

Abstract
Osteosarcoma is the most common type of primary bone tumor, and novel therapeutic approaches for this disease are urgently required. To identify effective agents, we screened a panel of Food and Drug Administration (FDA)‐approved drugs in AXT cells, our newly established mouse osteosarcoma line, and identified calcitriol as a candidate compound with therapeutic efficacy for this disease. Calcitriol inhibited cell proliferation in AXT cells by blocking cell cycle progression. From a mechanistic standpoint, calcitriol induced endoplasmic reticulum (ER) stress, which was potentially responsible for downregulation of cyclin D1, activation of p38 MAPK, and intracellular production of reactive oxygen species (ROS). Knockdown of Atf4 or Ddit3 restored cell viability after calcitriol treatment, indicating that the ER stress response was indeed responsible for the anti‐proliferative effect in AXT cells. Notably, the ER stress response was induced to a lesser extent in human osteosarcoma than in AXT cells, consistent with the weaker suppressive effect on cell growth in the human cells. Thus, the magnitude of ER stress induced by calcitriol might be an index of its anti‐osteosarcoma effect. Although mice treated with calcitriol exhibited weight loss and elevated serum calcium levels, a single dose was sufficient to decrease osteosarcoma tumor size in vivo. Our findings suggest that calcitriol holds therapeutic potential for treatment of osteosarcoma, assuming that techniques to diminish its toxicity could be established. In addition, our results show that calcitriol could still be safely administered to osteosarcoma patients for its original purposes, including treatment of osteoporosis.
Keywords: Calcitriol, cell cycle, endoplasmic reticulum stress, osteosarcoma, vitamin D
Osteosarcoma is the most common primary non‐hematogenous malignant tumor of bone in childhood and adolescence. At the early stage of the disease, the tumors grow rapidly, at first locally, followed by systemic dissemination. Although the combination of surgery and chemotherapy has improved the prognosis, more than 30% of patients do not survive over the long term, and the outcomes have not changed largely over the past 20 years.1, 2, 3 Therefore, novel therapeutic options are urgently required.
In a previous work, we generated a mouse model of osteosarcoma by overexpressing c‐MYC in bone marrow stromal cells derived from Ink4a/Arf knockout mice.4 Inoculation of these cells, designated AXT cells, into syngeneic C57BL/6 mice results in rapid formation of lethal tumors with metastatic lesions that faithfully mimic human osteoblastic osteosarcoma, providing a useful model for preclinical studies.5, 6, 7
We screened >1100 FDA‐approved drugs in AXT cells in vitro, 7 and identified calcitriol as a drug candidate with therapeutic potential against osteosarcoma. Calcitriol, [1,25(OH)2D3], the most biologically active hormonal form of vitamin D, is synthesized from 25(OH)D3 in the kidneys by the cytochrome P450 enzyme CYP27B1.8, 9, 10 Calcitriol regulates the balance of serum calcium and phosphate levels, which is critical for bone mineralization.8, 11 Accordingly, calcitriol has been used to treat bone diseases such as osteoporosis, rickets, and osteomalacia. Beyond its roles in bone metabolism, calcitriol exerts pleiotropic biological effects, including prevention and inhibition of cancer progression in multiple tumor types.8, 9, 10
Previous investigations revealed that vitamin D, including calcitriol, alone or in combination with other agents, exerts antineoplastic effects in osteosarcoma.12, 13, 14 However, the precise molecular mechanism underlying the anti‐tumor effect of calcitriol in osteosarcoma cells remains unknown. Furthermore, the anti‐tumor potential of calcitriol for the treatment of osteosarcoma has not been extensively investigated in animal models.
In this study, we examined the effects of calcitriol on osteosarcoma cells in vitro and in vivo using our syngeneic mouse model. Based on the resultant mechanistic insights, we propose that calcitriol could be applied to the treatment of osteosarcoma in the clinical setting if its toxicity can be successfully managed.
Materials and Methods
Cell culture
AXT cells, as well as Saos2, U2OS, and SJSA1 human osteosarcoma cells (American Type Culture Collection) were maintained under 5% CO2 at 37°C in IMDM (Thermo Fischer Scientific, Carlsbad, CA, USA) supplemented with 10% FBS.5
Reagents
Calcitriol and simvastatin were obtained from Cayman Chemical (Ann Arbor, MI, USA) and Combi‐Blocks (San Diego, CA, USA), respectively. Adriamycin was purchased from Kyowa Hakko Kirin (Tokyo, Japan). Calcitriol was reconstituted in ethanol at a stock concentration of 10 mM. Akt inhibitor X and p38MAPK inhibitor (BIRB796) were purchased from Merk Millipore (Darmstadt, Germany). Thapsigardin and N‐acetylcysteine (NAC) were from Sigma‐Aldrich (Munich, Germany).
Cell proliferation assay
Cell viability was measured as previously described.5, 6 Live and dead cells were identified with Trypan blue staining (Sigma‐Aldrich).
Tumor xenograft model
All animal care and procedures were performed in accordance with the guidelines of Hoshi University. To establish tumor xenografts, AXT cells (5 × 105) suspended in 100 μL of IMDM were injected subcutaneously and bilaterally into the flanks of 6‐week‐old female syngeneic C57BL/6 mice on day 0 (SLC, Shizuoka, Japan). The mice were then injected intraperitoneally with calcitriol once a day at a dose of 6.27 × 10−1 μg/mouse (31.4 μg/kg) or 2.09 × 10−2 μg/mouse (1 μg/kg) on days 4, 6, 9, 13, 16, 18, and 20. Calcitriol was diluted with normal saline in a volume of 100 μL. Twenty‐one days after cell inoculation, the mice were euthanized with a lethal dose of pentobarbital sodium (Tokyo Kasei Kogyo, Tokyo, Japan), and the tumors were subjected to analyses.
Serum calcium concentration
Serum was collected from mice, and calcium concentration was evaluated using the Calcium Detection Kit (Abcam, Cambridge, UK).
