

Abstract
Osteosarcoma is an aggressive malignant bone tumor that causes bone destruction. Although tumor‐specific replicating oncolytic adenovirus OBP‐301 induces an antitumor effect in an osteosarcoma tumor, it cannot prevent bone destruction. Zoledronic acid (ZOL) is a clinically available agent that inhibits bone destruction. In this study, we investigated the potential of combination therapy with OBP‐301 and ZOL against osteosarcomas with bone destruction. The antitumor activity of OBP‐301 and ZOL in monotherapy or combination therapy was assessed using three human osteosarcoma cell lines (143B, MNNG/HOS, SaOS‐2). The cytotoxic effect of OBP‐301 and/or ZOL was measured by assay of cell apoptosis. The effect of OBP‐301 and ZOL on osteoclast activation was investigated. The potential of combination therapy against tumor growth and bone destruction was analyzed using an orthotopic 143B osteosarcoma xenograft tumor model. OBP‐301 and ZOL decreased the viability of human osteosarcoma cells. Combination therapy with OBP‐301 and ZOL displayed a synergistic antitumor effect, in which OBP‐301 promoted apoptosis through suppression of anti‐apoptotic myeloid cell leukemia 1 (MCL1). Combination therapy significantly inhibited tumor‐mediated osteoclast activation, tumor growth and bone destruction compared to monotherapy. These results suggest that combination therapy of OBP‐301 and ZOL suppresses osteosarcoma progression via suppression of MCL1 and osteoclast activation.
Keywords: Adenovirus, bone destruction, osteosarcoma, telomerase, zoledronic acid
Osteosarcoma is the most common malignant bone tumor that occurs in teenagers or adolescents. Owing to recent advances in standard treatments such as pre‐ and post‐operative systemic chemotherapy and surgical resection, the 5‐year survival rate of osteosarcoma patients has improved to 60–78%.1 However, over 20% of osteosarcoma patients still die from tumor recurrence and distant metastasis. Bone destruction is a major obstacle in the treatment of patients with invasive osteosarcoma because highly aggressive osteosarcoma frequently invades within bone tissues, causes bone destruction though osteoclast activation and contributes to poor prognosis. Osteoclasts are mainly activated through the receptor activator of nuclear factor‐kappaB ligand (RANKL)‐RANK system, which further maintains the survival of osteoclasts.2 Recently, an osteoclast‐targeted therapy has been proposed as a novel treatment option for aggressive osteosarcoma with bone destruction.3 Therefore, the development of novel therapeutic strategies that target not only tumor growth but also bone destruction is required to improve the clinical outcome in patients with aggressive osteosarcomas.
As a novel therapeutic strategy for targeting tumor cells, we generated a telomerase‐specific replication‐competent oncolytic adenovirus, OBP‐301 (Telomelysin).4 OBP‐301 is a genetically engineered serotype 5 adenovirus, in which the human telomerase reverse transcriptase (hTERT) promoter drives the expression of the E1A and E1B genes linked to an internal ribosome entry site (IRES).4, 5 The hTERT promoter‐driven OBP‐301 replicates in telomerase‐positive tumor cells, but not in telomerase‐negative normal cells. We confirmed the antitumor effect of OBP‐301 against epithelial and mesenchymal types of malignant tumor cells in monotherapy4, 5, 6 and in combination therapy with radiation or chemotherapy.7, 8 Based on these preclinical studies, a phase I clinical study of OBP‐301, which was conducted in the United States on patients with advanced solid tumors, indicated that OBP‐301 was well tolerated by patients.9 However, in an orthotopic osteosarcoma xenograft tumor model, OBP‐301 could not efficiently inhibit osteosarcoma‐induced bone destruction.10 Thus, for clinical application of OBP‐301 in osteosarcoma patients with bone destruction, a novel therapeutic strategy for the suppression of both tumor growth and bone destruction is needed to improve the therapeutic potential of OBP‐301.
Zoledronic acid (ZOL) is a third‐generation bisphosphonate, which can inhibit bone destruction in patients with metastatic bone tumors11, 12, 13 or multiple myeloma.14 Moreover, ZOL has been shown to exert an antitumor effect against osteosarcoma cells.15, 16, 17 When ZOL was combined with chemotherapeutic agents, combination therapy showed a synergistic antitumor effect against osteosarcoma and prostate cancer cells.18, 19 A phase I clinical trial of ZOL in combination with standard chemotherapy against osteosarcoma patients has been conducted and ZOL was safe and feasible in combination with chemotherapy.20 Based on the inhibitory role of ZOL in osteosarcoma and bone destruction, we hypothesized that ZOL might enhance the therapeutic potential of OBP‐301 for aggressive osteosarcoma with bone destruction.
In the present study, we investigated the therapeutic potential of combination therapy of OBP‐301 and ZOL against osteosarcoma with bone destruction. Combination therapy with OBP‐301 and ZOL was assessed based on its effect on the viability of osteosarcoma cells, apoptosis induction, and osteoclast activation. Additionally, the in vivo antitumor effect of combination therapy and its effect on bone destruction status were assessed using an orthotopic osteosarcoma xenograft tumor model and a three‐dimensional computed tomography (3D‐CT) imaging system.
Materials and Methods
Cell lines
The human osteosarcoma cell line 143B and the mouse macrophage cell line RAW264.7 were purchased from the American Type Culture Collection (Manassas, VA, USA). The human osteosarcoma cell line MNNG/HOS was obtained from DS Pharma Biomedical (Osaka, Japan). The human osteosarcoma cell line SaOS‐2 was kindly provided by Dr. Satoru Kyo (Shimane University, Izumo, Japan). The normal human osteoblast NHOst cells and the human osteoclast OCP cells were purchased from Lonza (Walkersville, MD, USA). 143B and MNNG/HOS cells were maintained in Eagle's minimum essential medium containing 0.015 mg/mL 5‐bromo‐2′‐deoxyuridine and 1% nonessential amino acids, respectively. RAW264.7 and SaOS‐2 cells were maintained in Dulbecco's modified Eagle's medium. NHOst and OCP cells were maintained in One Normal Human Osteoblast Cell Medium BulletKit and in OCP Osteoclast Precursor Medium BulletKit, respectively. All media were supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin. 143B cells stably transfected with the green fluorescent protein (GFP) expression vector (143B‐GFP) and the firefly luciferase (Luc) expression vector (143B‐Luc) were established and maintained in medium containing Geneticin (Life Technologies, Tokyo, Japan).
Reagents
Zoledronic acid and mouse recombinant RANKL was purchased from Novartis Pharma (Tokyo, Japan) and Wako (Osaka, Japan), respectively.
