

Abstract
The fallopian tube epithelium (FTE) is one of the progenitor populations for high-grade serous ovarian cancer (HGSC). Loss of PAX2 is the earliest known molecular aberration in the FTE occurring in serous carcinogenesis followed by a mutation in p53. Pathological studies report consistent loss of PAX2 in benign lesions as well as serous tumors. In the current study, the combined loss of PAX2 and expression of the R273H p53 mutant protein in murine oviductal epithelial (MOE) cells enhanced proliferation and growth in soft agar in vitro but was insufficient to drive tumorigenesis in vivo. A serially passaged model was generated to investigate the role of aging, but was also insufficient to drive tumorigenesis. These models recapitulate early benign lesions and suggest that a latency period exists between loss of PAX2, p53 mutation, and tumor formation. Stathmin and fut8 were identified as downstream targets regulated by loss of PAX2 and mutation of p53 in MOE cells. Re-expression of PAX2 in PAX2-null human HGSC cells reduced cell survival via apoptosis. PTENshRNA negatively regulated PAX2 expression and stable re-expression of PAX2 in MOE:PTENshRNA cells significantly reduced proliferation and peritoneal tumor formation in athymic nude mice. PAX2 was determined to be a direct transcriptional target that was activated by wild-type p53, whereas mutant p53 inhibited PAX2 transcription in MOE cells. A small molecule screen using the proximal PAX2 promoter driving luciferase identified four small molecules that were able to enhance PAX2 mRNA expression in MOE cells. PAX2 re-expression in HGSC cells and PTEN-deficient oviductal tumors may have the potential to induce apoptosis. In summary, mutant p53 and PTEN loss negatively regulated PAX2 and PAX2 re-expression in HGSC cells induced cell death.
Keywords: PAX2, fallopian tube, high-grade serous ovarian cancer, oviduct, p53 mutation
INTRODUCTION
High-grade serous ovarian cancer (HGSC) is the most lethal gynecological malignancy affecting women worldwide and the fifth most common cause of cancer related death among U.S. women(1). One factor contributing to strategies to prevent or treat ovarian cancer involves the uncertainty surrounding the tissue of origin, which could be the ovarian surface epithelium (OSE) or the fallopian tube epithelium (FTE)(2, 3). Increasing evidence from high-risk populations demonstrates that specific pathological changes might represent a step-wise progression of high-grade serous cancer from the distal end of the fallopian tube(3–6). A SCOUT (secretory cell outgrowth) may be the earliest change observed in the FTE in the progression to HGSC. SCOUTs are clusters of ≥ 30 secretory cells that lack expression of the PAX2 protein(7). SCOUTs are thought to progress into “p53 signatures” after the p53 gene is mutated, which often stabilizes expression of the p53 protein. SCOUTs and p53 signatures are benign, but p53 signatures are hypothesized to progress into serous tubal intraepithelial carcinomas (STICs), which are malignant precursors to HGSC(8). These lesions are identified in fixed tissues making it difficult to confirm the molecular signaling and regulation of protein expression underlying lesion development. In order to accurately model fallopian tube-derived high-grade serous carcinogenesis; it is critical to determine the mechanism of precursor formation, and the signals that govern this process in living cell models.
Several FTE-derived cellular models of murine and human origin have been engineered with a variety of oncogenes to form tumors in mice that resemble human disease(9–11). A study using PAX8-TetON-Cre promoter to drive deletion of BRCA1 or 2, and PTEN plus mutation of P53 in combination as well as deletion of P53 together with PTEN loss in murine oviductal FTE cells formed invasive carcinomas that metastasized to the peritoneal cavity and immunohistochemical analysis revealed high similarity with human disease markers(9, 12). Another study conditionally silenced Dicer and PTEN using Amhr2-Cre to demonstrate that high-grade carcinomas develop in the FTE, with the primary epithelial tumors arising in the fallopian tube stroma(13). STICs reported from OVGP1-SV40 mice are a result of the viral oncoprotein, which inhibits activity of wild-type p53(14). Some of these models do not accurately recapitulate the pre-neoplastic lesions that harbor p53 mutations since p53 is either silenced or sequestered by SV40. A murine oviductal epithelium (MOE) cell line that expresses mutant p53 and is not immortalized by SV40, contributed to the pro-migratory in vitro behavior of mutant cells, but this was insufficient for tumor formation(15). Although these models mimic human disease and affirm FTE as the source of origin, they fail to address the role of SCOUTS, which often lack PAX2 protein expression. Interestingly, several high-grade carcinoma animal models have also been developed that have retained PAX2 expression in tumor samples (12, 16).
PAX2 belongs to the paired homeobox domain family and plays an important role during embryonic development(17). PAX2 is critical for the development of the urogenital tract, thyroid, kidney, inner ear, eye and central nervous system(18). PAX2 interacts with a nuclear protein containing BRCT domains, called Pax transactivation-domain interacting protein(19) which is responsible for methylation of histone H3 at lysine 4(20) and also plays a critical role in DNA damage repair(21). PAX2 is expressed in Müllerian derived tissues such as the FTE, but not in the OSE. PAX2 is absent in majority human HGSC cell lines as well as in several serous tumor samples(8, 22–24). Some ovarian cancer cell lines and tumor tissues retain PAX2 expression, the functional significance of which is yet to be determined. Re-expression of PAX2 in HGSC cells reduced proliferation(23), whereas PAX2 overexpression in an OSE-derived tumor model improved survival(25). Therefore, the purpose of this study was to understand the regulation of PAX2 and the mechanisms contributing to its loss in the FTE.
In this study, the functional significance of PAX2 loss in the fallopian tube cells, alone and in combination with a mutation in P53 was evaluated. In addition, this study sought to determine whether re-expression of PAX2 was able to reduce proliferation and tumor formation of fallopian tube-derived tumor models and in HGSC cell lines. In order to develop a strategy for increasing PAX2 expression and potentially reversing the formation of a SCOUT, a small molecule screen was performed to look for compounds that activate the proximal PAX2 promoter driving luciferase. Restored PAX2 expression holds potential use for both prevention and therapy of fallopian tube-derived high-grade serous tumors.
RESULTS
PAX2 expression in human serous tumor samples is associated with increased overall survival
In order to validate the clinical significance of PAX2 mRNA loss, microarray data from a Gene Expression Omnibus-GSE69429 was retrospectively analyzed by comparing PAX2 expression between normal FTE, STICs and invasive HGSC samples (n=6 per group)(26, 27). PAX2 expression progressively declined from STICs to HGSC when compared to normal FTE (Supplementary Fig. S1A). HGSC mRNA patient data was stratified to include stage III/IV serous tumor samples (n=956)(28). High PAX2 expression significantly correlated with increased overall survival (Fig. 1A). Although the p-value is significant, the proximity between the two survival curves suggests a minimal biological advantage to patients with PAX2 overexpression. Similarly, the TCGA dataset demonstrated that a subset of HGSC patients whose tumors had amplification in the PAX2 gene, survived longer than those without any alteration in PAX2 (Supplementary Fig. S1B).
Figure 1. Combination of PAX2 loss and TP53 mutation alters functional characteristics of MOE cells in vitro but is insufficient to drive tumorigenesis in vivo.
