

Abstract
It is well established that Notch functions as a transcriptional activator through the formation of a ternary complex that comprises Notch, Maml and CSL. This ternary complex then serves to recruit additional transcriptional cofactors that link to higher order transcriptional complexes. The mechanistic details of these events remain unclear. This report reveals that the Notch ternary complex can direct the formation of a repressor complex to terminate gene expression of select target genes. Herein, it is demonstrated that p19Arf and Klf4 are transcriptionally repressed in a Notch-dependent manner. Furthermore, results indicate that Notch recruits Polycomb Repressor Complex 2 (PRC2) and Lysine Demethylase 1 (KDM1A/LSD1) to these promoters, which leads to changes in the epigenetic landscape and repression of transcription. The demethylase activity of LSD1 is a pre-requisite for Notch-mediated transcriptional repression. In addition, a stable Notch transcriptional repressor complex is identified containing LSD1, PRC2 and the Notch ternary complex. These findings demonstrate a novel function of Notch and provides further insight into the mechanisms of Notch-mediated tumorigenesis.
Keywords: P19ARF, Notch, Repression, PRC2, LSD1, epigenetic marks
Introduction
Transcription factors mediate the cellular programs instructed by extracellular cues. These extracellular cues direct complex transcriptional cascades that result in the activation and repression of genes which govern all cellular processes including cell type specification, proliferation and differentiation (1–3). In many cases, there is a strict separation between transcription factors that assemble activation complexes and those that assemble repression complexes. In other cases, transcription factors can direct both activation and repression complexes based on complexes components and/or the epigenetic landscape, setting the context for transcriptional response (4,5). Notch is a receptor based transcription factor activated by ligand engagement through cell-to-cell contact. Notch signaling regulates key cellular functions such as proliferation, differentiation and apoptosis in a context-dependent manner. Deregulation of Notch activity is important in driving the neoplastic phenotype in many human malignancies (6–8). Upon activation, the Notch intracellular domain (Notchic) is released from the plasma membrane and translocates into the nucleus, where it forms a ternary complex with Mastermind (Maml) and CSL. This ternary complex then serves as a scaffold to recruit higher-order transcriptional machinery to initiate a downstream transcriptional cascade (9–12).
Although it is clear that Notch is a transcriptional activator, many expression-profiling experiments reveal that there are genes repressed by Notch activity (13–21). The mechanisms by which these genes are repressed by Notch signaling are underexplored, as it is thought that repression is an indirect mechanism. As part of the transcriptional cascade initiated by Notch, expression of the Hes/Hey family of transcriptional repressors is induced (22). In many cases, transcriptional repression directed by activation of Notch signaling is a secondary effect mediated by this family of transcriptional repressors (15,17,23). Examples of this mechanism include, among others, the regulation of PTEN (19). However, in many cases repression by Notch signaling cannot be attributed to Hes/Hey proteins and the mechanisms underlying these events are not known (16,18,20,21). This indicates that there are distinct mechanisms of gene repression upon activation of Notch signaling that contribute to the Notch-induced transcriptional cascade.
Previously, we reported that in Notch-induced T-cell leukemogenesis, Notch suppresses p53 activity through repression of Arf expression (24). However, the detailed mechanism by which Notch mediates Arf repression was not clear. In this study, we demonstrate that Notch directs transcriptional repression of Arf through direct binding of the Notch ternary complex to the Arf promoter region. Upon binding the Arf promoter region, Notch serves to recruit the polycomb repressive complex 2 (PRC2) in an LSD1-dependent manner. Recruitment of these factors leads to the enrichment of the repressive mark H3K27me3 and the loss of the active mark H3K4me3 and subsequent termination of Arf expression. In addition, we provide evidence for a Notch repressor complex bound to DNA that contains LSD1 and PRC2 and therefore, provide a novel mechanism of gene regulation by Notch. Importantly, we also observe this activity in the regulation of Klf4 by Notch indicating that direct repression of transcription by Notch is a common mechanism in Notch signaling. These data also provide rationale for the use of epigenetic therapy in combination with Notch inhibition for treating Notch-dependent cancers.
Materials and Methods
Cell culture
Primary lymphomas tumors and the lymphoma cell lines were collected and established in our laboratory as described previously between 2004 and 2005 (24). HPB-ALL, ALL-SIL and 293T cell lines were obtained from the ATCC (Manassas, VA, USA) and cultured under recommended condition for less than six months. Mouse embryonic fibroblasts (MEFs) were prepared from wild-type C57Bl/6 embryos at day E13.5 following a standard MEF isolation protocol (25) and were cultured in complete DMEM. Animal experiments were approved by the University of Miami Institutional Animal Care and Use Committee. The number of the passages of the cell lines used in this study are not exceed 15. All cell lines were maintained at 37 °C in 5% CO2 and tested for mycoplasma contamination.
