

Abstract
Piwi‐interacting RNAs (piRNAs), a novel class of small non‐coding RNAs, were first discovered in germline cells and are thought to silence transposons in spermatogenesis. Recently, piRNAs have also been identified in somatic tissues, and aberrant expression of piRNAs in tumor tissues may be implicated in carcinogenesis. However, the function of piR‐823 in colorectal cancer (CRC) remains unclear. Here, we first found that piR‐823 was significantly upregulated in CRC tissues compared with its expression in the adjacent tissues. Inhibition of piR‐823 suppressed cell proliferation, arrested the cell cycle in the G1 phase and induced cell apoptosis in CRC cell lines HCT116 and DLD‐1, whereas overexpression of piR‐823 promoted cell proliferation in normal colonic epithelial cell line FHC. Interestingly, Inhibition of piR‐823 repressed the expression of heat shock protein (HSP) 27, 60, 70. Furthermore, elevated HSPs expression partially abolished the effect of piR‐823 on cell proliferation and apoptosis. In addition, we further demonstrated that piR‐823 increased the transcriptional activity of HSF1, the common transcription factor of HSPs, by binding to HSF1 and promoting its phosphorylation at Ser326. Our study reveals that piR‐823 plays a tumor‐promoting role by upregulating phosphorylation and transcriptional activity of HSF1 and suggests piR‐823 as a potential therapeutic target for CRC.
Keywords: piRNA, colorectal cancer, non‐coding RNAs, heat shock protein, heat shock factor 1
Colorectal cancer (CRC), as a common gastrointestinal malignancy, is one of the leading causes of cancer‐related deaths worldwide and arises as a consequence of genetic mutations and epigenetic alterations.1 These changes activate oncogenes (KRAS)2 but inactivate tumor suppressor genes (p16 and TP53),3, 4 which endow normal cells with the capacity for infinite proliferation and self‐renewal, driving them to become cancer cells. Non‐coding RNAs have emerged as regulators of many biological processes and have participated in a wide range of human diseases, especially in cancers.5, 6 To date, the dysregulation of miRNAs and long non‐coding RNAs have been implicated in CRC development.7, 8
Piwi‐interacting RNAs (piRNAs), characterized by a 3‐terminal 2′‐O‐methylation, are a novel class of small non‐coding RNAs. piRNAs are named due to their exclusive association with the Piwi subfamily, but not the Ago subfamily, of the Argonaute (Ago) family.9, 10, 11, 12 piRNAs were originally perceived as germline‐specific and epigenetically silenced transposons by DNA methylation to maintain the integrity of the genome of germ cells.13, 14, 15 Recent studies have shown that piRNAs are present in human somatic tissues and involved in the development of a variety of tumor types.16, 17, 18, 19, 20, 21 These studies have revealed that piRNAs participate not only in epigenetic regulation but also in the post‐transcriptional regulation of gene expression,20, 22 further indicating the diverse and complex roles of piRNAs in tumors. Although there has been some progress in understanding the roles of piRNAs in cancers, additional work is needed to determine their functions in genome‐wide gene regulation at multiple levels among diverse cancer types.
piR‐823, a member of piRNAs, has been demonstrated to suppress gastric cacinogenesis23 but facilitate tumorigenesis in multiple myeloma by regulating de novo DNA methylation.22 Such opposite functions of piR‐823 in the above two cancer types aroused our curiosity about the role and the potential regulatory mechanisms of piR‐823 in CRC.
Heat shock proteins (HSPs), which are induced by stressful conditions, are essential for cell survival and are classified into the HSP27, HSP60, HSP70 and HSP90 families according to their molecular weight.24 Increasing evidence has shown that HSPs are significantly elevated in multiple cancer types24 and associated with increased cell proliferation rate, decreased apoptosis index, malignancy and poor prognosis.25, 26, 27 HSP expression is primarily regulated by a common transcription factor, heat shock factor 1 (HSF1), at the transcriptional level.28 Under non‐stressed conditions, HSF1 is mostly localized in the cytoplasm as an inactive monomer. Once activated, HSF1 forms a homotrimer that is capable of binding to heat shock elements (HSEs) in the promoter region of heat shock genes, translocates into the nucleus, and is then inducibly phosphorylated.29, 30, 31 Importantly, phosphorylation is a prerequisite for HSF1 to acquire transcriptional potency, in which phosphorylation at Ser326 is found to contribute to the activation of HSF1.32, 33 Extensive studies have shown that HSF1 is highly expressed in a wide range of human cancers and that a high level of HSF1 is a powerful driver of carcinogenesis, including CRC.34, 35, 36 These data indicate that HSF1 plays a critical role in tumorigenesis, angiogenesis, invasion and metastasis and may be a potential therapeutic target for cancers.37
Herein, we report for the first time that piR‐823 was significantly upregulated in CRC tissues. piR‐823 promoted proliferation and inhibited apoptosis. Moreover, piR‐823 upregulated HSPs expression by enhancing HSF1 transcriptional activity in colorectal carcinogenesis. These results suggest that piR‐823 contributes to tumorigenesis and reveal a novel mechanism by which piR‐823 modulates HSF1 transcriptional activity by regulating the post‐translational modification of HSF1.
Materials and Methods
Patients and samples
Fresh surgically resected tissue samples, including CRC tissues and matched adjacent tissues from 45 patients, were obtained from the Second Hospital of Hebei Medical University, Shijiazhuang, China. After excision, the tissues were immediately stored in RNAstore Reagent (Tiangen, Beijing, China). The clinical characteristics are presented in Table 1. This study was approved by the Ethics Committee of the Second Hospital of Hebei Medical University, and written informed consent was obtained from all patients.
Table 1.
Correlation of piR‐823expression in colorectal cancer tissues with clinicopathological characteristics
| Clinicopathological characteristics | No. patients | piR‐823 fold change (M ± QR)a | P‐value |
|---|---|---|---|
| All cases | 45 (100%) | ||
| Gender | |||
| Male | 30 (66.7%) | 2.24 ± 4.15 | 0.413 |
| Female | 15 (33.3%) | 3.19 ± 4.86 | |
| Age | |||
| ≥60 | 30 (66.7%) | 2.33 ± 5.81 | 0.904 |
| <60 | 15 (33.3%) | 2.69 ± 2.73 | |
| Tumor location | |||
| Colon | 21 (46.7%) | 2.33 ± 6.32 | 0.767 |
| Rectum | 24 (53.3%) | 2.60 ± 3.89 | |
| Differentiation | |||
| Well or Moderately | 34 (75.6%) | 2.10 ± 2.66 | 0.033 |
| Poorly | 11 (24.4%) | 5.33 ± 10.33 | |
| T stage | |||
| T1 or T2 | 9 (20.0%) | 2.03 ± 4.00 | 0.834 |
| T3 or T4 | 36 (80.0%) | 2.93 ± 4.23 | |
| Lymph node metastasis | |||
| No | 29 (64.4%) | 2.14 ± 3.75 | 0.308 |
| Yes | 16 (35.6%) | 3.22 ± 7.84 | |
| AJCC stage | |||
| I or II | 28 (62.2%) | 2.10 ± 3.36 | 0.210 |
| III or IV | 17 (37.8%) | 3.24 ± 6.63 | |
Compared with adjacent tissues. AJCC, American Joint Committee on Cancer; T, tumor.
