

Abstract
Adult T‐cell leukemia/lymphoma (ATL) is a peripheral T‐cell neoplasm with a dismal prognosis. It is caused by human T‐cell leukemia virus type‐1 (HTLV‐1) retrovirus. A long latency period from HTLV‐1 infection to ATL onset suggests that not only HTLV‐1 proteins, such as Tax and HBZ, but also additional genetic and/or epigenetic events are required for ATL development. Although many studies have demonstrated the biological functions of viral genes, alterations of cellular genes associated with ATL have not been fully investigated. Recently, a large‐scale integrated genetic analysis revealed the entire landscape of somatic aberrations in ATL. This neoplasm is characterized by frequent gain‐of‐function alterations in components of the T‐cell receptor/NF‐κB signaling pathway, including activating mutations in the PLCG1,PRKCB,CARD11 and VAV1 genes, and CTLA4‐CD28 and ICOS‐CD28 fusions. Importantly, molecules associated with immune surveillance, such as HLA‐A/B,CD58 and FAS, are affected recurrently. Among them, one notable lesion occurs as frequent structural variations that truncate the PD‐L1 3′‐untranslated region, leading to its overexpression. Other genetic targets include transcription factors (IRF4,IKZF2, and GATA3) and chemokine receptors (CCR4,CCR7 and GPR183), which are functionally relevant in normal T cells. A substantial proportion of ATL cases show widespread accumulation of repressive epigenetic changes, such as trimethylation of histone H3 lysine 27 and DNA hypermethylation of CpG islands, which coordinately modulate multiple pathways, including Cys2‐His2 zinc finger genes involved in silencing retroelements. Here we review the current understanding of the genetic/epigenetic aberrations in ATL, focusing on their relevance in its molecular pathogenesis.
Keywords: Adult T‐cell leukemia/lymphoma, human T‐cell leukemia virus type‐1, next‐generation sequencing, PD‐L1, T‐cell receptor/NF‐κB signaling
Adult T‐cell leukemia/lymphoma (ATL) is an aggressive peripheral T‐cell malignancy caused by human T‐cell leukemia virus type‐1 (HTLV‐1) infection.1, 2 Historically, a geographical clustering of leukemias in south‐west Japan led to the first description of this malignancy as a unique disease entity in the late 1970s.3 A few years later, HTLV‐1 was isolated as the exclusive causal agent of ATL in the USA and Japan, separately.4, 5 These landmark studies established HTLV‐1 as the first retrovirus directly linked to a human malignancy. At present, there are an estimated 5–10 million HTLV‐1 carriers worldwide, especially in endemic regions such as south‐west Japan, the Caribbean basin, Central and South America, and intertropical Africa.1, 6 HTLV‐1 transmission occurs primarily through breastfeeding, and approximately 6–7% of male and 2–3% of female HTLV‐1 carriers develop ATL after a latency period of 30–50 years from infection. These observations suggest that, although HTLV‐1‐derived proteins, such as Tax and HBZ, play central roles in ATL pathogenesis, additional genetic and/or epigenetic events are required for HTLV‐1‐infected cells to transform into ATL.2 However, most studies thus far have focused on viral proteins, while somatic alterations in ATL have not been fully elucidated, except for a few known targets, such as TP53, CDKN2A and FAS.7, 8, 9 Recently, a collaborative large‐scale genetic study delineated the entire portrait of genetic and epigenetic aberrations in ATL and identified a large number of novel mutational targets.10 Therefore, this review summarizes the recent progress on the genetic and epigenetic alterations in ATL, highlighting their roles in its molecular pathogenesis.
Overview of Adult T‐cell Leukemia/Lymphoma Genomes
A recent remarkable advance in HTLV‐1 biology is the comprehensive characterization of somatic alterations in ATL from an integrated genetic study comprising of whole‐exome, genome and transcriptome sequencing, as well as array‐based copy number and methylation analysis, followed by extensive validation in more than 400 ATL samples.10 This study showed that the mutation rate for ATL was relatively high compared to other hematologic malignancies, with an average of 2.3 mutations per megabase in coding regions. Despite the well‐known relationship between the activation of APOBEC deaminases and retroviral infection,11 APOBEC‐related TpC > T or G substitutions were rarely observed, but age‐related CpG > T substitutions were predominant.10 A total of 50 genes were shown to be significantly mutated, of which 13 genes were affected in more than 10% of cases. Copy number analysis identified 26 focal amplifications and 50 focal deletions, of which approximately one‐quarter contained recurrently mutated genes. ATL cases also exhibited many structural variations (SV), probably reflecting the underlying genomic instability. SV breakpoints (especially deletion breakpoints) were vastly overrepresented in common fragile sites (unstable genomic regions that tend to break under replication stress), including the NRXN3 (14q31.1), IMMP2L (7q31.1), DPYD (1p21.3) and FHIT (3p14.2) loci, although some of them recurrently affected driver genes that are biologically relevant in ATL pathogenesis (as detailed below).
Frequent Gain‐of‐function Alterations in TCR/NF‐κB Signaling
A hallmark of driver lesions in ATL is their strong enrichment in the molecules of TCR/NF‐κB signaling and their related or downstream pathways (Fig. 1).10 A notable feature in these pathways is the predominance of activating alterations (gain‐of‐function mutations and focal amplifications) found in the proximal components of TCR signaling (PLCG1, VAV1 and FYN), a co‐stimulatory receptor (CD28), more distal TCR signaling molecules belonging to the NF‐κB pathway (PRKCB and CARD11), and their downstream mediators involved in transcriptional regulation (IRF4) and cytoskeletal organization (RHOA). By contrast, loss‐of‐function mutations or deletions affect negative regulators of TCR/NF‐κB signaling, including CBLB, TRAF3, TNFAIP3 and CSNK1A1.