Retroviral Infections, qRT-PCR Analysis, nuclear extraction and Western Blotting
Retroviral Infections, qRT-PCR Analysis, nuclear extraction and Western Blotting were performed as described previously (26,27). Cell samples were collected 72h post-infection. The following antibodies were used in Western blotting: anti-Notch1 (1:1000, abcam), anti-CSL (1:1000; polyclonal), anti-MAML1 (1:1000, cell signaling), anti-LSD1 (1:1000, cell signaling) and anti-EZH2 (1:1000, cell signaling).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP was performed as described previously (28). Briefly, chromatin samples were immunoprecipitated with the following antibodies: rabbit IgG (Abcam), anti-Notch1 antibodies (Bethyl lab), anti-Maml1 (Cell signaling), anti-LSD1 (Cell signaling), anti-EZH2 (Cell siganling), anti-SUZ12 (Abcam), anti-H3K4me3 (Abcam) or anti-H3K27me3 (Abcam). DNA was cleaned using PCR purification kit (Qiagen) and target sequences were amplified by qPCR. Primer sequences are available upon request.
DNA affinity precipitation
The conjugation of biotinylated dsDNA and streptavidin agarose beads were performed as described previously (26). For DNA affinity precipitation experiments from 4084 nuclear extraction, the supernatant which contained nuclear protein fraction was incubated with DNA streptavidin beads (pre-blocked with 1mg/mL BSA and 100μg/mL sonicated salmon sperm DNA) for 1 h at 4°C on a rotator and beads were washed 3 times with non-supplemented DNA lysis buffer. Proteins bound to the beads were analyzed by SDS-PAGE and western blot using the appropriate antibodies.
Size exclusion chromatography
Size exclusion chromatography was performed as described previously (27). In brief, 10mg of the nuclear extracts from 4084 and 6780 are used. A 500μl portion of the sample was loaded onto the Superose 6 HR 10/30 resin column (GE healthcare life sciences) which was equilibrated with column buffer (150 mM NaCl, 40 mM HEPES [pH 7.4], 1 mM EDTA, 0.5 mM DTT, 5% glycerol, 0.001% NP-40) and collected into 350μl fractions; fractions were then subjected to western blotting analysis, or individual fractions were pooled for next step experiment.
DNA affinity chromatography
The Hitrap streptavidin column was washed with PBS and then introduced to previously annealed biotinylated dsDNA containing two high affinity CSL binding sites (2XCSL). Indicated samples containing 10ug/ml salmon sperm DNA were subjected to the column equilibrated with column buffer. The column was then eluted with column buffer containing 250mM or 350mM NaCl. Each sample was collected into 500μl fractions and concentrated accordingly to ideal volume.
Cell viability assay
Cell viability assays were performed with CellTiter-Glo (G7572, Promega), as described in the product manual.
Results
Notch represses Arf expression
Previously we reported that in a mouse model of Notch-driven T-cell lymphoma, p53 levels are suppressed by the repression of Arf expression (24). Consistent with this observation, in T-cell lymphoma cell lines 4084 and 3749 which are derived from the Top-Notch mouse, we observed lower Arf expression both at the mRNA and protein levels compared to the Myc-driven T-cell lymphoma cell line 6780 (29), which is derived from Tet-off Myc transgenic mouse (Figure 1A and 1B). To investigate whether the repression of Arf is directly regulated by Notch signaling, we ectopically expressed Notchic in primary MEFs and determined Arf mRNA and protein levels (Figure S1). Expression of Notchic in MEFs led to a dramatic reduction of Arf mRNA and protein compared to MEF infected with GFP control or mock. To demonstrate the specificity for the Arf locus, p16INK4a expression was analyzed and, no effects were observed on expression of p16INK4a (Figure 1C and 1D). Since Arf and p16INK4a share an overlapping locus with distinct regulatory regions, these data indicate that Notch specifically governs the transcriptional repression of Arf.
Figure 1. Notch represses Arf gene expression.
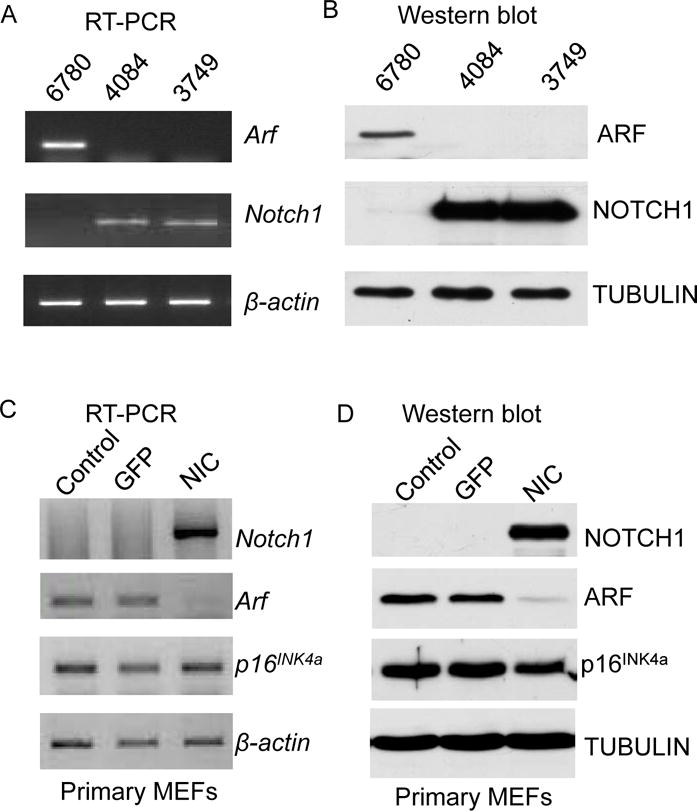
RT-PCR (A) and Western blot analysis (B) of Arf and Notch in mouse lymphoma cell-lines. RT-PCR (C) and Western Blot analysis (D) of Arf, p16INK4a and Notch in primary MEFs infected with GFP or Notchic (NIC) encoding vector. β-actin and Tubulin are used as loading controls.
