

Abstract
β‐Catenin is a central player in Wnt signaling, and activation of Wnt signaling is associated with cancer development. E‐cadherin in complex with β‐catenin mediates cell–cell adhesion, which suppresses β‐catenin‐dependent Wnt signaling. Recently, a tumor‐suppressive role for E‐cadherin has been reconsidered, as re‐expression of E‐cadherin was reported to enhance the metastatic potential of malignant tumors. To explore the role of E‐cadherin, we established an E‐cadherin‐expressing cell line, EC96, from AGS cells that featured undetectable E‐cadherin expression and a high level of Wnt signaling. In EC96 cells, E‐cadherin re‐expression enhanced cell proliferation, although Wnt signaling activity was reduced. Subsequent analysis revealed that nuclear factor‐κB (NF‐κB) activation and consequent c‐myc expression might be involved in E‐cadherin expression‐mediated cell proliferation. To facilitate rapid proliferation, EC96 cells enhance glucose uptake and produce ATP using both mitochondria oxidative phosphorylation and glycolysis, whereas AGS cells use these mechanisms less efficiently. These events appeared to be mediated by NF‐κB activation. Therefore, E‐cadherin re‐expression and subsequent induction of NF‐κB signaling likely enhance energy production and cell proliferation.
Keywords: Axin, E‐cadherin, mitochondria metabolism, NF‐κB, β‐Catenin mutation
E‐cadherin, which acts as a cell–cell adhesion molecule by connecting with cytoplasmic β‐catenin to form cadherin/catenin complexes,1 functions in epithelial differentiation.2 Decreased E‐cadherin expression and accompanying cell dissociation are frequently observed during the early stage of cancer cell development.3 Because E‐cadherin inhibits the invasion, or metastasis, of tumors and a deficiency in E‐cadherin decreases tumor differentiation, E‐cadherin is regarded as a tumor‐suppressive molecule.4 Understanding the control of E‐cadherin expression is important for understanding the development and carcinogenesis.5
β‐Catenin plays an important role in Wnt signaling, which is involved in malignant cancer development6 and mediates the connection between E‐cadherin and actin microfilaments.7 β‐Catenin is abundant in the cytoplasm and is degraded by the proteasome after phosphorylation by the Axin/glycogen synthase kinase 3β/adenomatous polyposis coli/casein kinase 1 destruction complex under normal conditions.8 Activation of the Wnt signaling pathway leads to disassembly of the β‐catenin destruction complex and translocation of β‐catenin from the cytoplasm to the nucleus, where it functions as a transcriptional activator for growth‐stimulating factors.9 Therefore, association of β‐catenin with E‐cadherin reduces the cytoplasmic β‐catenin level and negatively regulates the Wnt signaling pathway.10
Epithelial–mesenchymal transition (EMT) is a de‐differentiation program that occurs in the early stage of epithelial cancer and is frequently associated with reduced expression of E‐cadherin and dissociation of cell–cell junctions.11 Involvement of β‐catenin‐dependent Wnt signaling is the dynamic equivalent of EMT in colorectal cancer progression.12 Therefore, variations in the expression or structure of cadherin and catenin result in the impairment of adherens junctions (AJs), triggering of the EMT, and stimulation of cancer metastasis.11 However, E‐cadherin may function as more than a tumor suppressor in cancer development,4 and it may play an important role in the metastatic seeding of cancer cells and in the mesenchymal–epithelial transition (MET).13, 14 Many metastatic carcinomas are less de‐differentiated than the primary tumor from which they derived.15, 16 Membrane E‐cadherin staining in metastases but not in the primary cancer has also been reported.17 Therefore, the role of E‐cadherin in cancer metastasis is complex and requires further investigation.
Generally, to acquire ATP for energy, cancer cells preferentially use aerobic glycolysis instead of mitochondrial oxidative phosphorylation (OXPHOS). This glycolytic switching is frequently observed in the development of many malignant cancers.18, 19 For example, activation of Wnt signaling suppresses mitochondrial respiration by suppressing transcription of cytochrome c oxidase and inducing glycolytic switching in cancer cells.20 Although several studies have examined the association between E‐cadherin and cancer, few have investigated the association between E‐cadherin expression and cancer cell metabolism. Similar to its function in tumor development and progression, the function of E‐cadherin in energy metabolism in malignant cancer is also probably complex. Hypoxia‐inducible factor‐1α, which is a key molecule for glycolytic switching,21, 22 indirectly represses E‐cadherin expression and contributes to EMT.23 In contrast, E‐cadherin knockdown reduced glycolysis in inflammatory breast cancer cells, and E‐cadherin expression failed to induce hypoxia‐inducible factor‐1α expression in vivo in a xenograft model.24 These results indicate that E‐cadherin expression could play diverse roles in the energy metabolism of cancer cells.
The purpose of this study was to investigate the effect of E‐cadherin expression on the proliferation and energy metabolism of AGS gastric cancer cells with undetectable E‐cadherin expression and a β‐catenin mutation.
Materials and Methods
Cells, chemicals, and antibodies
AGS cell lines that were established from gastric cancer tissue were purchased from the Korean Cell Line Bank (Seoul, Korea) in 2003. Frozen aliquots of cells were thawed and tested for post‐freeze growth properties, morphology, and mycoplasma contamination prior to experiments. EC96 cells were derived from AGS cells after transfection of E‐cadherin cDNA, neomycin selection, and several rounds of single‐cell cloning. Establishment of EC96 cells was described previously.25 AGS and EC96 cells were maintained in DMEM supplemented with 10% FBS, penicillin, and streptomycin in a humidified atmosphere of 5% CO2.
Bay11‐7082 and triptolide were purchased from Invitrogen (Carlsbad, CA, USA) and MG132 from Calbiochem (San Diego, CA, USA). Specific antibodies for E‐cadherin and β‐catenin were obtained from BD Pharmingen (San Diego, CA, USA). Axin1, c‐myc, p‐IκBα, IκBα, nuclear factor‐κB (NF‐κB), Lamin A/C, GAPDH, and β‐actin were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA, USA).
Oxygen consumption rate (OCR), extracellular acidification rate (ECAR), and fuel flex assay
AGS and EC96 cells were plated at 20 000 cells/well in XF24 cell culture microplates (Seahorse Bioscience, North Billerica, MA, USA). Oxygen consumption rate (OCR) was measured on the following day using an XF24 analyzer (Agilent, Santa Clara, CA, USA). Cells were equilibrated for 1 h at 37°C in XF assay medium supplemented with 25 mM glucose, 4 mM L‐glutamine, and 1 mM sodium pyruvate (pH 7.4) before measurements. Oligomycin (2 μM), Carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP, 0.1 μM), antimycin A (1 μM), and rotenone (1 μM) were then consecutively added to each well. Extracellular acidification rate (ECAR) was measured on the day following culture using an XF24 analyzer. Cells were equilibrated for 1 h at 37°C in XF assay medium without supplements before measurement. Glucose (10 mM), oligomycin (2 μM), and 2‐deoxyglucose (2‐DG) (0.1 μM) were consecutively added to each well. Cells were equilibrated for 1 h at 37°C in XF assay medium supplemented with 25 mM glucose, 4 mM L‐glutamine, and 1 mM sodium pyruvate (pH 7.4) before measurements.