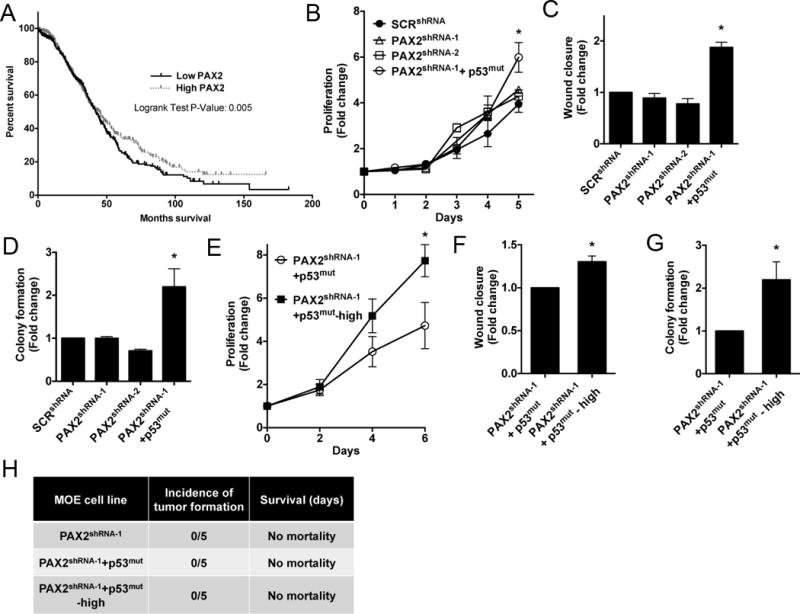
A) Human HGSC data mined from kmplot.com indicates serous tumors with high PAX2 (red line, n=478 patients) are associated with improved survival compared to other HGSC (black line, n=478 patients p-value = 0.005). B–D) Proliferation (day 5), migration (8 hours) and anchorage independent growth (day 14) of MOE:SCRshRNA, MOE:PAX2shRNA-1 and MOE:PAX2shRNA-1+p53mut. E–G) Proliferation (day 6), migration (8 hours) and anchorage independent growth (day 14) (respectively) of MOE:PAX2shRNA-1+p53mut and MOE:PAX2shRNA-1+p53mut-high. H) Athymic nude mice injected with MOE:PAX2shRNA-1, MOE:PAX2shRNA-1+p53mut and MOE:PAX2shRNA-1+p53mut-high cells demonstrated no tumorigenic growth in vivo at 6 months (n=5 mice per group).
Silencing PAX2 does not alter functional characteristics of murine fallopian tube epithelial cells
PAX2 is expressed by the human FTE cells, and its loss is thought to develop a pre-neoplastic lesion in the fallopian tube, called a SCOUT(7). MOE and MOSE cells were tested for PAX2 mRNA and protein expression levels by qPCR and western blot. PAX2 was abundantly expressed in the MOE cells and was absent in the MOSE cells (Supplementary Fig. S2A, S2B). In order to mimic SCOUTs, MOE cells were stably transfected with two unique shRNA sequences directed against PAX2 (PAX2shRNA-1, PAX2shRNA-2) and a scrambled control shRNA (SCRshRNA). The cells were validated to express less transcript and PAX2 protein (61% and 78% protein reduction in PAX2shRNA-1 and PAX2shRNA-2 respectively) (Supplementary Fig. S2C, S2D). MOE:PAX2shRNA-1 and MOE:PAX2shRNA-2 proliferation was not statistically different when compared to the MOE: SCRshRNA after 1, 2, 3, 4 or 5 days (Fig. 1B). PAX2 knockdown had no significant effect on migration of MOE cells compared to MOE:SCRshRNA when analyzed by scratch assay over 8 hours (Fig. 1C). At 14 days, there was no significant increase in colony formation observed in MOE:PAX2shRNA-1 and MOE:PAX2shRNA-2 cells compared to MOE:SCRshRNA (Fig. 1D). Therefore, the functional assays determined that loss of PAX2 by itself did not alter functional characteristics in vitro, which is consistent with their classification as benign lesions and suggests that additional molecular aberrations are necessary to drive tumorigenesis.
DNA binding mutation of p53 combined with loss of PAX2 in fallopian tube cells results in increased proliferation, migration and colony formation
SCOUTs are suggested to progress to p53 signatures when p53 is mutated. In order to mimic p53 signatures, MOE:PAX2shRNA-1 cells were stably transfected with the human mutant TP53R273H (p53mut), which is the most frequently identified mutation in HGSC. We previously reported that MOE cells stably expressing mutant p53 display enhanced migration(15). Overexpression of mutant p53 combined with the knockdown of PAX2 in MOE:PAX2shRNA-1+p53mut was validated by qPCR and western blot (Supplementary Fig. S2E, S2F). MOE:PAX2shRNA-1+p53mut cells proliferated more than the control (SCRshRNA) over 5 days (Fig. 1B). The addition of p53mut to MOE:PAX2shRNA-1 cells increased migration compared to MOE:SCRshRNA over 8 hours (Fig. 1C). However, this increase was comparable to the previously reported effect in MOE:p53mut alone, suggesting that PAX2 was not contributing to this change(29). MOE:PAX2shRNA-1+p53mut cells formed significantly more colonies compared to MOE:SCRshRNA (Fig. 1D). These results suggest that mutation of p53 when combined with loss of PAX2 enhanced in vitro transformation-like behavior, while changes in motility were largely related to only mutant p53.
Serial passaging of MOE cells harboring mutation of P53 and loss of PAX2 enhance in vitro growth and migration, but are insufficient to drive tumorigenesis in vivo
To determine the molecular changes necessary to drive MOE cells to form malignant STICs from p53 signatures, MOE:PAX2shRNA-1+p53mut cells (passages 18–30) were serially passaged to generate an “aged” model called MOE:PAX2shRNA-1+p53mut-high (passages 80–120). These “aged” cells showed significantly increased proliferation (Fig. 1E), migration (Fig. 1F) and colony formation (Fig. 1G) compared to MOE:PAX2shRNA-1+p53mut cells. To investigate the tumorigenic potential in vivo, these genetically engineered MOE lines were xenografted subcutaneously (s.c.) and intraperitoneally (i.p.) into athymic mice and tumor formation was monitored over a period of six months (n=5). Mice xenografted with MOE:PAX2shRNA-1, MOE:PAX2shRNA-1+p53mut, MOE:PAX2shRNA-1+p53mut-high and MOE:SCRshRNA did not display any evidence of tumor formation after six months (Fig. 1H). These data suggest that additional molecular events occur to progress a benign early lesion into STICs. These models recapitulate the benign characteristics of these lesions previously reported in fixed tissue samples.
