

Abstract
Purpose
Patients with ulcerative colitis (UC) are at increased risk for colorectal cancer, although mechanisms underlying neoplastic transformation are poorly understood. We sought to evaluate the role of microRNAs in neoplasia development in this high-risk population.
Experimental Design
Tissue from 12 controls, 9 UC patients without neoplasia, and 11 UC patients with neoplasia was analyzed. miRNA array analysis was performed and select miRNAs assayed by real-time PCR on the discovery cohort and a validation cohort. DNA methylation of miR-193a was assessed. Following transfection of miR-193a-3p, proliferation, IL17RD expression, and luciferase activity of the 3’UTR of IL17RD were measured. Tumor growth in xenografts as well as EGFR signaling were assessed in HCT116 cells expressing IL17RD with either a mutant 3’ untranslated region (UTR) or wild-type (WT) 3’UTR.
Results
miR-31, miR-34a, miR-106b, and miR-193a-3p were significantly dysregulated in UC-neoplasia and adjacent tissue. Significant down-regulation of miR-193a-3p was also seen in an independent cohort of UC-cancers. Changes in methylation of miR-193a or expression of pri-miR-193a were not observed in UC-cancer. Transfection of miR-193a-3p resulted in decreased proliferation, and identified IL17RD as a direct target of miR-193a-3p. IL17RD expression was increased in UC-cancers, and miR-193a-3p treatment decreased growth and EGFR signaling of HCT116 cells in xenografts expressing both IL17RD with WT 3’UTR compared to cells expressing IL17RD with mutant 3’UTR.
Conclusions
miR-193a-3p is down-regulated in UC-neoplasia, and its loss promotes carcinogenesis through up-regulation of IL17RD. These findings provide novel insight into inflammation-driven CRC and could suggest new therapeutic targets in this high-risk population.
Keywords: Inflammatory Bowel Disease, Ulcerative Colitis, miRNA, Dysplasia, Colon Cancer
Introduction
Patients with long-standing ulcerative colitis (UC) are at increased risk for colorectal cancer (CRC) (1,2). Although many mechanisms of carcinogenesis in IBD remain unknown, neoplastic lesions likely result from a combination of genetic alterations, inflammatory mediators, and activation of cell signaling pathways. Discovery of novel genomic mediators of these pathways could greatly facilitate risk stratification as well as treatment and chemoprevention strategies.
Gene expression is regulated in part by a class of non-coding RNAs, termed microRNAs (miRNAs). These small RNAs regulate gene expression by binding to the 3’ untranslated region (UTR) of messenger RNA (mRNA) and either inhibit protein translation or destabilize target mRNA (3). Changes in miRNA expression have been implicated in the pathogenesis of many cancer types, including colon cancer (4–7). miRNAs control the expression of signaling molecules and play important roles in cell growth (8). As such, growth promoting or cell death inhibiting miRNAs, if up-regulated, could act as gain-of-function oncogenes, whereas growth suppressing or cell death enhancing mRNAs, if down-regulated, could act as loss-of-function tumor suppressors (9).
Prior studies, including analyses from our group, indicate that the miRNA profile changes in chronic UC-associated inflammation (10–13). Several small studies have also demonstrated dysregulation of multiple miRNAs, including miR-31, miR-21, and miR-224 in IBD-associated neoplastic tissue (14–17). However, alterations in miRNA expression have not been examined globally in a large subset of patients with advanced UC-associated neoplasia, nor have miRNA changes been described in non-dysplastic tissue adjacent to a neoplastic lesion. In addition, there has been limited evaluation of the functional role of miRNAs dysregulated in inflammatory bowel disease (IBD)-carcinogenesis. In this study, we describe miRNA changes occurring in both IBD-neoplasia as well as adjacent non-dysplastic tissue and investigate the functional role of the tumor suppressor, miR-193a-3p, and a putative target which potentiates EGFR signaling, interleukin 17 receptor D (IL17RD), in ulcerative colitis-associated cancer.
Materials and Methods
Tissue
Discovery Cohort: The study was approved by the institutional review board at the University of Chicago (IRB# 11-0411 and 10-209A). Archived formalin-fixed paraffin-embedded tissues (FFPET) were obtained from normal controls and patients with UC who underwent a colectomy. Three groups of patients were examined: Normal Controls (N, n=12), UC patients without neoplasia (UC, n=9), and UC patients with neoplasia (n=11). From the UC patients with neoplasia, two separate tissues were analyzed, UC-associated neoplastic tissue (UCN, n=10; HGD-4, CRC-6), and non-dysplastic UC tissue adjacent to a neoplastic lesion (nUCaN, n=10). The tissue adjacent to the neoplastic lesion was obtained from the archived FFPET in closest proximity to the neoplastic lesion. Baseline demographics, site of sample collection, and medication usage for each group is presented in Supplementary Table 1. Additional information on the patient groups, inclusion, and exclusion criteria are included in the supplementary materials and methods.
Validation Cohort: Flash-frozen tissue was obtained by the University of Chicago Human Tissue Resource Center using mucosal stripping after surgical resection from 11 patients with sporadic colon cancer and 11 patients with UC-associated colon cancer. Comparison in this group was performed between normal-appearing tissue adjacent to sporadic colon cancers (normal controls, n=11), UC-associated cancer (n=11), and non-dysplastic mucosa adjacent to the UC-cancers (n=11). Individuals and samples were matched by age and colon location, respectively.
RNA and DNA extraction
RNA and DNA were extracted from FFPET and fresh frozen samples as described in the supplementary materials and methods.
Nanostring Analysis
miRNA analysis was performed using the Nanostring nCounter expression assay v1 per manufacturers specifications. Details regarding the analysis of array data are presented in the supplementary materials and methods.
Real-Time PCR
Quantitative real time PCR was performed to assess expression of miR-31, miR-34a, miR-106b, miR-193a-3p, miR-376, miR-497, pri-miR-193a, and IL17RD as detailed in the supplementary materials and methods. Results were normalized to beta actin, comparisons made between groups using the 2−ΔΔCT method (18), and significance calculated using the Student’s t-test.
Genomic Bisulfite Sequencing
DNA bisulfite treatment was carried out using the Qiagen Epitect Bisulfite Kit per manufacturer’s directions. Primers for miR-193a designed with methprimer software (L: TTTGAGGGATATTTAGAGTTT, R: AACCTAAAAAACAACCTAACC). PCR, subcloning of products, and sequencing were carried out as described in the supplementary materials and methods. Analysis and visualization of CpG dinucleotide methylation was performed using BISMA software (http://services.ibc.uni-stuttgart.de/BDPC/BISMA)(19)
Immunohistochemistry
Immunohistochemistry was carried out as previously described using antigen retrieval with a citrate buffer and primary antibody for IL17RD at a concentration of 1:15 (R & D Systems, Minneapolis, MN) (20).
Colonocyte Isolation
Colon biopsies (n=6–12/subject) were collected from the sigmoid colon and stored for up to 6 hrs at +40C in transport media which consists of Mg2+ and Ca2+ free PBS containing 50 IU/ml penicillin, 50 ug/ml streptomycin and 0.5 mg/ml gentamycin. Isolation of colonocyte and stromal fractions was performed as detailed in the supplementary materials and methods.
Cell Culture
HCT116, DLD-1, LoVo, and HT29 colon cancer cells, as well as human umbilical vein endothelial cells (HUVEC) and human colonic fibroblasts (CCD-18co) were obtained from ATCC. HCT116 luciferase expressing cells were obtained from Dr. Tong Chuan He (University of Chicago) for xenograft experiments. We routinely authenticate cell lines by short tandem repeat analysis (21). The most recent date of authentication was 2/2017 by the University of Arizona Genetics Core. miRNA transfection experiments were conducted on pre-confluent HUVEC and HCT116 cells. Cells were seeded on 6 well plates and transfected the following day with 6.25 nM and 12.5 nM mimics of mature miR-193a-3p (Life Technologies; assay ID: MC11123, catalog # 4464066) or control oligonucleotides (Life Technologies assay ID: AM17110, AM17111) and cells harvested 72 hrs later to examine protein and RNA expression.