Mitochondrial fuel flex was measured using an XF24 analyzer. Cells were equilibrated for 1 h at 37°C in XF assay medium supplemented with 25 mM glucose, 4 mM L‐glutamine, and 1 mM sodium pyruvate (pH 7.4) before measurements. A mixture of 2 μM UK5099 (glucose oxidation pathway inhibitor), 4 μM etomoxir (fatty acid oxidation inhibitor), and 3 μM Bis‐2‐(5‐phenylacetamido‐1,3,4‐thiadiazol‐2‐yl)ethyl sulfide (BPTES, glutamine oxidation pathway inhibitor) was then added to each well.
Transient transfection with siRNAs and stable knockdown cell line generation
To produce lentivirus expressing shRNA for pLKO‐shE‐cadherin or pLKO‐empty plasmids were co‐transfected with the lentivirus packaging plasmids (psPAX2, pMD2G, and VSV‐G) into 293T cells. The virus containing cell culture supernatant was harvested, filtered through a 0.22‐μm pore‐size filter and used to infect EC96 cells. To generate stable E‐cadherin knockdown cells, infected cells were selected by puromycin (2 μg/mL) for 1 month.
Specific siRNAs, targeting NF‐κB, and non‐specific control siRNAs were purchased from Santa Cruz Biotechnologies. For transient transfection, cells were seeded at a density of 5 × 104 cells/mL in antibiotic‐free medium, and siRNAs were transfected using the transfection reagent (Santa Cruz Biotechnology), according to the manufacturer's instructions. After incubation for 72 h, the cells were analyzed using BrdU or immunoblot assay.
Wnt reporter assay
Cells were seeded in a 12‐well plate and transfected in triplicate with the following plasmids: SuperTOP‐flash (500 ng) and pTK‐Renilla luciferase (50 ng). The transfected cells were assayed for luciferase activity using a GloMax‐multi Jr and the Dual Luciferase Reporter assay system (Promega, Madison, WI, USA) according to the manufacturer's protocols. Renilla luciferase activity was used to normalize TOP‐FLASH activity for transfection efficiency.
Assays and measurement of reactive oxygen species
The MTT, BrdU incorporation, and ATP assays were carried out using an MTT cell proliferation kit (Sigma‐Aldrich, St. Louis, MO, USA), BrdU cell Proliferation ELISA kit (Roche Applied Science, Indianapolis, IN, USA) and Luminescence ATP Detection Assay System (ATPlite; PerkinElmer, Waltham, MA, USA) according to the manufacturer's instructions, respectively. For reactive oxygen species (ROS) measurement, cells seeded on 96‐well microplates (104 cells/well) were incubated for 24 h, treated with 10 μM 2′,7′‐dichlorofluorescein diacetate (DCFH‐DA) (Sigma‐Aldrich) for 10 min at 37°C and then the fluorescence intensity was assessed by a multimode plate reader (EnSpire; PerkinElmer).
Detection of ROS, immunofluorescence, immunoblotting, and immunoprecipitation
Cells were treated with DCFH‐DA for 10 min, fixed with 4% para‐formaldehyde for 20 min, and washed with PBS, and the fluorescence intensity was assessed by a fluorescence microscope (Axioscope, Zeiss, Oberkochen, Germany). Immunofluorescence, immunoblot, and immunoprecipitation analyses were carried out as described previously.26
Quantitative RT‐PCR
Total RNA was isolated, and 1 μg was used for cDNA. The cDNAs were mixed with Power SYBR Green Master Mix and amplified on an ABI7500 (Applied Biosystems, Foster City, CA, USA). Primers used for the quantitative RT‐PCR of glucose transporter 1 (GLUT1), GLUT3, GLUT4, c‐myc, Axin2, cyclin D1, and β‐actin are listed in Table S1.
Statistical analysis
Student's t‐tests were applied for comparisons. Data are expressed as mean ± SD. The significance threshold was 5% (*P < 0.05; **P < 0.01; ***P < 0.001). All experiments were repeated three times.
Results
Effect of E‐cadherin expression on AGS cells
AGS cells showed E‐cadherin expression at undetectable levels and prominent nuclear β‐catenin expression (Fig. 1a,b), suggesting activation of Wnt signaling.27, 28 To determine the effects of restoration of E‐cadherin expression, we established AGS cells expressing high levels of E‐cadherin and tested five clones for E‐cadherin expression, cellular proliferation, and ATP production.25 Among those, AGS E‐cadherin1 was selected and named EC96 as this cell line showed clear increases in cell proliferation and ATP production than control parental AGS cells (Fig. S1). As expected, Wnt activity and nuclear β‐catenin were decreased in EC96 cells in comparison to AGS cells (Fig. 1c,d). In EC96 cells, β‐catenin expression was clearly observed at the cell–cell junctions, suggesting translocation of β‐catenin from the nucleus to the plasma membrane (Fig. 1d). β‐Catenin was immunoprecipitated from EC96 cells in association with E‐cadherin (Fig. 1e).
Figure 1.
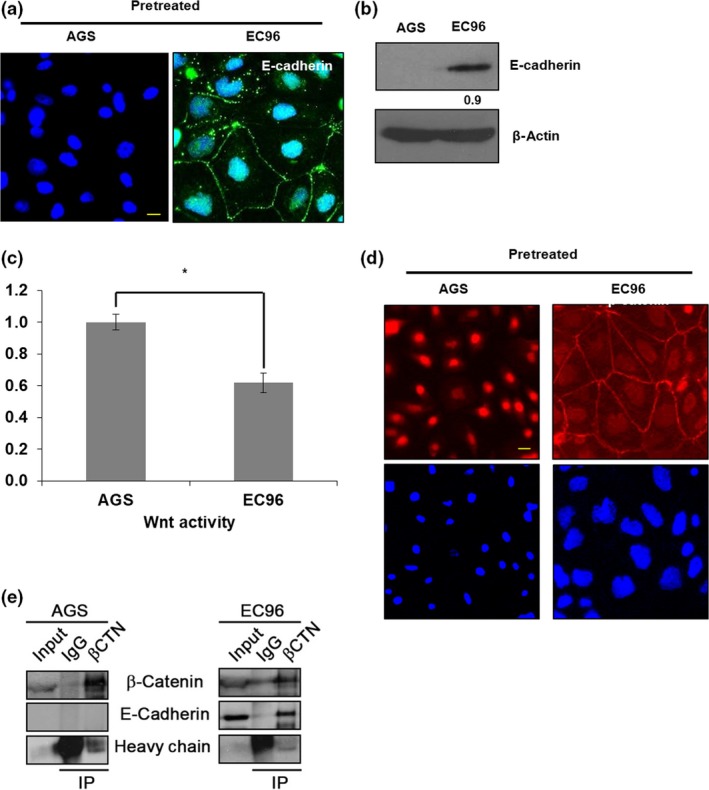
E‐cadherin expression suppresses Wnt signaling in AGS gastric cancer cells. (a) AGS and EC96 cells were subjected to immunofluorescence staining for E‐cadherin. Scale bar = 30 μm. (b) Cell lysates were subjected to immunoblotting analysis of E‐cadherin and β‐actin. Band intensity was normalized to β‐actin. (c) Wnt signaling activities were measured by dual luciferase reporter assay. *P < 0.05. (d) Cells were subjected to immunofluorescence staining for β‐catenin. Scale bar = 30 μm. (e) Cell lysates were precipitated using an anti‐β‐catenin antibody, and the levels of E‐cadherin and antibody‐bound proteins were determined by immunoblot analysis using anti‐β‐catenin (βCTN) and anti‐E‐cadherin antibodies. IP, immunoprecipitant.