Cell culture and transfection
The human normal colonic epithelial cell line FHC was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), and the human CRC cell lines HCT116 and DLD‐1 was obtained from Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). The FHC cells, HCT116 cells and DLD‐1 cells were cultured in DMEM/F12 Medium (HyClone, Logan, UT, USA), McCoy's 5A Medium (Sigma‐Aldrich, St. Louis, MO, USA) and RPMI 1640 medium (Gibco BRL, Rockville, MD, USA), respectively, supplemented with 10% FBS (Gibco BRL), 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in a 5% CO2 incubator. A chemically modified antisense sequence of piR‐823 (Ant‐823) was used to inhibit piR‐823 expression, and 2′‐O‐methoxy at 3′ end modified piR‐823 mimics (mimics‐823) was used to over‐express piR‐823. A non‐specific scrambled sequence was used as negative control (NC). They were synthesized by GenePharma Tech (Shanghai, China), and their sequences are shown in Table S1. Cells at 50% confluence were transfected with Ant‐823 or mimics‐823 using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions.
RNA isolation and real‐time PCR
Total RNA was extracted using miRcute miRNA isolation kit (Tiangen). For piR‐823 quantitative analysis, total RNA was reverse transcribed by miScript Plant RT Kit (Qiagen, Hilden, Germany), a kit specifically designed for small RNAs with 2′‐O‐Me modification at their 3′ end. Real‐time PCR was subsequently performed using a miScript SYBR Green PCR Kit (Qiagen) according to the manufacturer's instructions. The specific primer sequence for piR‐823 amplification was 5′‐AGCGTTGGTGGTATAGTGGT‐3′. The Hs_SNORD61_11 miScript Primer Assay (Qiagen) was used to normalize the levels of piR‐823. For mRNA quantitative analysis, total RNA was reverse transcribed into cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Rockford, IL, USA), and the cDNA was used for the real time PCR assay using Power SYBR Green PCR Master Mix (Life Technologies, Carlsbad, CA, USA). GAPDH was used as an internal control. The specific primers for HSP27, HSP60, HSP70, HSF1 and GAPDH are shown in Table S2. The relative expression was calculated using the method.
CCK‐8 assay and cell cycle assay
Cell proliferation was determined using a CCK‐8 kit (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. Briefly, CCK‐8 (10 μL/well) was added into cells treated according to designed groups and incubated for 2 h. And then the absorbance at 450 nm wavelength was read using a microplate reader (BioTek, Winooski, VT, USA). For the cell cycle distribution analysis, cells were transfected with 100 nM Ant‐823 for 48 h. Then, cells were washed in phosphate buffered saline (PBS), fixed with ice‐cold 70% ethanol and stained in PI/RNase Staining Solution (BD Biosciences, Franklin Lakes, NJ, USA). The samples were analyzed by a FACSVerse flow cytometer in combination with ModFit 3.0 software (BD Biosciences).
Colony formation assays
For the plate colony formation assays, cells were plated in a 6‐well plate and transfected with 100 nM Ant‐823. At 48 h after transfection, cells were re‐plated (2 × 103/well) in a 6‐well plate and cultured for 2 weeks. After being washed with PBS, fixed with methanol and stained with Giemsa, colonies were then counted. For soft agar assays, equal volumes of 1.2% agar and 2 × medium with 20% FBS was mixed and plated in a 6‐well plate (1.5 mL/well) as the underlayer. Next, the cells (2 × 103/well) transfected with 100 nM Ant‐823 for 48 h were suspended in 1 mL of mixture containing equal volume of 0.6% agar and 2 × medium with 20% FBS and then plated on top of 0.6% agar. The colony formation rate was calculated as follows: Number of colonies/Number of plated cells.
Apoptosis assays
Cell apoptosis was measured using the Annexin V:FITC Apoptosis Detection Kit (BD Biosciences) following the manufacturer's instructions. TUNEL technology was also employed to determine apoptosis using an In Situ Cell Death Detection Kit (Roche, Basel, Switzerland) according to the manufacturer's protocol. The apoptosis index is presented as the percentage of TUNEL‐positive cells that showed clear brown nuclear staining out of the total number of cells.
Western blot analysis
Cells were lysed using RIPA buffer (Sigma‐Aldrich) for total protein extraction and using NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) for cytoplasmic and nuclear protein extraction after being washed in PBS. Proteins were quantified using Pierce BCA Protein Assay Kit (Thermo Scientific) and denatured in SDS loading buffer by boiling for 10 min. Proteins from each sample were subjected to SDS–PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with 5% milk, the membrane was incubated with primary antibody overnight at 4°C, washed in TBST and incubated with fluorescence‐conjugated secondary antibodies (LI‐COR Biosciences, Lincoln, NE, USA) for 1 h at room temperature. The bands were visualized using an Odyssey infrared imaging system (LI‐COR Biosciences), and their intensities were quantified with ImageJ software (National Institutes of Health). The primary antibodies used in this study included anti‐cleaved caspase‐8, anti‐cleaved caspase‐9, anti‐cleaved caspase‐3, anti‐β‐actin, anti‐GAPDH (Cell Signaling Technology, Danvers, MA, USA), anti‐CyclinD1, anti‐CDK4, anti‐Cdc25C, anti‐CDK1, anti‐HSP27, anti‐HSP60, anti‐HSP70 (Abcam, Cambridge, UK), anti‐HSF1, anti‐pSer326HSF1 (Enzo Life Sciences, Farmingdale, NY, USA).
Human Apoptosis Antibody Array
The targets of piR‐823 were screened using the Human Apoptosis Antibody Array (RayBiotech, Norcross, GA, USA) following the manufacturer's instructions. The spots were detected using a chemiluminescence system (Bio‐Rad, Hercules, CA, USA), and the signal densities were analyzed with ImageJ software (National Institute of Health, Bethesda, MD, USA).