Figure 1.
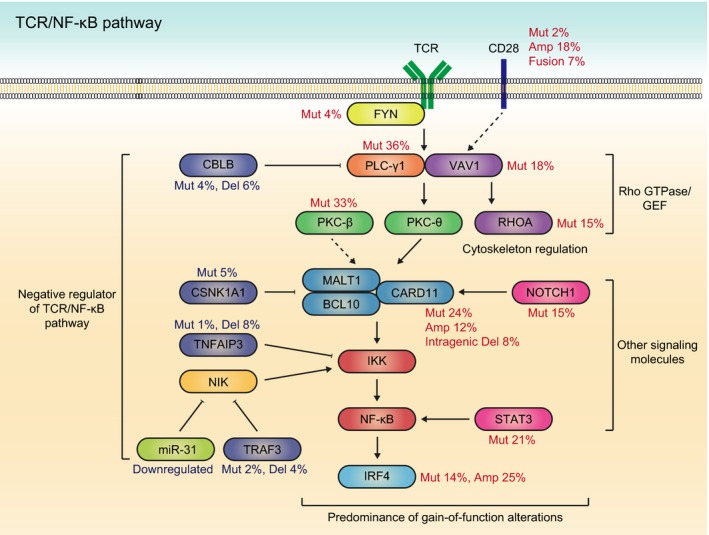
Predominance of gain‐of‐function mutations in the TCR/NF‐κB pathway. Frequent gain‐of‐function alterations in TCR/NF‐κB and other related pathways, as well as loss‐of‐function alterations of their negative regulators are observed in adult T‐cell leukemia/lymphoma (ATL). The major driver alterations are summarized with their frequencies. Amp, amplification; Del, deletion; Mut, mutation.
Among these, the most frequently altered gene is PLCG1 (36%), encoding phospholipase C γ‐1(PLC‐γ1), a key molecule in proximal TCR signaling.10 Upon TCR stimulation, PLC‐γ1 is activated through tyrosine phosphorylation and generates two second messengers: inositol 1,4,5‐trisphosphate (IP3) and diacylglycerol (DAG).12 Then, IP3 mobilizes intracellular calcium, which is essential for NFAT activation, and DAG stimulates protein kinase C (PKC), thereby mediating NF‐κB signaling. Similar to other subtypes of peripheral T‐cell lymphomas, ATL harbors several hotspot missense mutations, such as R48W, S345F, S520F, E1163K and D1165H, that have been shown to enhance downstream NFAT and NF‐κB activities.13, 14
The second most frequently mutated gene is PRKCB (33%), which encodes a member of the PKC family of proteins (PKC‐β), a major mediator downstream of PLC‐γ in antigen receptor signaling.10 In contrast to sporadically reported loss‐of‐function PKC mutations,15 more than 90% of PRKCB mutations in ATL are located within the highly conserved kinase domain, with a prominent hotspot at D427.10 In the absence of stimulatory signals, several autoinhibitory interactions, such as between the C1b domain and the NFD motif, maintain PKC‐β in an inactive form.16 PRKCB mutations are thought to destabilize these interactions, leading to enhanced PKC membrane translocation and downstream NF‐κB signaling.10 As PKC‐β and PKC‐θ have been implicated in B‐cell and T‐cell receptor signaling, respectively, the finding of recurrent PRKCB mutations, particularly those with a gain‐of‐function nature, in this T‐cell malignancy is rather unanticipated. Several lines of evidence have previously suggested a crucial but redundant role for PKC‐β, including NF‐κB transactivation, in the T‐cell lineage;17, 18 however, further studies are warranted to clarify the molecular pathogenesis mediated by their mutations.
CARD11, a cytoplasmic scaffolding protein required for both TCR and BCR‐induced NF‐κB activation, forms a signalosome complex with BCL10 and MALT1 and acts directly downstream of PKC.19 CARD11 mutations were first reported in activated B cell‐like diffuse large B cell lymphomas, in which most mutations resided in the coiled‐coil domain.20 In ATL, CARD11 mutations are more prevalent (24%) and are clustered, not only within the coiled‐coil domain, but also within the PKC‐responsive inhibitory domain, forming a hotspot at E626.10 Notably, recurrent small intragenic deletions targeting the identical inhibitory domain in ATL were identified by whole‐genome sequencing.10 These coiled‐coil and inhibitory domains interact with each other, rendering CARD11 in an inactive, closed conformation. Mutations and/or deletions in both domains have been shown to disrupt these interactions, leading to constitutive NF‐κB activation.10, 14, 20 Moreover, ATL cases frequently harbor amplification at 7p22 encompassing CARD11.10, 21 Taken together, CARD11 activation involves multiple mechanisms, such as mutations, intragenic deletions and copy number amplifications. Pairwise association analysis showed that CARD11 mutations frequently co‐occurred with PRKCB mutations, although few significant mutual exclusivity and co‐occurrence relationships were found between frequently mutated genes.10 This genetic relationship was confirmed by functional experiments showing a synergistic effect of both mutants on NF‐κB activation. These observations suggest that gain‐of‐function alterations in the multiple components of the TCR/NF‐κB pathway coordinately activate their downstream targets.