Immunoblot analysis
Cell lysate was prepared with 2× Laemmli sample buffer (Bio‐Rad, Hercules, CA, USA) supplemented with β‐mercaptoethanol. Immunoblot analysis was performed according to standard procedures5, 6, 15 using antibodies against the phosphorylated and total forms of p38 mitogen‐activated protein kinase (MAPK), AKT, mTOR, ERK1/2, S6 and eIF2α (Cell Signaling Technology, Danvers, MA, USA), as well as antibodies against PERK, IRE1α, ATF4, CHOP, vitamin D3 receptor (Cell Signaling Technology), cyclin D1 (Santa Cruz Biotechnology, Dallas, TX, USA) and α‐Tubulin (Sigma‐Aldrich). α‐Tubulin was used as a loading control. The antibody against phosphorylated form of PERK (Thr980) or (Thr982) was from Thermo Scientific or Abcam, respectively.
Detection of apoptotic cells by flow cytometry
Cells were collected, washed with ice‐cold phosphate‐buffered saline (PBS), suspended in PBS, and stained with propidium iodide and allophycocyanin‐conjugated annexin V (e‐Bioscience, Carlsbad, CA, USA). After incubation on ice in the dark for 1 h, the cells were assayed for apoptosis by flow cytometry on a FACSVerse (BD Biosciences, San Jose, CA, USA). Data were analyzed with the FlowJo software (Tree Star, Ashland, OR, USA).
Detection of ROS
After cells were treated with calcitriol, CellRox deep Red or Mitosox reagent (Thermo Fisher Scientific, Waltham, MA, USA) was added to monitor the level of total or mitochondrial reactive oxygen species (ROS), respectively. ROS level was evaluated by flow cytometry on a FACSVerse. At least 10 000 live cells were analyzed for each sample.
Detection of γH2AX positive cells
DNA damage was monitored using Alexa Fluor 647–conjugated γH2AX antibody and an isotype control antibody (Cell Signaling Technology). At least 10 000 live cells were analyzed by flow cytometry for each sample.
Cell cycle analysis
Cells were collected, fixed with 70% ethanol for 48 h at −20°C, washed twice with ice‐cold PBS, and stained with PBS containing 10 μg/mL propidium iodide and 20 μg/mL RNase. The cell cycle profile of 20 000 singlet cells was then analyzed by flow cytometry.
Reverse transcription (RT) and real‐time PCR analysis
Total RNA extraction, RT, and real‐time PCR analyses were performed as previously described using NucleoSpin RNA and prime script RTase (Takara, Shiga, Japan).5, 6 The sequences of PCR primers are provided in Table S1.
Knockdown of Ddit3 or Atf4 by siRNA
AXT cells in IMDM + 1% serum were subjected to reverse transfection on days 1 and 2 (24 h each), with siRNAs for Ddit3 or Atf4 or a control siRNA: BannoNegacon (siRNA Inc., Tokyo, Japan) conjugated with RNAi‐MAX reagent (Thermo Fisher Scientific). siRNAs were used at a concentration of 200 nM each. On day 3, cells were collected and subjected to cell proliferation assay and RNA collection. Before RNA collection from cells treated with siRNAs for Ddit3 and the respective control siRNA, the cells were exposed to 5 μM calcitriol for 8 h. Sequences of the sense and antisense strands of siRNAs are provided in Table S2.
Gene expression profiling
AXT cells were treated with 10 μM calcitriol for 11 h, and total RNA was subjected to gene expression analyses using the 3D‐DNA chip (Toray, Tokyo, Japan). To prepare RNA from tumors, AXT cells (1 × 106) were injected into mice on day 0 as described above. The mice were injected intraperitoneally with calcitriol at a dose of 6.25 × 10−1 μg/mouse on days 5, 8, 11, 13, 15, 18, 20, and 22. Twenty‐three days after cell inoculation, tumors were excised and subjected to RNA collection using a BioMasher (Nippi, Tokyo, Japan) and NucleoSpin RNA. RNA mixtures derived from four control mice or five calcitriol‐treated mice were used for gene expression analyses.
Statistical analysis
All assays were performed in triplicate, and quantitative data are expressed as means ± SD relative to the control value unless indicated otherwise. Data were analyzed with Student's t‐test, and a P‐value of <0.05 was considered statistically significant. (*, P < 0.05; **, P < 0.005; ***, P < 0.0005; NS, not significant).
Results
Calcitriol inhibits cell proliferation of AXT cells
To identify effective therapeutic drugs for osteosarcoma, we screened 1164 US Food and Drug Administration (FDA)–approved compounds all at the same concentration (1.92 μM), using a novel humanoid robot (Robotic Biology Institute, Tokyo, Japan).7 Calcitriol decreased the viability of AXT cells by 44% of the control level (data not shown). Calcitriol (1,25‐dihydroxycholecalciferol) is an active metabolite of vitamin D that is critically involved in bone metabolism.11, 16 Because osteosarcoma is an osteoblastic tumor, we investigated the therapeutic potency of calcitriol against this disease.
Calcitriol inhibited cell viability of AXT cells in a dose‐dependent manner, but the effects were less pronounced in the human osteosarcoma lines U2OS, SJSA1, and Saos2 (Fig. 1a). Notably, expression of vitamin D3 receptor could be detected at the protein level in both AXT and human osteosarcoma cells (Fig. 1b), indicating that the differences in the anti‐tumor effect of calcitriol among these lines cannot be attributed to the differences in the expression levels of vitamin D3 receptor.
Figure 1.
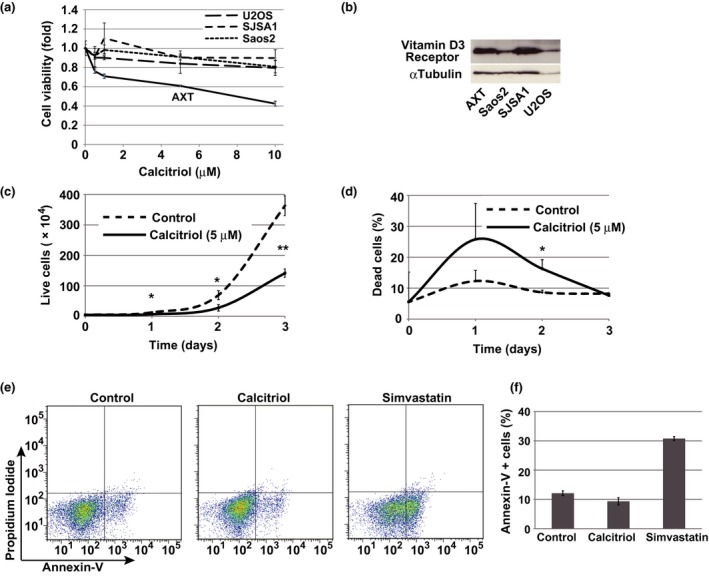
Calcitriol inhibits cell proliferation in AXT cells. (a) Viability of AXT, U2OS, Saos2 and SJSA1 cells treated with calcitriol for 2 days at the indicated concentrations. (b) Immunoblot analysis of vitamin D3 Receptor in indicated cells. (c, d) Number of viable AXT cells, identified by Trypan blue exclusion, was counted after exposure to 5 μM calcitriol for 2 days. Ratio of dead AXT cells is shown in (d). (e, f) Flow cytometric analysis of AXT cells treated with 10 μM calcitriol or 10 μM simvastatin for 24 h, and then stained with annexin V and propidium iodide. The corresponding percentages of annexin V+ cells were also determined (f).