Serially passaged MOE:PAX2shRNA-1+p53mut and human Type II SCOUTs share similar gene expression
A recent study compared human SCOUTs to HGSC using cDNA microarrays and validated that a subset of SCOUTS (Type II) had similar expression profiles to HGSC (22). Human tumor tissues and SCOUTS that do not express PAX2 protein likely have additional other genetic and molecular changes that could complicate RNA and protein expression analysis. MOE cells harboring mutation of TP53 or PAX2shRNA-1 or both, including the “aged” model represent a system with much less heterogeneity compared to human tissue samples for studying regulation of downstream targets of PAX2 and p53 that may contribute to tumor promotion. mRNA expression profiles in MOE derived cell line models were compared with expression data reported from human signatures. Nine targets (stmn1, fut8, mme, lef1, ezh2, runx2, rcn1, cox5a, ctnnb1) were selected from the data that compared human Type I vs. Type II SCOUTs. Targets were chosen based on expression similarity between Type II SCOUTs and tumor samples(22). Targets were investigated by qPCR in MOE:p53mut, MOE:PAX2shRNA-1, MOE:PAX2shRNA-1+p53mut and MOE:PAX2shRNA-1+p53mut-high cells (Fig. 2A, 2C, Supplementary Fig. S3A). Two targets were chosen for further investigation based on relevance to HGSC, FTE biology, and comparison with the TCGA: stathmin and fut8. Stathmin protein expression was upregulated by mutation of p53 and highly upregulated by the combination and in the “aged” line (Fig. 2B). Fut8 was upregulated by loss of PAX2, and was lost as the cells were serially passaged (Fig. 2D). Several studies have reported that stathmin is a pro-migratory gene that contributes to motility in HGSC(30, 31). To investigate the pro-migratory role of stathmin in MOE and its regulation by mutant p53, stathmin was transiently knocked down in MOE cells harboring PAX2 shRNA and mutant p53 and the aged model (Fig. 2E). Post 24-hour transfections with stathminshRNA, MOE cell models were subjected to scratch assays to measure wound closure over 12 hours. Downregulation of stathmin in MOE:PAX2shRNA-1+p53mut, including the aged model, demonstrated significant reduction in migration compared to cells transfected with control shRNA (Fig. 2F). These results suggest that stathmin may be regulated by mutant TP53 in the fallopian tube and could potentially contribute to increased migratory phenotype.
Figure 2. mRNA expression profiling of MOE cells harboring PAX2 loss and mutant TP53 in the FTE.
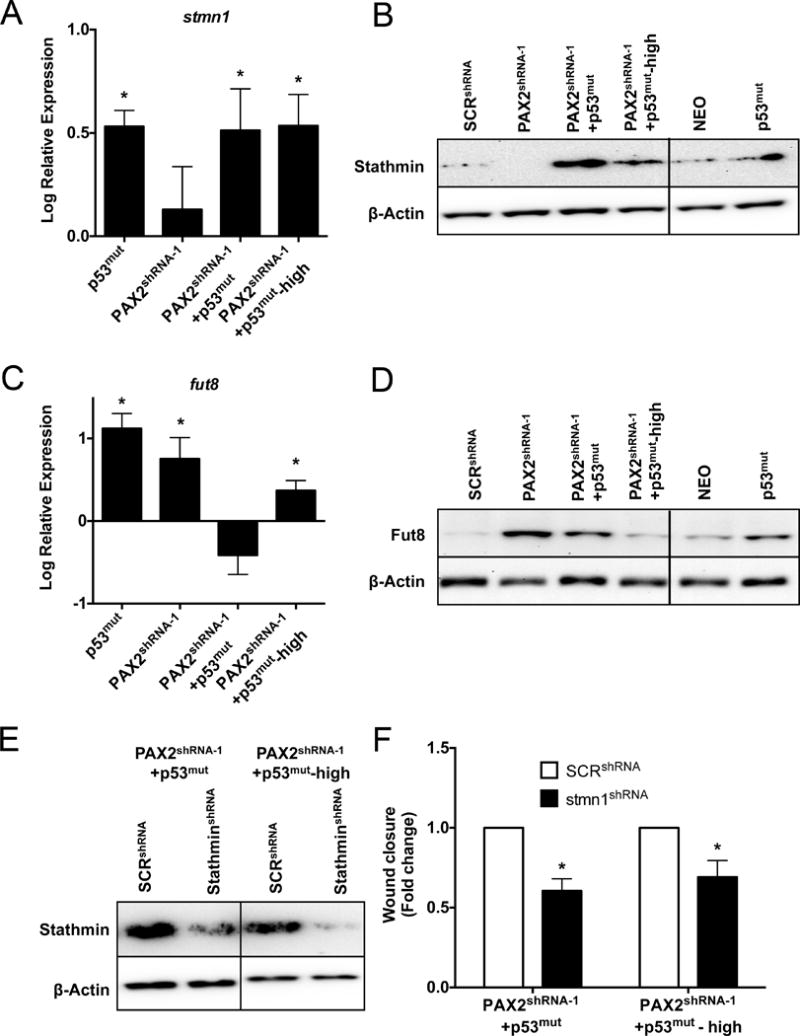
qPCR analysis of A) stmn1 and C) fut8 mRNA expression and western blot analysis of B) stathmin and D) fut8 protein expression in MOE:p53mut, MOE:PAX2shRNA-1, MOE:PAX2shRNA-1+p53mut and MOE:PAX2shRNA-1+p53mut-high cells. E) Western blot validating transient knockdown of stathmin in MOE:PAX2shRNA-1+p53mut and MOE:PAX2shRNA-1+p53mut-high cells. F) Migration assay (12 hours) comparing MOE:PAX2shRNA-1+p53mut and MOE:PAX2shRNA-1+p53mut-high cells transiently transfected with SCRshRNA and stathminshRNA.
Loss of PTEN regulates PAX2 and re-expression of PAX2 is sufficient to block tumorigenesis
Currently, there are no reported mechanisms for how PAX2 would be lost in fallopian tube cells. Joint loss of PAX2 and PTEN has been reported in endometrial carcinomas(32). MOE:PTENshRNA cells formed high-grade oviductal cancer when xenografted i.p. indicating that loss of PTEN in fallopian tube cells is sufficient to drive tumorigenesis in vivo (9). Analysis of PAX2 expression in MOE:PTENshRNA revealed a significant reduction of PAX2 mRNA (Fig. 3A) and protein (Fig. 3B) compared to scrambled shRNA control. To investigate the functional significance of PAX2 re-expression in PTEN silenced fallopian tube cells, MOE:PTENshRNA were stably transfected with a human PAX2 overexpression plasmid. Two clones (MOE:PTENshRNA+PAX2(Clone 1) and MOE:PTENshRNA+PAX2(Clone 2)) were generated and validated for PAX2 overexpression and PTEN knockdown (Supplementary Fig. S4A). Functional assays revealed that re-expression of PAX2 in PTENshRNA MOE cells significantly reduced PTENshRNA-induced proliferation (Fig. 3C) and migration (Fig. 3D). MOE:PTENshRNA+PAX2(Clone 1) and MOE:PTENshRNA+PAX2(Clone 2) cells were i.p. xenografted into nude mice. MOE:PTENshRNA formed metastatic disease at 153 ± 24 days. One out of the three mice xenografted with MOE:PTENshRNA+PAX2(Clone 1) cells developed metastatic disease on day 169 that had a similar presentation to MOE:PTENshRNA, while the remaining mice remained disease free. All the mice xenografted with MOE:PTENshRNA+PAX2(Clone 2) cells were disease free with no tumor formation (Fig. 3E, Supplementary Fig. S4B). Representative images of the gross tumor morphology of MOE:PTENshRNA grafted mice are shown in Supplementary Fig. S4C. H&E staining determined the tumors to be histologically similar to the previous study(9). Gross tumor morphology of one of the animals xenografted with MOE:PTENshRNA+PAX2(Clone 1) was similar to MOE:PTENshRNA. PAX2, PAX8 and WT-1 staining determined the PTENshRNA+PAX2(Clone 1) tumors to be of a similar pathological grade as MOE:PTENshRNA (Supplementary Fig. S4C) and with very low PAX2 protein staining. Representative H&E staining of reproductive tracts from each clone expressing PAX2 in MOE:PTENshRNA cells, demonstrated normal architecture of the uteri, oviduct and ovaries (data not shown). These results show that re-expressing PAX2 in PTENshRNA MOE cells attenuated and/or eliminated PTENshRNA-induced tumors and increased survival.