E‐Cadherin expression stimulates cellular proliferation through NF‐κB activation in AGS cells
β‐Catenin is associated with E‐cadherin at cell–cell adhesion junctions, which suppresses Wnt signaling activity and cell growth by reducing the cytoplasmic β‐catenin concentration.10 Unexpectedly, proliferation of EC96 cells was approximately 1.5‐fold higher than that of AGS cells (Fig. 2a,b). The increased proliferation of EC96 cells was not due to residual Wnt signaling activity because Wnt inhibitors (such as ICG‐001, inhibitor of beta‐catenin‐response transcription [iCRT], and NSC668036) exerted limited effects on EC96 cell proliferation (Fig. S2). EC96 cells were tolerant of inhibitors of other signaling pathways, with the exception of NF‐κB inhibitors. Treatment with NF‐κB inhibitors, such as MG132, triptolide, and Bay11‐7082, resulted in greater inhibition of the proliferation of EC96 cells in comparison to AGS cells (Fig. S2). Further analysis revealed that the proliferation of EC96 cells was inhibited by the NF‐κB inhibitors in a dose‐dependent manner (Fig. 2c). Immunoblot analysis showed an increase of c‐myc expression in accordance with the increase of the phosphorylation of IκBα in EC96 cells (Fig. 2d). Immunofluorescence staining and subcellular fractionation also revealed the nuclear translocation of NF‐κB (Fig. 2e,f), suggesting the activation of NF‐κB. Furthermore, treatment with inhibitors of NF‐κB signaling, such as Bay11‐7082 and triptolide, resulted in a decrease in the c‐myc level in EC96 cells (Fig. 2g). Both AGS and EC96 cells showed reduced expression of c‐myc in the case of MG132. Excess MG132 is cytotoxic, as it inhibits both the proteasome and multiple signaling pathways.29, 30 To determine whether restoration of E‐cadherin expression enhances c‐myc expression levels, AGS cells were transiently transfected with E‐cadherin cDNA; increased c‐myc expression was detected (Fig. S3). Considering that expression of c‐myc is also a target of Wnt signaling and that Wnt signaling activity is reduced in EC96 cells, the expression of other targets of Wnt signaling was analyzed by real‐time PCR; cyclin D1 and Axin2 mRNA levels were not increased (Fig. 2h). These results suggested that reduction of Wnt signaling activity downregulated cyclin D1 and Axin2, but activation of NF‐κB signaling sustained c‐myc expression in EC96 cells, resulting in an increase in cell proliferation.
Figure 2.
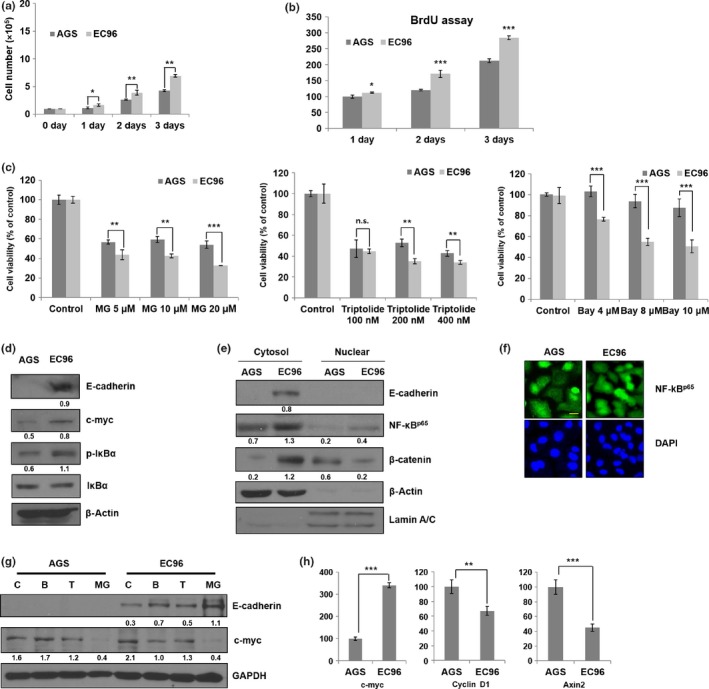
E‐cadherin expression enhances cell proliferation through the nuclear factor‐κB (NF‐κB) signaling pathway. (a) Gastric cancer cells were cultured for 24, 48, and 72 h and subjected to direct cell counting by Trypan blue dye exclusion staining. (b) Proliferation of AGS and EC96 cells over 24, 48 and 72 h subjected to BrdU assay. (c) Cells were treated with MG132 (5–20 μM), triptolide (100–400 nM), and Bay11‐7082 (4–10 μM) for 24 h, and cell viability was measured by MTT assay. (d) Cell lysates were subjected to immunoblot analysis for E‐cadherin, c‐myc, p‐IκBα, IκBα, and β‐actin. E‐cadherin and c‐myc band intensities were normalized to β‐actin and p‐IκBα band intensity was normalized to IκBα. (e) Cells were fractionated into cytosol and nucleus and then subjected to immunoblot analysis using the indicated antibodies. Cytosol protein band intensity was normalized to β‐actin and nuclear protein band intensity was normalized to Lamin A/C. (f) Cells were subjected to immunofluorescence staining for NF‐κB. Scale bar = 30 μm. (g) Total RNA extracted from AGS and EC96 cells subjected to RT‐PCR for c‐myc, cyclin D1, and Axin2 using the indicated gene‐specific primers. Averages of three independent experiments with error bars are presented. (h) Cells were treated with triptolide (200 nM), Bay11‐7082 (4 μM), and MG132 (5 μM) for 24 h and then subjected to immunoblot analysis for E‐cadherin, c‐myc, and GAPDH. Band intensity was normalized to GAPDH. *P < 0.05; **P < 0.01; ***P < 0.001. n.s., not significant.
E‐cadherin expression in AGS cells stimulates cellular energy metabolism
Because E‐cadherin expression increases proliferation of AGS cells, we explored the effects of E‐cadherin expression on the energy‐producing metabolism. EC96 cells showed a higher ATP concentration than AGS cells (Fig. 3a). To evaluate fuel utilization for ATP production by AGS and EC96 cells, fuel flex assays were carried out. Fuel dependency refers to the energy dependency of a target fuel throughout energy metabolism, and fuel capacity refers to the maximum energy utility of a particular energy source when other energy pathways are inhibited. Fuel flexibility is the ability to use another energy source when the preferred energy source is inhibited. Fuel flex assays showed that glucose capacity was high but dependency was low in AGS cells. Also, dependency on glutamine was high in AGS cells. However, glucose dependency was increased in EC96 cells, and glutamine dependency was decreased (Fig. 3b). These results show that EC96 cells were more dependent on glucose than were AGS cells, whereas AGS cells were more dependent on glutamine, indicating that the two cell lines use different energy sources.
Figure 3.