Among the members of the Rho GTPase family of proteins and their guanine nucleotide exchange factors (GEF) associated with the TCR/NF‐κB pathway, VAV1 and RHOA are recurrently mutated in 18 and 15% of ATL cases, respectively.10, 22 On recognition of its cognate antigen/major histocompatibility complex (MHC), TCR initiates a complex cascade of signaling events, resulting in the cytoskeletal reorganization and transcriptional upregulation required for T‐cell activation. This cascade involves rapid tyrosine phosphorylation of VAV1, which exerts its GEF activity toward RHOA, thereby resulting in TCR signaling activation.23 Although mutations of these genes are frequently found in other T‐cell lymphomas, such as peripheral T‐cell lymphoma‐not otherwise specified (PTCL‐NOS) and angioimmunoblastic T‐cell lymphoma (AITL), there are sharp differences in the mutational features among these PTCL. In ATL, VAV1 mutations are clustered at several hotspots in the acidic (E175), PH (K404), zinc‐finger (E556) and SH3 (R798 and R822) domains, whereas AITL and PTCL‐NOS primarily contain VAV1 in‐frame deletions and fusion genes with different partners. The latter alterations affect the C‐terminal SH3 domain and are shown to activate the TCR downstream signaling network.24 In contrast to RHOA G17V mutations characteristic of AITL, RHOA mutations in ATL are widely distributed across the entire gene, but largely clustered at the GTP‐binding domains, with C16 being a prominent hotspot.22 Intriguingly, depending on mutation type and position, these RHOA mutants have different, or even contrasting, functional consequences: C16R and A161P mutations, observed exclusively in ATL, behave as gain‐of‐function mutations, whereas G17V mutations act in a dominant‐negative manner.
Among the co‐signaling molecules of the B7‐CD28 family, CD28 and ICOS positively regulate, whereas CTLA4 negatively regulates TCR signaling.25 ATL cases harbor recurrent CTLA4‐CD28 and ICOS‐CD28 fusion genes (7%), where 5′ exons of CTLA4 or ICOS are fused with 3′ exons of CD28. These fusions are caused by tandem duplications of 2q33.2 segments containing CD28, CTLA4 and ICOS. These fusion proteins have the cytoplasmic domain of CD28 required for transmitting co‐stimulatory signals. In normal T cells, CD28 expression is suppressed and replaced by CTLA4 and ICOS expression after activation. In contrast, these fusions are expressed under the control of CTLA4 or ICOS promoters, enabling continuous or prolonged CD28 co‐stimulatory signaling by fusion proteins. In addition, as CTLA4 binds B7 ligands (CD80 and CD86) through the extracellular domain much more tightly than does CD28, part of the CTLA4‐CD28 fusion proteins are predicted to augment CD28 co‐signaling through enhanced ligand binding. In fact, these fusion proteins are reported to promote T‐cell proliferation.26 Together with recurrent high‐level amplifications and gain‐of‐function mutations, CD28 alterations account for one‐quarter of ATL patients. Besides CD28 co‐stimulatory signaling, a variety of other signal transduction molecules associated with the TCR/NF‐κB pathway are also altered in ATL, including the JAK‐STAT (STAT3) and NOTCH (NOTCH1) pathways. Therefore, a substantial number of genetic alterations in ATL converge on TCR/NF‐κB and its related pathways, reinforcing their pivotal roles in ATL pathogenesis.
Multiple Genetic Lesions Associated with Escape from Immune Surveillance
As HTLV‐1‐derived products confer high immunogenicity, escape from immune surveillance is likely to be critical in ATL development.2 In fact, immunogenic viral genes are silenced or even genetically inactivated in most cases, which would provide a fundamental strategy for evading the host immune system (as detailed below).10 Moreover, ATL cells frequently have genetic defects associated with immune response (Fig. 2). The most notable alteration among them is PD‐L1 SV disrupting the 3′‐UTR, thereby leading to PD‐L1 constitutive activation.27 Resulting from different types of SV, including tandem duplications, deletions, inversions and translocations, the 3′‐UTR disruption induces a marked increase in PD‐L1 expression, promoting tumor progression and immune evasion in vivo. Moreover, more than half of ATL cases have deteriorating mutations or focal deletions in the components of the MHC class 1 (HLA‐A, HLA‐B and B2M) and other molecules involved in T and natural killer cell‐mediated immune response (CD58 and FAS).10 Genetic inactivation of these genes is prevalently found in B‐cell lymphomas as well and shown to induce lymphomagenesis through impairment of immune surveillance.28, 29 In addition to their genetic alterations, ATL cells frequently show hypermethylation and transcriptional silencing of the MHC class 1 genes. Therefore, defects of antigen presentation through MHC class 1 abnormalities are mediated by multiple mechanisms, accounting for 90% of ATL cases.
Figure 2.
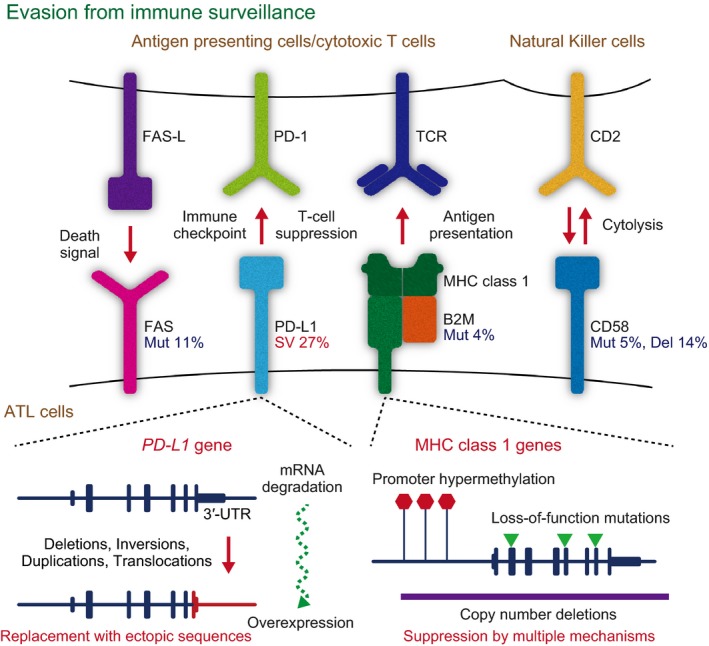
Multiple genetic lesions associated with immune evasion. A variety of molecules associated with immune evasion are altered in adult T‐cell leukemia/lymphoma (ATL). The commonly affected molecules and their ligands or receptors are shown with the frequencies of their alterations (upper). The genetic and epigenetic mechanisms underlying the alterations of PD‐L1 and major histocompatibility complex (MHC) class 1 genes are shown (lower). PD‐L1 overexpression is caused by structural variations (SV) inducing the 3′‐UTR truncation, such as deletions, inversions, duplications and translocations (lower left). MHC class 1 genes are inactivated by either nonsense and frameshift mutations (Mut), copy number deletions (Del) or hypermethylation of promoter CpG islands (lower right).