Figure 3. Loss of PTEN negatively regulates PAX2 and re-expression of PAX2 is sufficient to reduce PTENshRNA-induced tumorigenesis.
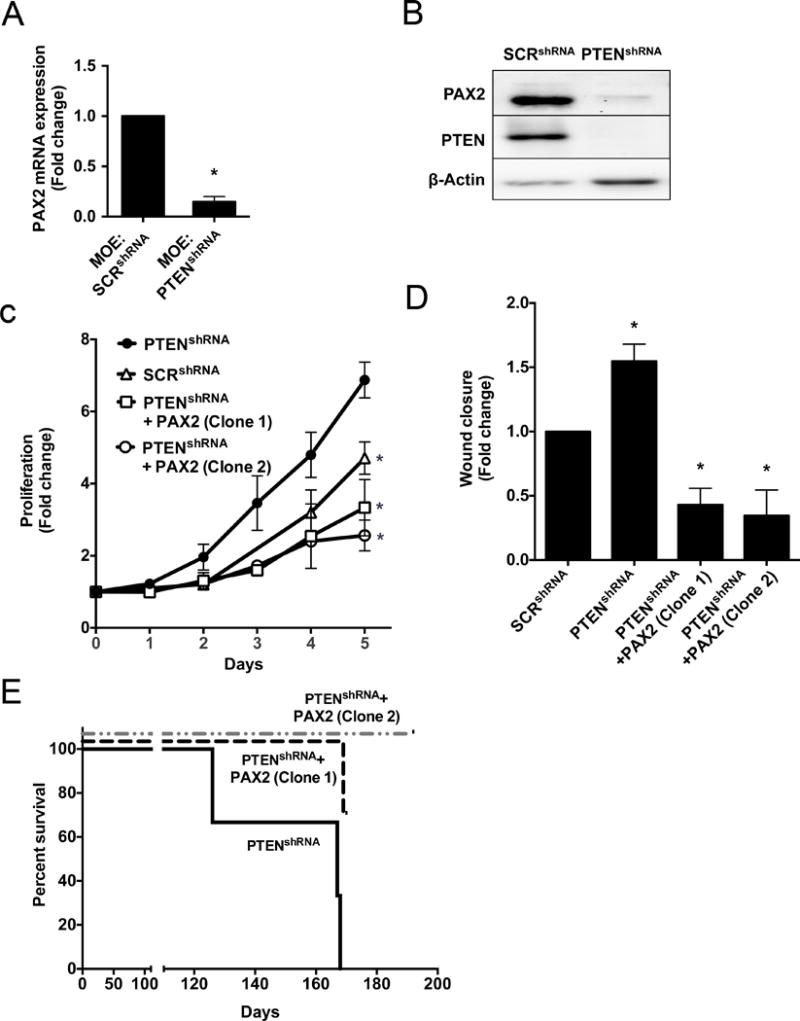
MOE:PTENshRNA cells expressed significantly lower PAX2 A) mRNA and B) protein compared to MOE:SCRshRNA control. MOE:PTENshRNA+PAX2 (Clone 1 and 2) reduced MOE:PTENshRNA-induced C) proliferation (day 5) and D) migration (hour 8). E) Kaplan-Meier survival plot demonstrating that PTENshRNA+PAX2 xenorafted mice survive longer (n=3).
PAX2 overexpression in human high-grade serous cancer cell lines reduces proliferation and migration via apoptosis
PAX2 expression is absent in benign precursors and malignant STICs as well as in HGSC (8). PAX2 is also absent in human HGSC cell lines (23). A panel of HGSC cell lines (OVCAR3, OVCAR4, KURAMOCHI and OVSAHO) lacked expression of PAX2 protein (Supplementary Fig. S5A). Attempts to stably express PAX2 in HGSC cells failed, likely due to cell death. Therefore, to determine the impact of re-introducing PAX2, HGSC cells (OVCAR3, OVCAR4, KURAMOCHI, OVSAHO) were transiently transfected with PAX2 or NEO (control) plasmid for 48 hours. PAX2 protein expression was confirmed by western blot (Supplementary Fig. S5B). PAX2 expression in the OVCAR3 and OVCAR4 cells resulted in significantly lower proliferation on day 3 (Fig. 4A) and reduced migration over 24 hours (Fig. 4B, Supplementary Fig. S5C) compared to those transfected with NEO. To determine the effect of PAX2 on apoptosis in OVCAR4 cells, transiently transfected cells were subjected to Annexin V/APC and PI staining. Flow cytometry analyses revealed that PAX2 transfected cells had significantly more cells in early and late stages of apoptosis (Fig. 4C). These data suggest that PAX2 re-expression in human cell lines may increase apoptosis.
Figure 4. PAX2 re-expression in human HGSC cell lines reduces proliferation and migration via increased apoptosis.
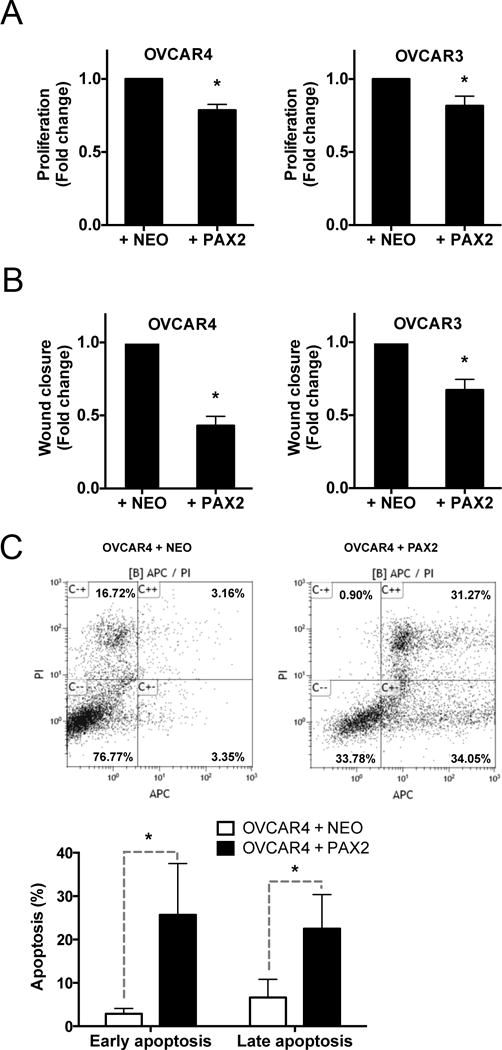
PAX2 transient expression in PAX2-null serous cancer cell lines resulted in significant reduction in A) proliferation (day 3) and B) migration (hour 8). C) PAX2 expression in OVCAR4 cells significantly increased early and late apoptosis compared to OVCAR4 cells treated with NEO (measured by flow cytometry) at hour 48.