To determine whether inhibition of AXT cell growth by calcitriol was caused by cell death, we counted live and dead cells using the Trypan blue exclusion test. After treatment with calcitriol, the number of live cells was markedly reduced, whereas the increase in the number of dead cells was less prominent (Fig. 1c,d). Consistent with these findings, apoptosis was not induced by calcitriol treatment (Fig. 1e,f). By contrast, the level of apoptosis is increased by simvastatin treatment.7
Together, these results indicated that calcitriol inhibits proliferation of AXT mouse osteosarcoma cells, but the effect was completely dependent on cell context.
Calcitriol inhibits cell cycle progression in AXT cells
Because calcitriol suppressed the growth in AXT cells, we next examined cell cycle status. Treatment of calcitriol induced accumulation of cells in G1, and significantly decreased the S‐phase fraction, in a dose‐dependent manner (Fig. 2a,b). The sub‐G1 fraction was slightly increased by treatment with 10 μM calcitriol, indicating that calcitriol can induce cell death at high concentrations. The protein level of cyclin D1 was decreased by calcitriol treatment, but the mRNA level was not affected (Fig. 2c,d), suggesting that this effect was not mediated by transcriptional regulation. In Saos2 or U2OS cells, calcitriol increased the G1 fraction, as in AXT cells, but the effect was much more modest (Fig. 2e–h). Thus, calcitriol blocks cell cycle progression in AXT cells, but its ability to induce cell death is much weaker.
Figure 2.
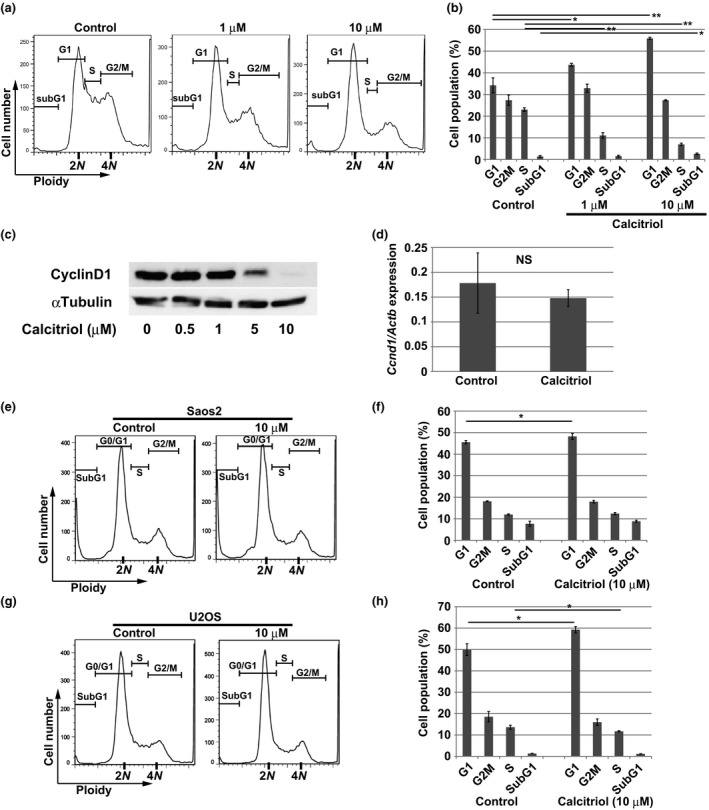
Calcitriol induces cell cycle arrest. (a, b) Flow cytometric analysis of DNA content in AXT cells treated with 1 or 10 μM calcitriol for 24 h. The size of each fraction of cells is shown in (b). (c) Immunoblot analysis of cyclinD1 in AXT cells treated with the indicated concentrations of calcitriol for 14 h. (d) RT and real‐time PCR analysis of Ccnd1 mRNA in AXT cells treated with 5 μM calcitriol for 12 h. Data are normalized against the amount of Actb mRNA, and are means ± SD of triplicates. (e–h) Flow cytometric analysis of DNA content in Saos2 (e) or U2OS (g) cells treated with 10 μM calcitriol for 48 h. The size of each fraction of cells is also shown in (f, h).
Accumulation of reactive oxygen species and activation of p38 MAPK in AXT cells upon calcitriol treatment
To investigate the intracellular events caused by calcitriol treatment, we evaluated several pathways, including kinase activation, by immunoblotting. Calcitriol induced activation of p38 MAPK in a concentration‐dependent manner (Fig. 3a).
Figure 3.