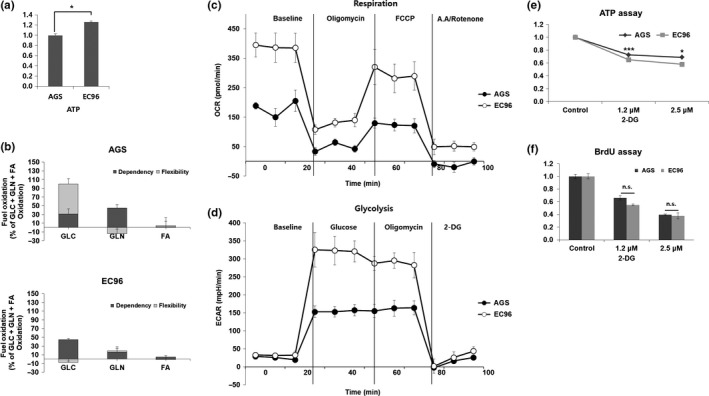
E‐cadherin expression enhances mitochondrial oxidative phosphorylation and glycolysis. (a,b) Gastric cancer cells were subjected to cellular ATP measurement (a) and fuel flex measurement (b). Fuel flex responses to 2 μM UK5099 (glucose oxidation pathway inhibitor), 4 μM etomoxir (fatty acid oxidation inhibitor), and 3 μM is‐2‐(5‐phenylacetamido‐1,3,4‐thiadiazol‐2‐yl)ethyl sulfideChange to Bis‐2‐(5‐phenylacetamido‐1,3,4‐thiadiazol‐2‐yl)ethyl sulfide (BPTES, glutamine oxidation pathway inhibitor) were measured. (c) Cells were plated in XF24 culture plates for 24 h using medium containing glucose, glutamine, and pyruvate. Oxygen consumption rate responses to oligomycin (2 μM), Carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazoneChange to Carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP, 0.1 μM), antimycin A (1 μM), and rotenone (1 μM) were measured. (d) Cells were plated in XF24 culture plates using nutrient‐free base medium. (e,f) Cells were subjected to cellular ATP measurement (e) and BrdU incorporation (f) by treatment with 2‐deoxyglucose (2‐DG; 0.1 μM) for 24 h. *P < 0.05; ***P < 0.001. n.s., not significant.
Glycolysis plays an important role in providing energy, and ATP production is mediated by oxygen‐independent glycolysis and oxygen‐dependent respiration, also known as mitochondrial OXPHOS. Therefore, we carried out cellular OCR and ECAR, which measure OXPHOS and glycolysis, respectively, using a Seahorse XF24 analyzer. Following growth in the presence of glucose, glutamine, and sodium pyruvate, the OCR of EC96 cells was higher than that of AGS cells, and the oligomycin‐induced reduction in the OCR was more prominent in EC96 cells than in AGS cells (Fig. 3c), suggesting greater utilization of OXPHOS. Under nutrient‐depleted conditions, the baseline OCR was also increased in EC96 cells compared to AGS cells (Fig. S4). The OCR decreased markedly following addition of glucose to EC96 cells, whereas AGS cells showed no such effect (Fig. S4). Under the same conditions, there was no difference in the basal ECAR of AGS and EC96 cells, but glucose addition substantially increased the ECAR in EC96 cells (Fig. 3d). These results suggested that EC96 cells generate energy using OXPHOS; however, the importance of glycolysis for energy production is increased under glucose‐rich conditions. Next, ATP and BrdU assays were undertaken to determine whether energy production through glucose metabolism is correlated with cellular energy production in EC96 cells. After treatment with 2‐DG, a glucose derivative, energy production was decreased, in a dose‐dependent manner, in EC96 cells compared to AGS cells (Fig. 3e). The BrdU assay indicated that 2‐DG treatment reduced the proliferation of EC96 cells (Fig. 3f). Therefore, E‐cadherin expression increased cell proliferation by enhancing glycolysis and OXPHOS.
E‐cadherin expression activates glucose metabolism through GLUT1 expression in AGS cells
We speculated that increased glucose metabolism would be associated with activation of a glucose transporter in EC96 cells because the expression of some glucose transporters is regulated by NF‐κB and c‐myc signaling.31, 32 Therefore, we measured the mRNA levels of the GLUT1, 3, and 4 glucose transporters, which have been reported to be expressed in cancers.33 The GLUT1, 3, and 4 mRNA levels were higher in EC96 cells, and that of GLUT1 showed the greatest increase (Fig. 4a). Also, triptolide treatment showed a decrease of GLUT1 expression in EC96 cells (Fig. 4b), suggesting that NF‐κB was involved in GLUT1 expression. To confirm the involvement of NF‐κB, knockdown experiments for NF‐κB were carried out. The results showed decreases in EC96 cell proliferation (Fig. 5a) and GLUT1 expression (Fig. 5b), consistent with the result of NF‐κB inhibitors (Figs. 2c,4b). Next, the OCR of EC96 cells was measured after knockdown of E‐cadherin using an E‐cadherin shRNA vector. The OCR increased in EC96 cells, but the OCR in E‐cadherin knockdown cells was similar to that in AGS cells (Fig. 6a). The OCR in cells transfected with the control vector was similar to that in non‐transfected EC96 cells. These results indicate that the measured differences in OCR were due to the E‐cadherin expression level (Fig. 6a). Furthermore, the expression levels of c‐myc and GLUT1 were substantially decreased in E‐cadherin knockdown cells (Fig. 6b). These results suggested that E‐cadherin expression led to increased expression of GLUT1 through NF‐κB signaling, which resulted in increased ATP production by increasing glucose uptake and metabolism (OXPHOS and glycolysis). These events enhance cell proliferation through NF‐κB signaling, which compensates for the E‐cadherin‐mediated suppression of Wnt signaling.
Figure 4.
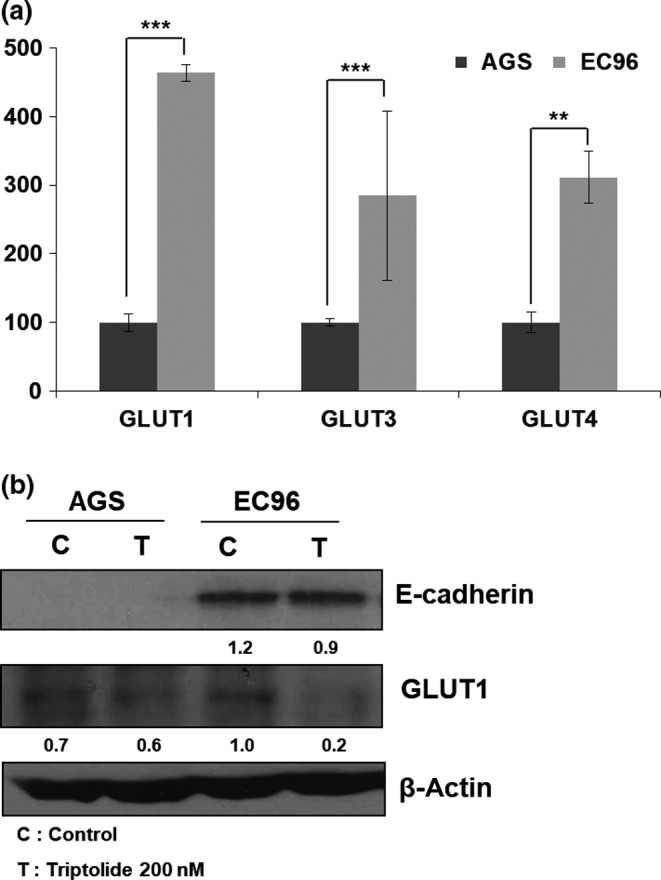
E‐cadherin expression enhances glucose transporter (GLUT) expression. (a) Total RNA extracted from AGS and EC96 gastric cancer cells were subjected to RT‐PCR with GLUT1‐, GLUT3‐, and GLUT4‐specific primers. Averages of three independent experiments with error bars are presented. *P < 0.05; **P < 0.01; ***P < 0.001. (b) Cells were treated with triptolide (200 nM) for 24 h, and the lysates were then subjected to immunoblot analysis for E‐cadherin, GLUT1, and β‐actin. Band intensity was normalized to β‐actin.