Immunofluorescence assay
Cells seeded on cover slips were transfected with 100 nM Ant‐823. At 24 h after transfection, cells were fixed in 4% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X‐100, diluted in PBS for 10 min and then blocked in 5% normal goat serum for 30 min at room temperature. The cells were incubated with rabbit anti‐HSF1 (Enzo Life Sciences) overnight at 4°C. After washing with PBS, the cells were incubated with the secondary antibody (FITC goat anti‐rabbit IgG; Beyotime Biotechnology, Shanghai, China) for 1 h at room temperature. After washing again, the slides were counterstained with 4′6′‐diamidino‐2‐phenylindole dihydrochloride (DAPI) (Sigma‐Aldrich) for 5 min. Images were acquired using a laser scanning confocal microscope (Olympus, Tokyo, Japan).
Luciferase reporter construct and dual‐luciferase reporter assay
HSF1 cDNA was cloned into XhoI and KpnI restriction sites of the GV230 vector (GeneChem, Shanghai, China), namely pGV230‐HSF1, and p(HSE)4‐TA‐Luc reporter plasmid was constructed on the basis of the GV238 vector (GeneChem) by inserting four copies of the palindromic HSE (5′‐GAT CTA GAA CGT TCT AGA ACG TTC TAG AAC GTT CTA‐3′) and a minimal TA promoter. 293T cells were seeded in 24‐well plates and co‐transfected with 0.5 μg of pGV230‐HSF1, 0.5 μg of p(HSE)4‐TA‐Luc, 50 pmol mimics‐823 or NC, along with 0.02 μg of pRL‐TK (Promega, Madison, WI, USA) as an internal control. The activities of firefly and Renilla luciferase were measured using the Dual‐Luciferase Reporter Assay System (Promega) at 24 h after co‐transfection.
RNA‐binding protein immunoprecipitation (RIP) assay
RIP assay was performed using the Magna RIP Kit (Millipore, Billerica, MA, USA) following the manufacturer's protocol. In brief, cells (2 × 107/sample) were lysed in RIP Lysis Buffer and then incubated with protein A/G magnetic beads coated with rabbit anti‐HSF1 (Enzo Life Sciences) or rabbit IgG antibody (Millipore) as a negative control. The precipitates were washed sufficiently with RIP Wash Buffer six times, followed by incubation with proteinase K at 55°C for 30 min to digest proteins. The RNAs bound with HSF1 were extracted using phenol:chloroform:isoamyl alcohol (pH = 4.3) and precipitated with ethanol. Purified RNAs were analyzed by real‐time PCR using the piR‐823‐specific primer.
Statistical analysis
Data represent the mean ± SD. The association of piR‐823 expression with patients’ clinicopathological features was evaluated by a nonparametric test, and the relative cell proliferation rate was analyzed by two‐way analysis of variance (anova). The difference between two groups was analyzed by Student's t‐test. For difference among multiple groups, one‐way anova was performed. Statistical analysis was performed using SPSS software version 17.0 (Chicago, IL, USA). P‐values of <0.05 were considered statistically significant.
Results
piR‐823 is significantly upregulated in CRC tissues
To identify whether piR‐823 was involved in colorectal tumorigenesis, we first detected piR‐823 expression in 45 pairs of CRC tissues and their corresponding adjacent tissues using real‐time PCR. As shown in Figure 1(a), compared with the adjacent tissues, piR‐823 was significantly upregulated in CRC tissues. We then investigated the association of piR‐823 expression with patients’ clinicopathological characteristics. piR‐823 expression did not correlate with gender, age, tumor location, tumor size, lymph node metastasis or AJCC stage but did correlate with the differentiation degree of tumors, with a higher expression found in poorly differentiated tumors (P < 0.05, Table 1). These results suggest that piR‐823 upregulation may play an important role in colorectal tumorigenesis.
Figure 1.
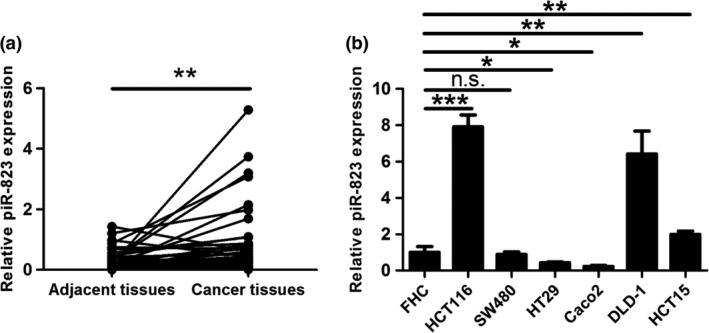
piR‐823 is upregulated in colorectal cancer. (a) piR‐823 expression normalized to SNORD61 in colorectal cancer and corresponding adjacent tissues (n = 45) was evaluated by real‐time PCR. **P < 0.01 vs adjacent tissues. (b) piR‐823 expression in human normal colonic epithelial cell line (FHC) and six colon cancer cell lines (HCT116, SW480, HT29, Caco2, DLD‐1 and HCT15) was assessed by real‐time PCR. Bar graphs represent the mean ± SD from three independent experiments, *P < 0.05, **P < 0.01, ***P < 0.001, n.s. indicates no significance vs FHC cells.
In addition, we also evaluated piR‐823 expression in human normal colonic epithelial cell line FHC and six CRC cell lines HCT116, SW480, HT29, Caco2, DLD‐1 and HCT15. We found that piR‐823 expression was upregulated in HCT116, DLD‐1 and HCT15 cells, especially in HCT116 and DLD‐1 cells, relative to its expression in FHC cells (Fig. 1b). Given that piR‐823 was upregulated in CRC tissues, HCT116 and DLD‐1 cells were selected as the research representatives of CRC cells.
piR‐823 promotes cell proliferation by modulating cell cycle progression
To evaluate the functional role of piR‐823 in cell proliferation, we inhibited piR‐823 by Ant‐823 (antisense sequence of piR‐823) in piR‐823 highly expressing CRC cell lines HCT116 and DLD‐1, and overexpressed piR‐823 by mimics‐823 in normal colonic epithelial cell line FHC. Compared with the negative control (NC) sequence, Ant‐823 led to a significantly decrease of piR‐823 expression in HCT116 and DLD‐1 cells, while mimics‐823 caused an increased piR‐823 expression in FHC cells at 24, 48 and 72 h (Fig. S1).