Recombinant adenovirus
The telomerase‐specific, conditionally replicating adenovirus OBP‐301 (Telomelysin), was previously constructed and characterized.4, 5
Cell viability assay
Human osteosarcoma and NHOst cells (1 × 103 cells) and OCP cells (1 × 104 cells), seeded on 96‐well plates, were treated with ZOL (0–10 μM) and/or were infected with OBP‐301 at a multiplicity of infection (MOI) of 0, 1, 5, 10, 50, 100, or 200 plaque forming units (PFU)/cell. Cell viability was determined on days 3 and 5 after treatment using the Cell Proliferation Kit II (Roche Molecular Biochemicals, Indianapolis, IN, USA) according to the manufacturer's protocol. The combined effect of ZOL and OBP‐301 was analyzed by calculating the combination index using CalcuSyn software (BioSoft, Cambridge, UK). The computation of the combination index was based on the methods of Chou.21
Flow cytometric analysis
The cells, treated with ZOL (0–10 μM), were labeled with mouse anti‐coxsackie and adenovirus receptor (CAR) monoclonal antibody (mAb; RmcB; Upstate Biotechnology, Lake Placid, NY, USA) for 30 min at 4°C. The cells were incubated with fluorescein isothiocyanate (FITC)‐conjugated rabbit anti‐mouse IgG second antibody (Zymed Laboratories, San Francisco, CA, USA) and were analyzed using flow cytometry (FACS Array; BD Bioscience, Franklin Lakes, NJ, USA). The mean fluorescence intensity (MFI) of CAR for each cell line was determined by calculating the difference between the MFI of antibody‐labeled and non‐labeled cells from three independent experiments.
The cells (1 × 105 cells), treated with ZOL and OBP‐301 for 72 h, were incubated for 20 min on ice in Cytofix/Cytoperm solution (BD Biosciences), were labeled with phycoerythrin (PE)‐conjugated rabbit anti‐active caspase‐3 mAb (BD Biosciences) for 30 min, and were then analyzed using a FACS array (BD Biosciences).
TUNEL staining
Human osteosarcoma 143B and MNNG/HOS cells (1.0 × 103 cells) and SaOS‐2 cells (5.0 × 103 cells/well), seeded on 8‐well glass plates, were treated with ZOL (10 μM) for 72 h. The cells were fixed in 1% paraformaldehyde for 10 min. After washing, 4′,6‐diamidino‐2‐phenylindole (DAPI; 1 μg/mL) and 0.1% TritonX‐100 solution were added and incubated for 10 min at room temperature. Then, cells were incubated with TdT reaction solution (1 μg terminal deoxynucleotidyle transferase [TdT] and 5 μg FITC‐conjugated deoxyuridine triphosphate [dUTP] in 1200 μL TdT buffer; Roche Diagnostics GmbH, Mannheim, Germany). The cells were then stained by Hoechst 33258 (10 mg/m) for 15 min at room temperature. The cells were then observed under a fluorescence microscope.
Western blot analysis
The cells (3 × 105 cells), seeded in 100‐mm dishes, were treated with ZOL (0–10 μM) and/or were infected with OBP‐301 (0–200 MOI) for 72 h. The cells (1 × 105 cells), seeded in 100‐mm dishes, were transfected with 10 nM MCL1 small interfering RNA (siRNA), or control siRNA (Applied Biosystems, Foster City, CA, USA) using HiPerfect transfection reagents (Qiagen, Valencia, CA, USA). Whole‐cell lysates were prepared in a lysis buffer (50 mM Tris‐HCl (pH 7.4), 150 mM NaCl, 1% Triton X‐100) containing a protease inhibitor cocktail (Complete Mini; Roche Applied Science, Mannheim, Germany). Proteins were electrophoresed on 8–15% sodium duodecyl sulfate polyacrylamide gels and were transferred to polyvinylidene difluoride membranes (Amersham, Hybond; GE Health Care, Buckinghamshire, UK). Blots were blocked with Blocking‐One (Nacalai Tesque, Kyoto, Japan) at room temperature for 30 min. The primary antibodies used were: rabbit anti‐poly (ADP‐ribose) polymerase (PARP) polyclonal antibody (pAb; Cell Signaling Technology, Danvers, MA, USA), anti‐Ad5 E1A mAb (BD Biosciences), rabbit anti‐E2F1 mAb, rabbit anti‐MCL1 mAb (Cell Signaling Technology), and mouse anti‐β‐actin mAb (Sigma‐Aldrich, St. Louis, MO, USA). The secondary antibodies used were: horseradish peroxidase‐conjugated antibodies against rabbit IgG (GE Healthcare, Buckinghamshire, UK), mouse IgG (GE Healthcare), or goat IgG (Chemicon International Inc., Temecula, CA, USA). Immunoreactive bands on the blots were visualized using enhanced chemiluminescence substrates (ECL Plus; GE Healthcare).
Co‐culture of 143B‐GFP cells and osteoclasts
A co‐culture system with 143B‐GFP and RAW264.7 cells in the presence of RANKL, which induces the differentiation of osteoclasts in RAW264.7 cells 22, was used. RAW264.7 cells (1 × 104 cells), seeded on a 96‐well plate, were treated with RANKL (50 ng/mL) for 72 h. Subsequently, after co‐culture with 143B‐GFP cells (1 × 103 cells), the cells were treated with ZOL (5 μM) and/or OBP‐301 (1 × 105 PFU) for 72 h. The number of 143B‐GFP cells was observed under a fluorescence microscope. Tartrate‐Resistant Acid Phosphatase (TRAP) staining was performed using a TRAP staining kit (Cosmo Bio, Tokyo, Japan) to confirm the number of TRAP‐positive osteoclasts.
In vivo orthotopic 143B‐Luc xenograft tumor model
Animal experimental protocols were approved by the Ethics Review Committee for Animal Experimentation of Okayama University School of Medicine (No. OKU‐2014464). 143B‐Luc cells (2 × 106 cells/site) were inoculated into the left tibias of female athymic nude mice aged 6 weeks (CLEA Japan, Tokyo, Japan). Mice were divided into four groups; mock, ZOL, OBP‐301, and combination treatment. One week after tumor inoculation, phosphate‐buffered saline (PBS) containing OBP‐301 (1 × 108 PFU) in a 20 μL volume was injected into the tumors. PBS containing ZOL (100 μg/kg body weight [BW]) in a 50 μL volume was intraperitoneally injected every week for three cycles. To monitor the tumor growth, the luciferase substrate luciferin (VivoGlo Luciferin; Promega, Madison, WI, USA) was intraperitoneally injected at a dose of 150 mg/kg BW. Images were collected once a week. Images were taken in the right decubitus position after luciferin injection with the Xenogen IVIS Lumina Imaging System (Caliper Life Sciences, Cheshire, UK). Photons emitted from the left knee region were quantified by using Xenogen Living Image Software (Caliper Life Science). Body weight was also measured at the same time as assessment of tumor growth.
Three‐dimensional computed tomography imaging
Bone destruction was assessed using 3D‐CT imaging (ALOKA Latheta LCT‐200; Hitachi Aloka Medical, Tokyo, Japan). The mice were set in the prone position in the CT scanner. A CT scan was performed at a slice of every 48 μm from the distal thigh to the ankle. The CT data were reconstructed and analyzed using AZE virtual place 99 software (AZE, Tokyo, Japan). Image quantification of bone volume was performed based on a radiodensity of Hounsfield units (HUs).