Figure 5.
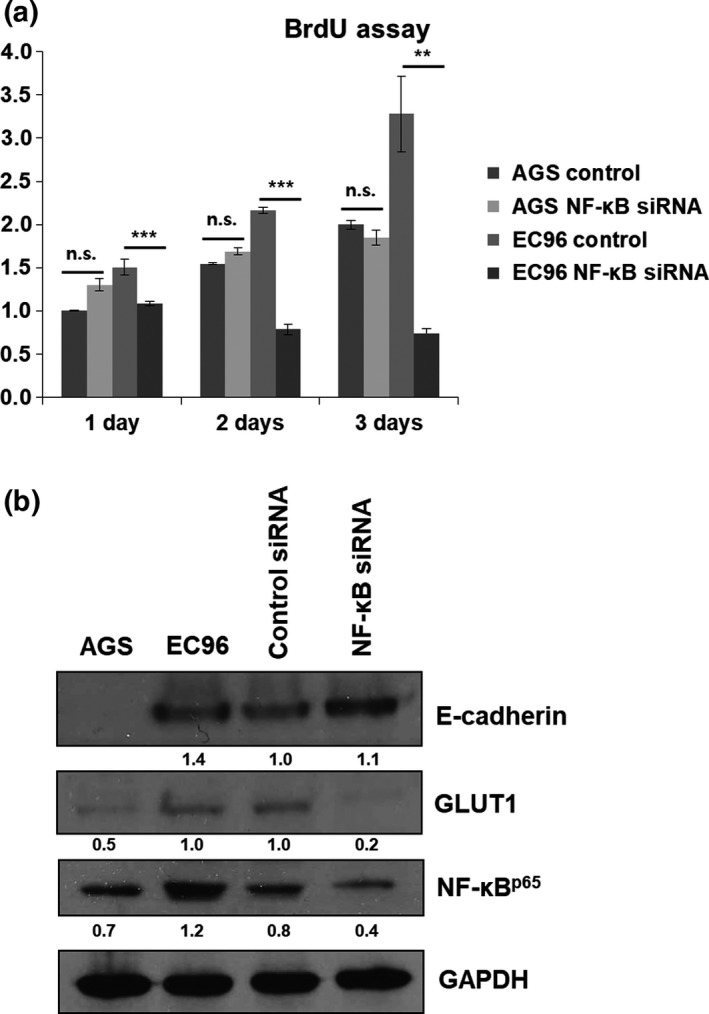
Nuclear factor‐κB (NF‐κB) knockdown reduces cell proliferation and glucose transporter 1 (GLUT1) expression in EC96 gastric cancer cells. (a) AGS and EC96 cells were transfected by scrambled (control) or NF‐κB siRNA and subjected to BrdU incorporation assay. **P < 0.01; ***P < 0.001. n.s., not significant. (b) AGS, EC96, EC96‐control siRNA, and EC96‐NF‐κB siRNA cells were subjected to immunoblot analysis for E‐cadherin, GLUT1, NF‐κB, and β‐actin. Band intensity was normalized to GAPDH.
Figure 6.
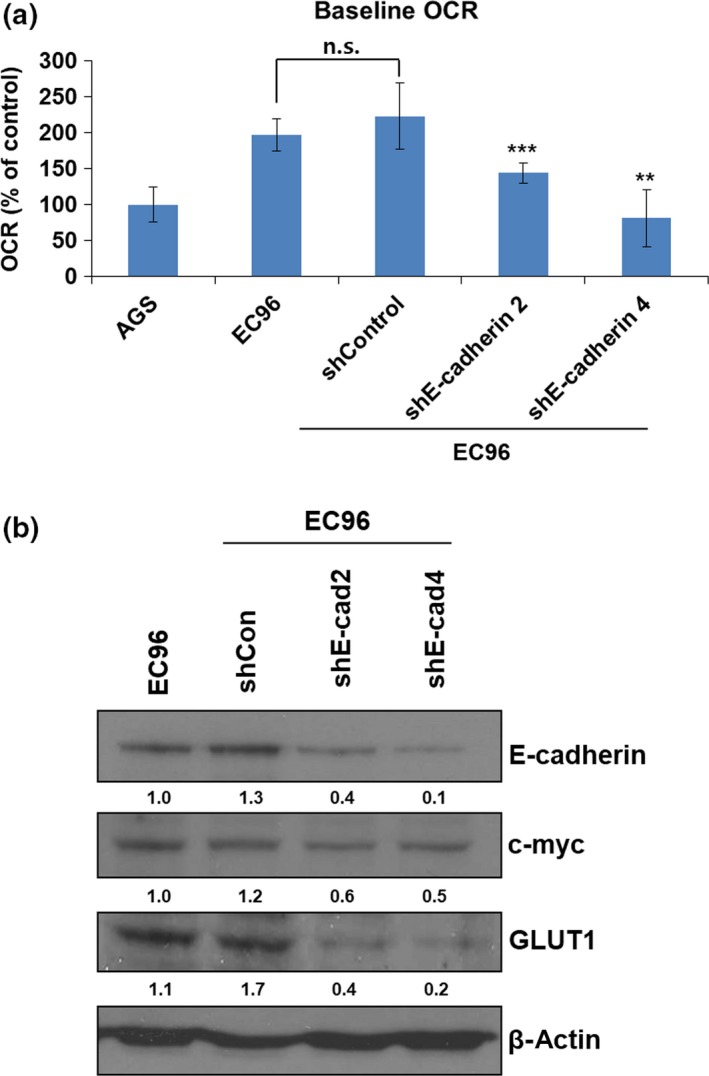
E‐cadherin knockdown reduces mitochondrial oxidative phosphorylation and glucose transporter 1 (GLUT1) expression in EC96 gastric cancer cells. (a) EC96‐, and shE‐cadherin transfected EC96 cells were seeded at 20 000 cells/well in XF24 culture plates for 24 h. The oxygen consumption rate (OCR) response to assay medium including glucose, glutamine, and sodium pyruvate. **P < 0.01; ***P < 0.001. n.s., not significant. (b) EC96‐ and sh‐Control and E‐cadherin‐knockdown EC96 cell lysates were subjected to immunoblot analysis for E‐cadherin, c‐myc, GLUT1, and β‐actin. Band intensity was normalized to β‐actin.