Preconfluent HCT116, LoVo, HT29, and DLD1 cells were treated with 0, 5 μM, or 10 μM 5-aza-2’-deoxycytidine daily for 5 days. The cells were harvested 24 hrs after the last treatment and pri-miR-193a as well as mature miR-193a-3p measured by qPCR.
Three seed matches for miR-193a-3p are contained in the 3’ untranslated region (UTR) of IL17RD at positions 128–134, 964–970, and 5378–5384. A 910 bp oligonucleotide flanking the first 2 seed sequences and a mutant construct, in which both seed sequences were deleted, were constructed by Origene (Rockville, MD) and inserted downstream from a luciferase encoding cDNA in the pMirTarget Destination Vectors (Origene) (Supplementary material and methods). HCT116 cells were co- transfected with the vector coding for firefly luciferase regulated by wildtype or mutant IL17RD 3’UTR together with a renilla luciferase regulated by a CMV to control for transfection efficiency. Cells were then transfected with a mature mimic of miR-193a-3p or scrambled probe (Life Sciences) and harvested 48 hours later. firefly luciferase activity regulated by IL17RD-3’UTR was normalized to renilla expression for each well. Eight wells for each condition were evaluated and mean expression between each group compared using a Student’s t-test.
To assess cellular proliferation, preconfluent (5000–7500 cells/96 well plate) HCT116, HT29, or CCD18co cells were transfected with 25 nM miR-193a-3p or control miRNA and proliferation assessed 48 hours later using a WST-1 proliferation assay. Raw optical density was obtained in 12 replicates, normalized, percent proliferation calculated as compared to a scrambled control miRNA, and significance assessed using a two-sample t-test.
hSEF (IL17RD) plasmid was obtained from Dr. Chang (Tsinghua University) (22). The plasmid was transiently transfected into HCT-116 or HT-29 cells and proliferation assessed by WST-1 proliferation assay 96 hrs later. In separate experiments, HT29 cells were transiently transfected with the IL17RD encoding plasmid, and 24 hrs later cells were either treated with 10 ng/ml of epidermal growth factor (EGF) or untreated. Cells were harvested for protein 15 and 30 min after EGF treatment. WST-1 proliferation assay was performed 72 hrs after EGF treatment of cells seeded on 12 well plates containing 2000-2500 cells/well.
IL17RD with either the 910 bp wild type 3’UTR or mutant 3’UTR on the 3’ end of IL17RD were stably transfected into HCT116 cells. A polyclonal population of IL17RD-3’UTR stable transfectants were untreated or treated with a 20nM of a scrambled probe or 5nM, 10nM, or 20nM of miR-193a-3p for 24 hours and harvested for protein extraction. In separate experiments, HCT116 cells expressing IL17RD with WT 3’UTR or mutant 3’UTR were transfected with similar doses of a miR-193a-3p antagomir (Life Technologies; assay ID: MH11123, catalog #4464088).
Animal Studies
Animal studies were approved by the University of Chicago Institutional Animal Care and Use Facility (#59771). Nod Scid Gamma (NSG−/−) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). A mixed population of 2.5 million HCT116 cells which were stably transfected with IL17RD with WT 3’UTR were injected in one flank and a comparable number of cells expressing IL17RD with mutant 3’UTR were injected into the opposite flank in 15 nine week old male mice. 10μg of mature miR-193a-3p was mixed with in-vivo jetPEI (Polyplus, Illkirch, France) for intraperitoneal injection per manufacturers specifications. The mice were treated with miR-193a-3p (n=10) or vehicle alone (n=5, jetPEI alone) on days 5 and 8 after implantation of HCT116 cells. Tumor growth was measured three times weekly for 3 weeks and the mice were sacrificed 25 days after implantation of tumors. Tumor volumes were compared at each time point using a Wilcoxon signed rank test. In separate experiments, 10μg of anti-miR-193a-3p was injected into 9-week old male and female NSG−/− mice (n=9) or vehicle (n=6) with HCT 116 cells containing IL17RD and the WT 3’UTR in one flank. Injections of anti-miR-193a-3p were initiated 12 days after tumor implantation and continued twice weekly for 3 weeks. Tumor growth was measured after the final injection (35 days after initial cell injection) and the animals were sacrificed 3 days later. Tumor volumes were compared between groups every 3 days using a Student’s t-test. Immunohistochemistry was performed for ki67 with a concentration of 1:500 (Thermo Scientific, Waltham, MA) on formalin fixed paraffin embedded tissues. Nuclear staining was assessed on these samples using ImmunoRatio software to evaluate cellular proliferation (23). Percent of positive staining was compared between tumors expressing the MT or WT 3’UTR in both vehicle and miR-193a-3p treated animals using a paired Student’s t-test.
Western Blotting
Protein extraction and Western blotting were performed as previously described (24). Primary antibodies against IL17RD, FLAG, pAKT, pERK, pERBB2 EGFR, pEGFR, and beta actin were used with a dilution of 1:500 for IL17RD (R and D systems), 1:1000 for FLAG (Clontech, Mountain View, CA),1:500 pAKT (Cell Signaling Technology, Danvers, MA), 1:500 for EGFR (Cell Signaling Technology), 1:100 for pEGFR (Santa Cruz Biotechnology, Dallas, TX), 1:2500 for pERK (Santa Cruz Biotechnology), 1:200 for pERBB2 (Santa Cruz Biotechnology) and 1:5000 for beta actin (Sigma Aldrich, St. Louis, MO). Mean expression between treatment group was compared using a Student’s t-test.
Results
Microarray analysis identifies 13 miRNAs which are significantly dysregulated in both UC-neoplasia and adjacent tissue compared to UC without neoplasia
Using RNA extracted from FFPET, we compared miRNA expression levels quantified with Nanostring technology among normal controls, UC without cancer, UC-associated neoplasia and mucosa adjacent to the neoplastic lesion. Following normalization, 11 samples were removed from the analysis as they demonstrated a ‘batch effect’, clustering together in 2-way unsupervised hierarchical clustering analysis. miRNA expression was analyzed from arrays in the remaining 30 patients, including 9 normal controls, 7 UC patients without neoplasia (UC), 7 UC-associated neoplastic tissues (UCN), and 7 non-dysplastic UC mucosa adjacent to a neoplastic lesion (nUCaN). As shown in the multidimensional scaling analysis in figure 1A, there was clustering by miRNA expression between UCN and UC (p=0.003) as well as between nUCaN and UC (p=0.056). Compared to normal controls, there were 39 significantly dysregulated miRNAs in UC patients without neoplasia, 10 significantly dysregulated miRNAs in UC neoplastic tissues, and 11 dysregluated miRNAs in nondysplastic UC tissue adjacent to a neoplastic lesion (LogFC>1 and p<0.01) (Figure 1C). Compared to UC without neoplasia, there were 59 miRNAs with a significant change in expression in UCN, and 18 miRNAs with a significant change in expression in nUCaN (Figure 1D, Supplementary data tables 2–6). Thirteen miRNAs were significantly dysregulated in both UCN and nUCaN compared to UC without neoplasia (Figure 1E).
Figure 1.