Genetic Aberrations in Transcription Factors Essential for Lymphocyte Function
Several transcription factors that are indispensable for lymphocyte activation and differentiation are recurrently altered in ATL. A remarkable target among them is IRF4, a member of the interferon regulatory factor family of transcription factors. IRF4 is a major downstream target of NF‐κB and is overexpressed in ATL cells.30 IRF4 mutations are frequently observed (14%) and clustered in the DNA‐binding domain, showing several hotspots, such as K59 and L70. Together with focal amplifications involving the IRF4 gene (6p25.3), these findings suggest that IRF4 alterations act in a gain‐of‐function manner.
Another commonly mutated transcription factor is GATA3, which is required for multiple steps of T‐cell differentiation in both developing thymocytes and mature T cells.31 Like other tumor suppressors, GATA3 is affected by nonsense and frameshift mutations distributed throughout the coding regions. Of note, there are hotspot mutations at the splice donor site of GATA3 exon 2 that cause intron retention and premature truncation. This suggests that these mutants confer altered protein function (possibly dominant‐negative) rather than haploinsufficiency of GATA3.
It is noteworthy that IKZF2 (or HELIOS), encoding a key transcription factor in T‐cell differentiation and activation, is exclusively affected by intragenic deletions, being one of the most common targets for genetic abnormalities in ATL (35%).10 Frequently affecting exons 5 and 6, these intragenic deletions cause abnormally spliced transcripts that lack the affected exons, and are thought to generate previously reported short isoforms of IKZF2.32 These abnormally spliced transcripts are shown to produce dominant‐negative forms against IKZF1 (or IKAROS) and IKZF2 with no DNA binding activity, which promote T‐cell growth in vitro, as well as induce T‐cell lymphomas in vivo.32, 33
Somatic Alterations in Chemokine Receptors
Another major category of molecules altered in ATL is chemokine receptors (CCR) associated with T‐cell trafficking, such as CCR4 (26–29%) and CCR7 (11%).10, 34 Both receptors are highly expressed in ATL cells and are implicated in ATL infiltration into other organs, such as lymphoid organs and skin.1 Most CCR4 and CCR7 mutations cause the truncation of the C‐terminal cytoplasmic domain, inducing increased surface receptor expression and impaired receptor internalization upon ligand stimulation. These mutations also enhance ligand‐induced chemotaxis and PI3K/AKT signaling, suggesting a gain‐of‐function nature for these mutations.10, 34 In contrast, another G protein‐coupled receptor, GPR183, is recurrently affected by loss‐of‐function mutations and focal deletions (28%). GPR183 encodes Epstein–Barr virus‐induced gene 2 (EBI2), a receptor for 7α,25‐dihydroxycholesterol (also called 7α,25‐OHC) and closely related oxysterols, that control positioning and cell fate determination of activated CD4+ T cells by regulating interaction with interleukin 2 (IL‐2)‐quenching dendritic cells.35 Given the constitutive expression of the IL‐2R‐α chain in ATL cells, GPR183 alterations may be involved in ATL leukemogenesis by modulating the IL‐2 signaling.
Widespread CpG Island DNA Hypermethylation in Adult T‐cell Leukemia/Lymphoma
DNA methylation analysis revealed that more than one‐third of ATL cases show widespread hypermethylation of CpG islands, termed as CpG island methylator phenotype (CIMP; Fig. 3). Intriguingly, in addition to MHC class 1 molecules, Cys2‐His2 (C2H2) zinc finger proteins are highly enriched in hypermethylated and silenced genes in ATL.10 Representing the largest class of putative human transcription factors, these proteins are proposed to bind and repress specific endogenous retroelements, ranging from currently active to ancient families.36, 37 Among them, approximately 50% of human C2H2 zinc finger proteins contain a Krüppel‐associated box (KRAB) domain. Such proteins are supposed to function through the interaction with its co‐factor, KAP1 (also known as TRIM28), that also has been implicated in silencing endogenous retroelements.36, 37 Furthermore, KRAB‐containing zinc finger proteins have been reported to potently block transcription from exogenous retroviruses, such as HTLV‐1.38 This notion is supported by a striking correlation in the number and evolutionary emergence of KRAB‐zinc finger genes and endogenous retroviruses within a wide range of vertebrate genomes.37 These observations suggest that ATL cells may avoid retroviral restriction activities by epigenetically silencing C2H2 zinc finger proteins.
Figure 3.