We first examined the effect of piR‐823 on cell proliferation. As shown using the CCK‐8 assay, the proliferation of HCT116 and DLD‐1 cells was significantly suppressed by Ant‐823 in a dose‐dependent manner (Fig. 2a). In keeping with this, overexpression of piR‐823 promoted the proliferation of FHC cells (Fig. 2b). The suppressive effect of Ant‐823 on cell proliferation was further verified by the plate colony formation assay and soft agar assay, which both showed that the colony formation rate was significantly decreased by Ant‐823 in HCT116 and DLD‐1 cells (Fig. 2c, d).
Figure 2.
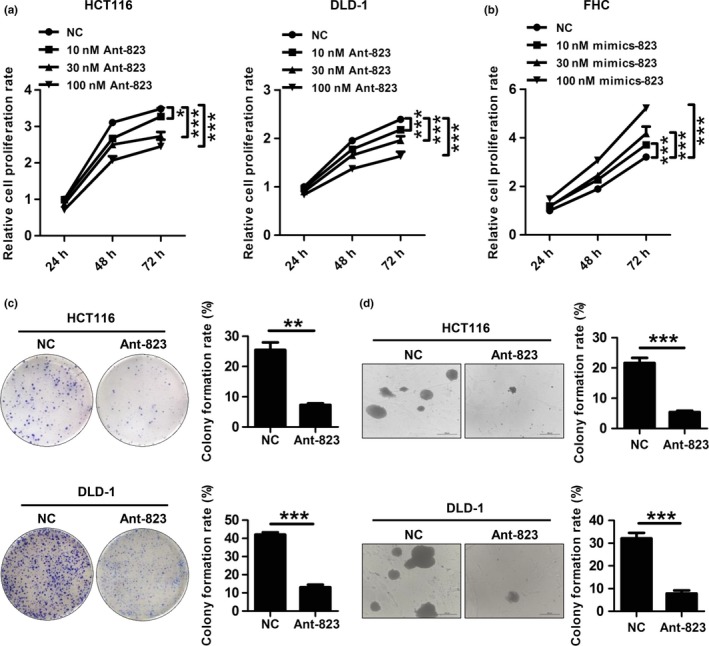
piR‐823 promotes cell proliferation. (a, b) Cell proliferation was examined by CCK‐8 assay in HCT116 and DLD‐1 cells transfected with Ant‐823 or NC (a), and normal colonic epithelial cell line FHC transfected with mimics‐823 or NC (b). (c, d) Cell proliferation was further measured by plate colony formation assay (c) and soft agar assay (d) in HCT116 and DLD‐1 cells. Bar graphs show the mean ± SD from three independent experiments, *P < 0.05, **P < 0.01, ***P < 0.001 vs NC.
To explore the mechanism by which Ant‐823 inhibited cell proliferation, we observed the effect of piR‐823 on CRC cell cycle progression by flow cytometry. Ant‐823‐transfected HCT116 and DLD‐1 cells both showed an increase in G1 phase population (41.9 ± 2.2% vs 26.1 ± 1.1%; 47.2 ± 1.3% vs 33.9 ± 1.8%), and a corresponding reduction in S phase (36.6 ± 0.5% vs 41.0 ± 0.6%; 34.1 ± 1.9% vs 43.1 ± 1.5%) and G2/M phase populations (21.8 ± 1.6% vs 32.7 ± 1.7%; 18.6 ± 0.6% vs 23.0 ± 1.3%) (Fig. 3a). Western blot analysis further confirmed that Ant‐823 inhibited the expression of the G1 phase regulators Cyclin D1 and CDK4 (Fig. 3b), but did not modulate the expression of the G2/M phase regulators Cdc25C and CDK1 (Fig. 3c) in both CRC cell lines. These data indicate that Ant‐823 blocks CRC cell cycle progression by inducing G1 phase arrest and suppressing the G1/S transition to inhibit cell proliferation.
Figure 3.
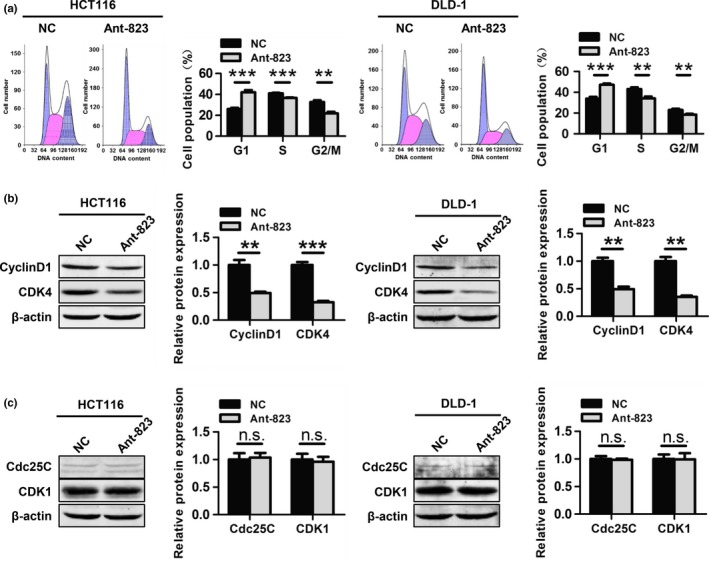
piR‐823 modulates cell cycle progression. (a) Cell cycle distribution was examined by flow cytometry in HCT116 and DLD‐1 cells transfected with Ant‐823 or NC. (b, c) Western blot analysis for G1 phase regulators Cyclin D1, CDK4 (b) and G2/M phase regulators Cdc25C, CDK1 (c) was performed in Ant‐823 or NC‐transfected HCT116 and DLD‐1 cells. Data represent the mean ± SD from three independent experiments, **P < 0.01, ***P < 0.001, n.s. indicates no significance vs NC.
piR‐823 inhibits cell apoptosis
Next, we evaluated whether piR‐823 modulated cell apoptosis in colorectal tumorigenesis. Compared with the NC, Ant‐823 significantly increased the percentage of apoptotic cells (Annexin V‐positive cells) in HCT116 cells (17.1 ± 0.7% vs 4.0 ± 0.5%) and DLD‐1 cells (22.7 ± 2.6% vs 5.0 ± 1.3%) (Fig. 4a). TUNEL technology showed similar data that Ant‐823 led to a remarkable increase of TUNEL‐positive cells in both HCT116 cells (59.8 ± 4.3% vs 13.0 ± 1.7%) and DLD‐1 cells (48.1 ± 7.2% vs 6.9 ± 1.5%) (Fig. 4b). Furthermore, the expressions of the active forms of apoptosis‐related proteins, including cleaved‐caspase‐8, cleaved‐caspase‐9 and cleaved‐caspase‐3, were significantly elevated in Ant‐823‐transfected HCT116 and DLD‐1 cells compared with those in NC‐transfected cells by western blot analysis (Fig. 4c). Taken together, these results show that piR‐823 plays a tumor‐promoting role in colorectal tumorigenesis.