Repression of Arf expression is a direct transcriptional effect of Notch and is accompanied by changes in the epigenetic landscape on the Arf promoter
We next sought to determine the mechanism by which Notch mediates transcriptional repression of Arf. The current model suggests that Notch drives a transcriptional network through the activation of gene expression. The HES and HEY proteins are canonical targets of Notch signaling and act as transcriptional repressors (22,23). Therefore, we reasoned that these proteins, among others, were plausible candidates for Notch-directed repression of Arf. However, neither overexpression nor siRNA mediated knock down of Hes/Hey members had any effect on Arf levels (Figure S2). Therefore, we thought that repression of Arf might be directly regulated by Notch.
In order to test this hypothesis, we carried out a chromatin immunoprecipitation (ChIP) experiment in primary lymphomas generated from TOP-Notchic mice. Using primers spanning approximately 7kb on the promoter region (Figure 2A) to investigate Notch binding, quantitative PCR analysis of the ChIP samples revealed that Notch binds to the Arf promoter primarily in the proximal region upstream of the transcription start site (Figure 2B). In order to determine the events subsequent to Notch binding, we examined epigenetic marks on the Arf promoter. In primary T-cell lymphomas derived from the Top-Notch mouse, the repressive mark H3K27me3 is more abundant compared to the active mark H3K4me3 (Figure 2C). This is consistent with the repressive state of the Arf locus. Similarly, we detected lower levels of H3K4me3 and higher levels of H3K27me3 on the Arf promoter in Notch driven T-cell lymphoma cell line 4084 compared to a Myc-driven T-cell lymphoma cell line 6780 (Figure 2D, 2E and Figure S3).
Figure 2. Down-regulation of Arf is a direct transcriptional effect of Notch and is accompanied by changes in the epigenetic landscape on the promoter.
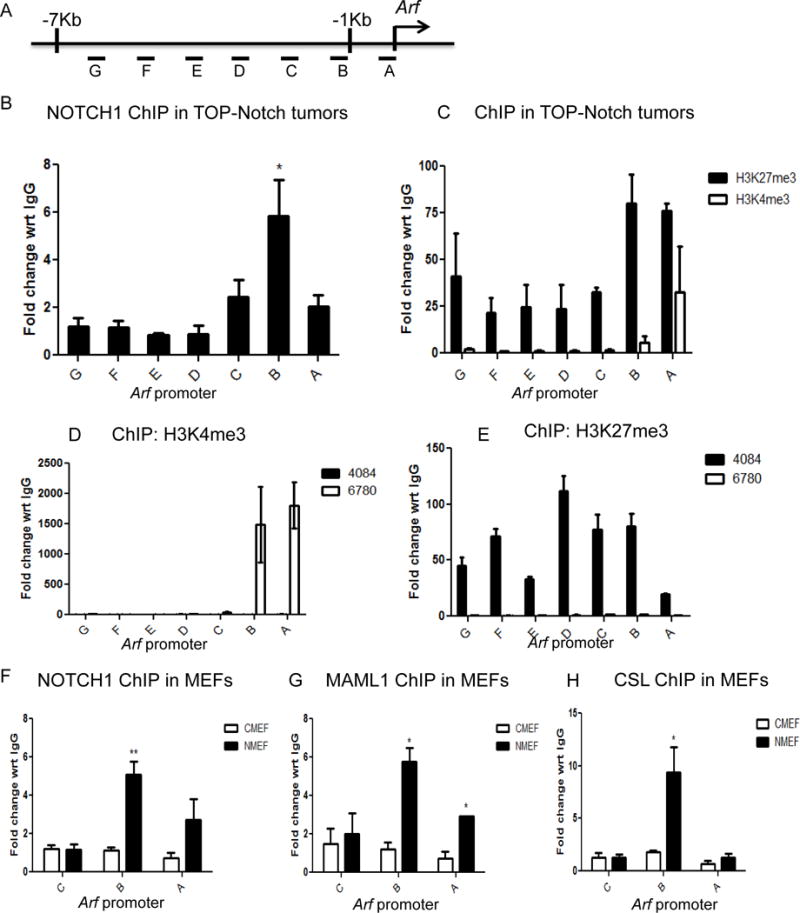
A. Schematic representation of the primers spanning the Arf promoter used for ChIP-PCR analysis. B. ChIP-PCR analysis showing Notch binding on the Arf promoter in primary mouse lymphoma tumors (three individual biological repeats). C. ChIP-PCR analysis depicting the amount of H3K4me3 and H3K27me3 binding on the Arf promoter in primary mouse lymphomas tumors. H3K4me3 (D) and H3K27me3 (E) binding in mouse lymphoma cell lines (4084 and 6780). In Notchic infected MEFs (NMEF) compare to control MEFs (CMEF), ChIP-PCR analysis showing: Notch (F), Maml1 (G) and CSL (H) binding in the proximal region on Arf promoter. Results are represented as fold change with respect to IgG. Data are shown as means ±SEM (n=3) *P≤0.05.