Histopathological analysis
Tumors including the knee joint were fixed in 10% neutralized formalin and embedded in paraffin blocks. Sections were made longitudinally along the tibia bone and were stained with hematoxylin/eosin to assess the tumor region. The formation of osteoclast cells was examined using a TRAP Staining Kit (Cosmo Bio) according to the manufacturer's protocol. Immunostaining with rabbit anti‐Ki67 mAb (Abcam, Cambridge, MA, USA) was used to detect proliferating tumor cells within tumor tissues. All sections were analyzed under a light microscope.
Statistical analysis
Data are expressed as mean ± SD. A one‐way anova followed by a Dunnett multiple‐group comparison test was used to compare differences between groups in in vivo experiments. Differences between groups were examined for statistical significance using Student's t‐test. P values <0.05 were considered statistically significant.
Results
In vitro cytopathic effect of OBP‐301 and ZOL against human osteosarcoma cells
To investigate the therapeutic potential of OBP‐301 and ZOL against osteosarcoma, we used three human osteosarcoma cell lines (143B, MNNG/HOS, SaOS‐2). Both OBP‐301 and ZOL decreased the viability of all cell lines in a time and dose dependent manner. OBP‐301 decreased the viability of MNNG/HOS and SaOS‐2 cells more efficiently than it decreased that of 143B cells, whereas ZOL decreased the viability of SaOS‐2 cells more efficiently than it decreased that of 143B and MNNG/HOS cells (Fig. 1a,b). These data indicated that 143B cells were relatively resistant to OBP‐301 and ZOL compared with MNNG/HOS and SaOS‐2 cells. We next examined the combined effect of OBP‐301 and ZOL on the viability of human osteosarcoma cells. All human osteosarcoma cells were concomitantly treated with OBP‐301 and ZOL at various doses for 5 days. Combination treatment of OBP‐301 and ZOL decreased the viability of all human osteosarcoma cells more efficiently than single treatment (Fig. 1c). Calculation of the combination index demonstrated a synergistic antitumor effect for the combination of OBP‐301 and ZOL in all human osteosarcoma cells, although a low concentration of ZOL in combination with OBP‐301 showed an antagonistic effect in the OBP‐301 and ZOL‐resistant 143B cells (Fig. 1d). These results indicate that combination therapy of OBP‐301 and ZOL is more effective antitumor therapy compared with monotherapy of OBP‐301 or ZOL.
Figure 1.
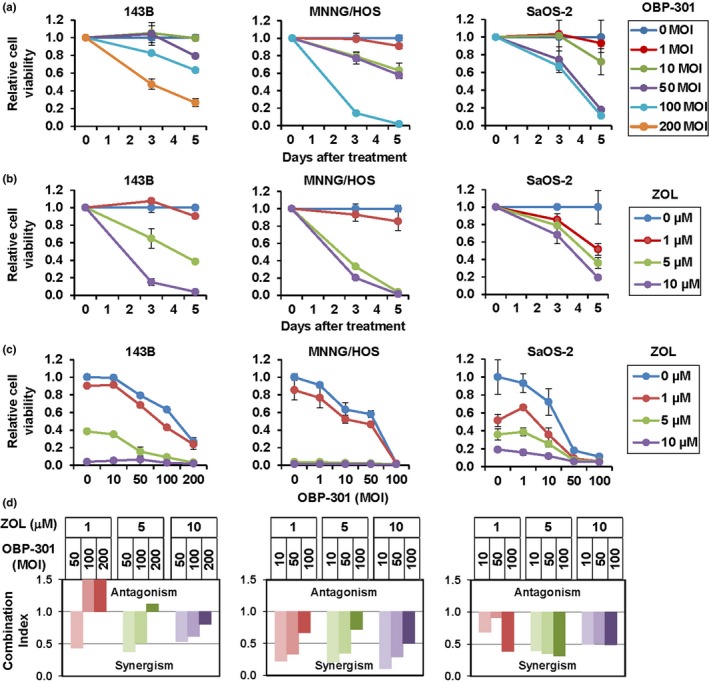
In vitro antitumor effect of OBP‐301 and/or zoledronic acid (ZOL) against human osteosarcoma cell lines. (a) Three human osteosarcoma cell lines (143B, MNNG/HOS, SaOS‐2) were infected with OBP‐301 and cell viability was assessed over 3 and 5 days using the XTT assay. Data are expressed as mean values ± SD (n = 3). (b) Cells were treated with ZOL and cell survival was quantified. (c) Cell viability was assessed 5 days after combined OBP‐301 and ZOL treatment. (d) The combination index was calculated using CalcuSyn software. Synergism and antagonism were defined as interaction indices of <1 and >1, respectively.
ZOL increases CAR expression and infection efficiency of OBP‐301
To explore the mechanism of the synergistic antitumor effect of the combination therapy with OBP‐301 and ZOL, we next addressed whether ZOL enhances CAR expression, infection efficiency or the replication rate of OBP‐301 in human osteosarcoma cells. Flow cytometric analysis demonstrated that ZOL treatment significantly increased CAR expression in all human osteosarcoma cells (Fig. 2a,b). Consistent with ZOL‐mediated CAR upregulation, the E1A copy number was significantly increased in the ZOL‐treated 143B and MNNG/HOS cells, but not in SaOS‐2 cells (Fig. 2c). However, there was no significant difference in the replication rate of OBP‐301 between non‐treated and ZOL‐treated osteosarcoma cells (Fig. 2c). These results indicate that the synergistic antitumor effect of the combination therapy is not due to enhancement of the OBP‐301‐mediated antitumor effect by ZOL.
Figure 2.
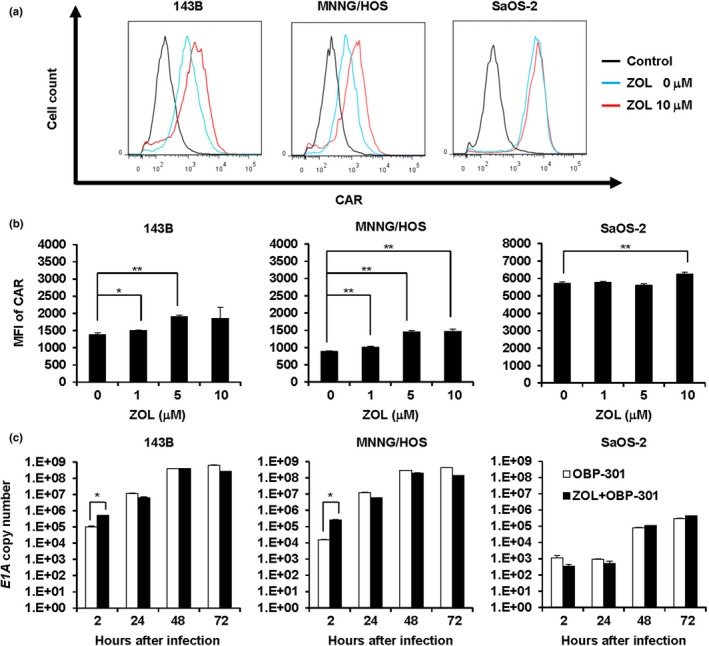
Zoledronic acid (ZOL)‐mediated enhancement of coxsackie and adenovirus receptor (CAR) expression and infection efficiency of OBP‐301. (a) Representative histogram of CAR expression in human osteosarcoma cells treated with ZOL. Cells were treated with ZOL for 72 h. The mean fluorescence intensity (MFI) of CAR expression was assessed by flow cytometric analysis. (b) Quantitative measurement of MFI in CAR expression. (c) Quantitative measurement of viral DNA replication. Cells were treated with ZOL (10 μM), and E1A copy number was analyzed using quantitative real‐time PCR. Data are expressed as mean values ± SD (n = 3). Statistical significance (* and **) was determined as P < 0.05 and P < 0.01, respectively.