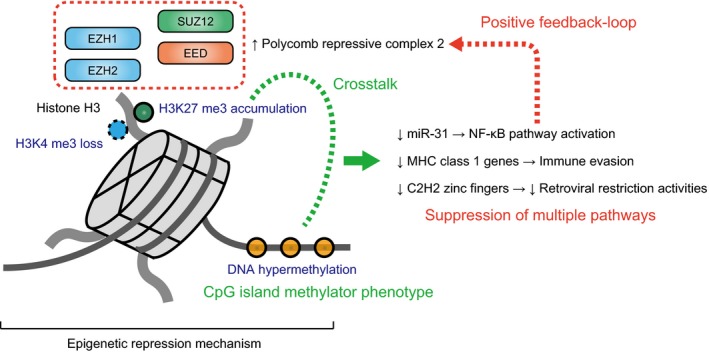
Multiple layers of epigenetic repression in adult T‐cell leukemia/lymphoma (ATL). Widespread accumulation of epigenetic repressive changes, such as H3K27 trimethylation (H3K27 me3) and DNA hypermethylation, are observed in ATL. H3K27 me3 accumulation is caused by polycomb repressive complex 2 activation and is usually associated with loss of H3K4 trimethylation (H3K4 me3). These epigenetic mechanisms are thought to coordinately cause NF‐κB pathway activation, MHC class 1 gene inactivation and C2H2 zinc finger gene suppression.
Extensive Accumulation of Repressive Histone Marks Caused by EZH1/2 Activation
The relevance of epigenetic repression in ATL is also supported by extensive accumulation of the trimethylation of histone H3 lysine 27 (H3K27), which is biologically linked to DNA methylation and jointly modulates gene expression (Fig. 3).39 This histone modification is usually accompanied by the loss of H3K4 methylation and is thought to be mediated by the aberrant upregulation of polycomb repressive complex (PRC) 2 proteins, such as EZH2, in ATL.40, 41 One of the potential mechanisms involved in EZH2 overexpression in ATL is the constitutive activation of the NF‐κB pathway, in which NF‐κB/Rel proteins have been shown to directly control EZH2 transcription.42 Together with EZH2, its homologue EZH1 is also highly expressed and simultaneously contributes to increased levels of H3K27 methylation in ATL. An interesting target of epigenetic repression is microRNA‐31 (miR‐31), for which suppression has been shown to induce NIK overexpression, leading to the constitutive activation of the noncanonical NF‐κB pathway.41 Therefore, in concert with genetic alterations, epigenetic abnormalities are involved in dysregulated TCR/NF‐κB signaling, contributing to ATL development.
HTLV‐1 Integration and Their Genetic Abnormalities
A fundamental feature of the ATL genome is HTLV‐1 proviral integration, which is clonal and widely distributed throughout the host genome, with a predisposition to transcriptionally active genomic regions.10, 43 Like other retroviruses, the HTLV‐1 proviral genome consists of gag, pol, and env genes, flanked by two long terminal repeats (LTR; Fig. 4). In addition, this genome includes a characteristic region, designated as pX, containing four partially overlapping open reading frames encoding accessory (p12, p13, p30 and Rex) and Tax proteins. These accessory proteins contribute to the establishment and maintenance of HTLV‐1 infection in vivo, whereas Tax is a transcriptional transactivator protein exerting pleiotropic activities.2 In addition to regulating viral transcription, this viral protein modulates cellular gene expression, especially for genes involved in T‐cell proliferation and activation, as in the NF‐κB and AP‐1 pathways.2 Importantly, Tax is sufficient to immortalize primary human T cells in vitro,44 and expression of the tax transgene under various promoters can induce malignant neoplasms, including T‐cell leukemia or lymphoma in vivo.45, 46, 47 However, tax transcripts are almost undetectable in most ATL cases. Genetic analyses of HTLV‐1 proviruses in ATL cells revealed several mechanisms to genetically and/or epigenetically silence tax expression: the accumulation of nonsense or frameshift mutations in tax gene is observed in approximately 10%,10, 48, 49 hypermethylation of 5′‐LTR CpG sites in 10–20%,49, 50 and varying lengths of deletions preferentially targeting 5′‐LTR in 20–30% of ATL cases.10, 51
Figure 4.
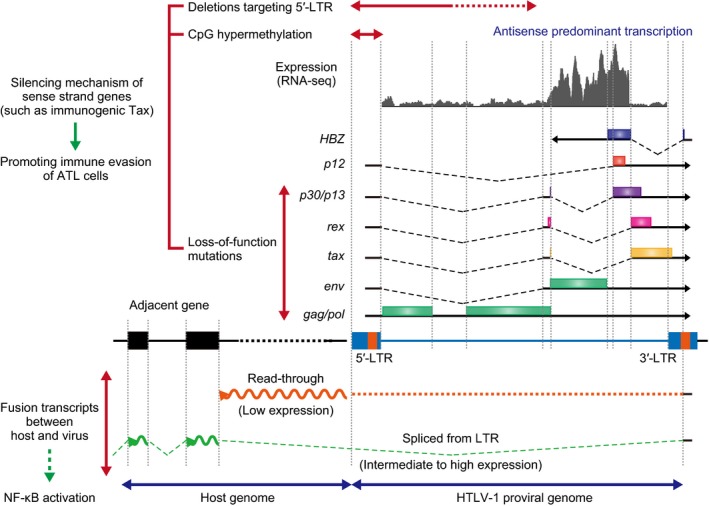
Human T‐cell leukemia virus type‐1 (HTLV‐1) proviral genome and transcription. HTLV‐1 proviral genome, sense and antisense transcripts, and abnormal fusion transcripts between viral and host sequences are shown. Sense strand genes, including tax, are frequently inactivated by deteriorating mutations and deletions and promoter hypermethylation. Viral transcripts predominantly originate in the antisense direction. Two different forms of fusion transcripts between host and viral sequences (read‐through and spliced transcripts) are frequently observed in adult T‐cell leukemia/lymphoma (ATL).