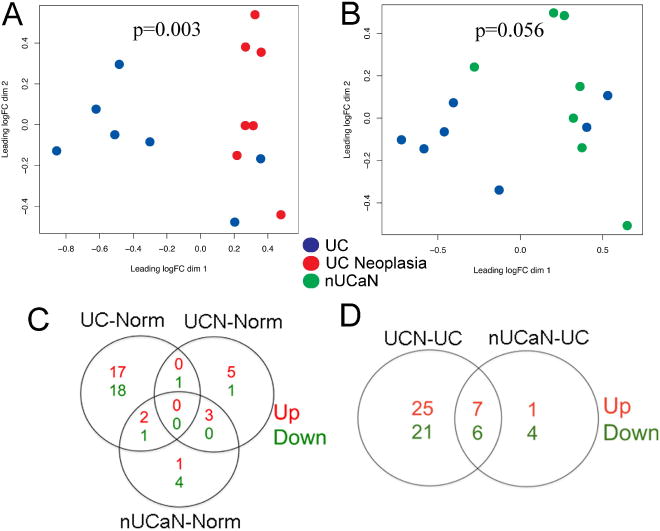
miRNA microarray analysis demonstrates differential expression of miRNAs in normal, UC, UC-neoplasia, and adjacent tissue. Clustering of miRNA expression using non-metric multidimensional scaling in A) UC vs. UC neoplasia, B) nUCaN vs. UC. Each circle represents a single subject in the indicated group. C) Venn diagram displaying the number of significantly up-regulated (red) and down-regulated (green) miRNAs between UC and Normal Controls, UCN and Normal Controls, as well as nUCaN and normal controls (LogFC>1, p<0.01). D) Venn diagram displaying the number of significantly dysregulated miRNAs between UCN and UC as well as nUCaN and UC (LogFC>1, p<0.01). E) miRNAs with a significant difference in expression (fold change >2, p value <0.05) in both UC-associated neoplasia as well as non-dysplastic tissue adjacent to a neoplastic lesion compared to UC without neoplasia.
Real-time PCR validates array findings that miR-31, miR-34a, miR-106b are up-regulated and miR-193a-3p is down-regulated in UC-associated neoplasia and adjacent tissue
Six miRNAs (miR-31, miR-34a, miR-106b, miR-193a-3p, miR-376c, and miR-476) which were significantly dysregulated in both UCN and nUCaN compared to UC without neoplasia and previously described to be dysregulated in other cancers were analyzed by qPCR in all samples (n=41). miR-31 demonstrated significant up-regulation in UCN and nUCaN compared to both normal controls and UC without neoplasia (UCN vs N: 9.7-fold, p<0.001; UCaN vs. N: 8.2-fold, p<0.001; UCN vs. UC: 4.7-fold, p=0.01; UCaN vs. UC: 4.0-fold, p=0.05). Similarly, miR-106b was significantly up-regulated in both UCN and UCaN compared to both normal controls and UC without neoplasia (UCN vs N: 1.8-fold, p=0.04; UCaN vs. N: 3.4-fold, p=0.005; UCN vs. UC: 2.2-fold, p=0.04; UCaN vs. UC: 4.1-fold, p=0.009). miR-34a was significantly up-regulated in UCN and nUCaN compared to UC without neoplasia (UCN vs. UC: 3.3-fold, p=0.03; UCaN vs. UC: 5.3-fold, p=0.006) although did not demonstrate a significant change in expression compared to normal controls (UCN vs. N: 1.2-fold, p=0.7; UCaN vs. N: 1.9-fold, p=0.2). In contrast to microarray findings, expression of miR-376c and miR-497 were not up-regulated in UCN by qPCR (figure 2A). miR-193a-3p expression was down-regulated 2.9-fold in UC (p=0.04), 3.3-fold in nUCaN (p=0.03), and 5.3-fold in UCN (p=0.003) compared to normal controls. Compared to UC without dysplasia, miR-193a-3p was down-regulated 1.2-fold in nUCaN (p=0.7) and 1.9-fold in UCN (p=0.05) (Figure 2B).
Figure 2.
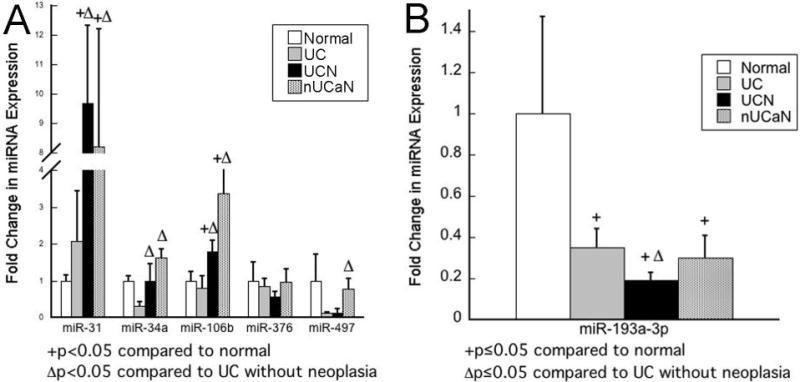
Real-time PCR results validate microarray finding that miR-31, miR-34a, miR-106b, and miR-193a-3p are differentially expressed in both UC-neoplasia and adjacent tissue compared to UC without neoplasia. miRNAs which were significantly dysregulated in both UC-neoplasia and adjacent mucosa compared to UC without neoplasia in the array analysis and had a previously described putative functional roles in carcinogenesis were quantified by real-time PCR. A) up-regulated miRNAs B) miR-193a-3p.
DNA Methylation of miR-193a is not increased in UC-associated colon cancer
Because miR-193a-3p was down-regulated in IBD-associated cancer and is located in a CpG island (Figure 3A), DNA methylation of the gene was analyzed. Bisulfite genomic sequencing was performed in 3 normal controls, 2 quiescent UC tissues, and 3 UC-associated cancers. As shown in figure 3B, there was a heterogeneous pattern of methylation across all samples without significant differences in total methylation between phenotypic groups (Normal: 62%, UC: 42%, UC-cancer: 46%). Likewise, there was no association between miRNA expression and percent methylation across these samples (Figure 3B).
Figure 3.
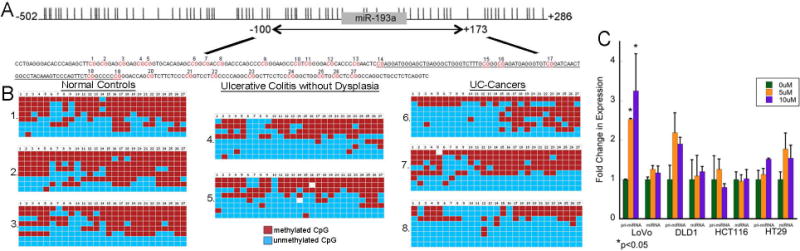
miR-193a does not have increased DNA methylation in UC-associated neoplasia. A) Genomic structure of miR193a-3p. Location of miR-193a (gray rectangle) with respect to CpG sites (vertical lines) and region examined by bisulfite genomic sequencing. The analyzed genomic region had 27 CpG dinucleotides as shown in red. The genomic sequence of pre-miR-193a is underlined. B) Bisulfite genomic sequencing in eight subjects, including three normal controls, two UC without dysplasia, and three UC-associated cancers. Red boxes represent methylated CpG sites and blue boxes unmethylated sites. C) Real time PCR of pri-miR-193a and miR-193a-3p following treatment with 0, 5μM, or 10 μM of 5-aza-2’-deoxycytidine daily for 5 days in the indicated colon cancer cell lines (*p <0.05).
In vitro, pri-miR-193a expression was significantly increased following treatment with 5-AZA in LoVo cells, although there were no differences in mature miR-193a-3p expression. In contrast, 5-AZA did not induce changes in expression of pri-miR-193a or mature miR-193a-3p in HCT116, DLD-1, or HT29 cells (Figure 3C).