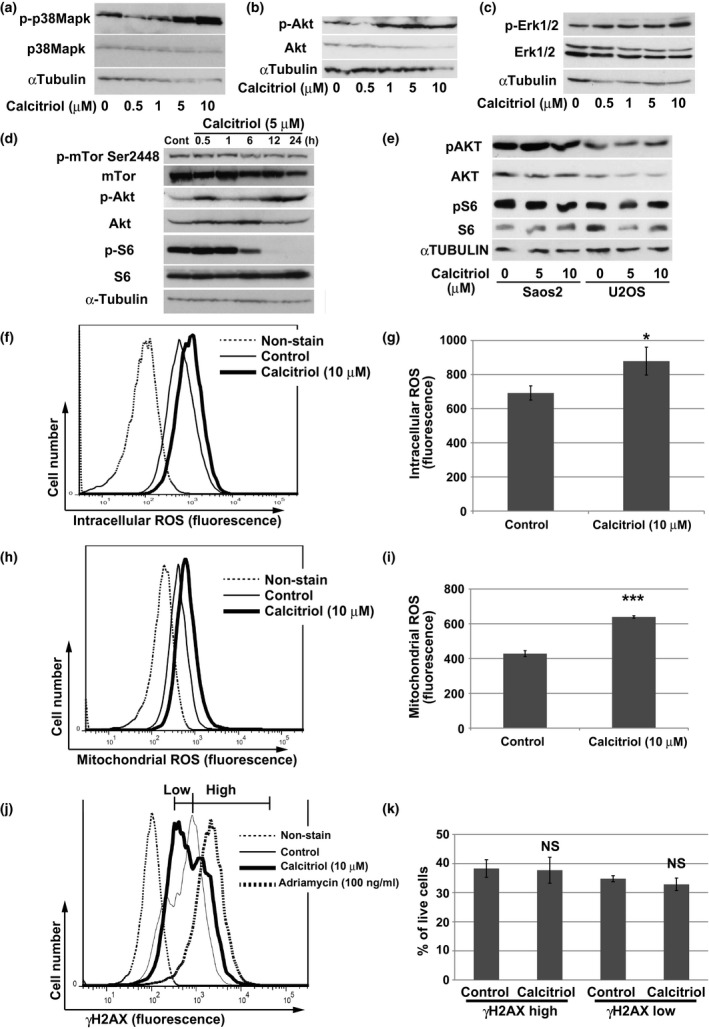
Activation of p38 MAPK by calcitriol treatment in AXT cells. (a) Immunoblot analysis of total or phosphorylated (p‐) forms of p38MAPK in AXT cells treated with the indicated concentrations of calcitriol for 14 h. (b) Immunoblot analysis of total or phosphorylated (p‐) forms of Akt in AXT cells treated with the indicated concentrations of calcitriol for 14 h. (c) Immunoblot analysis of total or phosphorylated (p‐) forms of Erk1/2 in AXT cells treated with the indicated concentrations of calcitriol for 22 h. (d) Immunoblot analysis of total or phosphorylated (p‐) forms of mTor, Akt, and S6 in AXT cells treated with 5 μM calcitriol for the indicated times. (e) Immunoblot analysis of total or phosphorylated (p‐) forms of AKT and S6 in Saos2 and U2OS cells treated with 5 or 10 μM calcitriol for 19 h. (f–i) Flow cytometric analyses of intracellular (f) or mitochondrial ROS (h) in AXT cells after treatment with 10 μM calcitriol for 24 h or 21 h, respectively. Quantitation of ROS levels in each case, as geometric means of triplicates, is shown in (g, i). (j, k) Representative histograms (j) and quantitation (k) of AXT cells strongly or weakly positive for γ‐H2AX, as determined by flow cytometric analyses, after treatment with 10 μM calcitriol or 100 ng/mL adriamycin for 24 h.
Likewise, the level of the phosphorylated form of AKT was elevated upon treatment with calcitriol, whereas the levels of phospho‐Erk1/2 were not affected (Fig. 3b,c). Intriguingly, phosphorylation of S6, an indicator of translational processing of mRNA,17, 18 was markedly attenuated by calcitriol treatment, whereas activation of mTor was unaffected (Fig. 3d). None of these cellular events were observed in human cell lines (Fig. 3e).
The production of intracellular reactive oxygen species (ROS) induces activation of p38 MAPK.19, 20 The intracellular ROS level was significantly elevated in AXT cells after calcitriol treatment (Fig. 3f,g). In addition, the mitochondrial ROS level was elevated by calcitriol, suggesting that at least some fraction of intracellular ROS was derived from mitochondria (Fig. 3h,i). Because excessive production of intracellular ROS might induce DNA damage, resulting in anti‐tumor activity,20, 21 we examined the amount of phosphorylated histone H2AX (γ‐H2AX), a marker of DNA damage.22 The proportion of AXT cells that were strongly positive for γ‐H2AX, as determined by flow cytometry, was elevated after treatment with adriamycin (Fig. 3j), used as a positive control for DNA damage. By contrast, the proportions of strongly and weakly positive cells did not differ significantly between calcitriol‐treated and control cells (Fig. 3j,k). These findings suggested that ROS production induced by calcitriol does not cause DNA damage. To further evaluate the influence of activation of p38 MAPK and elevation of ROS, we performed combined treatment of calcitriol with a p38 MAPK inhibitor or NAC. Both reagents failed to recover the viability attenuated by calcitriol (Fig.S1).
Therefore, activation of p38 MAPK or ROS production is not responsible for growth inhibition by calcitriol in AXT cells.
Calcitriol induced endoplasmic reticulum stress in AXT cells
To acquire further insight into the molecular mechanisms underlying inhibition of cell growth by calcitriol, we performed gene expression analyses. For this purpose, we treated AXT cells with or without 10 μM calcitriol for 11 h. Among the genes whose expression was upregulated more than fivefold in the calcitriol‐treated cells, we identified 14 genes that were also upregulated after administration of calcitriol in vivo (Table 1). One of these was Cyp24a1, which encodes a mitochondrial protein involved in the degradation of calcitriol8, 9, 10 (Table 1 and Fig. 4a). Notably, the expression of Ddit3, which encodes Chop, a key molecule induced during endoplasmic reticulum (ER) stress,23, 24 was markedly elevated after calcitriol treatment. Ddit4 and Gadd45a, which are also implicated in the ER stress response, were likewise upregulated25, 26, 27, 28, 29 (Table 1). Therefore, we focused our investigation on the ER stress pathway. Expression of Atf4 and Ddit3/Chop was increased by calcitriol treatment at both the mRNA and protein levels (Fig. 4b–d). In addition, calcitriol treatment increased expression of Ire1α and phosphorylation of eIf2α (Fig. 4d), both of which are indicators of the ER stress response.23, 24, 30 As the upstream event of these molecules, phosphorylation of Perk was assessed. Phosphorylation level at Thr980 of Perk was not strongly elevated by calcitriol compared to thapsigardin, a well‐known inducer of ER stress,25 although phosphorylation level was clearly enhanced after 30 min treatment of calcitriol and ATF4 expression was equally induced by calcitriol and thapsigardin (Fig. 4e). Notably, Thr982 of Perk was strongly phosphorylated by calcitriol treatment. Treatment of calcitriol or thapsigardin might induce phosphorylation of Perk at different residues. Thus, calcitriol treatment induced ER stress in AXT cells.