To recapitulate the molecular mechanism, we performed ChIP experiments in primary MEFs that ectopically expressed Notchic (NMEF). When Notchic is expressed in primary MEFs, we readily detect the occupancy of Notch, Maml and CSL on the Arf locus in comparison to control-infected MEF cells (CMEF) (Figure 2F, G and H). These data demonstrate that the Notch ternary complex directly localizes to the Arf locus.
Taken together, these results indicate that repression of Arf transcription is a direct transcriptional effect of Notch. Notch mediated repression is through the binding of Notch ternary complex to the Arf promoter, which induces the reduction of active mark H3K4me3 and the accumulation of repressive mark H3K27me3.
Notch recruits polycomb repressive complex-2 (PRC2) to the Arf promoter
Since an enrichment of H3K27me3 was detected on the Arf promoter, we next investigated the putative mediators responsible for the methylation of H3K27. One of the well-studied histone methyl transferases that methylates H3K27 is Polycomb repressor complex - 2 (PRC2) (30–34). To investigate the involvement of PRC2 in Notch-mediated transcriptional repression, we first examined the mRNA levels of its core components - Ezh2 and Suz12 - in lymphoma cells. Cell lines that have higher levels of activated Notch (4084 and NA1) express greater levels of both factors compared to the Myc-driven lymphoma cell line 6780, which has no detectable Notch activity (Figure S3A and S3B). In primary MEFs, Notchic expression also results in increased transcriptional levels of Ezh2 and Suz12 (Figure 3A). We then conducted ChIP experiments to investigate whether PRC2 is co-localized to the Arf promoter together with the Notch ternary complex. In Notchic infected MEFs, occupancy of the proximal Arf promoter by EZH2 and SUZ12 mirrors the occupancy by Notchic (Figure 3B and 3C). In contrast, EZH2 and SUZ12 were not detected on the Arf locus in control infected MEFs. These data indicate that PRC2 localizes to the Arf locus in a Notch-dependent manner. Moreover, when Notchic infected MEFs are treated with the EZH2 inhibitor EPZ-6438, the transcriptional activity of Arf is rescued from Notch repression (Figure 3D) (35,36). In comparison, ChIP analysis on the Hey2 promoter, which is transcriptionally activated by Notch, reveals that the ternary complex but not the PRC2 complex is readily detected (Figure 3E). This data clearly indicates that Notch-mediated PRC2 recruitment leading to transcriptional repression is a specific and distinct Notchevent.
Figure 3. Notch recruits polycomb repressive complex-2 (PRC2) to the Arf promoter.
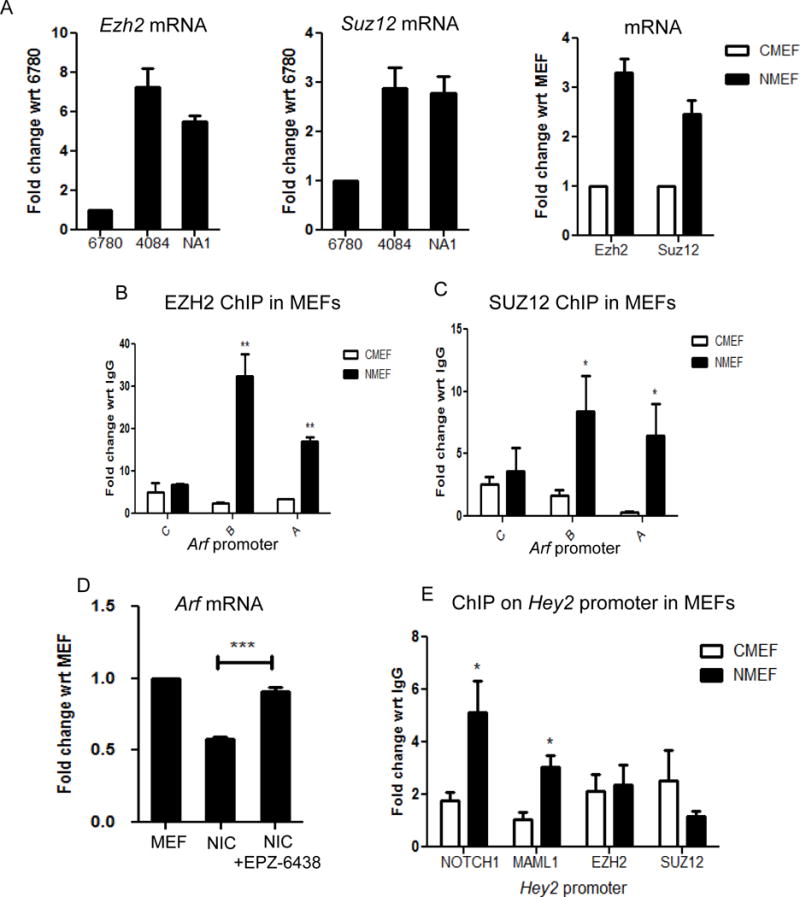
A. qRT-PCR analysis of Ezh2 and Suz12 in mouse lymphoma cell lines and primary MEFs infected with control and Notchic retrovirus. ChIP-PCR analysis showing EZH2 (B) and SUZ12 (C) binding to the Arf promoter in control and Notchic infected MEFs. Results are expressed as fold change with respect to IgG. Shown are means ±SEM (n=3) D. qRT-PCR analysis of Arf in primary MEFs, Notchic infected MEFs and Notchic infected MEFs further treated with EPZ-6438 (5μM, 48h). Data are shown as means±SEM (n=3). E. ChIP-PCR analysis showing Notch, Maml, EZH2 and SUZ12 binding level on the Hey2 promoter in control and Notchic infected MEFs. Results are expressed as fold change with respect to IgG. Shown are means ±SEM (n=3). *P≤0.05, **P≤0.01, ***P≤0.001.