Expression of the primary transcript of miR-193a is unchanged although miR-193a-3p is significantly down-regulated in a validation set of UC-cancers
pri-miR-193a and miR-193a-3p were assessed by qPCR in an independent cohort of normal controls, UC-associated colon cancers, and non-dysplastic mucosa adjacent to UC-associated cancers (n=11/group). As shown in figure 4A, there were no differences in primary transcript expression levels of pri-miR-193a in UC-cancer (p=0.49) or adjacent tissue (p=0.75) compared to normal controls. There was a significant decrease, however, in expression of mature miR-193a-3p in UC-cancers (5.6-fold, p=0.03) and adjacent non-dysplastic tissue (4.8-fold, p=0.05) compared to normal controls. These findings paired with the methylation analysis support the conclusion that down-regulation of miR-193a-3p in IBD-cancer involves post-transcriptional dysregulation.
Figure 4.
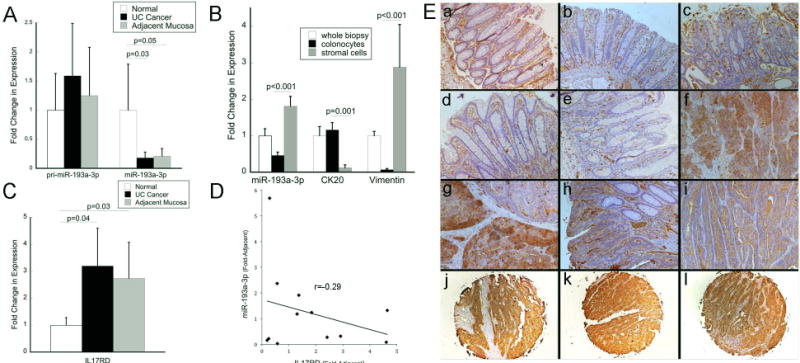
miR-193a-3p is down-regulated and IL17RD up-regulated in UC-associated cancer A) Expression of the primary transcript of miR-193a and mature miR-193a-3p in an independent validation cohort of UC-associated cancers and adjacent tissue as compared to normal controls (n=11/group) B) Expression of miR-193a-3p, in whole colon biopsies, the fractionated colonocytes, and stroma from the same normal control patients (n=13). Vimentin, which is highly expressed in colonic fibroblasts, and CK20 which is expressed in colonocytes but not stromal cells, were measured as cell purity controls. C) Expression of IL17RD in UC-associated cancers and adjacent tissue as compared to normal controls (n=7/group). D) Correlation between miR-193a-3p and IL17RD transcripts. Individual sample expression of miR-193a-3p as compared to expression IL17RD with logarithmic trend line in IBD-cancers and adjacent tissue (fold change of expression as compared to normal controls). Samples included had expression data available for both miR-193a-3p and IL17RD (n=12, UC-cancer=6, adjacent tissue=6). E) Immunohistochemistry of IL17RD at 10× magnification [a: Normal Control; b: Quiescent UC; c: Active UC; d,e: Non-dysplastic mucosa adjacent to a cancer; f,g: UC-cancer; h,i: Sporadic colon cancer; j,k,l: Representative images from UC-cancers in tissue array demonstrating positive staining in epithelial cancer cells
Relative expression of miR-193a-3p is higher in stromal cells than colonocytes in normal controls
In order to explore the cell specificity of miR-193a-3p, we isolated colonocytes from stromal cells in biopsies obtained from colonoscopy in 13 normal controls. To confirm separation of stromal cells from colonocytes, we measured expression of vimentin, which is specific for stromal cells, and CK20, which is specific for colonocytes (Figure 4B). miR193a-3p was detected in both isolated colonocytes and stromal cells. Compared to whole biopsy specimens, however, miR193a-3p was enriched in isolated stromal cells and decreased in isolated colonocytes (p<0.001) (Figure 4B).
IL17RD transcript and protein expression are increased in UC-associated neoplasia and discordant to expression of miR-193a-3p
As IL17RD has the second highest weighted context++ score as a target of miR-193a-3p in TargetScan (www.targetscan.org) and we theorized it has an oncogenic role in colitis-associated cancer, we evaluated transcript and protein expression of IL17RD in UC and UC-associated neoplasia (25). Real time PCR was carried out in normal controls, UC-associated cancers, and non-dysplastic mucosa adjacent to the cancer from samples in the validation cohort (n=7 samples/group). Six of the seven UC-associated cancers had active inflammation in the surrounding mucosa. Compared to normal controls, IL17RD was increased in UC-associated cancer 3.2-fold (p=0.04), and in non-dysplastic mucosa adjacent to the cancers 2.7-fold (p=0.03) (Figure 4C). In subjects who had expression of miR-193a-3p and IL17RD analyzed in the same sample, there was an inverse correlation between miR-193a-3p and IL17RD (R2=-0.23) (Figure 4D). By immunohistochemistry, IL17RD expression was present in malignant colonocytes in IBD-associated cancers, whereas expression was seen predominately in endothelial and other stromal cells in normal controls, active UC, and quiescent UC. In a tissue array of 24 UC-associated cancers, 19/24 had strong positive staining and 5/24 had weak positive staining (figure 4E).
miR-193a-3p decreases colon cancer cell proliferation and directly targets IL17RD
To examine the growth phenotype of miR-193a-3p, HT-29, HCT-116 colon cancer cells, and CCD-18co colonic fibroblasts (a stromal cellular component) were transfected with mature mimic of miR-193a-3p and proliferation was assessed. As shown in figure 5A, up-regulation of miR-193a-3p significantly decreased proliferation in both colon cancer cell lines as well as cultured fibroblasts. At 48 hours, miR-193a-3p up-regulation resulted in a 34% decrease in proliferation of HCT116 (p<0.001), 19.5% decrease in proliferation of HT-29 cells (p=0.002), and 14.1% decrease in proliferation of CCD-18co cells (p=0.03).
Figure 5.
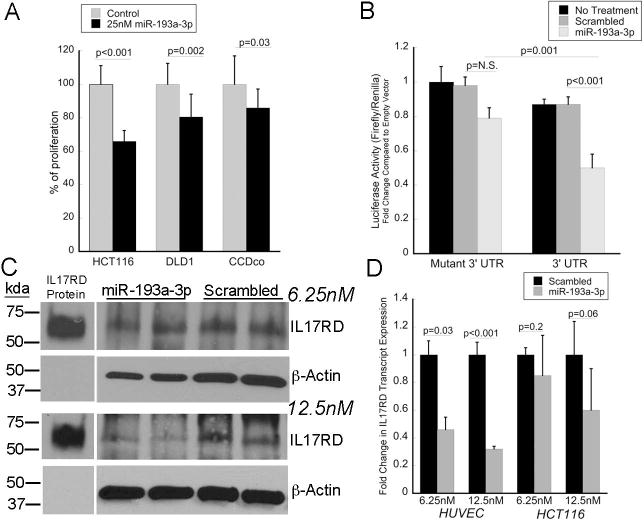
miR-193a-3p decreases cellular proliferation and targets IL-17RD. A) Cellular proliferation at 48 hours in the indicated cell lines following transfection with 25 nM of miR-193a-3p or scrambled control. B) Activity of luciferase regulated by mutated or wild type IL17RD 3’-UTR following transfection with mature miR-193a-3p or a scrambled control oligonucleotide. Results are expressed as the ratio of firefly to renilla luciferase activity and normalized to empty vector. C). Expression of IL17RD by Western blot following transfection with 6.25 nM or 12.5 nM of miR-193a-3p in HUVEC cells. D) RNA transcript expression of IL17RD following transfection of 6.25 nM or 12.5 nM of IL17RD in HUVEC or HCT116 cells.