Table 1.
Comparative gene expression analysis in AXT cells with or without calcitriol treatment
| Gene symbol | Gene name | In vitro | In vivo |
|---|---|---|---|
| Fold | Fold | ||
| Cyp24a1 | Cytochrome P450, family 24, subfamily a, polypeptide 1 | 127.50 | 3.02 |
| Akr1b7 | Aldo‐keto reductase family 1, member B7 | 13.89 | 1.97 |
| Avil | Advillin | 13.54 | 1.19 |
| Gdpd5 | Glycerophosphodiester phosphodiesterase domain containing 5 | 10.56 | 3.74 |
| Ddit3 | DNA‐damage inducible transcript 3 | 10.40 | 1.62 |
| Vdr | Vitamin D receptor | 7.61 | 2.71 |
| Ddit4 | DNA‐damage‐inducible transcript 4 | 7.59 | 1.14 |
| Csn3 | Casein kappa | 7.34 | 1.15 |
| Gpr141 | G protein‐coupled receptor 141 | 7.21 | 1.03 |
| Tmem100 | Transmembrane protein 100 | 5.93 | 2.32 |
| Gadd45a | Growth arrest and DNA‐damage‐inducible 45 alpha | 5.69 | 1.68 |
| Plek | Pleckstrin | 5.64 | 1.45 |
| V1rh15 | Vomeronasal 1 receptor, H15 | 5.20 | 1.40 |
| Gdf15 | Growth differentiation factor 15 | 5.14 | 1.14 |
Figure 4.
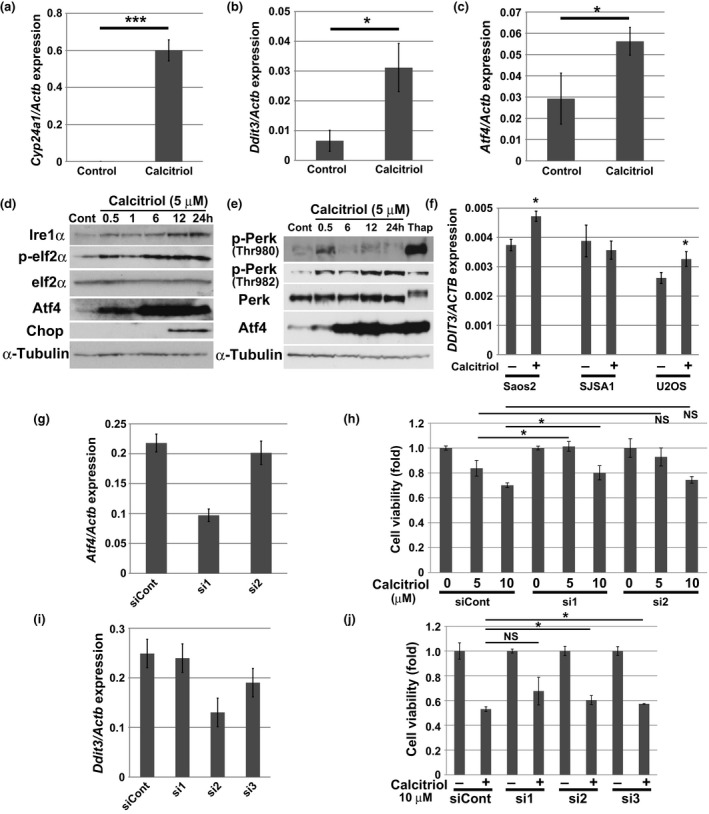
Induction of ER stress by calcitriol treatment. (a–c) RT and real‐time PCR analysis of Cyp24a1, Ddit3, or Atf4 mRNA in AXT cells treated with 5 μM calcitriol for 12 h. Data are normalized against the amount of Actb mRNA, and are means ± SD of triplicates. (d) Immunoblot analysis of Ire1α and total or phosphorylated (p‐) forms of eIf2α, Atf4 and Chop in AXT cells treated with 5 μM calcitriol for the indicated times. (e) Immunoblot analysis of total or phosphorylated (p‐) forms of Perk and Atf4 in AXT cells treated with 5 μM calcitriol for the indicated times or with 1 μM thapsigardin (Thap) for 1.5 h. (f) RT and real‐time PCR analysis of DDIT3 mRNA in Saos2, SJSA1, and U2OS cells treated with 5 μM calcitriol for 12 h. Data are normalized against the amount of ACTB mRNA, and are means ± SD of triplicates. (g) RT and real‐time PCR analysis of Atf4 in AXT cells transfected with control siRNA and Atf4‐targeted (si1, si2) siRNAs. Data are normalized against the amount of Actb mRNA, and are means ± SD of triplicates. (h) Cell viability of siRNA‐transfected AXT cells as in (f) treated with calcitriol for 24 h. (i) RT and real‐time PCR analysis of Ddit3 in AXT cells transfected with control siRNA and Ddit3‐targeted (si1, si2, si3) siRNAs. Before RNA collection, cells were exposed to 5 μM calcitriol for 8 h. Data are normalized against the amount of Actb mRNA, and are means ± SD of triplicates. (j) Viability of siRNA‐transfected AXT cells as in (h) treated with calcitriol for 24 h.
By contrast, although the expression of DDIT3 was significantly increased by calcitriol treatment in Saos2 or U2OS cells, the upregulation was less prominent in human osteosarcoma cells than in AXT cells, consistent with the magnitude of growth inhibition (Fig. 4f). Atf4, which is induced by ER stress, acts in a cell context‐dependent manner to promote transcription of downstream target genes involved in recovery from ER stress or induction of apoptosis.23, 24, 30, 31 To clarify the role of Atf4 in AXT cells, we knocked down Atf4 using siRNA (Fig. 4g,h). Treatment with siRNA sequence 1 (si1), which exerted a potent knockdown effect, significantly restored cell viability attenuated by calcitriol. Knockdown of Ddit3 also slightly but significantly restored cell viability; the small magnitude of the rescue effect may have been a consequence of the low efficiency of the knockdown (Fig. 4i,j).
Collectively, these findings show that calcitriol treatment induced the ER stress response in AXT cells, leading to suppression of cell viability.