Discussion
E‐cadherin mediates epithelial cell–cell adhesion to form AJs. Together with tight junctions, AJs limit the access of growth factors to the basolateral spaces and breakage of cell–cell adhesion to allow the basolateral surface of cells to access growth factor and enhance the proliferation and migration of epithelial cells.33 Therefore, E‐cadherin plays antiproliferative and antitumorigenic roles, and downregulation of E‐cadherin expression at the cell surface is regarded as a hallmark of carcinoma EMT and tumor cell mesenchymal invasion in the initial step of cancer metastasis.1, 2, 3 Re‐expression of E‐cadherin occurs following the metastasis of breast carcinoma cells to the liver, lung, and brain, suggesting an important role in the metastatic seeding of disseminated carcinomas.34, 35 In this study, to investigate the role of E‐cadherin in cancer EMT, E‐cadherin was ectopically expressed in malignant gastric cancer AGS cells to know phenotypic changes. For this purpose, AGS cells appeared appropriate because of elevated Wnt signaling activity due to a mutation in β‐catenin and low level of E‐cadherin expression (Fig. 1).
Initially, we speculated that E‐cadherin expression reduces Wnt signaling activity and cell proliferation. Although Wnt signaling activity was decreased with reduction of nuclear β‐catenin (Fig. 1) and cell proliferation was enhanced (Fig. 2a,b), c‐myc expression was increased in both EC96 cells and transiently E‐cadherin‐expressing AGS cells compared to AGS cells. These results led us to speculate that the c‐myc targeting signal pathway might be induced by E‐cadherin expression, leading to an increase in cell proliferation. The proliferation and c‐myc expression of EC96 cells were suppressed by treatment with NF‐κB inhibitors (Fig. 2c,h). Nuclear factor‐κB is present in the cytoplasm bound to IκB, which inhibits the nuclear translocation of NF‐κB. However, phosphorylation of IκB by IKK releases NF‐κB, which then translocates to the nucleus and activates transcription of various genes, including c‐myc, which is involved in cellular proliferation, movement, and survival.32 Consistent with this paradigm, IκB phosphorylation was increased and intranuclear NF‐κB movement was stimulated in EC96 cells. Therefore, our results suggested that E‐cadherin expression stimulates extranuclear β‐catenin translocation, resulting in the inhibition of Wnt signaling and the compensatory stimulation of NF‐κB nuclear translocation. β‐Catenin can form complexes with NF‐κB, which represses the transcriptional activity of NF‐κB against target genes (such as FAS and TRAF1) in colon and breast cancer.36 Generally, E‐cadherin binding elicits canonical survival signals, and MAPK and Akt signaling have been investigated extensively in this regard.37, 38, 39 In this study, inhibitors of MAPK and Akt/mTOR were less effective against EC96 than AGS cells (Fig. S2).
Cancer cells are different from most normal cells in terms of energy metabolism. Although mitochondrial OXPHOS is a highly efficient energy production system, many cancer cells metabolize glucose by aerobic glycolysis. To support their high rates of proliferation, cancer cells avidly take up glucose for aerobic glycolysis and/or metabolize additional nutrients, such as glutamine.18, 19 Considering that downregulation of E‐cadherin and cell dissociation is an initial event for cancer development,4 re‐expression of E‐cadherin in AGS cells is expected to revert carcinoma EMT and release suppression of OXPHOS. Our results revealed that E‐cadherin expression enhanced ATP production and an increased utility of OXPHOS (Fig. 3c). It was observed that Wnt signaling suppresses mitochondrial respiration and induces glycolytic switching.20 Also, one of the β‐catenin destruction complexes, Axin1, is present in the mitochondria of HeLa cells, where it influences the activity of mitochondria complex IV and inhibits mitochondrial ATP synthesis.40 In this study, Axin1 was expressed in the mitochondria of AGS cells and, at a lower level, in the mitochondria of EC96 cells (Fig. S5A). Furthermore, β‐catenin and E‐cadherin binding to Axin1 was detected by immunoprecipitation in EC96 cells (Fig. S4B). These observations suggest that suppression of OXPHOS, which is frequently observed in cancer EMT, is reduced by E‐cadherin expression.
E‐cadherin expression also enhanced glycolysis (Fig. 3d). Malignant cancer cells tend to show a higher rate of glycolysis and glucose absorption; these processes are controlled by glucose transport proteins, such as GLUT.41 Glucose transporter 1 is overexpressed in many malignant tumors42, 43 and a high level of GLUT1 expression was suggested as a predictor of a poor prognosis.44 The mRNA levels of GLUT1, GLUT3, and GLUT4 increased, and the increase in that of GLUT1 was most pronounced (Fig. 4a). Oxidative stress is involved in controlling glucose transporter expression, and GLUT1 mRNA levels increase with increasing oxidative stress.45 Myc expression is associated with glycolysis and GLUT1 expression.31 In this study, E‐cadherin knockout and inhibition of NF‐κB signaling reduced c‐myc and GLUT1 expression in EC96 cells (Figs. 4, 5), suggesting that E‐cadherin expression enhances cancer‐related metabolism. In addition, EC96 cells showed increases of cellular ROS production (Fig. S6). Reactive oxygen species, primarily produced by mitochondrial OXPHOS, are involved in various cellular signaling pathways and, at proper concentrations, activate transcription factors, such as NF‐κB or AP‐1, leading to cellular proliferation.46 Therefore, it is possible that E‐cadherin expression led to ROS production, resulting in NF‐κB signaling.
Additionally, myc expression is involved in glutamine consumption.47 EC96 cells showed decreased dependence on glutamine as an energy source, suggesting that glutamine use and GLUT1 expression are regulated by different mechanisms. E‐cadherin is involved in cancer‐associated MET at distant metastatic sites. E‐cadherin re‐expression and MET occur during metastatic seeding, and these events are influenced by various cancer‐associated cells in metastatic microenvironments.35 After excluding the influence of various factors present in the tumor microenvironment, we investigated the effects of E‐cadherin re‐expression on cancer cells before metastatic seeding. Our results showed that E‐cadherin re‐expression induces the reverse of the EMT (as evidenced by enhanced OXPHOS), and also enhances the malignant phenotype (as shown by increased glucose transport and glycolysis), suggesting the requirement of the cancer‐associated microenvironment for further cell fate determination to reverse EMT or MET.
In conclusion, this study showed that E‐cadherin re‐expression increases ATP production by glycolysis and mitochondrial OXPHOS, which stimulates cellular proliferation. These cellular changes may be involved through the activation of NF‐κB, increase of GLUT1 expression, and intracellular glucose absorption, sequentially.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Cell proliferation and ATP production is increased by E‐cadherin expression in AGS gastric cancer cells. (A) AGS and E‐cadherin expressed AGS cell lysates subjected to immunoblotting analysis using E‐cadherin and β‐actin. AGS and E‐cadherin‐expressing AGS cells were subjected to cell proliferation (B) and cellular ATP (C) measurement. *P < 0.05; **P < 0.01; ***P < 0.001. n.s., not significant.