We next carried out cell culture studies to investigate regulation of IL17RD by miR-193a-3p. To assess a direct interaction between miR-193a-3p and IL17RD, a 910bp oligonucleotide containing 2 predicted binding sites for the miR-193a-3p seed sequences in the 3’UTR of IL17RD and a mutant IL17RD 3’UTR with the seed complement sequences deleted were cloned into a luciferase vector regulated by IL17RD 3’UTR. Compared to a scrambled oligonucleotide control, miR-193a-3p decreased expression of luciferase regulated by wildtype 3’UTR construct by 42% (p<0.001). Luciferase activity was also decreased 37% with transfection of miR-193a-3p in cells containing the wildtype 3’UTR compared to the mutated 3’UTR (p=0.001). In untreated cells, luciferase activity was decreased 13% in cells containing luciferase regulated by IL17RD wild-type 3’ UTR compared to the mutated 3’ UTR, likely related to endogenous miR-193a-3p activity in these colon cancer cells (p=0.02) (Figure 5B).
HCT116 and HUVEC cells were also transfected with 6.25 nM or 12.5 nM mature mimic of miR-193a-3p or a scrambled RNA oligonucleotide and cells were harvested 72 hours later to measure IL17RD protein and gene expression. HUVEC cells were selected because of high baseline expression of IL17RD (26). Following transfection with 6.25 nM in HUVEC, there was a decrease in IL17RD RNA expression (2.1-fold; p=0.03), but no differences in IL17RD protein expression compared to the scrambled control. With transfection of 12.5 nM of mature miR-193a-3p, there was downregulation of both IL17RD protein (2.5-fold) and transcript expression (3.1-fold; p<0.001) (Figure 5C). In HCT116 cells, baseline IL17RD protein expression was too low to detect a band on Western blot. Similar to the HUVEC cells, however, IL17RD RNA expression in HCT116 was minimally decreased (1.2-fold, p=0.2) following transfection with 6.25 nM of the miRNA although there was a greater decrease in expression following transfection with 12.5 nM miR193a-3p (1.7-fold, p=0.06) (Figure 5D).
IL17RD increases EGF-induced colon cancer cell proliferation and EGFR signaling
In previous studies, IL17RD prevented degradation of EGFR and potentiated EGF signaling in HEK293T human embryonic kidney cells (27). As such, we explored the effect of IL17RD over-expression on proliferation of colon cancer cells with or without EGF treatment. Without EGF treatment, IL17RD over-expression resulted in a decrease in cell proliferation at 96 hours in HT-29 cells (12.1%, p<0.001), which have a p53 mutation, and no change in proliferation of HCT116 cells, which do not carry a p53 mutation (Supplementary Figure 1A). In contrast, in colon cancer cells that were treated with EGF, IL17RD significantly increased proliferation of both HT-29 cells (16%, p<0.001) and HCT116 cells (53%, p<0.001) 96 hrs following transfection (Supplementary Figure 1B). We also examined pAKT, pERK pEGFR, and pan-EGFR in EGF-treated cells transfected with a plasmid coding for IL17RD or an empty vector (EV) plasmid. In HT-29 cells transfected with IL17RD, EGFR and ERK were activated 15 minutes after treatment of EGF, and AKT was activated by 30 minutes (Supplementary Figure 1C and 1D). As expected short-term EGF treatment led to a decrease of pan-EGFR in both IL17RD transfected and control cells, although EGF-treated IL17RD and controls were not different with respect to pan-EGFR (Supplementary Figure 1C). However, IL17RD transfection into HT29 cells not treated EGF exhibited a trend towards an increase in total EGFR expression compared to cells treated with an empty vector 96 hours after transfection of IL17RD (p=0.1) (Supplementary Figure 1E).
miR-193a-3p regulates IL17RD controlled EGFR signaling
To explore the interaction between miR-193a-3p, IL17RD, and EGFR signaling, HCT116 cells stably transfected with the WT 3’UTR and IL17RD or the mutant 3’UTR and IL17RD were treated with miR-193a-3p. As shown in figure 6A and 6B, miR-193a-3p resulted in significant down-regulation of pAKT and pERK in cells expressing the WT 3’UTR at 5nM, 10nM, and 20nM. In addition, there was down-regulation of pERBB2 with treatment of 20nM of miR-193a-3p in cells expressing the WT 3’UTR. In contrast, expression of pAKT and pERK were increased in cells expressing the WT 3’UTR and IL17RD compared to those expressing the mutant 3’UTR and IL17RD following treatment with a miR-193a-3p antagomir (Figure 6C and 6D). We did not observe differences in EGFR expression between WT 3’UTR and mutant 3’UTR expressing cells following treatment with miR-193a-3p or its inhibitor.
Figure 6.
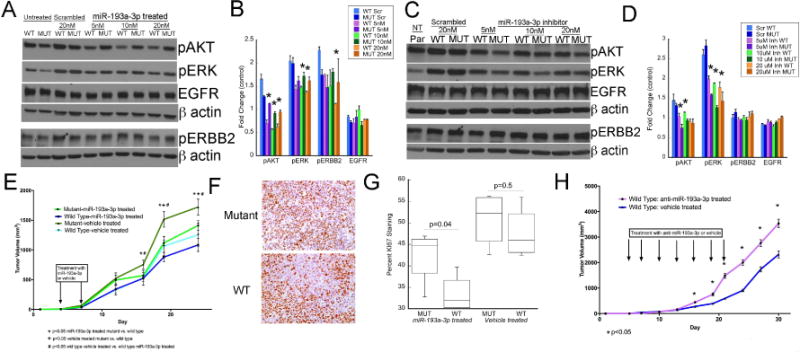
IL17RD mediates the impact of miR-193a-3p on tumor growth and EGFR signaling A) Western blots demonstrating protein expression following transfection with the indicated doses of miR-193a-3p or a scrambled probe in HCT116 cells stably transfected with a vector containing IL17RD with wildtype 3’UTR or IL17RD with mutant 3’UTR. Images represent one of two blots using the same conditions B) Quantitation of protein expression following transfection with the indicated doses of miR-193a-3p or a scrambled probe in HCT116 cells stably transfected with a vector containing IL17RD with wildtype 3’UTR or IL17RD with mutant 3’UTR. *p<0.05. C) Western blots demonstrating protein expression in parental HCT116 cells (par) or following treatment with a scrambled probe or miR-193a-3p inhibitor (Inh) at the indicated doses in HCT116 cells stably transfected with a vector containing IL17RD with wildtype 3’UTR or IL17RD with mutant 3’UTR. Images represent one of two blots using the same conditions D) Quantitation of protein expression following transfection with the indicated doses of a miR-193a-3p antagomir or a scrambled probe in HCT116 cells stably transfected with a vector containing IL17RD with wildtype 3’UTR or IL17RD with mutant 3’UTR. *p<0.05. E) Time course of tumor growth of HCT116 cells stably transfected with a vector coding for IL17RD with wildtype 3’UTR or mutant 3’UTR with mutations in the seed binding sites for IL17RD and treated with intraperitoneal injection of miR-193a-3p (n=10 mice containing one cell line in each flank) or vehicle (n=5). F) Representative images of ki67 staining of tumor xenografts of cells expressing IL17RD with wild type or mutant 3’UTR in mice treated with miR-193a-3p. G) Quantification of KI67 staining from tumors consisting of HCT116 cells expressing IL17RD with wild-type 3’UTR. Mice were treated with miR-193a-3p or vehicle as indicated (n=5/group). H) Tumor volume by day in tumor xenografts containing HCT116 cells stably transfected with a vector containing IL17RD and a wildtype 3’UTR treated with intraperitoneal injection of anti-miR-193a-3p (n=9) or vehicle (n=6).
miR-193a-3p suppression of colon cancer growth is mediated through IL17RD
To explore the interaction between miR-193a-3p, IL17RD, and tumor growth, HCT116 cells were stably transfected with the plasmid coding for IL17RD controlled by either wild type or mutant 3’ UTR for the IL17RD gene. NSG−/− mice were injected on contralateral flanks with cells expressing IL17RD regulated by either wild-type or mutant 3’UTR. Intraperitoneal treatment of mice with miR-193a-3p significantly decreased growth of tumors expressing IL17RD with wild type 3’UTR compared to tumors in vehicle treated mice expressing IL17RD with WT 3’UTR or tumors in mice expressing IL17RD with mutant 3’UTR (Figure 6E). Similarly, in miRNA treated mice cellular proliferation was significantly decreased in tumors expressing IL17RD with wild-type 3’UTR compared to tumors with cells expressing IL17RD with mutant 3’UTR (Figure 6F and 6G). In separate experiments, intraperitoneal injection of anti-miR-193a-3p resulted in significantly increased growth of tumors expressing wild-type 3’ UTR of IL17RD (Figure 6H).