OBP‐301 enhances ZOL‐induced apoptosis
Recent reports have shown that ZOL induces apoptosis in osteosarcoma cells.15, 16, 17 Moreover, we recently showed a chemosensitizing effect of OBP‐301 in human malignant tumor cells.22 To further explore the mechanism underlying the synergistic antitumor effect of the combination therapy, we assessed whether OBP‐301 enhances ZOL‐mediated apoptosis induction. The apoptosis‐inducing effect of ZOL against osteosarcoma cells was examined by analysis of apoptotic cells and expression of cleaved PARP using TUNEL staining and western blot analysis, respectively. These analyses indicated that ZOL induced the percentage of apoptotic cells and increased expression of cleaved PARP in all of the human osteosarcoma cells (Fig. 3a,b and Fig. S1). Next, to evaluate if OBP‐301 promotes ZOL‐induced apoptosis, human osteosarcoma cells were treated with ZOL and OBP‐301 for 5 days. Compared with the expression level of cleaved PARP and active caspase‐3 in the non‐treated control or in cells treated with monotherapy, the combination of ZOL and OBP‐301 significantly increased the expression of cleaved PARP and active caspase‐3 in human osteosarcoma cells (Fig. 3c,d). These results indicate that OBP‐301 promotes ZOL‐mediated apoptosis induction.
Figure 3.
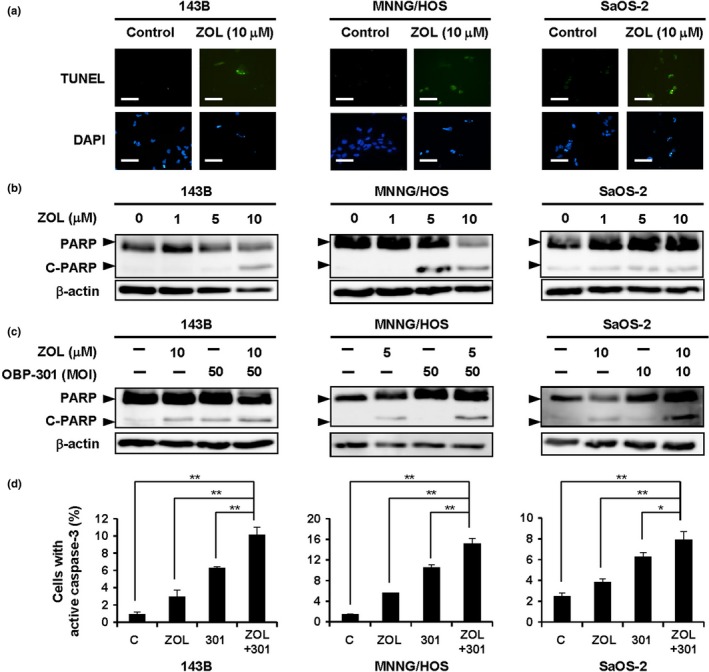
OBP‐301‐mediated enhancement of zoledronic acid (ZOL)‐induced apoptosis. (a) Cells were treated with ZOL (10 μM) for 72 h. Apoptotic cells were visualized by TUNEL staining and were analyzed under a fluorescence microscope. White scale bar, 100 μm. (b) Lysates of cells treated with the indicated concentrations of ZOL for 72 h were analyzed by Western blotting for PARP and cleaved PARP (C‐PARP). β‐actin was assayed as a loading control. (c) Cells were treated with ZOL and/or OBP‐301 for 72 h. (d) The percentage of cells with active caspase‐3 was quantified using flow cytometric analysis. Data are expressed as mean values ± SD (n = 3). Statistical significance (* and **) was determined as P < 0.05 and P < 0.01, respectively.
MCL1 suppression is critical for enhancement of ZOL‐induced apoptosis by OBP‐301
We previously reported that OBP‐301 activates expression of the transcription factor E2F1 in human cancer cells.23 Croxton et al. previously reported that E2F1 inhibits the expression of the anti‐apoptotic BCL2 family member, myeloid cell leukemia 1 (MCL1), whose depletion resulted in the induction of apoptosis.24 To evaluate if inhibition of MCL1 expression was involved in the underlying mechanism by which OBP‐301 mediated the enhancement of ZOL‐induced apoptosis, we investigated the effect of OBP‐301 on MCL1 expression. OBP‐301 increased the expression of adenoviral E1A and E2F1, whereas it decreased the expression of MCL1 in all human osteosarcoma cells (Fig. 4a). Furthermore, in 143B and MNNG/HOS cells, the combination of OBP‐301 and ZOL induced apoptosis (PARP cleavage), which was associated with upregulation of E1A and E2F1 and downregulation of MCL1, more efficiently compared to monotherapy with ZOL or OBP‐301 (Fig. 4b). To confirm the functional role of MCL1 suppression in ZOL‐mediated apoptosis induction, we further assessed the effect of MCL1 knockdown by RNA interference on ZOL‐induced apoptosis. MCL1 siRNA suppressed MCL1 expression and resulted in the enhancement of ZOL‐induced apoptosis in 143B and MNNG/HOS cells compared to control siRNA (Fig. 4c). These results suggest that OBP‐301 enhances ZOL‐mediated apoptosis induction via MCL1 suppression.
Figure 4.
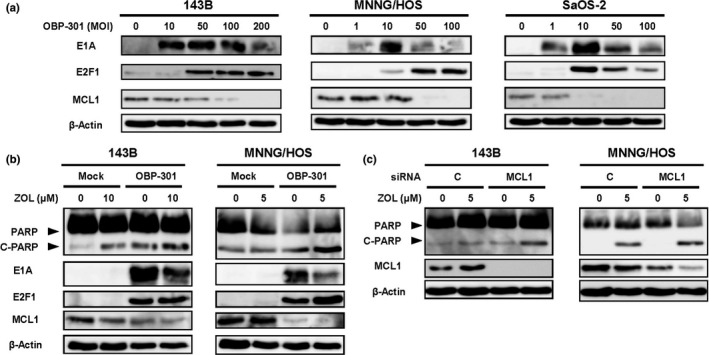
MCL1 suppression is involved in the OBP‐301‐enhancemet of zoledronic acid (ZOL)‐induced apoptosis. (a) Cells were infected with OBP‐301 for 72 h. Cell lysates were then analyzed by Western blotting for E1A, E2F1, and MCL1. (b) Cells were treated with OBP‐301 and/or ZOL for 72 h. Cell lysates were then analyzed by Western blotting for PARP, cleaved PARP (C‐PARP), E1A, E2F1, and MCL1. (c) Cells were transfected with 10 nmol/L MCL1 siRNA or control siRNA and were treated with ZOL (5 μM). Cell lysates were then analyzed by Western blotting for PARP, and MCL1. β‐actin was assayed as a loading control.