HTLV‐1 expresses another oncogenic viral gene, the HTLV‐1 basic leucine zipper factor (HBZ), encoded by the minus strand of the provirus.52 Interestingly, HBZ gene products are known to have different functions depending on their molecular form. HBZ protein inhibits the Tax‐mediated transactivation of viral transcription from the 5′‐LTR by interacting with various cellular proteins, such as JUN and CREB‐2.2 Conversely, HBZ RNA promotes proliferation of ATL cells, thereby contributing to ATL lymphomagenesis.53 Moreover, CD4+ T cell‐specific expression of the HBZ transgene induces T‐cell lymphoma as well as systemic inflammation in mice.53, 54 In contrast to sense strand genes and its promoter 5′‐LTR, the HBZ gene and 3′‐LTR remain intact in almost all cases. Generally, viral transcripts predominantly originate in antisense direction, while sense strand transcription is largely repressed (Fig. 4). In particular, HBZ is universally expressed in ATL cells, whereas tax expression is almost completely abrogated in most cases, as noted above. These findings suggest that antisense strand genes, transcribed from the 3′‐LTR, have a crucial role in the maintenance of ATL.
Of interest in the viral transcriptome is the generation of two different types of fusion transcripts between host and viral sequences: read‐through transcripts and spliced transcripts (Fig. 4).10 In almost all cases, antisense viral transcripts do not terminate in the 5′‐LTR, but read through it into the juxtaposed host genome. These read‐through transcripts generally extend for <20 kb, albeit at low expression levels. When proviral integration occurs within intronic regions of coding genes, aberrantly spliced fusion transcripts between the LTR and the affected genes are observed. These spliced fusion transcripts are more closely associated with integration in the antisense rather than the sense direction, and frequently are accompanied by upregulation of the affected gene expression. In addition, similar spliced fusion transcripts are occasionally detected between HBZ and an exon of a highly expressed host gene adjacent to the proviral integration site. Given that a recent study shows that antisense transcripts containing the LTR region activate the NF‐κB pathway,55 these new aberrant fusion transcripts may contribute to the pathogenesis of ATL; however, their precise role remains to be determined.
Conclusion
The recent advent of high‐throughput sequencing technologies has unraveled the unexpected complexity of cellular gene alterations in ATL, many of which coordinately activate the TCR/NF‐κB pathway. Among these, multiple frequently altered genes belong to the Tax interactome, a molecular network that the Tax protein directly interacts with and/or deregulates,56 even though Tax itself is no longer expressed, or even genetically perturbed in most cases. Also significant are frequent genetic and epigenetic aberrations of essential molecules associated with antigen presentation and immune evasion. Therefore, it is supposed that ATL cells develop alternative oncogenic mechanisms by acquiring genetic alterations in the Tax‐related pathway, while escaping from immune surveillance by modulating both cellular and viral genes.
A conspicuous feature of the ATL genome is a predominance of gain‐of‐function mutations, including components of the TCR/NF‐κB pathway and chemokine receptors. Among these molecules, an antibody targeting CCR4 (mogamulizumab) has demonstrated potent efficacy against ATL patients, although the relationship between its activity and CCR4 mutations remains unknown.57, 58 These observations provide a theoretical rationale for developing molecularly‐targeted agents for these genes with recurrent gain‐of‐function mutations. In particular, PKC‐β and CARD11 are promising targets, as several small molecule inhibitors against these and/or related molecules are currently available.19, 59 Clinical trials assessing novel agents that can potentially target other genetic and/or epigenetic alterations of ATL, including bortezomib (NF‐κB inhibitor), nivolumab (anti‐PD‐1 antibody) and DS‐3201b (EZH1/2 inhibitor), are ongoing. In particular, nivolumab holds great promise for ATL patients with PD‐L1 3′‐UTR disruption, given its excellent efficacy against Hodgkin lymphoma, in which PD‐L1/PD‐L2 genetic alterations are frequently observed.60, 61 Thus, identification of PD‐L1 3′‐UTR disruption may constitute a diagnostic marker to identify patients most likely to benefit from immune checkpoint therapy. Therefore, these arguments not only provide therapeutic implications of genetic alterations, but also suggest the relevance of genetic profiling, which could refine patient classification and stratification to provide better therapeutic options in ATL. Together, our findings offer novel insights into the molecular basis of ATL, which can be exploited for further therapeutic and diagnostic development to improve the management of ATL patients.
Disclosure Statement
K.K. owns stock in Asahi Genomics. The remaining author declares no relevant competing financial interests.
Acknowledgments
This work was supported by a Grant‐in‐Aid from the Japan Agency for Medical Research and Development (Practical Research for Innovative Cancer Control [15Ack0106014h0002] and Medical Research and Development Programs Focused on Technology Transfer [15im0210102h0001]) and a Grant‐in‐Aid for Scientific Research (KAKENHI 16H06249).
Cancer Sci 108 (2017) 1719–1725
Funding Information
Grant‐in‐Aid for Scientific Research (KAKENHI 16H06249) Japan Agency for Medical Research and Development (15Ack0106014h0002, 15im0210102h0001).