Discussion
This report highlights the importance of miRNAs in the pathogenesis of IBD-associated colon cancer. We identified several miRNAs that were dysregulated in IBD-associated neoplasia which may contribute causally to cancer development in this high-risk population. In addition, this study established that changes in miRNAs occur in non-dysplastic mucosa adjacent to a neoplastic lesion as a ‘field effect”. We also demonstrated down-regulation of miR-193a-3p in IBD-associated neoplasia, validated changes in an independent cohort, and identified a possible key mechanism of carcinogenesis in IBD involving IL17RD. Although miR-193a-3p has been reported as down-regulated in several cancers, this is the first study to report down-regulation of miR-193a-3p in IBD-associated neoplasia.
Previous miRNA analyses in IBD-associated cancers have focused on the up-regulation of miR-31, miR-21, miR-155, miR-224, let-7e, miR-17, and miR-122 (14–17,28). In concordance with these studies, we confirmed up-regulation of miR-31 in UC-associated neoplasia. The fact that we did not identify changes in several other miRNAs found to be dysregulated in prior studies may be related to differences in patient populations and methodological differences including miRNA platforms. Our investigations had the advantage of controlling for inflammation and location of tissue biopsies among sample groups as well as having a more homogeneous patient population that included only ulcerative colitis patients. In addition to demonstrating changes in miR-31, we also identified significant up-regulation miR-34a and miR-106b, as well as down-regulation of miR-193a-3p in IBD-associated neoplasia and adjacent tissue. miR-106b, like miR-31, has been ascribed an oncogenic role in colon cancer previously (29,30). In contrast to our findings in IBD-cancer, however, previous studies indicate that miR-34a is decreased in a subset of sporadic colon cancers (31). As the impact of miR-34a on cancer cell proliferation is dependent on p53 and its expression is associated with that of p53 (32), the up-regulation observed in neoplasia in our analysis may be linked to the tumor suppressor gene status. However, we did not directly measure p53 expression in our cohort. In this study, we chose to focus mechanistic investigations on miR-193a-3p given the fact that it may be integral to bowel inflammation and is known to function as a tumor suppressor in other cancers, but had not been investigated functionally in colorectal cancer (33–35). Adding to the strength of our studies, we confirmed and thereby validated changes of miR-193a-3p in an independent cohort. Although miR-193a-3p has not been reported as down-regulated in previous studies examining IBD-neoplasia, a direct real time PCR analysis of this miRNA was not performed in the majority of prior analyses (16,28) (15).
miR-193a is located on chromosome 17. During processing, the hairpin precursor miRNA is cleaved into mature miR-193a-3p and miR-193a-5p. miR-193a-3p has been reported as down-regulated in breast, medullary thyroid cancer, as well as non-small cell and squamous cell lung cancer (34–38). Although there is limited data on expression of this miRNA in colon cancer, a previous study demonstrated that overexpression of miR-193a decreased cell proliferation and tumor growth in HT-29 and HCT-116 tumor xenograft experiments (39). We confirmed the antiproliferative effects of miR-193a-3p in HCT116 tumor xenografts further supporting the tumor suppressor role of this miRNA. Additional support for the role of miR-193a-3p in IBD comes from a recent publication, which demonstrated that down-regulation of this miRNA occurs in active UC and intracolonic delivery of miR-193a-3p ameliorated dextran sodium sulfate-induced colitis (33).
miR-193a is located in a CpG island. In other cancer types, including leukemia, oral cancer, and non-small cell lung cancer, hypermethylation at the DNA loci of miR-193a has been associated with decreased expression of this miRNA (40–42). In contrast to other cancers, we did not identify changes in methylation associated with decreased expression of this miRNA in IBD-associated neoplasia. In fact, expression of the primary transcript of miR-193a was not different between normal controls and IBD-associated cancer. These results indicate that down-regulation of this miRNA in IBD-associated cancer occurs post-transcriptionally, most likely secondary to dysregulation of processing or stability of the mature miRNA in neoplastic tissue.
IL17RD (aka hSEF) is a predicted target miR-193a-3p based on three possible binding sites in the 3’ UTR of IL17RD that complement the miRNA seed sequence. Prior studies have concluded that IL17RD has several mechanistic properties, including functioning as an orphan receptor to potentiate IL17 signaling (43). Interestingly, IL17RD has been described to inhibit tumorigenesis in vitro through antagonistic effects on fibroblast growth factor (FGF) signaling and inhibition of MAP kinase (MAPK) signaling (22,26,44). In contrast to this tumor suppressing mechanism, Ren et al demonstrated a dual role of IL17RD whereby IL17RD potentiates epidermal growth factor (EGF)-mediated MAPK signaling in the presence of EGF, whereas it exerts opposing effects in the presence of FGF (27). Our findings support this mechanism as basal cell proliferation was inhibited by IL17RD in the absence of EGF treatment, but IL17RD transfection increased cell proliferation in the presence of EGF. In addition, in cells stably transfected with a plasmid coding for IL17RD with wild type 3’UTR miR-193a-3p decreased pAKT, pERK, and pERBB2 in vitro and reduced tumor xenograft growth in vivo compared to cells expressing IL17RD with mutant 3’UTR. Taken together, our data support the hypothesis that by preventing EGFR degradation and enhancing EGFR signaling, IL17RD up-regulation mediated by loss of miR-193a-3p may be an important mechanism driving colon cancer growth in patients with IBD.
Only one previous study has examined the impact of IL17RD in colon cancer (45). The authors of this in vitro study demonstrated that RAS down-regulation of IL17RD in colon cancers results in aberrant nuclear localization of MEK 1 and 2 leading to carcinogenesis. They also reported low baseline levels of IL17RD in colon cancer cell lines, adenomas, and invasive carcinoma (45). Although we found similar low baseline expression of IL17RD in HCT116 cells and low immunohistochemical staining in normal controls, we identified markedly elevated IL17RD expression in malignant colonocytes in IBD and sporadic colon cancers. This discrepancy may be explained from the fact that the previous analysis only examined one cancer, and we demonstrate that in some cancers staining is minimal or patchy. The finding that IL17RD up-regulation in the presence of EGF promotes cellular proliferation, combined with demonstration that IL17RD is up-regulated in a large number of IBD-cancers as assessed by both real time PCR and immunohistochemistry, supports our contention of its oncogenic role in IBD-associated colon cancer.
Although we were able to demonstrate concomitant down-regulation of miR-193a-3p in association with up-regulation of IL17RD in the same tissues, the cell-specific localization of miR-193a-3p regulation of IL17RD remains uncertain. While miR-193a-3p is expressed in the epithelial cell, expression levels are higher in stroma cells of normal colons. By immunohistochemistry, IL17RD is expressed primarily in the stromal cells in normal controls, although over-expression of IL17RD in IBD-cancer appears to be predominantly in malignant colonocytes. For this reason, we hypothesize that colonocyte miR-193a-3p downregulation contributes to IL17RD up-regulation in epithelial cells (colonocytes) of IBD-cancers. Other factors are also likely driving IL17RD up-regulation in cancer cells. In this regard, stromal cell- and colonocyte-specific regulation of IL17RD by miR193a-3p will require further study.