The combination of OBP‐301 and ZOL inhibits tumor cell proliferation and osteoclast activation
A recent report showed that RANKL induces osteoclast differentiation in RAW264.7 cells.25 Osteoclasts are activated and maintained by osteosarcoma cells in the process of osteosarcoma‐induced bone destruction.3 To investigate whether the combination of OBP‐301 and ZOL suppresses the viability of osteoclasts in a tumor microenvironment, we co‐cultured RANKL‐treated RAW264.7 cells with 143B‐GFP osteosarcoma cells (Fig. 5a). We then analyzed the effect of OBP‐301 and/or ZOL treatment on the viability of the 143B‐GFP osteosarcoma cells and of the induced TRAP‐positive RAW264.7 osteoclasts. OBP‐301 significantly reduced the viability of 143B‐GFP cells regardless of whether it was combined with or without ZOL (Fig. 5b,c). In contrast, the number of TRAP‐positive osteoclasts was significantly inhibited by the combination of OBP‐301 and ZOL compared to control or OBP‐301 treatment alone (Fig. 5b,c). Furthermore, to determine the effect of OBP‐301 and ZOL against normal cells in the bone microenvironment, normal human osteoclasts and osteoblasts were treated with ZOL or OBP‐301 for 5 days. ZOL decreased the viability of osteoclasts, but not of osteoblasts, whereas OBP‐301 did not affect the viability of either osteoclasts or osteoblasts (Fig. 5d). These results suggest that combination therapy with OBP‐301 and ZOL suppresses the viability of osteoclasts and osteosarcoma cells without affecting the viability of osteoblasts.
Figure 5.
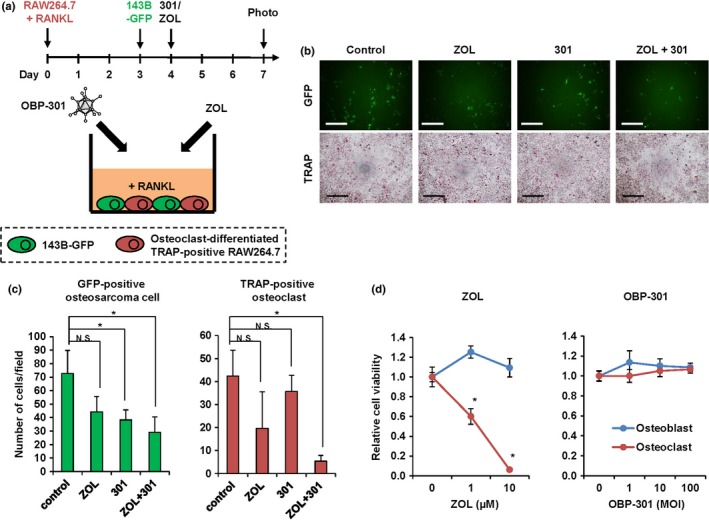
The effect of zoledronic acid (ZOL) and OBP‐301 against osteosarcoma cells and osteoclasts in a bone tumor microenvironment. (a) Scheme of the co‐culture of 143B‐GFP cells and osteoclast‐differentiated RAW264.7 cells. RAW264.7 cells were differentiated to osteoclasts by adding RANKL (50 ng/mL). (b) Representative photographs of GFP‐positive and TRAP‐positive (osteoclasts) cells in the co‐culture system after treatment with control, ZOL (5 μM), OBP‐301 (1 × 105 PFU), or both ZOL and OBP‐301. White scale bar, 200 μm. Black scale bar, 500 μm. (c) The number of GFP‐positive osteosarcoma cells and TRAP‐positive osteoclasts following treatment. (d) The viability of normal human osteoblasts and osteoclasts 5 days after treatment with ZOL or OBP‐301. Data are expressed as mean values ± SD (n = 3). Statistical significance (*) was determined as P < 0.05. N.S., not significant.
The combination of OBP‐301 and ZOL inhibits tumor growth and bone destruction
Finally, to assess the effect of OBP‐301 and ZOL on xenograft tumor growth and tumor‐mediated bone destruction, we used an orthotopic 143B‐Luc xenograft osteosarcoma tumor model. OBP‐301 or PBS was injected into the tumors and ZOL was intraperitoneally injected every week for three cycles. The combination of OBP‐301 and ZOL significantly suppressed tumor growth, which was evaluated by luciferase activity, when compared to control group (Fig. 6a). There was no significant difference in the mean body weight of mice in each treatment group (Fig. S2). Immunohistochemical analysis demonstrated that combination therapy with OBP‐301 and ZOL significantly decreased the percentage of Ki67‐positive proliferating cells within tumor tissues compared to control (Fig. 6b). In contrast, 3D‐CT indicated that the combination of OBP‐301 and ZOL significantly prevented the reduction in bone volume that was seen in control and OBP‐301 treatment alone (Fig. 6c). Moreover, TRAP staining demonstrated that the number of osteoclasts in the tibial metaphysis was significantly suppressed by ZOL or by the combination of OBP‐301 and ZOL (Fig. 6d). These results suggest that combination therapy with OBP‐301 and ZOL efficiently inhibits osteosarcoma progression and bone destruction through suppression of osteoclast activation.
Figure 6.
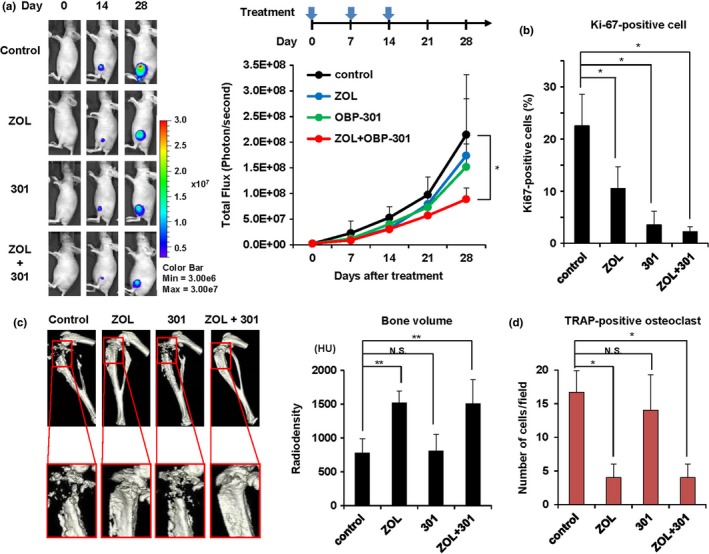
Combination therapy with zoledronic acid (ZOL) and OBP‐301 inhibits tumor growth and bone destruction in an orthotopic 143B‐Luc osteosarcoma xenograft model. (a) Seven days after intratibial inoculation of 143B‐Luc cells (2.0 × 106 cells/site), ZOL (100 μg/kg) was intraperitoneally injected and OBP‐301 (1 × 108 PFUs) was injected into the tumor on days 0, 7, and 14. Six mice were used for each group. Representative images of mice treated with control, ZOL, OBP‐301, or both ZOL and OBP‐301, on days 0, 14, and 28. (b) The percentage of Ki67‐positive cells in tumor tissue. (c) Representative 3D‐CT pictures of the left tibia bone at 28 days after treatment with control, ZOL, OBP‐301, or both ZOL and OBP‐301. Bone volume was assessed as total Hounsfield units (HU) from the proximal tibia to 5 mm distal to the sagittal slice. (d) The number of TRAP‐positive osteoclasts in tumor tissue following treatment. Data are expressed as mean values ± SD (n = 6 for each group). Statistical significance (* and **) was determined as P < 0.05 and P < 0.01, respectively. N.S., not significant.