Wild-type p53 directly binds to and activates PAX2 promoter whereas mutation in p53 significantly reduces p53 occupancy and expression of PAX2 in FTE
Since p53 had previously been reported to regulate PAX2 in the kidney, luciferase and chromatin immunoprecipitation (ChIP) assays were performed in MOE cells. MOE cells with the murine PAX2 promoter driving luciferase expression revealed that wild-type p53 increased PAX2 promoter activity. In contrast, mutant p53 significantly repressed PAX2 promoter activation (Fig. 5A). In silico analysis revealed two putative binding sites of p53 on the murine PAX2 promoter. Chromatin from MOE and MOE:p53mut cells were prepared for immunoprecipitation assays. Isolated chromatin was precipitated using IgG and p53 antibodies. The p53 occupancy on GAPDH promoter vs. PAX2 promoter was used to compare degree of enrichment relative to input. Wild-type p53 had a higher occupancy on the PAX2 promoter compared to IgG indicating that p53 can directly bind to the PAX2 promoter (Fig. 5B). The R273H mutation of p53 significantly abrogated occupancy on the PAX2 promoter (Fig. 5B). PCR primer sets (Supplementary Table S4) were designed against the p53-PAX2 promoter binding region, a non-target region (Fig. 5C), MDM2 (positive control for p53 occupancy Fig. 5D) and ATF (positive control for mutant p53 occupancy, Fig. 5E). These data suggest that mutant P53 represses whereas wild-type p53 activates PAX2 transcriptional activity by directly regulating the promoter.
Figure 5. PAX2 is a direct transcriptional target of wild-type p53 and mutant p53 represses the PAX2 promoter in MOE cells.
A) Wild-type p53 activates whereas p53mut represses PAX2 transcriptional promoter activation as measured by luciferase assay. B) Chromatin Immunoprecipitation (ChIP) analysis revealed that wild-type p53 enhanced whereas p53mut significantly reduced p53 occupancy on the PAX2 promoter. Statistical differences (p≤0.05) are between groups labeled a and b. ChIP analysis demonstrates wild-type p53 occupancy on C) Non-target control and D) MDM2, mutant p53 occupancy on the E) ATF promoter.
Identification of small molecules that can re-activate PAX2 expression in fallopian tube epithelial cells
Bioinformatic analyses of the TCGA indicates that PAX2 is not methylated as a mechanism for repression in HGSC and that it is rarely mutated(33). Therefore, transcriptional repression or lack of activation could be one mechanism responsible for the loss of PAX2. Identifying molecules that can restore PAX2 expression could provide therapeutic avenues to slow progression of pre-neoplastic lesions or even reverse them. A screening assay with the 4.1 kb murine PAX2 promoter driving luciferase construct was used to investigate if small molecules could increase expression of PAX2. The murine and human PAX2 promoters share 80% sequence homology(34). Prestwick library was used at 10 μM for 10 hours to find molecules that stimulate PAX2 expression in MOE cells(35). The primary assay revealed that the top 2% of the compounds increased PAX2 promoter activity by 193% ± 23% (Mean ± SD) compared to cells treated with DMSO (Supplementary Fig. S6A). Upon re-testing, 12 out of 24 compounds showed consistent activity (Fig. 6A). A secondary biological screen revealed that 4 out of 12 compounds activated the endogenous PAX2 mRNA transcript in MOE cells (Supplementary Fig. S6B). In response to these data, relevance in HGSC and published reports, future investigation was focused on determining the effect of luteolin on MOE cells. Luteolin increased PAX2 protein and mRNA expression in MOE cells at 10 hours post incubation (Fig. 6B). Luteolin significantly reduced proliferation and migration in MOE cells (Fig. 6C). Luteolin failed to alter PAX2 mRNA and protein expression in MOE: PTENshRNA cells (Supplementary Fig. S6D). Future investigations will focus on evaluating the role of luteolin in combination with existing treatments as well as its effect in HGSC cell lines. Using a human promoter for the luteolin experiments could accurately reflect the direct correlation between luteolin and induction of PAX2 which will also be tested in the future.
Figure 6. Biological screen to identify small molecules that can enhance activation of the proximal PAX2 promoter in MOE cells.
A) Top 12 compounds that consistently activated PAX2 promoter activity upon re-testing by primary biological assay. Luteolin increased PAX2 B) mRNA and protein in MOE cells compared to DMSO. Functionally, luteolin significantly decreased C) proliferation (day 3) and migration (hour 24) in MOE cells.
DISCUSSION
The aim of this study was to identify the functional role and regulation of PAX2 in normal fallopian tube epithelium and high-grade serous ovarian cancer. Studies published so far have shown divergent roles of PAX2 depending on the tumor histotype(23–25, 36, 37). Consistent with pathological observations, published microarray data indicates that PAX2 was significantly downregulated in STIC and HGSC tissues compared to normal FTE(27). A small subset of TCGA patients with amplification in the PAX2 gene, had a longer disease-free interval before relapse. The present study utilized MOE cells that were altered to knockdown PAX2 and express the R273H mutant of p53. Although, PAX2 loss alone did not have a significant effect on MOE characteristics, combining it with a p53 mutation resulted in functional changes in vitro, but was not sufficient to drive tumor formation. Re-expression of PAX2 in fallopian tube-derived tumor models reduced tumor burden and a series of small molecules was identified that enhanced PAX2 mRNA expression in MOE cells.
Several studies have demonstrated that the fallopian tube is likely one of the sources of origin of HGSC(9, 11, 13, 16, 38). However, no models of SCOUTs or of fallopian tube cells lacking PAX2 have been reported. A study by Perets et. al. showed that PAX2 was present in the tumors derived from transgenic animal models(12). Similarly, another high-grade carcinoma animal model presented with higher PAX2 expression as the cells became tumorigenic(16). These data along with those in this report suggest that loss of PAX2 alone is not sufficient for tumor progression from fallopian tube cells. The lack of tumorigenicity in vivo of the SCOUT and p53 signature cellular models resonated with their benign pathological designation. Serial passaging of the MOE cells harboring both alterations resulted in an increase in all functional characteristics. These data are consistent with a previous study performed in serially passaged fallopian tube cells that indicate that aging is sufficient to induce functional alterations(16). In contrast to the tumorigenic model of serially passaging MOE cells on a CD1 background, the current model with PAX2 knocked down and p53 mutated did not form tumors after serial passaging. These data suggest that loss of PAX2 and mutation of p53 are not sufficient to drive tumorigenesis. In addition, it suggests that a latency period may exist between SCOUTs, p53 signatures, and tumor formation that may increase the opportunity for intervention or prevention.
