing polyethylene glycol‐coated liposome improves antitumor activity in a murine colorectal tumor model. Cancer Sci 2010; 101: 2470–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abu Lila AS, Kizuki S, Doi Y, Suzuki T, Ishida T, Kiwada H. Oxaliplatin encapsulated in PEG‐coated cationic liposomes induces significant tumor growth suppression via a dual‐targeting approach in a murine solid tumor model. J Control Release 2009; 137: 8–14. [DOI] [PubMed] [Google Scholar]
- 9. Abu‐Lila A, Suzuki T, Doi Y, Ishida T, Kiwada H. Oxaliplatin targeting to angiogenic vessels by PEGylated cationic liposomes suppresses the angiogenesis in a dorsal air sac mouse model. J Control Release 2009; 134: 18–25. [DOI] [PubMed] [Google Scholar]
- 10. Yang C, Liu HZ, Fu ZX, Lu WD. Oxaliplatin long‐circulating liposomes improved therapeutic index of colorectal carcinoma. BMC Biotechnol 2011; 11: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lesterhuis WJ, Haanen JB, Punt CJ. Cancer immunotherapy—Revisited. Nat Rev Drug Discov 2011; 10: 591–600. [DOI] [PubMed] [Google Scholar]
- 12. Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 2011; 71: 4809–20. [DOI] [PubMed] [Google Scholar]
- 13. Tongu M, Harashima N, Monma H et al Metronomic chemotherapy with low‐dose cyclophosphamide plus gemcitabine can induce anti‐tumor T cell immunity in vivo . Cancer Immunol Immunother 2013; 62: 383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vincent J, Mignot G, Chalmin F et al 5‐Fluorouracil selectively kills tumor‐associated myeloid‐derived suppressor cells resulting in enhanced T cell‐dependent antitumor immunity. Cancer Res 2010; 70: 3052–61. [DOI] [PubMed] [Google Scholar]
- 15. Fujimoto H, Sangai T, Ishii G et al Stromal MCP‐1 in mammary tumors induces tumor‐associated macrophage infiltration and contributes to tumor progression. Int J Cancer 2009; 125: 1276–84. [DOI] [PubMed] [Google Scholar]
- 16. Bartlett GR. Colorimetric assay methods for free and phosphorylated glyceric acids. J Biol Chem 1959; 234: 469–71. [PubMed] [Google Scholar]
- 17. Ishida T, Wang X, Shimizu T, Nawata K, Kiwada H. PEGylated liposomes elicit an anti‐PEG IgM response in a T cell‐independent manner. J Control Release 2007; 122: 349–55. [DOI] [PubMed] [Google Scholar]
- 18. Garrido F, Ruiz‐Cabello F, Cabrera T et al Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today 1997; 18: 89–95. [DOI] [PubMed] [Google Scholar]
- 19. Chang CL, Hsu YT, Wu CC et al Immune mechanism of the antitumor effects generated by bortezomib. J Immunol 2012; 189: 3209–20. [DOI] [PubMed] [Google Scholar]
- 20. Suzuki E, Sun J, Kapoor V, Jassar AS, Albelda SM. Gemcitabine has significant immunomodulatory activity in murine tumor models independent of its cytotoxic effects. Cancer Biol Ther 2007; 6: 880–5. [DOI] [PubMed] [Google Scholar]
- 21. Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre‐treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer 2010; 102: 115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid‐derived suppressor cells in tumor‐bearing mice. J Leukoc Biol 2012; 91: 167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakamura H, Abu Lila AS, Nishio M et al Intra‐tumor distribution of PEGylated liposome upon repeated injection: no possession by prior dose. J Control Release 2015; 220: 406–13. [DOI] [PubMed] [Google Scholar]
- 24. Tesniere A, Schlemmer F, Boige V et al Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010; 29: 482–91. [DOI] [PubMed] [Google Scholar]
- 25. Huang X, Huang G, Song H, Chen L. Preconditioning chemotherapy with paclitaxel and cisplatin enhances the antitumor activity of cytokine induced‐killer cells in a murine lung carcinoma model. Int J Cancer 2011; 129: 648–58. [DOI] [PubMed] [Google Scholar]
- 26. Kang TH, Mao CP, Lee SY et al Chemotherapy acts as an adjuvant to convert the tumor microenvironment into a highly permissive state for vaccination‐induced antitumor immunity. Cancer Res 2013; 73: 2493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee Y, Auh SL, Wang Y et al Therapeutic effects of ablative radiation on local tumor require CD8 + T cells: changing strategies for cancer treatment. Blood 2009; 114: 589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hazama S, Nakamura Y, Tanaka H et al A phase II study of five peptides combination with oxaliplatin‐based chemotherapy as a first‐line therapy for advanced colorectal cancer (FXV study). J Transl Med 2014; 12: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Depletion of CD8+ T cells attenuated the antitumor effect of liposomal l‐OHP in C26 tumor‐bearing immunocompetent BALB/c mice.