Discussion
Osteosarcomas show an aggressive phenotype with bone destruction. Since the dissemination of osteosarcoma cells into bone tissues contributes to bone destruction, tumor recurrence, and poor prognosis, the prevention of bone destruction is a main obstacle to improving the clinical outcome of osteosarcoma patients. In this study, we demonstrated that combination therapy with OBP‐301 and ZOL showed a synergistic antitumor effect, which was mainly due to enhancement of apoptosis. OBP‐301 promoted ZOL‐induced apoptosis through suppression of anti‐apoptotic MCL1 expression in osteosarcoma cells, whereas ZOL suppressed the viability of osteoclasts. Furthermore, combination therapy with OBP‐301 and ZOL significantly suppressed both tumor growth and bone destruction compared to the control group. Thus, combination therapy of tumor‐targeted OBP‐301 and bone‐targeted ZOL is a promising antitumor strategy for improvement of the therapy of bone‐invasive osteosarcoma.
Combination therapy with OBP‐301 and ZOL showed a synergistic antitumor effect, in which apoptosis induction was significantly and synergistically enhanced. We and our collaborators recently demonstrated that the antitumor effect of OBP‐301 in human osteosarcoma cells was mediated through induction of apoptosis‐ and autophagy‐related cell death.6, 10, 26 ZOL has a therapeutic potential to induce apoptosis in human osteosarcoma cells, as previously shown in other reports.15, 16, 17 Regarding the underlying mechanism by which OBP‐301 enhances ZOL‐induced apoptosis, OBP‐301 caused the suppression of anti‐apoptotic MCL1 expression, which resulted in enhancement of apoptosis. Although oncolytic adenoviruses have been previously shown to suppress MCL1 expression,27, 28 the precise underlying mechanism of virus‐mediated MCL1 suppression remains unclear. Adenoviral E1A accumulation is associated with a reduction in MCL1 expression in adenovirus‐infected cells.29 Since E1A induces E2F1 activation30 and E2F1 overexpression suppresses MCL1 expression,24, 31 E1A‐mediated E2F1 activation would be a critical factor for OBP‐301‐mediated MCL1 downregulation. Moreover, E2F1 overexpression has been shown to sensitize human osteosarcoma U2OS cells to chemotherapeutic agents.32 We recently showed that, in combination therapy with OBP‐301 and chemotherapeutic agents, OBP‐301 suppressed MCL1 expression via E2F1‐dependent upregulation of MCL1‐targeting microRNA‐29, and subsequently enhanced chemotherapy‐induced apoptosis in human osteosarcoma cells.33 A clinical study of combination therapy of ZOL with chemotherapeutic agents has recently been evaluated in aggressive osteosarcoma patients.20 Therefore, a multimodal antitumor therapy with OBP‐301, ZOL, and chemotherapy may be a promising antitumor strategy for strong induction of the apoptosis of aggressive osteosarcomas through MCL1 suppression.
Osteosarcoma‐mediated bone destruction is mainly caused by osteoclast activation.3 Aggressive osteosarcoma cells promote osteoclast activation and bone resorption activity through the production of RANKL and cytokines.3, 34, 35 Since RANKL has a crucial role in the induction, activation, and survival of osteoclasts,3 pharmacological RANKL blockade is expected to be a promising therapeutic strategy against aggressive osteosarcoma tumors.36 ZOL has been shown to inhibit RANKL production in osteosarcoma cells.34 However, osteosarcoma cells further produce some osteotropic factors, including parathyroid hormone and prostaglandin, which induce the osteoblast‐mediated production of RANKL and subsequent osteoclast activation.3 Although OBP‐301 and ZOL did not affect the viability of osteoblasts, OBP‐301 indirectly inhibits the activation of osteoclast and osteoblast by inducing tumor‐specific cell death and ablating the RANKL and cytokine network. In contrast, ZOL directly inhibits the survival of osteoclasts probably through the induction of apoptosis.37, 38 Thus, the combination treatment of OBP‐301 and ZOL may be a novel promising strategy to suppress tumor‐dependent osteoclast activation and bone destruction of osteosarcomas with a bone‐invasive phenotype.
Zoledronic acid significantly increased the expression level of CAR in human osteosarcoma cells. Consistent with this effect of ZOL, we previously found that histone deacetylase inhibitor,39 radiation,7 and valproic acid40 also increase the expression level of CAR in human cancer cells. Although the infection efficiency of OBP‐301 was significantly increased in 143B and MNNG/HOS cells at 2 h after infection, ZOL could not enhance the replication of OBP‐301 during 72 h after infection. Regarding the replication of hTERT‐driven OBP‐301, we recently showed that adenoviral E1A accumulation increases the expression of hTERT mRNA via hTERT promoter activation, which leads to the enhancement of hTERT‐driven OBP‐301 replication in human bone and soft tissue sarcoma cells.6 Thus, although ZOL enhances CAR expression and the infection efficiency of OBP‐301 in human osteosarcoma cells, the replication of hTERT‐driven OBP‐301 may be mainly enhanced by adenoviral E1A accumulation rather than by ZOL.
It is worth noting that adenoviral E1A accumulation is suppressed after treatment with OBP‐301 and ZOL. E1A protein expression was decreased by treatment with high concentration of OBP‐301 compared to low concentration of OBP‐301 (Fig. 4a). Adenoviral late genes have been suggested to suppress the E1A expression during the late phase of adenovirus infection.41 In contrast, ZOL treatment suppressed the E1A protein expression in combination therapy (Fig. 4b). Recent report has also shown slightly decreased expression of E1A protein in human mesothelioma cells after treatment with replication‐competent adenovirus Ad‐delE1B55 and ZOL.42 However, the molecular mechanism underlying the ZOL‐mediated E1A suppression remains unclear. Further experiment would be warranted to investigate the role of ZOL in the accumulation of adenoviral E1A protein during adenovirus infection.
Bone destruction is caused not only by primary bone tumors but also by metastatic bone tumors. Bone metastasis is one of the most common complications in patients with advanced breast and prostate cancers. ZOL is a clinically available bone‐sparing agent that is used to treat patients with bone metastases.43 Although ZOL inhibits tumor‐mediated osteoclast activation, the formation of bone metastases was not prevented by ZOL in a prostate cancer bone metastasis model.44 Regarding the underlying mechanism of bone metastasis, metastatic tumor cells can display a dormancy that causes them to be refractory to conventional anticancer therapy and to form metastatic foci.45 We recently demonstrated that OBP‐301 efficiently eliminates dormant cancer stem‐like cells with sphere‐forming capacity and resistance to chemoradiotherapy through modulation of the cell cycle.46 If OBP‐301 has therapeutic potential against bone metastasis‐initiating dormant cancer cells, then combination therapy of OBP‐301 and ZOL may be further beneficial for the improvement of clinical outcome in advanced cancer patients with metastatic bone tumors.