Fig. S2. Effects of various small molecule inhibitors of signaling pathways on AGS and EC96 gastric cancer cell proliferation. Cell viability was measured by MTT assay after treatment with the following inhibitors: ICG‐001 (10 μM), inhibitor of beta‐catenin‐response transcription (iCRT) (25 μM), NSC668036 (10 μM) as Wnt signaling inhibitors; thiadiazolidinones (TDZD, 10 μM) as glycogen synthase kinase 3β inhibitor; LY294002 (25 μM) as PI3K/Akt inhibitor; U0126 (25 μM), PD90859 (2.5 μM) as MAPK inhibitors; A23187 (5 μM) as Ca2+ ionophore; rapamycin (20 μM) as mTOR inhibitor; cryptotanshinone (2.5 μM) as Stat3 inhibitor; SP600125 (5 μM) as JNK inhibitor; and MG132 (5 μM), triptolide (200 nM), and Bay11‐7082 (8 μM) as nuclear factor‐κB inhibitors. *P < 0.05; **P < 0.01; ***P < 0.001.
Fig. S3. Effect of E‐cadherin expression on c‐myc expression. Lysates from AGS‐, EC96‐, and E‐cadherin‐transfected cells were subjected to immunoblotting analysis for E‐cadherin, nuclear factor‐κB (NF‐κB), c‐myc, survivin, and GAPDH. Band intensity was normalized to GAPDH.
Fig. S4. E‐cadherin increased basal oxygen consumption rate (OCR) levels. Cells were incubated on XF24 culture plates for 24 h using substrate‐free base medium. The kinetic OCR responses of AGS and EC96 cells to glucose (10 mM) oligomycin (2 μM), and 2‐deoxyglucose (2‐DG; 0.1 μM) were measured.
Fig. S5. Analysis of Axin expression in mitochondria. (A) Cells were fractionated into cytosol and mitochondria and subjected to immunoblot analysis for the indicated proteins. (B) Cells were cultured for 24 h, and proteins were immunoprecipitated using an anti‐Axin1 antibody and subjected to immunoblot analysis for Axin1, E‐cadherin, and β‐catenin.
Fig. S6. E‐cadherin increased cellular reactive oxygen species levels. (A) Cells were incubated with 10 μM 2′,7′‐dichloro‐dihydro‐fluorescein diacetate (DCFH‐DA), and the fluorescence intensity was assessed by a luminometer. (B) Cells were incubated with 10 μM 2′,7′‐dichloro‐dihydro‐fluorescein diacetate (DCFH‐DA), and the fluorescence intensity was assessed by a fluorescence microscope.
Table S1. Primer sequences for quantitative RT‐PCR.
Acknowledgement
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF‐2015R1D1A1A09056775).
Cancer Sci 108 (2017) 1769–1777
Funding Information
National Research Foundation of Korea, (Grant/Award Number: ‘NRF‐2015R1D1A1A09056775′)
References
- 1. Debruyne P, Vermeulen S, Mareel M. The role of the E‐cadherin/catenin complex in gastrointestinal cancer. Acta Gastroenterol Belg 1999; 62: 393–402. [PubMed] [Google Scholar]
- 2. Berx G, Strumane K, Comijn J et al E‐cadherin controlling epithelial differentiation in human carcinomas. Nat Genet 2001; 27: 43. [Google Scholar]
- 3. Pecina‐Slaus N. Tumor suppressor gene E‐cadherin and its role in normal and malignant cells. Cancer Cell Int 2003; 3(1): 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene 2008; 27: 6920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jaggi M, Johansson SL, Baker JJ et al Aberrant expression of E‐cadherin and beta‐catenin in human prostate cancer. Urol Oncol 2005; 23: 402–6. [DOI] [PubMed] [Google Scholar]
- 6. Oloumi A, McPhee T, Dedhar S. Regulation of E‐cadherin expression and beta‐catenin/Tcf transcriptional activity by the integrin‐linked kinase. Biochem Biophys Acta 2004; 1691(1): 1–15. [DOI] [PubMed] [Google Scholar]
- 7. Hong S, Troyanovsky RB, Troyanovsky SM. Binding to F‐actin guides cadherin cluster assembly, stability, and movement. J Cell Biol 2013; 201(1): 131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stamos JL, Weis WI. The beta‐catenin destruction complex. Cold Spring Harb Perspect Biol 2013; 5(1): a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. MacDonald BT, Tamai K, He X. Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17(1): 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Su YJ, Chang YW, Lin WH et al An aberrant nuclear localization of E‐cadherin is a potent inhibitor of Wnt/beta‐catenin‐elicited promotion of the cancer stem cell phenotype. Oncogenesis 2015; 4: e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15: 178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Basu S, Haase G, Ben‐Ze'ev A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Research 2016: doi: 10.12688/f1000research.7579.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Auersperg N, Pan J, Grove BD et al E‐cadherin induces mesenchymal‐to‐epithelial transition in human ovarian surface epithelium. Proc Natl Acad Sci USA 1999; 96: 6249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yao D, Dai C, Peng S. Mechanism of the mesenchymal‐epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res 2011; 9(12): 1608–20. [DOI] [PubMed] [Google Scholar]
- 15. Kowalski PJ, Rubin MA, Kleer CG. E‐cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res 2003; 5: R217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yates CC, Shepard CR, Stolz DB et al Co‐culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E‐cadherin. Br J Cancer 2007; 96: 1246–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Imai T, Horiuchi A, Shiozawa T et al Elevated expression of E‐cadherin and alpha‐, beta‐, and gamma‐catenins in metastatic lesions compared with primary epithelial ovarian carcinomas. Hum Pathol 2004; 35: 1469–76. [DOI] [PubMed] [Google Scholar]
- 18. Zheng J. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation (Review). Oncol Lett 2012; 4: 1151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dang CV. Links between metabolism and cancer. Genes Dev 2012; 26: 877–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee SY, Jeon HM, Ju MK et al Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Can Res 2012; 72: 3607–17. [DOI] [PubMed] [Google Scholar]
- 21. Lu H, Forbes RA, Verma A. Hypoxia‐inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem 2002; 277: 23111–5. [DOI] [PubMed] [Google Scholar]
- 22. Zhou W, Choi M, Margineantu D et al HIF1alpha induced switch from bivalent to exclusively glycolytic metabolism during ESC‐to‐EpiSC/hESC transition. EMBO J 2012; 31: 2103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krishnamachary B, Zagzag D, Nagasawa H et al Hypoxia‐inducible factor‐1‐dependent repression of E‐cadherin in von Hippel‐Lindau tumor suppressor‐null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Can Res 2006; 66: 2725–31. [DOI] [PubMed] [Google Scholar]
- 24. Chu K, Boley KM, Moraes R et al The paradox of E‐cadherin: role in response to hypoxia in the tumor microenvironment and regulation of energy metabolism. Oncotarget 2013; 4: 446–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim SM, Kim R, Ryu JH et al Multinuclear giant cell formation is enhanced by down‐regulation of Wnt signaling in gastric cancer cell line, AGS. Exp Cell Res 2005; 308(1): 18–28. [DOI] [PubMed] [Google Scholar]
- 26. Shin JH, Kim HW, Rhyu IJ et al Axin expression reduces staurosporine‐induced mitochondria‐mediated cell death in HeLa cells. Exp Cell Res 2012; 318: 2022–33. [DOI] [PubMed] [Google Scholar]