LSD1, a histone demethylase, is required for the assembly of the Notch repressor complex
In addition to increased levels of the repressive mark H3K27me3, there is a significant decrease in the levels of the active mark H3K4me3 on the Arf promoter in Notch-induced T-cell lymphoma (Figure 2D); therefore, we sought to determine the enzyme that directs this epigenetic change. LSD1 is a histone demethylase, which has been shown to account for demethylation of H3K4 in many cases (37). ChIP experiments performed in Notchic infected primary MEFs revealed that Notch induces the binding of LSD1 to the Arf promoter compared to control MEFs that show no enrichment. These data demonstrate that LSD1 colocalizes with the Notch ternary complex and PRC2 on the Arf promoter (Figure 4A). Moreover, when Notchic-infected MEFs were treated with the LSD1 inhibitor Tranylcypromine (TCP), repression of Arf transcription by Notch was blocked (Figure 4B) (36,38). We next examined the occupancy of these factors on the Arf locus in Notchic MEF cells treated with TCP. Compared to Notchic MEF cells treated with vehicle, lower occupancy levels of LSD1 and Notch on the Arf promoter were observed in the TCP treated cells (Figure 4C and D). Consequently, the binding of EZH2 and repressive mark H3K27me3 to the promoter are also decreased, consistent with the reduction of Notch binding activity (Figure 4E and F). Western blot analysis indicates that there is no difference in expression of Notch1, LSD1 and EZH2 in Notchic MEFs and the TCP-treated Notchic MEFs (Figure 4G). Therefore, the lower occupancy levels observed on the Arf promoter are not a result of the protein loss. Collectively, these data indicate that the demethylation activity of LSD1 is a pre-requisite for the Notch repressor complex formation.
Figure 4. LSD1, a histone demethylase, is required for the assembly of the Notch repressor complex.
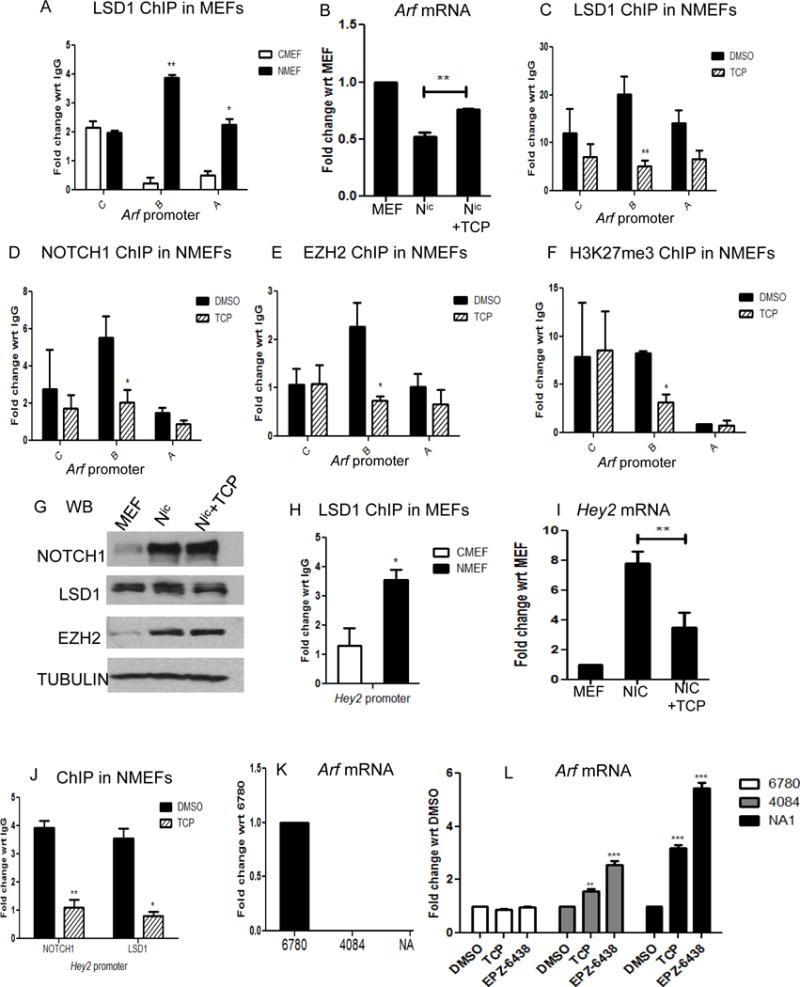
A. ChIP-PCR analysis showing LSD1 binding to the Arf promoter in control and Notchic infected MEFs. B. qRT-PCR analysis of Arf in primary MEFs, Notchic infected MEFs and Notchic infected MEFs further treated with TCP (10μM, 48h). Shown are means±SEM (n=3), **P<0.01. C,D,E and F. ChIP-PCR analysis of Notch1(C), LSD1(D), EZH2 (E) and H3K27me3 (F) binding to Arf promoter between control and TCP treatment group. G. Western Blot analysis revealed protein levels of Notch1, LSD1 and EZH2 in primary MEFs, Notchic infected MEFs and TCP treated Notchic infected MEFs. Tubulin is used as loading controls. H. ChIP-PCR analysis showing LSD1 binding to the Hey2 promoter in control and Notchic infected MEFs. I. qRT-PCR analysis of Hey2 in primary MEFs, Notchic infected MEFs and Notchic infected MEFs further treated with TCP (10μM, 48h). Shown are means±SEM (n=3), **P<0.01. J. ChIP-PCR analysis of Notch and LSD1 binding to the Hey2 promoter between control and TCP treatment group. K. qRT-PCR analysis of Arf in lymphoma cell lines 6780, 4084 and NA1. L. qRT-PCR analysis of Arf in the three lymphoma Cell lines treated with LSD1 inhibitor TCP (10μM, 48h) or EZH2 inhibitor EPZ-6438 (5μM, 48h). ChIP results are represented as fold change with respect to IgG. Data are shown as means ±SEM (n=3) *P≤0.05, **P<0.01.