In conclusion, we identified several dysregulated miRNAs, including down-regulation of the tumor suppressor miR-193a-3p, in IBD-associated colon cancer and adjacent non-dysplastic mucosa. In addition, we were able to demonstrate a novel mechanism of carcinogenesis whereby loss of miR-193a-3p up-regulates expression of IL-17RD. While we focused our investigations on miR-193a-3p, it is likely that additional miRNA regulated pathways are important in inflammation associated colon cancer. Although miRNAs have not been widely used clinically for therapeutics to date, several in vivo delivery approaches have been developed and are currently being investigated in clinic trials (46,47). Using these or alternative methods, the miR-193a-3p-IL17RD pathway is a potential target for future chemopreventive approaches in this high-risk population.
Supplementary Material
Translational Relevance.
Approximately 1.6 million Americans have Inflammatory bowel disease (IBD), which is comprised of Crohn’s disease and ulcerative colitis. Patients with long-standing colonic inflammation related to IBD have a high-risk of developing colonic neoplasia. Although cancer in IBD differs from sporadic colon cancer in clinical presentation, progression from dysplasia, and outcome, few studies have mechanistically examined the pathogenesis of neoplasia in IBD using human samples. In this study, we investigated miRNA expression in UC-associated cancer and report that loss of the tumor suppressor, miR-193a-3p, promotes colon carcinogenesis in patients with UC. These findings uncover a novel mechanism of inflammation-associated colon cancer that has the potential to be used as a target for chemoprevention and treatment in this high-risk population.
Acknowledgments
Funding NIH K08DK090152 (JP), NIH P40K42086 (University of Chicago DDRCC)
Abbreviations
- UC
Ulcerative colitis
- CRC
colorectal cancer
- miRNA
microRNA
- mRNA
messenger RNA
- UTR
untranslated region
- IBD
inflammatory bowel disease
- EGFR
epidermal growth factor receptor
- IL17RD
interleukin-17 receptor protein D
- qPCR
quantitative real time reverse transcription-polymerase chain reaction
- pri-miRNA
primary miRNA
- RNA
ribonucleic acid
- DNA
deoxyribonucleic acid
- cDNA
complementary DNA
- FFPET
formalin fixed paraffin embedded tissue
- 5-AZA
5-aza-2’-deoxycytidine
- CpG
5’-cytosine-phosphate-guanine-3’
- hSEF
human similar expression to fibroblast growth factor
- NSG
Nod Scid Gamma
- pAKT
phosphorylated protein kinase B
- pERK
phosphorylated extracellular regulated kinase
- pERBB2
phosphorylated Erb-B2 receptor tyrosine kinase
Footnotes
Author Contributions:
Study Concept: JP, DTR, JHK, MB
Funding: JP
Bioinformatics analysis: YH
Real Time PCR and nanostring array: KM, UD
Immunohistochemistry: XC, AA, AIC, HH
Histologic interpretation: JH
In vitro studies: KM, RM, FB, ZD
Animal Studies: UD, KM, HH, ZD
Data Analysis: JP, YH, UD, KM, MB
Drafting of Manuscript: JP
Critical review of the manuscript JP, KM, UD, DTR, JHK, JH, MB
Approval of final version of the manuscript: All authors
Conflicts of interest related to work: None
Microarray Data: Available at Geo DataSets. Accession GSE68306
References
- 1.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48(4):526–35. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rutter MD, Saunders BP, Wilkinson KH, Rumbles S, Schofield G, Kamm MA, et al. Thirty-year analysis of a colonoscopic surveillance program for neoplasia in ulcerative colitis. Gastroenterology. 2006;130(4):1030–8. doi: 10.1053/j.gastro.2005.12.035. doi:S0016-5085(05)02562-X [pii] 10.1053/j.gastro.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5. doi: S0092867404000455 [pii] [DOI] [PubMed] [Google Scholar]
- 4.Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299(4):425–36. doi: 10.1001/jama.299.4.425. doi: 299/4/425 [pii] 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schepeler T, Reinert JT, Ostenfeld MS, Christensen LL, Silahtaroglu AN, Dyrskjot L, et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008;68(15):6416–24. doi: 10.1158/0008-5472.CAN-07-6110. doi: 68/15/6416 [pii] 10.1158/0008-5472.CAN-07-6110. [DOI] [PubMed] [Google Scholar]
- 6.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–61. doi: 10.1073/pnas.0510565103. doi: 0510565103 [pii] 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michael MZ, SM OC, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1(12):882–91. [PubMed] [Google Scholar]
- 8.Liu W, Mao SY, Zhu WY. Impact of tiny miRNAs on cancers. World J Gastroenterol. 2007;13(4):497–502. doi: 10.3748/wjg.v13.i4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5. doi: 10.1038/nature02871. doi: 10.1038/nature02871 nature02871 [pii] [DOI] [PubMed] [Google Scholar]
- 10.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104(5):1604–9. doi: 10.1073/pnas.0610731104. doi: 0610731104 [pii] 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu F, Zikusoka M, Trindade A, Dassopoulos T, Harris ML, Bayless TM, et al. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology. 2008;135(5):1624–35e24. doi: 10.1053/j.gastro.2008.07.068. doi: S0016-5085(08)01408-X [pii] 10.1053/j.gastro.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 12.Pekow JR, Dougherty U, Mustafi R, Zhu H, Kocherginsky M, Rubin DT, et al. miR-143 and miR-145 are downregulated in ulcerative colitis: Putative regulators of inflammation and protooncogenes. Inflamm Bowel Dis. 2011 doi: 10.1002/ibd.21742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pekow JR, Kwon JH. MicroRNAs in inflammatory bowel disease. Inflamm Bowel Dis. 2011 doi: 10.1002/ibd.21691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olaru AV, Selaru FM, Mori Y, Vazquez C, David S, Paun B, et al. Dynamic changes in the expression of MicroRNA-31 during inflammatory bowel disease-associated neoplastic transformation. Inflamm Bowel Dis. 2011;17(1):221–31. doi: 10.1002/ibd.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanaan Z, Rai SN, Eichenberger MR, Barnes C, Dworkin AM, Weller C, et al. Differential microRNA expression tracks neoplastic progression in inflammatory bowel disease-associated colorectal cancer. Hum Mutat. 2012;33(3):551–60. doi: 10.1002/humu.22021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svrcek M, El-Murr N, Wanherdrick K, Dumont S, Beaugerie L, Cosnes J, et al. Overexpression of microRNAs-155 and 21 targeting mismatch repair proteins in inflammatory bowel diseases. Carcinogenesis. 2013;34(4):828–34. doi: 10.1093/carcin/bgs408. [DOI] [PubMed] [Google Scholar]
- 17.Olaru AV, Yamanaka S, Vazquez C, Mori Y, Cheng Y, Abraham JM, et al. MicroRNA-224 negatively regulates p21 expression during late neoplastic progression in inflammatory bowel disease. Inflammatory bowel diseases. 2013;19(3):471–80. doi: 10.1097/MIB.0b013e31827e78eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. doi: 10.1006/meth.2001.1262 S1046-2023(01)91262-9 [pii] [DOI] [PubMed] [Google Scholar]