Figure 4.
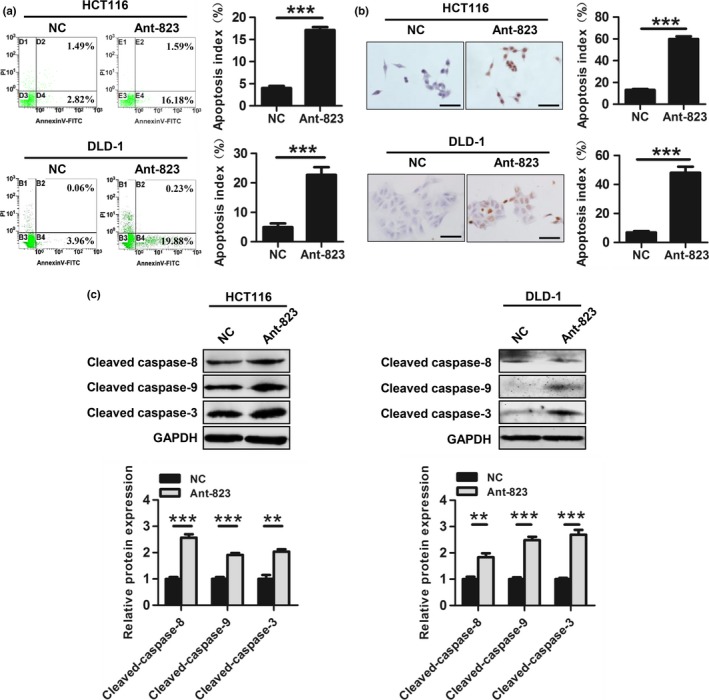
piR‐823 inhibits cell apoptosis. (a, b) Cell apoptosis of Ant‐823 or NC‐transfected HCT116 and DLD‐1 cells was determined by Annexin V/PI staining (a) and TUNEL technology (b). Scale bar, 50 μm. (c) Activated apoptosis‐related proteins caspase‐8, caspase‐9 and caspase‐3 were assessed by western blot analysis in HCT116 and DLD‐1 cells. All values are the mean ± SD from three independent experiments, **P < 0.01, ***P < 0.001 vs NC.
piR‐823 regulates cell proliferation and apoptosis by HSPs
We then searched for the targets of piR‐823 using the Human Apoptosis Antibody Array. As shown in Figure 5(a), piR‐823 inhibition resulted in a remarkable reduction of HSP27, HSP60, HSP70, HTRA and IGFBP5 levels in HCT116 cells. Interestingly, three of these five proteins were HSPs. We further confirmed the above data by real‐time PCR and western blot analysis, showing that HSP27, HSP60 and HSP70 were all significantly downregulated by Ant‐823 at both the mRNA and protein levels (Fig. 5b,c). However, the mRNA and protein levels of HTRA and IGFBP5 showed no difference between these two groups (data not shown).
Figure 5.
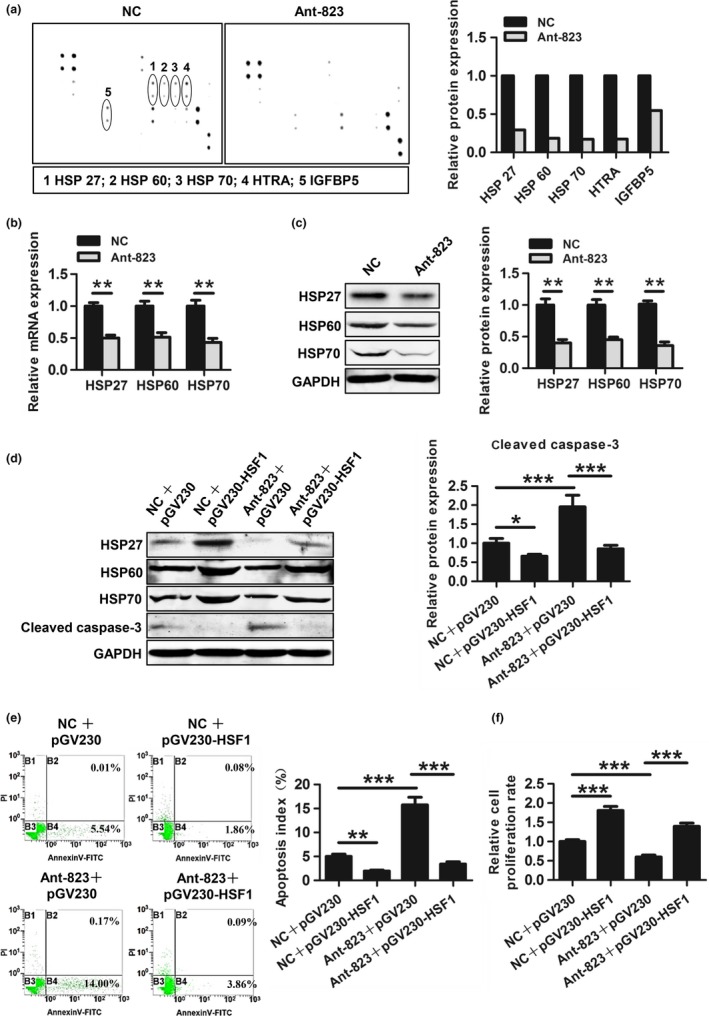
piR‐823 regulates cell proliferation and apoptosis by HSPs. (a) Human Apoptosis Antibody Array was performed in HCT116 cells transfected with Ant‐823 or NC. Bar graphs show the differential expression of protein related to apoptosis. (b, c) mRNA levels (b) and protein levels (c) of HSP27, HSP60 and HSP70 was validated in HCT116 cells. (d) The expression of HSP27, HSP60, HSP70 and activated caspase‐3 was evaluated by Western blot analysis in HCT116 cells co‐transfected with indicated RNA and plasmid. (e) Cell apoptosis of HCT116 cells co‐transfected with indicated RNA and plasmid was assessed by Annexin V/PI staining. (f) Cell proliferation of HCT116 cells co‐transfected with indicated RNA and plasmid was detected by CCK‐8 assay. Data are presented as mean ± SD from three independent experiments, *P < 0.05, **P < 0.01, ***P < 0.001 vs indicated groups.