References
- 1. Ishitsuka K, Tamura K. Human T‐cell leukaemia virus type I and adult T‐cell leukaemia‐lymphoma. Lancet Oncol 2014; 15: e517–26. [DOI] [PubMed] [Google Scholar]
- 2. Matsuoka M, Jeang KT. Human T‐cell leukaemia virus type 1 (HTLV‐1) infectivity and cellular transformation. Nat Rev Cancer 2007; 7: 270–80. [DOI] [PubMed] [Google Scholar]
- 3. Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. Adult T‐cell leukemia: clinical and hematologic features of 16 cases. Blood 1977; 50: 481–92. [PubMed] [Google Scholar]
- 4. Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T‐cell lymphoma. Proc Natl Acad Sci USA 1980; 77: 7415–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T‐cell leukemia and its implication in the disease. Proc Natl Acad Sci USA 1982; 79: 2031–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV‐1 infection. Front Microbiol 2012; 3: 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamada Y, Hatta Y, Murata K et al Deletions of p15 and/or p16 genes as a poor‐prognosis factor in adult T‐cell leukemia. J Clin Oncol 1997; 15: 1778–85. [DOI] [PubMed] [Google Scholar]
- 8. Sakashita A, Hattori T, Miller CW et al Mutations of the p53 gene in adult T‐cell leukemia. Blood 1992; 79: 477–80. [PubMed] [Google Scholar]
- 9. Tamiya S, Etoh K, Suzushima H, Takatsuki K, Matsuoka M. Mutation of CD95 (Fas/Apo‐1) gene in adult T‐cell leukemia cells. Blood 1998; 91: 3935–42. [PubMed] [Google Scholar]
- 10. Kataoka K, Nagata Y, Kitanaka A et al Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet 2015; 47: 1304–15. [DOI] [PubMed] [Google Scholar]
- 11. Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol 2008; 26: 317–53. [DOI] [PubMed] [Google Scholar]
- 12. Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol 2013; 13: 257–69. [DOI] [PubMed] [Google Scholar]
- 13. Vaque JP, Gomez‐Lopez G, Monsalvez V et al PLCG1 mutations in cutaneous T‐cell lymphomas. Blood 2014; 123: 2034–43. [DOI] [PubMed] [Google Scholar]
- 14. Vallois D, Dobay MP, Morin RD et al Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T‐cell‐derived lymphomas. Blood 2016; 128: 1490–502. [DOI] [PubMed] [Google Scholar]
- 15. Antal CE, Hudson AM, Kang E et al Cancer‐associated protein kinase C mutations reveal kinase's role as tumor suppressor. Cell 2015; 160: 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leonard TA, Rozycki B, Saidi LF, Hummer G, Hurley JH. Crystal structure and allosteric activation of protein kinase C betaII. Cell 2011; 144: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thuille N, Wachowicz K, Hermann‐Kleiter N et al PKCθ/β and CYLD are antagonistic partners in the NFκB and NFAT transactivation pathways in primary mouse CD3+ T lymphocytes. PLoS One 2013; 8: e53709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lutz‐Nicoladoni C, Thuille N, Wachowicz K, Gruber T, Leitges M, Baier G. PKCα and PKCβ cooperate functionally in CD3‐induced de novo IL‐2 mRNA transcription. Immunol Lett 2013; 151: 31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thome M, Charton JE, Pelzer C, Hailfinger S. Antigen receptor signaling to NF‐kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb Perspect Biol 2010; 2: a003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lenz G, Davis RE, Ngo VN et al Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008; 319: 1676–9. [DOI] [PubMed] [Google Scholar]
- 21. Oshiro A, Tagawa H, Ohshima K et al Identification of subtype‐specific genomic alterations in aggressive adult T‐cell leukemia/lymphoma. Blood 2006; 107: 4500–7. [DOI] [PubMed] [Google Scholar]
- 22. Nagata Y, Kontani K, Enami T et al Variegated RHOA mutations in adult T‐cell leukemia/lymphoma. Blood 2016; 127: 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kataoka K, Ogawa S. Variegated RHOA mutations in human cancers. Exp Hematol 2016; 44: 1123–9. [DOI] [PubMed] [Google Scholar]
- 24. Abate F, da Silva‐Almeida AC, Zairis S et al Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T‐cell lymphomas. Proc Natl Acad Sci USA 2017; 114: 764–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol 2013; 13: 227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ungewickell A, Bhaduri A, Rios E et al Genomic analysis of mycosis fungoides and Sezary syndrome identifies recurrent alterations in TNFR2. Nat Genet 2015; 47: 1056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kataoka K, Shiraishi Y, Takeda Y et al Aberrant PD‐L1 expression through 3′‐UTR disruption in multiple cancers. Nature 2016; 534: 402–6. [DOI] [PubMed] [Google Scholar]
- 28. Afshar‐Sterle S, Zotos D, Bernard NJ et al Fas ligand‐mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat Med 2014; 20: 283–90. [DOI] [PubMed] [Google Scholar]
- 29. Challa‐Malladi M, Lieu YK, Califano O et al Combined genetic inactivation of beta2‐Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 2011; 20: 728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramos JC, Ruiz P Jr, Ratner L et al IRF‐4 and c‐Rel expression in antiviral‐resistant adult T‐cell leukemia/lymphoma. Blood 2007; 109: 3060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ho IC, Tai TS, Pai SY. GATA3 and the T‐cell lineage: essential functions before and after T‐helper‐2‐cell differentiation. Nat Rev Immunol 2009; 9: 125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Asanuma S, Yamagishi M, Kawanami K et al Adult T‐cell leukemia cells are characterized by abnormalities of Helios expression that promote T cell growth. Cancer Sci 2013; 104: 1097–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z, Swindle CS, Bates JT, Ko R, Cotta CV, Klug CA. Expression of a non‐DNA‐binding isoform of Helios induces T‐cell lymphoma in mice. Blood 2007; 109: 2190–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakagawa M, Schmitz R, Xiao W et al Gain‐of‐function CCR4 mutations in adult T cell leukemia/lymphoma. J Exp Med 2014; 211: 2497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL‐2‐quenching dendritic cells. Nature 2016; 533: 110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Najafabadi HS, Mnaimneh S, Schmitges FW et al C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat Biotechnol 2015; 33: 555–62. [DOI] [PubMed] [Google Scholar]