Anti‐tumor effect of calcitriol in vivo
To determine whether calcitriol might possess antitumor activity against AXT cells in vivo, we injected these cells into syngeneic C57BL/6 mice, and then treated the animals with calcitriol by intraperitoneal injection. Calcitriol was administered at 1 or 31.4 μg/kg, seven times in total, and both treatment protocols induced significant decreases in primary tumor weight (Fig. 5a,b). Cyp24a1 was upregulated in a dose‐dependent manner in tumors from mice subjected to calcitriol treatment (Fig. 5c), suggesting that calcitriol acted directly on tumors. Atf4 was significantly upregulated in tumors from mice receiving the high‐dose regimen (Fig. 5d). Calcitriol also upregulated Ddit3, but the effect was not statistically significant (Fig. 5e). Thus, as in the in vitro experiments, calcitriol induced ER stress in AXT cells in vivo. Mice treated with 1 μg/kg or with 31.4 μg/kg calcitriol exhibited body weight loss of 15.5% and 30.9%, respectively, relative to control mice (Fig. 5f), and the serum calcium level was increased by calcitriol treatment (Fig. 5g), suggesting that these doses were toxic to some extent; however all mice were alive and showed no signs of exhaustion.
Figure 5.
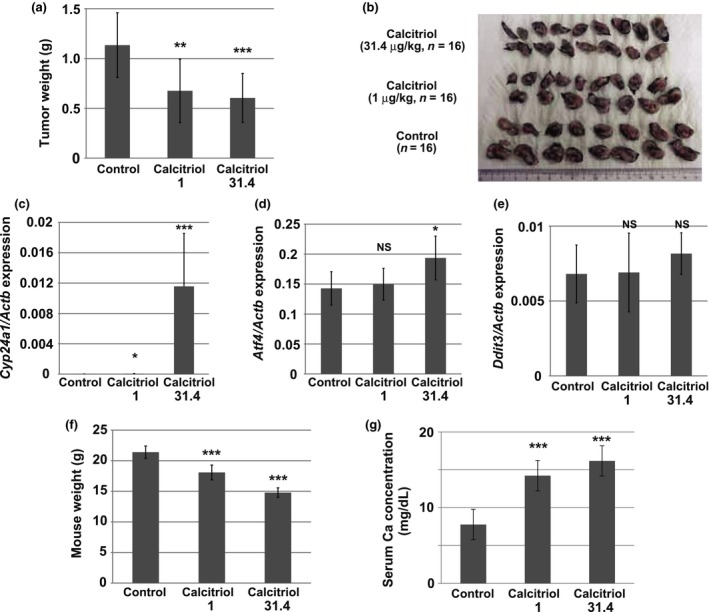
Anti‐tumor activity of calcitriol in vivo. (a, b) Weight of AXT cell‐derived tumors developed in control mice or mice injected intraperitoneally with calcitriol (1 or 31.4 μg/kg). Data are means ± SD (n = 16); P‐values indicate comparison with control (Student's t‐test). The excised tumors are shown in (b). (c–e) RT and real‐time PCR analysis of Cyp24a1 (c), Atf4 (d) and Ddit3 (e) in tumors derived from control mice or mice injected intraperitoneally with calcitriol (1 or 31.4 μg/kg). Data are normalized by the amount of Actb mRNA and are means ± SD (n = 8); P‐values indicate comparison with control (Student's t‐test). (f) Weight of control mice or mice injected intraperitoneally with calcitriol (1 or 31.4 μg/kg). Data are means ± SD (n = 8); P‐values indicate comparison with control (Student's t‐test). (g) Serum Ca concentration of control mice or mice injected intraperitoneally with calcitriol (1 or 31.4 μg/kg). Data are means ± SD (n = 8); P‐values indicate comparison with control (Student's t‐test).
Collectively, these findings demonstrate that calcitriol exerts an anti‐osteosarcoma effect in vivo, and could represent a potential therapeutic option if its toxicity could be effectively managed.
Discussion
In this study, we performed a multidrug screen in AXT cells, which identified calcitriol as a candidate therapeutic agent against osteosarcoma. Calcitriol exerted a strong anti‐proliferative effect in AXT cells, whereas its efficacy in human osteosarcoma cells was less potent. Notably, the effect of calcitriol was not correlated with the expression level of vitamin D3 receptor (Fig. 1a,b). The expression level of CYP24A1, which is transcriptionally upregulated by calcitriol,8, 9, 10 in human osteosarcoma cells; Saos2, SJSA1 and U2OS, was extremely low even after calcitriol treatment (data not shown), although all of the cells express a substantial level of vitamin D3 receptor (Fig. 1b). Therefore, the intensity of signals via the vitamin D3 receptor might not be correlated with its expression level. A previous study reported that expression of vitamin D receptor varies among human osteosarcoma specimens, but is not significantly correlated with tumor grade.32
Previous studies of various malignancies showed that calcitriol exerts its anti‐tumor effect by directly inducing cell death, including apoptosis, or by modulating the tumor environment in vivo.8, 9, 10, 33, 34, 35 By contrast, the anti‐proliferative effect in AXT cells by calcitriol was mainly attributable to induction of cell cycle arrest rather than cell death (Fig. 2). Despite the growth inhibition, phosphorylation of AKT was elevated in AXT cells upon calcitriol treatment (Fig. 3). PI3K and AKT are activated in hematopoietic cells during differentiation in response to calcitriol.36, 37 A previous report suggested that AKT activation accompanied by ER stress could block the induction of apoptosis mediated by CHOP.38 Notably, combined treatment of calcitriol and an AKT inhibitor to AXT cells slightly but significantly increased Annexin V‐positive apoptotic cells, while each reagent alone did not induce apoptosis significantly (Fig.S2a,b). In addition, AXT cells were more sensitive to the AKT inhibitor under the presence of calcitriol (Fig.S2c). Therefore, induction of CHOP after treatment of calcitriol might work to attenuate viability of AXT cells, while at the same time AKT activation might counteract the effect. AXT cells lack p16Ink4a/p19Arf and harbor mutant p53 (5, data not shown). These findings suggest that the response to calcitriol is completely dependent on the molecular background in malignant cells.