- 27. Mao J, Fan S, Ma W et al Roles of Wnt/beta‐catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis 2014; 5: e1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Busche S, Kremmer E, Posern G. E‐cadherin regulates MAL‐SRF‐mediated transcription in epithelial cells. J Cell Sci 2010; 123(Pt 16): 2803–9. [DOI] [PubMed] [Google Scholar]
- 29. Fujita T, Washio K, Takabatake D et al Proteasome inhibitors can alter the signaling pathways and attenuate the P‐glycoprotein‐mediated multidrug resistance. Int J Cancer 2005; 117: 670–82. [DOI] [PubMed] [Google Scholar]
- 30. Yuan BZ, Chapman JA, Reynolds SH. Proteasome Inhibitor MG132 Induces Apoptosis and Inhibits Invasion of Human Malignant Pleural Mesothelioma Cells. Transl Oncol 2008; 1(3): 129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Osthus RC, Shim H, Kim S et al Deregulation of glucose transporter 1 and glycolytic gene expression by c‐Myc. J Biol Chem 2000; 275: 21797–800. [DOI] [PubMed] [Google Scholar]
- 32. Sommermann TG, O'Neill K, Plas DR et al IKKbeta and NF‐kappaB transcription govern lymphoma cell survival through AKT‐induced plasma membrane trafficking of GLUT1. Can Res 2011; 71: 7291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harris TJ, Peifer M. Adherens junction‐dependent and ‐independent steps in the establishment of epithelial cell polarity in Drosophila. J Cell Biol 2004; 167(1): 135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chao YL, Shepard CR, Wells A. Breast carcinoma cells re‐express E‐cadherin during mesenchymal to epithelial reverting transition. Mol Cancer 2010; 9: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wells A, Yates C, Shepard CR. E‐cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin Exp Metas 2008; 25: 621–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Deng J, Miller SA, Wang HY et al Beta‐catenin interacts with and inhibits NF‐kappa B in human colon and breast cancer. Cancer Cell 2002; 2: 323–34. [DOI] [PubMed] [Google Scholar]
- 37. Conacci‐Sorrell M, Simcha I, Ben‐Yedidia T et al Autoregulation of E‐cadherin expression by cadherin‐cadherin interactions: the roles of beta‐catenin signaling, Slug, and MAPK. J Cell Biol 2003; 163: 847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang HG, Jenabi JM, Zhang J et al E‐cadherin cell‐cell adhesion in ewing tumor cells mediates suppression of anoikis through activation of the ErbB4 tyrosine kinase. Can Res 2007; 67: 3094–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reddy P, Liu L, Ren C et al Formation of E‐cadherin‐mediated cell‐cell adhesion activates AKT and mitogen activated protein kinase via phosphatidylinositol 3 kinase and ligand‐independent activation of epidermal growth factor receptor in ovarian cancer cells. Mol Endocrinol (Baltimore, Md) 2005; 19: 2564–78. [DOI] [PubMed] [Google Scholar]
- 40. Shin JH, Kim HW, Rhyu IJ et al Axin is expressed in mitochondria and suppresses mitochondrial ATP synthesis in HeLa cells. Exp Cell Res 2016; 340(1): 12–21. [DOI] [PubMed] [Google Scholar]
- 41. Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 2005; 202: 654–62. [DOI] [PubMed] [Google Scholar]
- 42. Koch A, Lang SA, Wild PJ et al Glucose transporter isoform 1 expression enhances metastasis of malignant melanoma cells. Oncotarget 2015; 6: 32748–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oh S, Kim H, Nam K et al Glut1 promotes cell proliferation, migration and invasion by regulating epidermal growth factor receptor and integrin signaling in triple‐negative breast cancer cells. BMB Rep 2016; 50: 132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun HW, Yu XJ, Wu WC et al GLUT1 and ASCT2 as Predictors for Prognosis of Hepatocellular Carcinoma. PLoS ONE 2016; 11: e0168907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kozlovsky N, Rudich A, Potashnik R et al Transcriptional activation of the Glut1 gene in response to oxidative stress in L6 myotubes. J Biol Chem 1997; 272: 33367–72. [DOI] [PubMed] [Google Scholar]
- 46. Lian S, Xia Y, Khoi PN et al Cadmium induces matrix metalloproteinase‐9 expression via ROS‐dependent EGFR, NF‐small ka, CyrillicB, and AP‐1 pathways in human endothelial cells. Toxicology 2015; 338: 104–16. [DOI] [PubMed] [Google Scholar]
- 47. Miller DM, Thomas SD, Islam A et al c‐Myc and cancer metabolism. Clin Cancer Res 2012; 18: 5546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Cell proliferation and ATP production is increased by E‐cadherin expression in AGS gastric cancer cells. (A) AGS and E‐cadherin expressed AGS cell lysates subjected to immunoblotting analysis using E‐cadherin and β‐actin. AGS and E‐cadherin‐expressing AGS cells were subjected to cell proliferation (B) and cellular ATP (C) measurement. *P < 0.05; **P < 0.01; ***P < 0.001. n.s., not significant.
Fig. S2. Effects of various small molecule inhibitors of signaling pathways on AGS and EC96 gastric cancer cell proliferation. Cell viability was measured by MTT assay after treatment with the following inhibitors: ICG‐001 (10 μM), inhibitor of beta‐catenin‐response transcription (iCRT) (25 μM), NSC668036 (10 μM) as Wnt signaling inhibitors; thiadiazolidinones (TDZD, 10 μM) as glycogen synthase kinase 3β inhibitor; LY294002 (25 μM) as PI3K/Akt inhibitor; U0126 (25 μM), PD90859 (2.5 μM) as MAPK inhibitors; A23187 (5 μM) as Ca2+ ionophore; rapamycin (20 μM) as mTOR inhibitor; cryptotanshinone (2.5 μM) as Stat3 inhibitor; SP600125 (5 μM) as JNK inhibitor; and MG132 (5 μM), triptolide (200 nM), and Bay11‐7082 (8 μM) as nuclear factor‐κB inhibitors. *P < 0.05; **P < 0.01; ***P < 0.001.
Fig. S3. Effect of E‐cadherin expression on c‐myc expression. Lysates from AGS‐, EC96‐, and E‐cadherin‐transfected cells were subjected to immunoblotting analysis for E‐cadherin, nuclear factor‐κB (NF‐κB), c‐myc, survivin, and GAPDH. Band intensity was normalized to GAPDH.
Fig. S4. E‐cadherin increased basal oxygen consumption rate (OCR) levels. Cells were incubated on XF24 culture plates for 24 h using substrate‐free base medium. The kinetic OCR responses of AGS and EC96 cells to glucose (10 mM) oligomycin (2 μM), and 2‐deoxyglucose (2‐DG; 0.1 μM) were measured.
Fig. S5. Analysis of Axin expression in mitochondria. (A) Cells were fractionated into cytosol and mitochondria and subjected to immunoblot analysis for the indicated proteins. (B) Cells were cultured for 24 h, and proteins were immunoprecipitated using an anti‐Axin1 antibody and subjected to immunoblot analysis for Axin1, E‐cadherin, and β‐catenin.
Fig. S6. E‐cadherin increased cellular reactive oxygen species levels. (A) Cells were incubated with 10 μM 2′,7′‐dichloro‐dihydro‐fluorescein diacetate (DCFH‐DA), and the fluorescence intensity was assessed by a luminometer. (B) Cells were incubated with 10 μM 2′,7′‐dichloro‐dihydro‐fluorescein diacetate (DCFH‐DA), and the fluorescence intensity was assessed by a fluorescence microscope.
Table S1. Primer sequences for quantitative RT‐PCR.