Interestingly, on the Hey2 promoter, we also detect LSD1 binding, which is induced by Notch (Figure 4H). To address the specificity of LSD1 recruitment to repressed genes, we analyzed the effect of TCP treatment on Hey2 expression. Similar to the results obtained for the repression of Arf expression by Notch, TCP also blocked Notch in transcriptional activation. That is, the treatment of Notchic-infected MEFs with TCP resulted in a decrease in transcription of Hey2 and loss of Notch and LSD1 occupancy from the promoter (Figure 4I and J). Taken together, this data indicate that LSD1 is required for both activation and repression by Notch.
The Notch driven lymphoma cell line NA1 and 4084 has undetectable levels of Arf mRNA compared to the Myc-driven lymphoma cell line 6780 (Figure 4K). When NA1 and 4084 cells are treated with TCP and EPZ-6438, transcriptional activity is released from Notch-directed repression. In contrast, no effect on Arf transcription is observed in 6780 cells treated with either TCP or EPZ-6438 (Figure 4L). Moreover, both inhibitors only affect NA1 cell line viability but not 6780 cell line (Figure S4). Similar has been observed in human leukemia cell line ALL-SIL that Arf transcriptional level is elevated by both inhibitors (Figure S5). Taken together, the inhibition of LSD1 or EZH2 contributes to the rescue of ARF expression in Notchic-expressing MEFs and Notch-driven T-cell lymphoma cell lines, indicating that both LSD1 and EZH2 activities are critical for Notch-directed transcriptional repression of Arf expression.
Transcriptional repression is likely a common mechanism of Notch-directed transcriptional programming
To extend the Notch repressor paradigm, we sought to investigate other genes repressed by Notch. Previous studies in mouse GI tract, human colon cancer cell lines and human esophageal adenocarcinoma cells have shown lower levels of KLF4 expression when Notch activity is high (20,21). Consistent with these studies, Klf4 expression in Notch-driven T-cell lymphoma cell lines and Notchic-infected MEFs is relatively low when compared to 6780 cells and control-infected MEFs (Figure 5A and 5B). To determine if Klf4 is repressed directly by Notch as in the case of Arf, we performed ChIP analysis on the Klf4 promoter. As observed with the Arf locus, the Notch ternary complex and PRC2 are present on the Klf4 locus in Notchic-infected MEFs and absent in control-infected MEFs (Figure 5C). When Notch expressing MEFs are treated with LSD1 or EZH2 inhibitors, the transcriptional levels of Klf4 are similarly rescued from Notch directed repression (Figure 5B). These data reveal that the recruitment of PRC2 complex by Notch is likely an essential mechanism for Notch-mediated gene repression and is a more general component of a Notch-induced transcriptional cascade.