Calcitriol induced ER stress in AXT cells (Fig. 4). Endoplasmic reticulum stress has not been extensively characterized as a mechanistic cause of the cytotoxicity of calcitriol, although a recent study described this phenomenon in breast cancer.39 Endoplasmic reticulum stress results in inhibition of protein translation via phosphorylation of eIF2α,23, 24, 30, 31, 40 causing rapid downregulation of cyclin D1 at the protein level and inhibition of the G1‐to‐S transition.41 In addition, the ROS production and activation of p38 MAPK detected in AXT cells after calcitriol treatment (Fig. 3) could also be initiated by ER stress.42, 43 Our further investigations indicated that the ROS were derived at least in part from mitochondria and were not associated with DNA damage (Fig. 3). Therefore, all of the cellular events in AXT cells resulting from calcitriol treatment might be attributable to ER stress.
Endoplasmic reticulum stress can exert opposing effects on malignant cells, either promoting tumor growth or inducing cell death.23, 24, 30, 31 However, because knockdown of Atf4 or Ddit3 in AXT cells restored cell viability, we conclude that the ER stress response accompanying the induction of ER stress by calcitriol is responsible for the drug's anti‐proliferative effect in AXT cells. In human osteosarcoma lines, DDIT3 was slightly upregulated by calcitriol, consistent with the more modest inhibition of proliferation in comparison with AXT cells (Figs 1a,4f). Therefore, the strength of ER stress induced by calcitriol treatment might serve as an index of the anti‐osteosarcoma effect, and the levels of DDIT3 or ATF4 expression might be used as biomarkers of the efficacy of calcitriol in osteosarcoma.
Calcitriol also exerted an anti‐tumor effect in vivo by inducing ER stress (Table 1, Fig. 5d). However, the drug's toxicity, reflected by body weight loss and hypercalcemia (Fig. 5f,g), is a problem that cannot be ignored in a clinical context. Therefore, an accurate monitoring system capable of extracting appropriate clinical cases, as well as techniques that enable the activation of ER stress by calcitriol in a tumor‐specific manner, would be required for the use of calcitriol to treat human osteosarcoma. If these obstacles could be overcome, the drug could represent a potent therapeutic option. In addition, given that our results showed that the progression of osteosarcoma was not promoted by calcitriol, the drug could still be safely used for its original pharmaceutical purposes (e.g., treatment of osteoporosis) in osteosarcoma patients.
Disclosure Statement
H. Saya reports receiving commercial research grants from Daiichi Sankyo Co., Ltd., Eisai Co., Ltd., Nihon Noyaku Co., Ltd., Pola Pharma Inc. and AQUA Therapeutics Co., Ltd. T Shimizu reports receiving a research grant form Takeda Science Foundation. No potential conflicts of interest were disclosed by the other authors.
Supporting information
Fig. S1. NAC or BIRB796 did not recover the viability of AXT cells attenuated by calcitriol.
Fig. S2. Inhibition of AKT activation increased the apoptotic cells induced by calcitriol.
Table S1. Sequences of primers and predicted product sizes for real‐time RT‐PCR analysis.
Table S2. Sense and antisense strands of siRNAs.
Acknowledgments
We thank I. Ishimatsu for technical assistance and M. Sato for help with preparation of the manuscript. This work was supported by funding from Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to H.S.), by KAKENHI grant from Japan Society for the Promotion of Science (JSPS) (to T.S. #15K06845), by the Supported Program for the Strategic Research Foundation at Private Universities of the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT) (T.S.), and by the Takeda Science Foundation (T.S.).
Cancer Sci 108 (2017) 1793–1802
Funding information
KAKENHI grant from Japan Society for the Promotion of Science (JSPS), (Grant / Award Number: ‘15K06845’) Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Supported Program for the Strategic Research Foundation at Private Universities of the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT), Takeda Science Foundation.
References
- 1. Chou AJ, Geller DS, Gorlick R. Therapy for osteosarcoma: where do we go from here? Paediatr Drugs 2008; 10: 315–27. [DOI] [PubMed] [Google Scholar]
- 2. Ritter J, Bielack SS. Osteosarcoma. Ann Oncol 2010; 21(Suppl 7): vii320–5. [DOI] [PubMed] [Google Scholar]
- 3. Jaffe N. Osteosarcoma: review of the past, impact on the future. The American experience. Cancer Treat Res 2009; 152: 239–62. [DOI] [PubMed] [Google Scholar]
- 4. Shimizu T, Ishikawa T, Sugihara E et al c‐MYC overexpression with loss of Ink4a/Arf transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene 2010; 29: 5687–99. [DOI] [PubMed] [Google Scholar]
- 5. Shimizu T, Sugihara E, Yamaguchi‐Iwai S et al IGF2 preserves osteosarcoma cell survival by creating an autophagic state of dormancy that protects cells against chemotherapeutic stress. Cancer Res 2014; 74: 6531–41. [DOI] [PubMed] [Google Scholar]
- 6. Yamaguchi SI, Ueki A, Sugihara E et al Synergistic antiproliferative effect of imatinib and adriamycin in platelet‐derived growth factor receptor‐expressing osteosarcoma cells. Cancer Sci 2015; 106: 875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamel WA, Sugihara E, Nobusue H et al Simvastatin‐induced apoptosis in osteosarcoma cells: a key role of RhoA‐AMPK/p38 MAPK signaling in antitumor activity. Mol Cancer Ther 2017; 16: 182–92. [DOI] [PubMed] [Google Scholar]
- 8. Feldman D, Krishnan AV, Swami S, Giovannucci E, Feldman BJ. The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer 2014; 14: 342–57. [DOI] [PubMed] [Google Scholar]
- 9. Bikle DD. Vitamin D and cancer: the promise not yet fulfilled. Endocrine 2014; 46: 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diaz L, Diaz‐Munoz M, Garcia‐Gaytan AC, Mendez I. Mechanistic effects of calcitriol in cancer biology. Nutrients 2015; 7: 5020–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holick M. Vitamin D deficiency. N Engl J Med 2007; 357: 266–81. [DOI] [PubMed] [Google Scholar]
- 12. Thompson L, Wang S, Tawfik O et al Effect of 25‐Hydroxyvitamin D3 and 1 α,25 Dihydroxyvitamin D3 on differentiation and apoptosis of human osteosarcoma cell lines. J Orthop Res 2012; 30: 831–44. [DOI] [PubMed] [Google Scholar]