- 37. Feschotte C, Gilbert C. Endogenous viruses: insights into viral evolution and impact on host biology. Nat Rev Genet 2012; 13: 283–96. [DOI] [PubMed] [Google Scholar]
- 38. Wolf D, Goff SP. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 2009; 458: 1201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10: 295–304. [DOI] [PubMed] [Google Scholar]
- 40. Sasaki D, Imaizumi Y, Hasegawa H et al Overexpression of enhancer of zeste homolog 2 with trimethylation of lysine 27 on histone H3 in adult T‐cell leukemia/lymphoma as a target for epigenetic therapy. Haematologica 2011; 96: 712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamagishi M, Nakano K, Miyake A et al Polycomb‐mediated loss of miR‐31 activates NIK‐dependent NF‐kappaB pathway in adult T cell leukemia and other cancers. Cancer Cell 2012; 21: 121–35. [DOI] [PubMed] [Google Scholar]
- 42. Fujikawa D, Nakagawa S, Hori M et al Polycomb‐dependent epigenetic landscape in adult T‐cell leukemia. Blood 2016; 127: 1790–802. [DOI] [PubMed] [Google Scholar]
- 43. Gillet NA, Malani N, Melamed A et al The host genomic environment of the provirus determines the abundance of HTLV‐1‐infected T‐cell clones. Blood 2011; 117: 3113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Akagi T, Ono H, Shimotohno K. Characterization of T cells immortalized by Tax1 of human T‐cell leukemia virus type 1. Blood 1995; 86: 4243–9. [PubMed] [Google Scholar]
- 45. Grossman WJ, Kimata JT, Wong FH, Zutter M, Ley TJ, Ratner L. Development of leukemia in mice transgenic for the tax gene of human T‐cell leukemia virus type I. Proc Natl Acad Sci USA 1995; 92: 1057–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hasegawa H, Sawa H, Lewis MJ et al Thymus‐derived leukemia‐lymphoma in mice transgenic for the Tax gene of human T‐lymphotropic virus type I. Nat Med 2006; 12: 466–72. [DOI] [PubMed] [Google Scholar]
- 47. Ohsugi T, Kumasaka T, Okada S, Urano T. The Tax protein of HTLV‐1 promotes oncogenesis in not only immature T cells but also mature T cells. Nat Med 2007; 13: 527–8. [DOI] [PubMed] [Google Scholar]
- 48. Furukawa Y. Existence of escape mutant in HTLV‐I tax during the development of adult T‐cell leukemia. Blood 2001; 97: 987–93. [DOI] [PubMed] [Google Scholar]
- 49. Takeda S, Maeda M, Morikawa S et al Genetic and epigenetic inactivation of tax gene in adult T‐cell leukemia cells. Int J Cancer 2004; 109: 559–67. [DOI] [PubMed] [Google Scholar]
- 50. Koiwa T, Hamano‐Usami A, Ishida T et al 5′‐long terminal repeat‐selective CpG methylation of latent human t‐cell leukemia virus type 1 provirus in vitro and in vivo. J Virol 2002; 76: 9389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miyazaki M, Yasunaga J, Taniguchi Y, Tamiya S, Nakahata T, Matsuoka M. Preferential selection of human T‐cell leukemia virus type 1 provirus lacking the 5′ long terminal repeat during oncogenesis. J Virol 2007; 81: 5714–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gaudray G, Gachon F, Basbous J, Biard‐Piechaczyk M, Devaux C, Mesnard JM. The complementary strand of the human t‐cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down‐regulates viral transcription. J Virol 2002; 76: 12813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV‐I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA 2006; 103: 720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Satou Y, Yasunaga J, Zhao T et al HTLV‐1 bZIP factor induces T‐cell lymphoma and systemic inflammation in vivo. PLoS Pathog 2011; 7: e1001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kinpara S, Ito S, Takahata T et al Involvement of double‐stranded RNA‐dependent protein kinase and antisense viral RNA in the constitutive NFkappaB activation in adult T‐cell leukemia/lymphoma cells. Leukemia 2014; 29: 1425–9. [DOI] [PubMed] [Google Scholar]
- 56. Boxus M, Twizere JC, Legros S, Dewulf JF, Kettmann R, Willems L. The HTLV‐1 Tax interactome. Retrovirology 2008; 5: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamamoto K, Utsunomiya A, Tobinai K et al Phase I study of KW‐0761, a defucosylated humanized anti‐CCR4 antibody, in relapsed patients with adult T‐cell leukemia‐lymphoma and peripheral T‐cell lymphoma. J Clin Oncol 2010; 28: 1591–8. [DOI] [PubMed] [Google Scholar]
- 58. Ishida T, Joh T, Uike N et al Defucosylated anti‐CCR4 monoclonal antibody (KW‐0761) for relapsed adult T‐cell leukemia‐lymphoma: a multicenter phase II study. J Clin Oncol 2012; 30: 837–42. [DOI] [PubMed] [Google Scholar]
- 59. Mackay HJ, Twelves CJ. Targeting the protein kinase C family: are we there yet? Nat Rev Cancer 2007; 7: 554–62. [DOI] [PubMed] [Google Scholar]
- 60. Roemer MG, Advani RH, Ligon AH et al PD‐L1 and PD‐L2 Genetic alterations define classical hodgkin lymphoma and predict outcome. J Clin Oncol 2016; 34: 2690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ansell SM, Lesokhin AM, Borrello I et al PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015; 372: 311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]