Figure 5. Assembly of the Notch repressor complex on the Klf4 promoter.
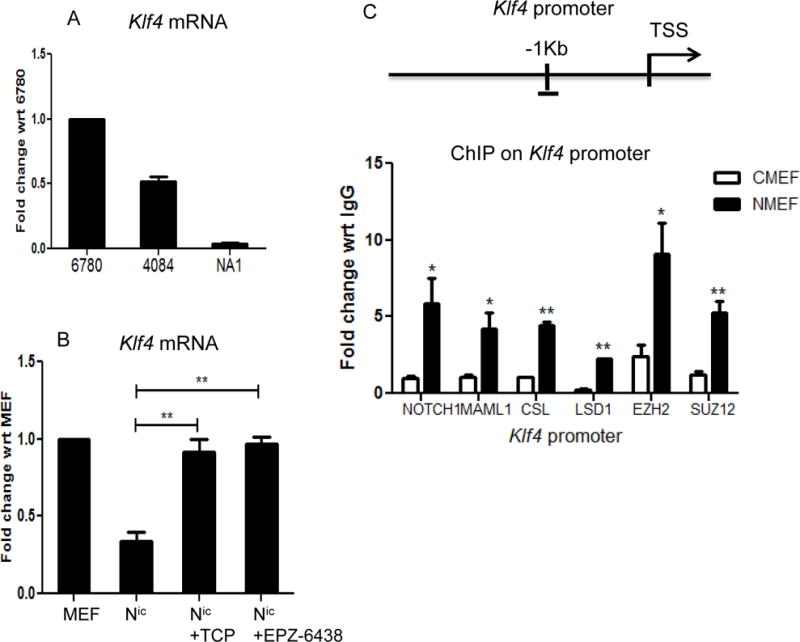
A. qRT-PCR analysis of Klf4 in lymphoma cell lines 6780, 4084 and NA1. B. qRT-PCR analysis of Klf4 in primary MEFs, Notchic infected MEFs and Notchic infected MEFs treated with TCP (10μM) or EPZ-6438 (5μM) for 48 hours. C. Schematic representation of the CSL binding region on the Klf4 promoter and ChIP analysis of Notch, Maml1, EZH2 and SUZ12 on that region. Results are expressed as fold change with respect to IgG. Data are shown as means±SEM (n=3), *P≤0.05, **P≤0.01.
Identification of a Notch transcriptional repressor complex
We provide compelling evidence that Notch directs the transcriptional repression of Arf by the recruitment of the epigenetic enzymes EZH2 and LSD1 accompanied by changes in histone methylation. In order to determine if Notch in fact forms a stable transcriptional repression complex on the Notch ternary complex scaffold, we carried out a DNA affinity precipitation assay using oligonucleotides, harboring CSL binding sites (CSL) conjugated to agarose beads and nuclear lysates derived from 4084 cells. DNA oligonucleotides harboring mutant CSL binding sites (Mut) were used as a control for specificity. When nuclear lysates from 4084 cells were subjected to the CSL DNA affinity precipitation we can readily detect the Notch ternary complex along with LSD1 and EZH2. However, in the CSL mutant pull-down neither the Notch ternary complex nor EZH2 and LSD1 are isolated (Figure 6A). These results indicate that the Notch ternary complex is associated with EZH2 and LSD1 and furthermore, that the binding of this complex is CSL-dependent.
Figure 6. Identification of the CSL-dependent Notch transcriptional repressor complex.
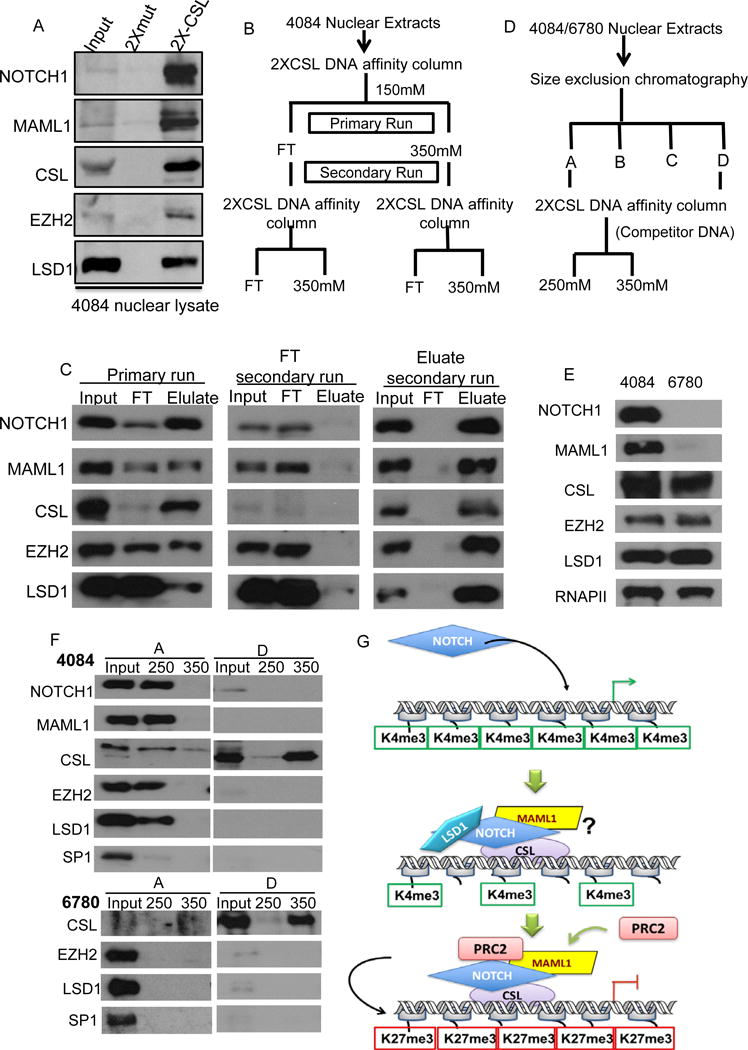
A. DNA affinity precipitation analysis using nuclear lysate from 4084 cell line. B. Purification scheme for reloading the flow-through (FT) and the eluate back to the 2XCSL column. C. According to the purification scheme shown in B, 4084 nuclear lysate was subjected to a 2X-CSL affinity binding column twice, Notch, Maml1, CSL, EZH2 and LSD1 proteins in FT (flow-through) and eluate were visualized by Western blot analysis. Input represents 1% of the total protein loaded onto the column, FT and Eluate represent 4% of the total collection. D. The 2-step purification scheme for 4084 nuclear extracts. E. In nuclear lysate from 4084 compared to 6780, protein levels of Notch, Maml1, CSL, EZH2 and LSD1 were determined by Western blot analysis, RNA polymerase II was used as loading control. F. Section A and section D from 4084 and 6780 fractionation were subjected to 2X-CSL affinity binding column and eluted with column buffer containing 250mM and 350mM NaCl. Notch, Maml, CSL, EZH2 and LSD1 proteins were visualized by Western blotting. Sp1 was used as negative control. Input represents 1% of the total protein loaded onto the column, 10% of the eluate was loaded as sample. G. The transcriptional status of the target promoters is active prior to Notch activation. The binding of Notch ternary complex recruits co-repressors including, LSD1 and EZH2 resulting in changes in epigenetic landscape and leads to the transcriptional repression of target genes.