Gene expression analysis of MOE cells harboring altered PAX2 and mutant p53 expression revealed stathmin and fut8 to be regulated by PAX2 in the FTE. Our results suggest that serially passaged MOE cells with PAX2 knockdown and p53 mutation resemble Type II SCOUTs. The similarity between the present models and benign SCOUTs may further explain the lack of tumorigenicity seen in vivo. Stathmin is a microtubule destabilizing protein that is overexpressed in HGSC(30). Consistent with stathmin’s established role in influencing migration in other tumor types, knockdown of stathmin in MOE:PAX2shRNA-1+p53mut cells and it’s “aged” variant, significantly reduced migration(39). Investigators have shown that stathmin knockdown has anti-tumorigenic effects in animal models, making it be a favorable drug target in several tumor types(40, 41). As per the TCGA, patients with STMN1 gene amplification demonstrated a significantly lower overall survival compared to patients without the alteration (Supplementary Fig. S3B)(42). In agreement with the TCGA data, fut8 is amplified in serially passaged MOE: PAX2shRNA-1+p53mut. Fut8 protein is highly expressed in MOE:PAX2shRNA-1 cells and this increased expression is progressively inhibited when combined with p53 mutation. Interestingly, a recently published undifferentiated high-grade carcinoma mouse model that expressed PAX2 and a p53 splice variant(16), failed to express fut8. In contrast, MOE cells harboring mutation of p53 and overexpression of KRASG12V, that formed poorly differentiated carcinomas in vivo(15), did not express PAX2, but expressed fut8 (Supplementary Fig. S6C). These data further support that loss of PAX2 influences fut8 expression in murine models of fallopian-derived tumors. Further investigation of aberrant pathway modulation downstream of PAX2 can assist to inform upon benign to malignant transformation.
Consistent with endometrial cancers, MOE cells harboring PTEN loss had less PAX2 and formed high-grade carcinomas(9). Stable re-expression of PAX2 in PTEN-lacking MOE cells reduced tumor burden that is otherwise induced by PTEN loss. If the mice xenografted with MOE: PTENshRNA+PAX2 cells were allowed to survive longer, it is possible that tumors may form but after a longer time period, which was not examined in this study. Re-introduction of PAX2 in HGSC cells reduced cell viability(23), similar to what is shown in this study using HGSC cells. Interestingly, multiple attempts to create a stable HGSC cell line with PAX2 overexpression led to cell death. Intriguingly, the ability to successfully develop stable PAX2 variants in MOE:PTENshRNA cells in contrast to human HGSC cell lines suggests that additional mutations, cell of origin, or species variations may impact the role of PAX2 in cancer cells. PAX2 overexpression in HGSC cells that lacked its expression confirmed that restoring PAX2 can induce apoptosis. A modest decrease in proliferation and migration upon PAX2 overexpression in HGSC cells suggests that introduction of PAX2 alone may have less impact as cells become malignant and accrue multiple changes in tumor signaling. Future studies will focus on whether strategies to restore PTEN or block AKT and PI3K may contribute to reduced tumor burden partially through enhanced expression of PAX2.
If early putative lesions of FTE-derived HGSC lose PAX2, and PAX2 continues to be lost in many tumor samples, then restoration of PAX2 might provide a route for disease prevention. Using a cell-based screening assay, luteolin was identified to be a potential “hit” that can increase PAX2 expression in MOE cells. Luteolin is a flavonoid that has anti-cancer properties and has the potential to slow tumor growth(43). Mechanistically, luteolin is known to inhibit NFκB transcription and activate c-Jun(44). NFκB has putative binding sites on the PAX2 promoter whereas c-Jun phosphorylates PAX2 that increases PAX2-dependent transcription(45).
Wild-type p53 was able to activate whereas mutant p53 repressed PAX2 promoter activity in MOE cells. Similar results were reported in mouse embryonic kidney cells where a ChIP-Seq analysis revealed multiple peaks of p53 occupancy on the PAX2 promoter(46). In light of these results, what is unclear is whether elimination of repression from mutant p53 is critical to enhance PAX2 in cancer cells. Treatment of HGSC cells with luteolin failed to demonstrate an increase in PAX2. It may be particularly interesting to combine luteolin with a mutant p53-repressing molecule (e.g. NSC59984) or an AKT inhibitor (e.g. MK2206) to observe their additive effect on PAX2 activation(47).
In summary, this is the first study to outline the functional role and regulation of PAX2 in the murine oviductal epithelial cells and HGSC. Combining mutant p53 with PAX2 loss was critical to observe functional changes, which could explain how early lesions form in the FTE before progressing to serous tumors. Our data also revealed that PTEN loss and mutation of p53 contributes to loss of PAX2 in MOE cells. These data suggest a dynamic interplay between PTEN, PAX2 and p53 manifested in human samples. These findings also imply that strategies to restore PTEN may contribute to reduced tumor burden partially through enhanced expression of PAX2. A novel proof-of-concept small molecule screen, that could enhance expression of key proteins lost during progression, such as PAX2, is presented in this study. Overall, understanding the molecular basis of precursor lesion development is critical to early detection strategies that may help to halt serous cancer progression.
MATERIALS AND METHODS
Animals
All animals were treated in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and the established Institutional Animal Use and Care protocol at the University of Illinois at Chicago. All xenograft experiments were performed as described previously(9, 15).
Cell culture
MOE and HGSC cell lines (OVCAR3, OVCAR4, KURAMOCHI, OVSAHO) were cultured as previously described(48, 49). All lines were authenticated by STR analysis at the University of Illinois at Chicago in May 2015. Cell lines were transfected using TransIT LT1™ with plasmids containing the gene of interest (Supplementary Table S1) as described previously(9).
Immunohistochemistry (IHC)
Tissues were prepared for immunohistochemistry or hematoxylin and eosin stain as described in past publications(9, 50). Images were acquired on a Nikon Eclipse E600 microscope using a DS-Ri1 digital camera and NIS Elements software. Antibody information – Supplementary Table S2.
Cell viability assay
MOE cells were seeded into 96-well plates at 1.0 × 103 cells per well. Proliferation was measured using sulforhodamine B (SRB) colorimetric assay and analysis was completed using a Biotek Synergy 2 microplate reader as described previously(15, 51).
Soft agar colony assay
Soft agar colony formation assay was performed as previously described(15). Images were captured on day 14 in culture. Colonies were counted at 4× magnification using ImageJ software.
Wound healing assay
Cells were seeded at 1.0 × 105 cells per well and assay was performed as described previously(15, 51). Pictures were taken at 0 and 8 or 12 or 24 hours after scratching using an AmScope MU900 with Toupview software (AmScope, Irvine, CA).
Western blot analyses
Protein lysate (30μg) was run on SDS-PAGE and transferred to nitrocellulose membrane. Blots were blocked with 5% milk or BSA in TBS-T and probed at 4°C overnight with primary antibodies (Supplementary Table S2). Anti-rabbit HRP-linked secondary antibody was used for 30 min in blocking buffer. Membranes were incubated and developed as described previously(15).
RNA isolation, cDNA synthesis and RT-PCR
RNA extraction was performed as described in(15). iScript™ cDNA synthesis kit (BioRad, Hercules, CA) and SYBR green (Roche, Madison, WI) were used according to manufacturer’s instructions. All qPCR runs were performed on the ABI ViiA7 (Life Technologies, San Diego, CA).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation was performed as described previously(52). Briefly, for each pulldown, 30 μl Dynabeads protein G were incubated with normal rabbit IgG (Cell signaling, 1:100) and p53 (Santa Cruz, 1:100) overnight at 4°C. Buffer recipes listed in Supplementary Table S3.
High Throughput Screen (HTS)
The UIC HTS core facility provided a library (Prestwick) of 1200 FDA approved drugs. Primary assay: MOE cells were transiently transfected with the PAX2 promoter driving luciferase expression and β-Galactosidase (18 hours). Cells were seeded in 96-well plates at 7.5 × 103 cells/well and incubated with drugs (10 μM) for 10 hours at 37°C in a 5% CO2 incubator. Luciferase readout was measured as described previously(53). Secondary assay: MOE cells were treated with 10 μM of “hits”. Cells were lysed after 10 hours to determine PAX2 mRNA (qPCR) and protein (western blot analyses).