To confirm whether piR‐823 modulated cell proliferation and apoptosis by HSPs, rescue experiment for HSPs was performed in HCT116 cells. To over‐express HSP27, HSP60 and HSP70, an over‐expression plasmid of their common transcription factor HSF1 (pGV230‐HSF1) was transfected into HCT116 cells. Western blot analysis showed that exogenous expression of HSF1 induced the expressions of HSP27, HSP60 and HSP70 compared with empty vector (pGV230) (Fig. 5d). Elevated HSPs expression inhibited cell apoptosis and partially abrogated Ant‐823‐dependent apoptosis determined by the activity of caspase‐3 (Fig. 5d) and Annexin V/PI staining (Fig. 5e). Meanwhile, CCK‐8 assay showed that HSP upregulation enhanced cell proliferation and partially rescued the inhibitory effect of Ant‐823 on proliferation (Fig. 5f).
piR‐823 regulates the transcriptional activity but not the expression of HSF1
Since piR‐823 regulated HSPs expression at the transcriptional level, we speculated that piR‐823 might regulate their transcription factor HSF1. We first explored whether piR‐823 modulated HSF1 expression and found that the mRNA and protein levels of HSF1 showed no significant difference between Ant‐823 transfected cells and NC‐transfected cells (Fig. 6a,b). Therefore, we next examined the effect of piR‐823 on the transcriptional activity of HSF1 using a dual‐luciferase reporter assay. As shown in Figure 6(c), overexpression of piR‐823 increased luciferase activity relative to that observed in the NC group.
Figure 6.
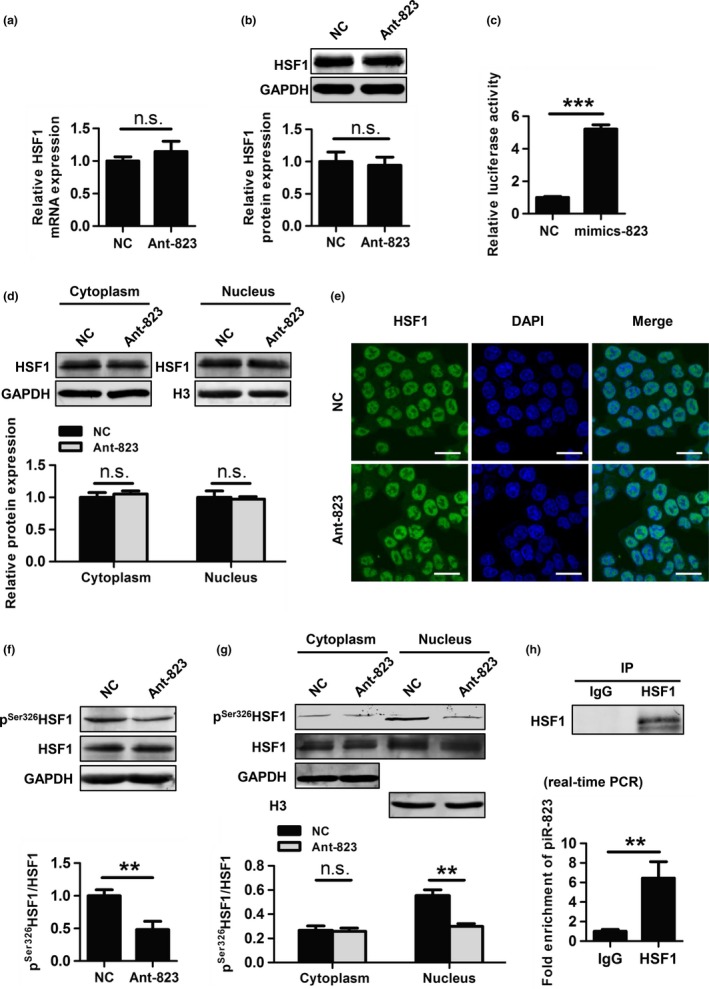
piR‐823 regulates HSF1 activity by binding to HSF1 and modulating its phosphorylation at Ser326. (a, b) mRNA levels (a) and protein levels (b) of HSF1 was evaluated in HCT116 cells transfected with Ant‐823 or NC. (c) The transcriptional activity of HSF1 was assessed by dual‐luciferase reporter assay in HCT116 cells. (d, e) Nuclear translocation of HSF1 in HCT116 cells was evaluated by western blot analysis of cytoplasmic and nuclear extractions (d) and immunofluorescence staining for HSF1 (green) (e). Nuclear DNA was stained with DAPI (blue). Scale bar, 20 μm. (f) The expression of pS er326 HSF1 in HCT116 cells was assessed by western blot analysis. (d) Cellular localization of pS er326 HSF1 in HCT116 cells was determined by western blot analysis. (h) The interaction of piR‐823 with HSF1 in HCT116 cells was assessed by RIP assay using anti‐HSF1 or IgG antibody as a control. The eluted RNA was analyzed by real‐time PCR. Immunoprecipitation efficiency was assessed by western blot analysis. Data are shown as the mean ± SD from three independent experiments, **P < 0.01, ***P < 0.001, n.s. indicates no significance vs indicated groups.
piR‐823 regulates HSF1 phosphorylation at Ser326
Because the complete activation of HSF1 requires its nuclear translocation and phosphorylation at Ser326, we first determined the effect of piR‐823 on the nuclear translocation of HSF1 in HCT116 cells. Compared with NC, Ant‐823 did not significantly change the HSF1 cellular location (Fig. 6d). A similar result was observed using an immunofluorescence assay (Fig. 6e). Both western blot analysis and immunofluorescence assay indicated that piR‐823 did not modulate the nuclear translocation of HSF1.
We next observed whether piR‐823 played a role in regulating the phosphorylation at Ser326 of HSF1 by western blot analysis. As shown in Figure 6(f), the inhibition of piR‐823 resulted in a lower level of pSer326HSF1 than NC.
To determine the cellular localization of pSer326HSF1, equal amounts of cytoplasmic and nuclear protein were used for western blot analysis, which showed that pSer326HSF1 was predominantly localized in the nucleus of HCT116 cells with or without Ant‐823 treatment. Moreover, the inhibition of piR‐823 only led to a marked reduction of nuclear pSer326HSF1 level (Fig. 6g).
piR‐823 binds to HSF1
RIP assay was used to determine whether there was an interaction between piR‐823 and HSF1. We found that piR‐823 was significantly more enriched with the HSF1 antibody than with the IgG antibody (Fig. 6h). All together, these data indicate that piR‐823 regulates HSF1 activity through interacting with HSF1 and modulating its phosphorylation at Ser326.