- 19.Rohde C, Zhang Y, Reinhardt R, Jeltsch A. BISMA–fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics. 2010;11:230. doi: 10.1186/1471-2105-11-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pekow J, Dougherty U, Huang Y, Gometz E, Nathanson J, Cohen G, et al. Gene Signature Distinguishes Patients with Chronic Ulcerative Colitis Harboring Remote Neoplastic Lesions. Inflammatory bowel diseases. 2013 doi: 10.1097/MIB.0b013e3182802bac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reid Y, Storts D, Riss T, Minor L. Authentication of Human Cell Lines by STR DNA Profiling Analysis. In: Sittampalam GS, Coussens NP, Nelson H, Arkin M, Auld D, Austin C, et al., editors. Assay Guidance Manual Bethesda (MD) 2004. [Google Scholar]
- 22.Xiong S, Zhao Q, Rong Z, Huang G, Huang Y, Chen P, et al. hSef inhibits PC-12 cell differentiation by interfering with Ras-mitogen-activated protein kinase MAPK signaling. J Biol Chem. 2003;278(50):50273–82. doi: 10.1074/jbc.M306936200. [DOI] [PubMed] [Google Scholar]
- 23.Tuominen VJ, Ruotoistenmaki S, Viitanen A, Jumppanen M, Isola J. ImmunoRatio: a publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67. Breast Cancer Res. 2010;12(4):R56. doi: 10.1186/bcr2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pekow JR, Dougherty U, Mustafi R, Zhu H, Kocherginsky M, Rubin DT, et al. miR-143 and miR-145 are downregulated in ulcerative colitis: putative regulators of inflammation and protooncogenes. Inflammatory bowel diseases. 2012;18(1):94–100. doi: 10.1002/ibd.21742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4 doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang RB, Ng CK, Wasserman SM, Komuves LG, Gerritsen ME, Topper JN. A novel interleukin-17 receptor-like protein identified in human umbilical vein endothelial cells antagonizes basic fibroblast growth factor-induced signaling. J Biol Chem. 2003;278(35):33232–8. doi: 10.1074/jbc.M305022200. [DOI] [PubMed] [Google Scholar]
- 27.Ren Y, Cheng L, Rong Z, Li Z, Li Y, Zhang X, et al. hSef potentiates EGF-mediated MAPK signaling through affecting EGFR trafficking and degradation. Cellular signalling. 2008;20(3):518–33. doi: 10.1016/j.cellsig.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ludwig K, Fassan M, Mescoli C, Pizzi M, Balistreri M, Albertoni L, et al. PDCD4/miR-21 dysregulation in inflammatory bowel disease-associated carcinogenesis. Virchows Archiv : an international journal of pathology. 2013;462(1):57–63. doi: 10.1007/s00428-012-1345-5. [DOI] [PubMed] [Google Scholar]
- 29.Cekaite L, Rantala JK, Bruun J, Guriby M, Agesen TH, Danielsen SA, et al. MiR-9, -31, and -182 deregulation promote proliferation and tumor cell survival in colon cancer. Neoplasia. 2012;14(9):868–79. doi: 10.1593/neo.121094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu S, Dong TS, Dalal SR, Wu F, Bissonnette M, Kwon JH, et al. The microbe-derived short chain fatty acid butyrate targets miRNA-dependent p21 gene expression in human colon cancer. PLoS One. 2011;6(1):e16221. doi: 10.1371/journal.pone.0016221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A. 2007;104(39):15472–7. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao J, Li N, Dong Y, Li S, Xu L, Li X, et al. miR-34a-5p suppresses colorectal cancer metastasis and predicts recurrence in patients with stage II/III colorectal cancer. Oncogene. 2015;34(31):4142–52. doi: 10.1038/onc.2014.348. [DOI] [PubMed] [Google Scholar]
- 33.Dai X, Chen X, Chen Q, Shi L, Liang H, Zhou Z, et al. MicroRNA-193a-3p Reduces Intestinal Inflammation in Response to Microbiota via Down-regulation of Colonic PepT1. J Biol Chem. 2015;290(26):16099–115. doi: 10.1074/jbc.M115.659318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Y, Li X, Yang Q, Wang X, Zhou Y, Jiang T, et al. The role of microRNA in human lung squamous cell carcinoma. Cancer Genet Cytogenet. 2010;200(2):127–33. doi: 10.1016/j.cancergencyto.2010.03.014. doi: S0165-4608(10)00147-0 [pii] 10.1016/j.cancergencyto.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 35.Tahiri A, Leivonen SK, Luders T, Steinfeld I, Ragle Aure M, Geisler J, et al. Deregulation of cancer-related miRNAs is a common event in both benign and malignant human breast tumors. Carcinogenesis. 2014;35(1):76–85. doi: 10.1093/carcin/bgt333. [DOI] [PubMed] [Google Scholar]
- 36.Uhlmann S, Mannsperger H, Zhang JD, Horvat EA, Schmidt C, Kublbeck M, et al. Global microRNA level regulation of EGFR-driven cell-cycle protein network in breast cancer. Mol Syst Biol. 2012;8:570. doi: 10.1038/msb.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu T, Li J, Yan M, Liu L, Lin H, Zhao F, et al. MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/IK3R3/mTOR/S6K2 signaling pathway. Oncogene. 2014 doi: 10.1038/onc.2013.574. [DOI] [PubMed] [Google Scholar]
- 38.Santarpia L, Calin GA, Adam L, Ye L, Fusco A, Giunti S, et al. A miRNA signature associated with human metastatic medullary thyroid carcinoma. Endocrine-related cancer. 2013;20(6):809–23. doi: 10.1530/ERC-13-0357. [DOI] [PubMed] [Google Scholar]
- 39.Iliopoulos D, Rotem A, Struhl K. Inhibition of miR-193a expression by Max and RXRalpha activates K-Ras and PLAU to mediate distinct aspects of cellular transformation. Cancer Res. 2011;71(15):5144–53. doi: 10.1158/0008-5472.CAN-11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao XN, Lin J, Li YH, Gao L, Wang XR, Wang W, et al. MicroRNA-193a represses c-kit expression and functions as a methylation-silenced tumor suppressor in acute myeloid leukemia. Oncogene. 2011;30(31):3416–28. doi: 10.1038/onc.2011.62. [DOI] [PubMed] [Google Scholar]
- 41.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68(7):2094–105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 42.Heller G, Weinzierl M, Noll C, Babinsky V, Ziegler B, Altenberger C, et al. Genome-wide miRNA expression profiling identifies miR-9-3 and miR-193a as targets for DNA methylation in non-small cell lung cancers. Clin Cancer Res. 2012;18(6):1619–29. doi: 10.1158/1078-0432.CCR-11-2450. [DOI] [PubMed] [Google Scholar]
- 43.Rong Z, Wang A, Li Z, Ren Y, Cheng L, Li Y, et al. IL-17RD (Sef or IL-17RLM) interacts with IL-17 receptor and mediates IL-17 signaling. Cell research. 2009;19(2):208–15. doi: 10.1038/cr.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren Y, Cheng L, Rong Z, Li Z, Li Y, Li H, et al. hSef co-localizes and interacts with Ras in the inhibition of Ras/MAPK signaling pathway. Biochem Biophys Res Commun. 2006;347(4):988–93. doi: 10.1016/j.bbrc.2006.06.193. [DOI] [PubMed] [Google Scholar]
- 45.Duhamel S, Hebert J, Gaboury L, Bouchard A, Simon R, Sauter G, et al. Sef downregulation by Ras causes MEK1/2 to become aberrantly nuclear localized leading to polyploidy and neoplastic transformation. Cancer Res. 2012;72(3):626–35. doi: 10.1158/0008-5472.CAN-11-2126. [DOI] [PubMed] [Google Scholar]
- 46.Chapman CG, Pekow J. The emerging role of miRNAs in inflammatory bowel disease: a review. Therap Adv Gastroenterol. 2015;8(1):4–22. doi: 10.1177/1756283X14547360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christopher AF, Kaur RP, Kaur G, Kaur A, Gupta V, Bansal P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect Clin Res. 2016;7(2):68–74. doi: 10.4103/2229-3485.179431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.