Flow cytometry
OVCAR4 cells were transiently transfected with NEO or PAX2 plasmid for 48 hours followed by trypsinization. Flow cytometry analysis was performed as described as described previously(54, 55).
Statistical analyses
All data are represented as mean ± standard error. Statistical analysis was carried out using Prism software (GraphPad, La Jolla, CA). All conditions were tested in three replicates in at least triplicate experiments. Statistical significance was determined by Student’s t-test, or one-way ANOVA with Dunnett’s post-hoc test. *p ≤ 0.05 was considered significant.
Supplementary Material
Acknowledgments
This work was supported by NIH R21 CA208610-01 (J.E.B.) and the Office of the Director, National Institutes of Health (OD) & National Center for Complementary & Integrative Health (NCCIH) (T32AT007533) (M.D.). We thank Dr. Barbara Vanderhyden (University of Ottawa) for kindly providing MOE and MOSE cells, Dr. Kwong Wong (University of Texas, M.D. Anderson Cancer Center) for the NEO and PAX2 plasmids, Dr. Gregory Dressler (University of Michigan) for the PAX2-Luc plasmid, Dr. Kiira Ratia (University of Illinois at Chicago, HTS Core) for providing compounds for screening.
Financial Support: American Cancer Society
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
SUPPLEMENTARY INFORMATION
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Auersperg N. The origin of ovarian carcinomas: a unifying hypothesis. Int J Gynecol Pathol. 2011;30(1):12–21. doi: 10.1097/PGP.0b013e3181f45f3e. [DOI] [PubMed] [Google Scholar]
- 3.Piek JM, van Diest PJ, Zweemer RP, Jansen JW, Poort-Keesom RJ, Menko FH, et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J Pathol. 2001;195(4):451–6. doi: 10.1002/path.1000. [DOI] [PubMed] [Google Scholar]
- 4.Kindelberger DW, Lee Y, Miron A, Hirsch MS, Feltmate C, Medeiros F, et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am J Surg Pathol. 2007;31(2):161–9. doi: 10.1097/01.pas.0000213335.40358.47. [DOI] [PubMed] [Google Scholar]
- 5.Medeiros F, Muto MG, Lee Y, Elvin JA, Callahan MJ, Feltmate C, et al. The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am J Surg Pathol. 2006;30(2):230–6. doi: 10.1097/01.pas.0000180854.28831.77. [DOI] [PubMed] [Google Scholar]
- 6.Shaw PA, Rouzbahman M, Pizer ES, Pintilie M, Begley H. Candidate serous cancer precursors in fallopian tube epithelium of BRCA1/2 mutation carriers. 2009;22(9):1133–8. doi: 10.1038/modpathol.2009.89. [DOI] [PubMed] [Google Scholar]
- 7.Chen EY, Mehra K, Mehrad M, Ning G, Miron A, Mutter GL, et al. Secretory cell outgrowth, PAX2 and serous carcinogenesis in the Fallopian tube. J Pathol. 2010;222(1):110–6. doi: 10.1002/path.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehra K, Mehrad M, Ning G, Drapkin R, McKeon FD, Xian W, et al. STICS, SCOUTs and p53 signatures; a new language for pelvic serous carcinogenesis. Front Biosci (Elite Ed) 2011;3:625–34. doi: 10.2741/e275. [DOI] [PubMed] [Google Scholar]
- 9.Eddie SL, Quartuccio SM, Oh E, Moyle-Heyrman G, Lantvit DD, Wei JJ, et al. Tumorigenesis and peritoneal colonization from fallopian tube epithelium. Oncotarget. 2015;6(24):20500–12. doi: 10.18632/oncotarget.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jazaeri AA, Bryant JL, Park H, Li H, Dahiya N, Stoler MH, et al. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia. 2011;13(10):899–911. doi: 10.1593/neo.11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karst AM, Levanon K, Drapkin R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc Natl Acad Sci U S A. 2011;108(18):7547–52. doi: 10.1073/pnas.1017300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell. 2013;24(6):751–65. doi: 10.1016/j.ccr.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc Natl Acad Sci U S A. 2012;109(10):3921–6. doi: 10.1073/pnas.1117135109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherman-Baust CA, Kuhn E, Valle BL, Shih Ie M, Kurman RJ, Wang TL, et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J Pathol. 2014;233(3):228–37. doi: 10.1002/path.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quartuccio SM, Karthikeyan S, Eddie SL, Lantvit DD, Oh E, Modi DA, et al. Mutant p53 expression in fallopian tube epithelium drives cell migration. Int J Cancer. 2015;137(7):1528–38. doi: 10.1002/ijc.29528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Endsley MP, Moyle-Heyrman G, Karthikeyan S, Lantvit DD, Davis DA, Wei JJ, et al. Spontaneous Transformation of Murine Oviductal Epithelial Cells: A Model System to Investigate the Onset of Fallopian-Derived Tumors. Front Oncol. 2015;5:154. doi: 10.3389/fonc.2015.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dressler GR, Woolf AS. Pax2 in development and renal disease. Int J Dev Biol. 1999;43(5):463–8. [PubMed] [Google Scholar]
- 18.Dressler GR, Deutsch U, Chowdhury K, Nornes HO, Gruss P. Pax2, a new murine paired-box-containing gene and its expression in the developing excretory system. Development. 1990;109(4):787–95. doi: 10.1242/dev.109.4.787. [DOI] [PubMed] [Google Scholar]
- 19.Lechner MS, Levitan I, Dressler GR. PTIP, a novel BRCT domain-containing protein interacts with Pax2 and is associated with active chromatin. Nucleic Acids Res. 2000;28(14):2741–51. doi: 10.1093/nar/28.14.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel SR, Kim D, Levitan I, Dressler GR. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell. 2007;13(4):580–92. doi: 10.1016/j.devcel.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu J, Prindle MJ, Dressler GR, Yu X. PTIP regulates 53BP1 and SMC1 at the DNA damage sites. J Biol Chem. 2009;284(27):18078–84. doi: 10.1074/jbc.M109.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ning G, Bijron JG, Yamamoto Y, Wang X, Howitt BE, Herfs M, et al. The PAX2-null immunophenotype defines multiple lineages with common expression signatures in benign and neoplastic oviductal epithelium. J Pathol. 2014;234(4):478–87. doi: 10.1002/path.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song H, Kwan SY, Izaguirre DI, Zu Z, Tsang YT, Tung CS, et al. PAX2 Expression in Ovarian Cancer. Int J Mol Sci. 2013;14(3):6090–105. doi: 10.3390/ijms14036090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quick CM, Ning G, Bijron J, Laury A, Wei TS, Chen EY, et al. PAX2-null secretory cell outgrowths in the oviduct and their relationship to pelvic serous cancer. Mod Pathol. 2012;25(3):449–55. doi: 10.1038/modpathol.2011.175. [DOI] [PubMed] [Google Scholar]
- 25.Al-Hujaily EM, Tang Y, Yao DS, Carmona E, Garson K, Vanderhyden BC. Divergent Roles of PAX2 in the Etiology and Progression of Ovarian Cancer. Cancer Prev Res (Phila) 2015;8(12):1163–73. doi: 10.1158/1940-6207.CAPR-15-0121-T. [DOI] [PubMed] [Google Scholar]