- 13. Engel N, Adamus A, Schauer N et al Synergistic action of genistein and calcitriol in immature osteosarcoma MG‐63 cells by SGPL1 up‐regulation. PLoS One 2017; 12: e0169742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garimella R, Tadikonda P, Tawfik O, Gunewardena S, Rowe P, Van Veldhuizen P. Vitamin D impacts the expression of Runx2 target genes and modulates inflammation, oxidative stress and membrane vesicle biogenesis gene networks in 143B osteosarcoma cells. Int J Mol Sci 2017; 18: E642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ueki A, Shimizu T, Masuda K et al Up‐regulation of Imp3 confers in vivo tumorigenicity on murine osteosarcoma cells. PLoS One 2012; 7: e50621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anderson PH, Lam NN, Turner AG et al The pleiotropic effects of vitamin D in bone. J Steroid Biochem Mol Biol 2013; 136: 190–4. [DOI] [PubMed] [Google Scholar]
- 17. Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70S6K. EMBO J 1997; 12: 3693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peterson RT, Schreiber SL. Translation control: connecting mitogens and the ribosome. Curr Biol 1998; 8: R248–50. [DOI] [PubMed] [Google Scholar]
- 19. Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. BioFactors 2003; 17: 287–96. [DOI] [PubMed] [Google Scholar]
- 20. Murakami S, Noguchi T, Takeda K, Ichijo H. Stress signaling in cancer. Cancer Sci 2007; 98: 1521–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsuzuki T, Nakatsu Y, Nakabeppu Y. Significance of error‐avoiding mechanisms for oxidative DNA damage in carcinogenesis. Cancer Sci 2007; 4: 465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WN. DNA double‐stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273: 5858–68. [DOI] [PubMed] [Google Scholar]
- 23. Yadav RK, Chae SW, Kim HR, Chae HJ. Endoplasmic reticulum stress and cancer. J Cancer Prev 2014; 19: 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress‐activated cell reprogramming in oncogenesis. Cancer Discov 2015; 5: 586–97. [DOI] [PubMed] [Google Scholar]
- 25. Whitney ML, Jefferson LS, Kimball SR. ATF4 is necessary and sufficient for ER stress‐induced upregulation of REDD1 expression. Biochem Biophys Res Commun 2009; 379: 451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin HO, Seo SK, Woo SH et al Activating transcription factor 4 and CCAAT/enhancer‐binding protein‐β negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radic Biol Med 2009; 46: 1158–67. [DOI] [PubMed] [Google Scholar]
- 27. Kimball S, Jefferson LS. Induction of REDD1 gene expression in the liver in response to endoplasmic reticulum stress is mediated through a PERK, eIF2α phosphorylation, ATF4‐dependent cascade. Biochem Biophys Res Commun 2012; 427: 485–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kubota K, Lee DH, Tsuchiya M et al Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. J Biol Chem 2005; 24: 23194–202. [DOI] [PubMed] [Google Scholar]
- 29. Tan HK, Muhammad TS, Tan ML. 14‐Deoxy‐11,12‐didehydroandrographolide induces DDIT3‐dependent endoplasmic reticulum stress‐mediated autophagy in T‐47D breast carcinoma cells. Toxicol Appl Pharmacol 2016; 300: 55–69. [DOI] [PubMed] [Google Scholar]
- 30. Mollereau B, Manie S, Napoletano F. Getting the better of ER stress. J Cell Commun Signal 2014; 8: 311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yusta B, Baggio LL, Estall JL et al GLP‐1 receptor activation improves β cell function and survival following induction of endoplasmic reticulum stress. Cell Metab 2006; 4: 391–406. [DOI] [PubMed] [Google Scholar]
- 32. Gallagher R, Keighley J, Tancabelic J et al Clinicopathologic correlation of vitamin D receptor expression with retinoid X receptor and MIB‐1 expression in primary and metastatic osteosarcoma. Ann Diagn Pathol 2012; 16: 323–9. [DOI] [PubMed] [Google Scholar]
- 33. Blutt SE, McDonnell TJ, Polek TC, Weigel NL. Calcitriol‐induced apoptosis in LNCaP cells is blocked by overexpression of Bcl‐2. Endocrinology 2000; 141: 10–7. [DOI] [PubMed] [Google Scholar]
- 34. Chen PT, Hsieh CC, Wu CT et al 1α,25 Dihydroxyvitamin D3 inhibits esophageal squamous cell carcinoma progression by reducing IL6 signaling. Mol Cancer Ther 2015; 14: 1365–75. [DOI] [PubMed] [Google Scholar]
- 35. Chung I, Han G, Seshadri M et al Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor‐derived endothelial cells and tumor angiogenesis in vivo. Cancer Res 2009; 69: 967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y, Zhang J, Studzinski GP. AKT pathway is activated by 1,25 Dihydroxyvitamin D3 and participates in its anti‐apoptotic effect and cell cycle control in differentiating HL60 cells. Cell Cycle 2006; 5: 447–51. [DOI] [PubMed] [Google Scholar]
- 37. Hmama BZ, Nandan D, Sly L, Knutson KL, Herrera‐Velit P, Reiner NE. 1α,25 ‐Dihydroxyvitamin D3‐induced myeloid cell differentiation is regulated by a vitamin D receptor‐Phosphatidylinositol 3‐Kinase signaling complex. J Exp Med 1999; 190: 1583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress‐induced cell death. J Biol Chem 2004; 279: 49420–9. [DOI] [PubMed] [Google Scholar]
- 39. Haddur E, Ozkaya AB, Ak H, Aydin HH. The effect of calcitriol on endoplasmic reticulum stress response. Biochem Cell Biol 2015; 93: 268–71. [DOI] [PubMed] [Google Scholar]
- 40. McConkey DJ. The integrated stress response and proteotoxicity in cancer therapy. Biochem Biophys Res Commun 2017; 482: 450–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell 2005; 16: 5493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and oxidative stress in the pathogenesis of non‐alcoholic fatty liver disease. Free Radic Res 2015; 49: 1405–18. [DOI] [PubMed] [Google Scholar]
- 43. Zeeshan HMA, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci 2016; 17: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. NAC or BIRB796 did not recover the viability of AXT cells attenuated by calcitriol.
Fig. S2. Inhibition of AKT activation increased the apoptotic cells induced by calcitriol.
Table S1. Sequences of primers and predicted product sizes for real‐time RT‐PCR analysis.
Table S2. Sense and antisense strands of siRNAs.