In conclusion, we demonstrated that combination therapy with OBP‐301 and ZOL exerted a synergistic antitumor effect by promoting apoptotic cell death via OBP‐301‐mediated MCL1 suppression. Furthermore, ZOL prevented bone destruction through inhibition of osteoclast activation. These data suggest that this combination therapy would have great therapeutic potential as a novel dual targeted strategy against highly aggressive osteosarcoma with bone destruction. The administration route of OBP‐301 is limited to the local intratumoral injection; however, the recent approval of oncolytic herpesvirus, talimogene laherparepvec (T‐VEC), for melanoma patients supports the clinical relevance of local administration.47 Further clinical studies are warranted to investigate the tolerability and efficacy of this dual‐targeted combination therapy in aggressive osteosarcoma patients.
Disclosure Statement
Y. Urata is President & CEO of Oncolys BioPharma, Inc., the manufacturer of OBP‐301 (Telomelysin). H. Tazawa and T. Fujiwara are consultants of Oncolys BioPharma, Inc. The other authors disclosed no potential conflicts of interest.
Supporting information
Fig. S1. Quantitative analysis of ZOL‐induced apoptotic cells in human osteosarcoma cells.
Fig. S2. Chronological change in the body weight of 143B‐Luc tumor‐bearing mice treated with OBP‐301 and/or ZOL.
Acknowledgements
We thank Dr Satoru Kyo (Shimane University) for providing the SaOS‐2 cells, Dr Ken Takeda for helpful discussion and Takeshi Ieda and Tomoko Sueishi for their excellent technical support.
Cancer Sci 108 (2017) 1870–1880
Funding Information
This study was supported in part by grants from the Ministry of Education, Science, and Culture, Japan (to T. Fujiwara; No. 25293283, T. Ozaki; No. 25293323, T. Kunisada; No. 25462333, K. Sugiu; No. 15K10446, H. Tazawa: No. 25462057) and by the National Cancer Center Research and Development Fund (to T. Ozaki; 23‐A‐10).
References
- 1. Messerschmitt PJ, Garcia RM, Abdul‐Karim FW, Greenfield EM, Getty PJ. Osteosarcoma. J Am Acad Orthop Surg 2009; 17: 515–27. [DOI] [PubMed] [Google Scholar]
- 2. Tanaka S, Nakamura K, Takahasi N, Suda T. Role of RANKL in physiological and pathological bone resorption and therapeutics targeting the RANKL‐RANK signaling system. Immunol Rev 2005; 208: 30–49. [DOI] [PubMed] [Google Scholar]
- 3. Akiyama T, Dass CR, Choong PF. Novel therapeutic strategy for osteosarcoma targeting osteoclast differentiation, bone‐resorbing activity, and apoptosis pathway. Mol Cancer Ther 2008; 7: 3461–9. [DOI] [PubMed] [Google Scholar]
- 4. Kawashima T, Kagawa S, Kobayashi N et al Telomerase‐specific replication‐selective virotherapy for human cancer. Clin Cancer Res 2004; 10: 285–92. [DOI] [PubMed] [Google Scholar]
- 5. Hashimoto Y, Watanabe Y, Shirakiya Y et al Establishment of biological and pharmacokinetic assays of telomerase‐specific replication‐selective adenovirus. Cancer Sci 2008; 99: 385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sasaki T, Tazawa H, Hasei J et al Preclinical evaluation of telomerase‐specific oncolytic virotherapy for human bone and soft tissue sarcomas. Clin Cancer Res 2011; 17: 1828–38. [DOI] [PubMed] [Google Scholar]
- 7. Kuroda S, Fujiwara T, Shirakawa Y et al Telomerase‐dependent oncolytic adenovirus sensitizes human cancer cells to ionizing radiation via inhibition of DNA repair machinery. Cancer Res 2010; 70: 9339–48. [DOI] [PubMed] [Google Scholar]
- 8. Liu D, Kojima T, Ouchi M et al Preclinical evaluation of synergistic effect of telomerase‐specific oncolytic virotherapy and gemcitabine for human lung cancer. Mol Cancer Ther 2009; 8: 980–7. [DOI] [PubMed] [Google Scholar]
- 9. Nemunaitis J, Tong AW, Nemunaitis M et al A phase I study of telomerase‐specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol Ther 2010; 18: 429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hasei J, Sasaki T, Tazawa H et al Dual programmed cell death pathways induced by p53 transactivation overcome resistance to oncolytic adenovirus in human osteosarcoma cells. Mol Cancer Ther 2013; 12: 314–25. [DOI] [PubMed] [Google Scholar]
- 11. He M, Fan W, Zhang X. Adjuvant zoledronic acid therapy for patients with early stage breast cancer: an updated systematic review and meta‐analysis. J Hematol Oncol 2013; 6: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Orita Y, Sugitani I, Toda K, Manabe J, Fujimoto Y. Zoledronic acid in the treatment of bone metastases from differentiated thyroid carcinoma. Thyroid 2011; 21: 31–5. [DOI] [PubMed] [Google Scholar]
- 13. Hatoum HT, Lin SJ, Guo A, Lipton A, Smith MR. Zoledronic acid therapy impacts risk and frequency of skeletal complications and follow‐up duration in prostate cancer patients with bone metastasis. Curr Med Res Opin 2011; 27: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alegre A, Gironella M, Bailen A, Giraldo P. Zoledronic acid in the management of bone disease as a consequence of multiple myeloma: a review. Eur J Haematol 2014; 92: 181–8. [DOI] [PubMed] [Google Scholar]
- 15. Evdokiou A, Labrinidis A, Bouralexis S, Hay S, Findlay DM. Induction of cell death of human osteogenic sarcoma cells by zoledronic acid resembles anoikis. Bone 2003; 33: 216–28. [DOI] [PubMed] [Google Scholar]
- 16. Kubista B, Trieb K, Sevelda F et al Anticancer effects of zoledronic acid against human osteosarcoma cells. J Orthop Res 2006; 24: 1145–52. [DOI] [PubMed] [Google Scholar]
- 17. Dass CR, Choong PF. Zoledronic acid inhibits osteosarcoma growth in an orthotopic model. Mol Cancer Ther 2007; 6: 3263–70. [DOI] [PubMed] [Google Scholar]
- 18. Moriceau G, Ory B, Mitrofan L et al Zoledronic acid potentiates mTOR inhibition and abolishes the resistance of osteosarcoma cells to RAD001 (Everolimus): pivotal role of the prenylation process. Cancer Res 2010; 70: 10329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karabulut B, Erten C, Gul MK et al Docetaxel/zoledronic acid combination triggers apoptosis synergistically through downregulating antiapoptotic Bcl‐2 protein level in hormone‐refractory prostate cancer cells. Cell Biol Int 2009; 33: 239–46. [DOI] [PubMed] [Google Scholar]