Discussion
piRNAs have been identified outside germline cells and have been found to regulate protein‐coding genes expression beyond transposons, suggesting that piRNAs have more extensive roles than previously supposed.38, 39 Recent evidence has shown that piRNAs are aberrantly expressed in a variety of cancers and correlate with tumorigenesis, including gastric cancer, multiple myeloma, liver cancer and bladder cancer.18, 21, 22, 23 Our data showed that piR‐823 was significantly upregulated in CRC tissues. Moreover, piR‐823 promoted cell proliferation by regulating cell cycle progression and suppressed cell apoptosis, indicating that piR‐823 contributed to colorectal tumorigenesis and might be a new therapeutic target for CRC.
Notably, a previous study demonstrated that piR‐823 is downregulated and plays a tumor‐suppressive role in gastric cancer,23 whereas our results reveal a tumor‐promoting role of piR‐823 in CRC. These data may appear contradictory, but they imply that piRNAs play different roles in different cancer types, further suggesting the complex roles of piRNAs in cancers.
Piwil2, the human Piwi homolog, has been shown to contribute to tumorigenesis by activating the Stat3/Bcl‐xl/Cyclin D1 signaling pathway, indirectly affecting cell cycle progression.40 Piwil2 suppresses p53 expression, which can induce cyclin‐dependent kinase inhibitor (CDKI) expression and regulate G1 phase progression.41 These data indicate that Piwil2 plays a critical role in cell cycle regulation. Because piRNAs function by association with the Piwi family, it is therefore probable that piRNAs also regulate the cell cycle in tumorigenesis. Consistent with these data, we found that the inhibition of piR‐823 induced G1 phase arrest and suppressed the expression of the G1 phase regulators Cyclin D1 and CDK4, thereby inhibiting cell proliferation and promoting cell apoptosis in CRC.
Mechanism analysis revealed that piR‐823 promoted the expression of HSP27, HSP60, HSP70 concurrently by increasing the transcriptional activity of their common transcription factor HSF1 that was demonstrated to be overexpressed in CRC and to play a potent role in colorectal carcinogenesis.36 We next found that piR‐823 modulated the transcriptional activity of HSF1 by regulating the phosphorylation at Ser326 but not its nuclear translocation. Interestingly, the RIP assay showed an interaction between piR‐823 and HSF1. Collectively, these results clearly indicate that piR‐823 is involved in HSF1 transcriptional activity regulation by binding to HSF1 and modulating its phosphorylation at Ser326.
In the past few years, the functional mechanism of piRNAs in tumors has been preliminarily elucidated, and most of these studies suggested an epigenetic regulation role. In addition, a few studies also revealed that piRNAs regulate mRNA expression through binding to the 3′UTR of mRNA.18, 20 However, all of these studies focused on the regulation of gene expression. Here, we were surprised to find that piRNAs regulated the post‐translational modification and activity of HSF1 via interaction with this protein, which provides new insight into the mechanism of piRNAs in carcinogenesis.
Is this mechanism an exception? A recent study appears to provide the answer. This study showed that piRNA‐like‐163 regulated p‐ERM activity through directly interacting with p‐ERM in lung cancer cells.42 Thus, it may be a universal mechanism that piRNAs regulate the functional activity of proteins. It should be emphasized that we found that pSer326HSF1 was primarily localized in the nucleus, and piR‐823 only regulated the nuclear levels of pSer326HSF1. These data suggest that piR‐823 functions in the nucleus, consistent with previous studies indicating that piRNAs exert epigenetic silencing function in the nucleus.43, 44 Nevertheless, the exact mechanisms of how piRNAs identify their target proteins and how piRNAs regulate the phosphorylation of target proteins remain unclear. Additionally, it is also unclear whether piRNAs regulate other modifications of target proteins, such as acetylation, methylation and ubiquitination. Therefore, further work should be performed to address these issues.
In summary, we first demonstrated that piR‐823 was upregulated in CRC tissues, and piR‐823 promoted colorectal carcinogenesis by enhancing cell proliferation and suppressing cell apoptosis. In addition, the molecular mechanism by which piR‐823 exerted an oncogenic function involved the interaction with HSF1 to promote its phosphorylation at Ser326, inducing the activation of HSF1. However, in this study, we mainly focused on a specific piRNA, and this piRNA may not be representative of the global piRNA population. It is therefore necessary to determine the global piRNA expression profile in CRC, which will provide insight into the role of piRNAs in carcinogenesis.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. The expression of piR‐823 was downregulated by Ant‐823 and upregulated by mimics‐823.
Table S1. The sequences for Ant‐823, mimics‐823 and scrambled sequence.
Table S2. Primer pairs for real‐time PCR analysis of different gene.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81570546 and 81602529) and the Key Basic Research Foundation of Applied Basic Research Program of Hebei Province, China (14967717D).
Cancer Sci 108 (2017) 1746–1756
Funding Information
National Natural Science Foundation of China (81570546 and 81602529); Key Basic Research Foundation of Applied Basic Research Program of Hebei Province, China (14967717D).