- 26.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto Y, Ning G, Howitt BE, Mehra K, Wu L, Wang X, et al. In vitro and in vivo correlates of physiological and neoplastic human Fallopian tube stem cells. J Pathol. 2015:1096–9896. doi: 10.1002/path.4649. Electronic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8(12):e82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quartuccio SM, Karthikeyan S, Eddie SL, Lantvit DD, Óh E, Modi DA, et al. Mutant p53 expression in fallopian tube epithelium drives cell migration. 2015;137(7):1528–38. doi: 10.1002/ijc.29528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karst AM, Levanon K, Duraisamy S, Liu JF, Hirsch MS, Hecht JL, et al. Stathmin 1, a marker of PI3K pathway activation and regulator of microtubule dynamics, is expressed in early pelvic serous carcinomas. Gynecol Oncol. 2011;123(1):5–12. doi: 10.1016/j.ygyno.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novak M, Lester J, Karst AM, Parkash V, Hirsch MS, Crum CP, et al. Stathmin 1 and p16(INK4A) are sensitive adjunct biomarkers for serous tubal intraepithelial carcinoma. Gynecol Oncol. 2015;139(1):104–11. doi: 10.1016/j.ygyno.2015.07.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monte NM, Webster KA, Neuberg D, Dressler GR, Mutter GL. Joint loss of PAX2 and PTEN expression in endometrial precancers and cancer. Cancer Res. 2010;70(15):6225–32. doi: 10.1158/0008-5472.CAN-10-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stayner CK, Cunliffe HE, Ward TA, Eccles MR. Cloning and characterization of the human PAX2 promoter. J Biol Chem. 1998;273(39):25472–9. doi: 10.1074/jbc.273.39.25472. [DOI] [PubMed] [Google Scholar]
- 35.Patel SR, Dressler GR. Expression of Pax2 in the intermediate mesoderm is regulated by YY1. Dev Biol. 2004;267(2):505–16. doi: 10.1016/j.ydbio.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 36.Roh MH, Yassin Y, Miron A, Mehra KK, Mehrad M, Monte NM, et al. High-grade fimbrial-ovarian carcinomas are unified by altered p53, PTEN and PAX2 expression. Mod Pathol. 2010;23(10):1316–24. doi: 10.1038/modpathol.2010.119. [DOI] [PubMed] [Google Scholar]
- 37.Monte NM, Webster KA, Neuberg D, Dressler GR, Mutter GL. Joint loss of PAX2 and PTEN expression in endometrial precancers and cancer. 2010;70(15):6225–32. doi: 10.1158/0008-5472.CAN-10-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levanon K, Ng V, Piao HY, Zhang Y, Chang MC, Roh MH, et al. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene. 2010;29(8):1103–13. doi: 10.1038/onc.2009.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, Spessotto P, et al. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell. 2005;7(1):51–63. doi: 10.1016/j.ccr.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 40.Alli E, Bash-Babula J, Yang JM, Hait WN. Effect of stathmin on the sensitivity to antimicrotubule drugs in human breast cancer. Cancer Res. 2002;62(23):6864–9. [PubMed] [Google Scholar]
- 41.Rana S, Maples PB, Senzer N, Nemunaitis J. Stathmin 1: a novel therapeutic target for anticancer activity. Expert Rev Anticancer Ther. 2008;8(9):1461–70. doi: 10.1586/14737140.8.9.1461. [DOI] [PubMed] [Google Scholar]
- 42.Integrated genomic analyses of ovarian carcinoma. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin Y, Shi R, Wang X, Shen HM. Luteolin, a flavonoid with potential for cancer prevention and therapy. Curr Cancer Drug Targets. 2008;8(7):634–46. doi: 10.2174/156800908786241050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JS, Jobin C. The flavonoid luteolin prevents lipopolysaccharide-induced NF-kappaB signalling and gene expression by blocking IkappaB kinase activity in intestinal epithelial cells and bone-marrow derived dendritic cells. Immunology. 2005;115(3):375–87. doi: 10.1111/j.1365-2567.2005.02156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai Y, Lechner MS, Nihalani D, Prindle MJ, Holzman LB, Dressler GR. Phosphorylation of Pax2 by the c-Jun N-terminal kinase and enhanced Pax2-dependent transcription activation. J Biol Chem. 2002;277(2):1217–22. doi: 10.1074/jbc.M109663200. [DOI] [PubMed] [Google Scholar]
- 46.Saifudeen Z, Liu J, Dipp S, Yao X, Li Y, McLaughlin N, et al. A p53-Pax2 pathway in kidney development: implications for nephrogenesis. PLoS One. 2012;7(9):e44869. doi: 10.1371/journal.pone.0044869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang S, Zhou L, Hong B, van den Heuvel AP, Prabhu VV, Warfel NA, et al. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015;75(18):3842–52. doi: 10.1158/0008-5472.CAN-13-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.King SM, Quartuccio SM, Vanderhyden BC, Burdette JE. Early transformative changes in normal ovarian surface epithelium induced by oxidative stress require Akt upregulation, DNA damage and epithelial-stromal interaction. Carcinogenesis. 2013;34(5):1125–33. doi: 10.1093/carcin/bgt003. [DOI] [PubMed] [Google Scholar]
- 49.Mitra AK, Davis DA, Tomar S, Roy L, Gurler H, Xie J, et al. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol Oncol. 2015;138(2):372–7. doi: 10.1016/j.ygyno.2015.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.King SM, Hilliard TS, Wu LY, Jaffe RC, Fazleabas AT, Burdette JE. The impact of ovulation on fallopian tube epithelial cells: evaluating three hypotheses connecting ovulation and serous ovarian cancer. Endocr Relat Cancer. 2011;18(5):627–42. doi: 10.1530/ERC-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hilliard TS, Modi DA, Burdette JE. Gonadotropins activate oncogenic pathways to enhance proliferation in normal mouse ovarian surface epithelium. Int J Mol Sci. 2013;14(3):4762–82. doi: 10.3390/ijms14034762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vasudevan D, Hickok JR, Bovee RC, Pham V, Mantell LL, Bahroos N, et al. Nitric Oxide Regulates Gene Expression in Cancers by Controlling Histone Posttranslational Modifications. Cancer Res. 2015;75(24):5299–308. doi: 10.1158/0008-5472.CAN-15-1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sukerkar PA, MacRenaris KW, Townsend TR, Ahmed RA, Burdette JE, Meade TJ. Synthesis and biological evaluation of water-soluble progesterone-conjugated probes for magnetic resonance imaging of hormone related cancers. Bioconjug Chem. 2011;22(11):2304–16. doi: 10.1021/bc2003555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodgers LH, Oh E, Young AN, Burdette JE. Loss of PAX8 in high-grade serous ovarian cancer reduces cell survival despite unique modes of action in the fallopian tube and ovarian surface epithelium. Oncotarget. 2016 doi: 10.18632/oncotarget.9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen WL, Pan L, Kinghorn AD, Swanson SM, Burdette JE. Silvestrol induces early autophagy and apoptosis in human melanoma cells. BMC cancer. 2016;16:17. doi: 10.1186/s12885-015-1988-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.