- 20. Goldsby RE, Fan TM, Villaluna D et al Feasibility and dose discovery analysis of zoledronic acid with concurrent chemotherapy in the treatment of newly diagnosed metastatic osteosarcoma: a report from the Children's Oncology Group. Eur J Cancer 2013; 49: 2384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58: 621–81. [DOI] [PubMed] [Google Scholar]
- 22. Fujiwara T, Kagawa S, Tazawa H. Synergistic interaction of telomerase‐specific oncolytic virotherapy and chemotherapeutic agents for human cancer. Curr Pharm Biotechnol 2012; 13: 1809–16. [DOI] [PubMed] [Google Scholar]
- 23. Tazawa H, Yano S, Yoshida R et al Genetically engineered oncolytic adenovirus induces autophagic cell death through an E2F1‐microRNA‐7‐epidermal growth factor receptor axis. Int J Cancer 2012; 131: 2939–50. [DOI] [PubMed] [Google Scholar]
- 24. Croxton R, Ma Y, Song L, Haura EB, Cress WD. Direct repression of the Mcl‐1 promoter by E2F1. Oncogene 2002; 21: 1359–69. [DOI] [PubMed] [Google Scholar]
- 25. Cuetara BL, Crotti TN, O'Donoghue AJ, McHugh KP. Cloning and characterization of osteoclast precursors from the RAW264.7 cell line. In Vitro Cell Dev Biol Anim 2006; 42: 182–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li G, Kawashima H, Ogose A et al Efficient virotherapy for osteosarcoma by telomerase‐specific oncolytic adenovirus. J Cancer Res Clin Oncol 2011; 137: 1037–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wirth T, Kuhnel F, Fleischmann‐Mundt B et al Telomerase‐dependent virotherapy overcomes resistance of hepatocellular carcinomas against chemotherapy and tumor necrosis factor‐related apoptosis‐inducing ligand by elimination of Mcl‐1. Cancer Res 2005; 65: 7393–402. [DOI] [PubMed] [Google Scholar]
- 28. You L, Wang Y, Jin Y, Qian W. Downregulation of Mcl‐1 synergizes the apoptotic response to combined treatment with cisplatin and a novel fiber chimeric oncolytic adenovirus. Oncol Rep 2012; 27: 971–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cuconati A, Mukherjee C, Perez D, White E. DNA damage response and MCL‐1 destruction initiate apoptosis in adenovirus‐infected cells. Genes Dev 2003; 17: 2922–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bagchi S, Raychaudhuri P, Nevins JR. Adenovirus E1A proteins can dissociate heteromeric complexes involving the E2F transcription factor: a novel mechanism for E1A trans‐activation. Cell 1990; 62: 659–69. [DOI] [PubMed] [Google Scholar]
- 31. Ma Y, Croxton R, Moorer RL Jr, Cress WD. Identification of novel E2F1‐regulated genes by microarray. Arch Biochem Biophys 2002; 399: 212–24. [DOI] [PubMed] [Google Scholar]
- 32. Hofland K, Petersen BO, Falck J, Helin K, Jensen PB, Sehested M. Differential cytotoxic pathways of topoisomerase I and II anticancer agents after overexpression of the E2F‐1/DP‐1 transcription factor complex. Clin Cancer Res 2000; 6: 1488–97. [PubMed] [Google Scholar]
- 33. Osaki S, Tazawa H, Hasei J et al Ablation of MCL1 expression by virally induced microRNA‐29 reverses chemoresistance in human osteosarcomas. Sci Rep 2016; 6: 28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohba T, Cole HA, Cates JM et al Bisphosphonates inhibit osteosarcoma‐mediated osteolysis via attenuation of tumor expression of MCP‐1 and RANKL. J Bone Miner Res 2014; 29: 1431–45. [DOI] [PubMed] [Google Scholar]
- 35. Costa‐Rodrigues J, Fernandes A, Fernandes MH. Reciprocal osteoblastic and osteoclastic modulation in co‐cultured MG63 osteosarcoma cells and human osteoclast precursors. J Cell Biochem 2011; 112: 3704–13. [DOI] [PubMed] [Google Scholar]
- 36. Chen Y, Di Grappa MA, Molyneux SD et al RANKL blockade prevents and treats aggressive osteosarcomas. Sci Transl Med 2015; 7: 317ra197. [DOI] [PubMed] [Google Scholar]
- 37. Sudhoff H, Jung JY, Ebmeyer J, Faddis BT, Hildmann H, Chole RA. Zoledronic acid inhibits osteoclastogenesis in vitro and in a mouse model of inflammatory osteolysis. Ann Otol Rhinol Laryngol 2003; 112: 780–6. [DOI] [PubMed] [Google Scholar]
- 38. Tai TW, Su FC, Chen CY, Jou IM, Lin CF. Activation of p38 MAPK‐regulated Bcl‐xL signaling increases survival against zoledronic acid‐induced apoptosis in osteoclast precursors. Bone 2014; 67: 166–74. [DOI] [PubMed] [Google Scholar]
- 39. Watanabe T, Hioki M, Fujiwara T et al Histone deacetylase inhibitor FR901228 enhances the antitumor effect of telomerase‐specific replication‐selective adenoviral agent OBP‐301 in human lung cancer cells. Exp Cell Res 2006; 312: 256–65. [DOI] [PubMed] [Google Scholar]
- 40. Watanabe Y, Hashimoto Y, Kagawa S et al Enhanced antitumor efficacy of telomerase‐specific oncolytic adenovirus with valproic acid against human cancer cells. Cancer Gene Ther 2012; 19: 767–72. [DOI] [PubMed] [Google Scholar]
- 41. Fessler SP, Young CSH. Control of adenovirus early gene expression during the late phase of infection. J Virol 1998; 72: 4049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiang Y, Zhong B, Kawamura K et al Combination of a third generation bisphosphonate and replication‐competent adenoviruses augments the cytotoxicity on mesothelioma. BMC Cancer 2016; 16: 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ibrahim A, Scher N, Williams G et al Approval summary for zoledronic acid for treatment of multiple myeloma and cancer bone metastases. Clin Cancer Res 2003; 9: 2394–9. [PubMed] [Google Scholar]
- 44. Hung TT, Chan J, Russell PJ, Power CA. Zoledronic acid preserves bone structure and increases survival but does not limit tumour incidence in a prostate cancer bone metastasis model. PLoS One 2011; 6: e19389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weidle UH, Birzele F, Kollmorgen G, Ruger R. Molecular mechanisms of bone metastasis. Cancer Genomics Proteomics 2016; 13: 1–12. [PubMed] [Google Scholar]
- 46. Yano S, Tazawa H, Hashimoto Y et al A genetically engineered oncolytic adenovirus decoys and lethally traps quiescent cancer stem‐like cells in S/G2/M phases. Clin Cancer Res 2013; 19: 6495–505. [DOI] [PubMed] [Google Scholar]
- 47. Andtbacka RH, Kaufman HL, Collichio F et al Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015; 33: 2780–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Quantitative analysis of ZOL‐induced apoptotic cells in human osteosarcoma cells.
Fig. S2. Chronological change in the body weight of 143B‐Luc tumor‐bearing mice treated with OBP‐301 and/or ZOL.