References
- 1. Migliore L, Migheli F, Spisni R, Coppede F. Genetics, cytogenetics, and epigenetics of colorectal cancer. J Biomed Biotechnol 2011; 2011: 792362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bogaert J, Prenen H. Molecular genetics of colorectal cancer. Ann Gastroenterol 2014; 27: 9–14. [PMC free article] [PubMed] [Google Scholar]
- 3. Goto T, Mizukami H, Shirahata A et al Aberrant methylation of the p16 gene is frequently detected in advanced colorectal cancer. Anticancer Res 2009; 29: 275–7. [PubMed] [Google Scholar]
- 4. Iacopetta B, Russo A, Bazan V et al Functional categories of TP53 mutation in colorectal cancer: results of an International Collaborative Study. Ann Oncol 2006; 17: 842–7. [DOI] [PubMed] [Google Scholar]
- 5. Esteller M. Non‐coding RNAs in human disease. Nat Rev Genet 2011; 12: 861–74. [DOI] [PubMed] [Google Scholar]
- 6. Heyns M, Kovalchuk O. Non‐coding RNAs including miRNAs, piRNAs, and tRNAs in human cancer. Oncotarget 2015; 6: 23055–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bongiorni MG, Soldati E, Arena G et al Multicenter clinical evaluation of a new SSIR pacemaker. Pacing Clin Electrophysiol 1992; 15: 1798–803. [DOI] [PubMed] [Google Scholar]
- 8. Luo J, Xu L, Jiang Y et al Expression profile of long non‐coding RNAs in colorectal cancer: a microarray analysis. Oncol Rep 2016; 35: 2035–44. [DOI] [PubMed] [Google Scholar]
- 9. Aravin A, Gaidatzis D, Pfeffer S et al A novel class of small RNAs bind to MILI protein in mouse testes. Nature 2006; 442: 203–7. [DOI] [PubMed] [Google Scholar]
- 10. Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline‐specific class of small RNAs binds mammalian Piwi proteins. Nature 2006; 442: 199–202. [DOI] [PubMed] [Google Scholar]
- 11. Grivna ST, Beyret E, Wang Z, Lin H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev 2006; 20: 1709–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lau NC, Seto AG, Kim J et al Characterization of the piRNA complex from rat testes. Science 2006; 313: 363–7. [DOI] [PubMed] [Google Scholar]
- 13. Aravin AA, Sachidanandam R, Girard A, Fejes‐Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 2007; 316: 744–7. [DOI] [PubMed] [Google Scholar]
- 14. Aravin AA, Bourc'his D. Small RNA guides for de novo DNA methylation in mammalian germ cells. Genes Dev 2008; 22: 970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuramochi‐Miyagawa S, Watanabe T, Gotoh K et al DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 2008; 22: 908–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martinez VD, Vucic EA, Thu KL et al Unique somatic and malignant expression patterns implicate PIWI‐interacting RNAs in cancer‐type specific biology. Sci Rep 2015; 5: 10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheng J, Guo JM, Xiao BX et al piRNA, the new non‐coding RNA, is aberrantly expressed in human cancer cells. Clin Chim Acta 2011; 412: 1621–5. [DOI] [PubMed] [Google Scholar]
- 18. Chu H, Hui G, Yuan L et al Identification of novel piRNAs in bladder cancer. Cancer Lett 2015; 356: 561–7. [DOI] [PubMed] [Google Scholar]
- 19. Hashim A, Rizzo F, Marchese G et al RNA sequencing identifies specific PIWI‐interacting small non‐coding RNA expression patterns in breast cancer. Oncotarget 2014; 5: 9901–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peng L, Song L, Liu C et al piR‐55490 inhibits the growth of lung carcinoma by suppressing mTOR signaling. Tumour Biol 2016; 37: 2749–56. [DOI] [PubMed] [Google Scholar]
- 21. Law PT, Qin H, Ching AK et al Deep sequencing of small RNA transcriptome reveals novel non‐coding RNAs in hepatocellular carcinoma. J Hepatol 2013; 58: 1165–73. [DOI] [PubMed] [Google Scholar]
- 22. Yan H, Wu QL, Sun CY et al piRNA‐823 contributes to tumorigenesis by regulating de novo DNA methylation and angiogenesis in multiple myeloma. Leukemia 2015; 29: 196–206. [DOI] [PubMed] [Google Scholar]
- 23. Cheng J, Deng H, Xiao B et al piR‐823, a novel non‐coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Lett 2012; 315: 12–7. [DOI] [PubMed] [Google Scholar]
- 24. Lianos GD, Alexiou GA, Mangano A et al The role of heat shock proteins in cancer. Cancer Lett 2015; 360: 114–8. [DOI] [PubMed] [Google Scholar]
- 25. Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci 2006; 31: 164–72. [DOI] [PubMed] [Google Scholar]
- 26. Murphy ME. The HSP70 family and cancer. Carcinogenesis 2013; 34: 1181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beere HM. “The stress of dying”: the role of heat shock proteins in the regulation of apoptosis. J Cell Sci 2004; 117: 2641–51. [DOI] [PubMed] [Google Scholar]
- 28. Pirkkala L, Nykanen P, Sistonen L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J 2001; 15: 1118–31. [DOI] [PubMed] [Google Scholar]
- 29. Sorger PK. Heat shock factor and the heat shock response. Cell 1991; 65: 363–6. [DOI] [PubMed] [Google Scholar]
- 30. Sarge KD, Murphy SP, Morimoto RI. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA‐binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol 1993; 13: 1392–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baler R, Dahl G, Voellmy R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol Cell Biol 1993; 13: 2486–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guettouche T, Boellmann F, Lane WS, Voellmy R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem 2005; 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cotto JJ, Kline M, Morimoto RI. Activation of heat shock factor 1 DNA binding precedes stress‐induced serine phosphorylation. Evidence for a multistep pathway of regulation. J Biol Chem 1996; 271: 3355–8. [DOI] [PubMed] [Google Scholar]
- 34. Mendillo ML, Santagata S, Koeva M et al HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012; 150: 549–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang S, Tu K, Fu Q et al Multifaceted roles of HSF1 in cancer. Tumour Biol 2015; 36: 4923–31. [DOI] [PubMed] [Google Scholar]
- 36. Cen H, Zheng S, Fang YM, Tang XP, Dong Q. Induction of HSF1 expression is associated with sporadic colorectal cancer. World J Gastroenterol 2004; 10: 3122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Whitesell L, Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets 2009; 13: 469–78. [DOI] [PubMed] [Google Scholar]
- 38. Esposito T, Magliocca S, Formicola D, Gianfrancesco F. piR_015520 belongs to Piwi‐associated RNAs regulates expression of the human melatonin receptor 1A gene. PLoS One 2011; 6: e22727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rajasethupathy P, Antonov I, Sheridan R et al A role for neuronal piRNAs in the epigenetic control of memory‐related synaptic plasticity. Cell 2012; 149: 693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee JH, Schutte D, Wulf G et al Stem‐cell protein Piwil2 is widely expressed in tumors and inhibits apoptosis through activation of Stat3/Bcl‐XL pathway. Hum Mol Genet 2006; 15: 201–11. [DOI] [PubMed] [Google Scholar]
- 41. Lu Y, Zhang K, Li C et al Piwil2 suppresses p53 by inducing phosphorylation of signal transducer and activator of transcription 3 in tumor cells. PLoS One 2012; 7: e30999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mei Y, Wang Y, Kumari P et al A piRNA‐like small RNA interacts with and modulates p‐ERM proteins in human somatic cells. Nat Commun 2015; 6: 7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Siomi MC, Sato K, Pezic D, Aravin AA. PIWI‐interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol 2011; 12: 246–58. [DOI] [PubMed] [Google Scholar]
- 44. Ishizu H, Nagao A, Siomi H. Gatekeepers for Piwi‐piRNA complexes to enter the nucleus. Curr Opin Genet Dev 2011; 21: 484–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The expression of piR‐823 was downregulated by Ant‐823 and upregulated by mimics‐823.
Table S1. The sequences for Ant‐823, mimics‐823 and scrambled sequence.
Table S2. Primer pairs for real‐time PCR analysis of different gene.