In order to purify the Notch-repressor complex, we utilized CSL-DNA affinity FPLC. We applied 4084 nuclear lysate to a CSL-DNA affinity column, washed extensively and eluted bound proteins using 350mM NaCl. In this analysis, both EZH2 and LSD1 co-elute with the Notch ternary complex, consistent with the batch DNA affinity precipitation. To demonstrate that these coeluted proteins were complexed we reloaded the eluate onto the CSL-DNA affinity column and observed that EZH2 and LSD1 coeluted with Notch again. In this experiment, no proteins were detected in the flow-through (FT). In contrast, if we reload the primary FT, which is largely depleted of CSL, we observe EZH2, LSD1 along with unbound Notch and Maml in the FT and no proteins detected in the eluate. Moreover, the input lane in the FT reload clearly shows the loss of CSL from the primary run (Figure 6B and 6C). These data support the hypothesis that a stable Notch repression complex can be purified.
To further prove the existence of a Notch repressor complex, we employed a two-step chromatography scheme and compared nuclear extracts from 4084 and 6780 cells (Figure 6D). Western blot analysis of the nuclear lysates from both cell lines demonstrate that unlike 4084 cells, NOTCH and MAML protein levels are undetectable in 6780, whereas other components including CSL are equivalent (Figure 6E). Nuclear lysates from 4084 and 6780 were fractionated by size exclusion chromatography on a Superose 6 column. Based on migration profiles (Figure S6) we prepared two sets of pooled fractions. Pool A refers to the largest complexes, which contain the higher-molecular-weight Notch-containing complexes (A); and pool D which refers to the smallest complex (D), in which monomeric CSL is enriched. Pools A and D were then separately subjected to CSL DNA affinity FPLC followed by stepwise elution at 250 mM NaCl and 350 mM NaCl. Analysis of the pool A derived from 4084 cells revealed that EZH2 and LSD1 co-elute with the Notch ternary complex under 250mM NaCl condition whereas the negative control SP1 failed to co-elute (Figure 6F). As described above, monomeric CSL in the pool D eluted at 350 mM NaCl (Figure S7). Analysis of pool D derived from 6780 cell lysates reveals that monomeric CSL is readily purified, however, no CSL, EZH2 or LSD1 were detected in pool A (Figure 6F). Samples containing the pool A/250 mM eluate were analyzed by LC-MS/MS. Comparison of 4084 and 6780 purifications by MS analysis showed significant enrichment of PRC2 and LSD1 from 4084 lysates along with the Notch ternary complex but no enrichment from 6780-derived lysates (Table 1). These data indicate that co-purification of PRC2 and LSD1 is dependent on the presence of the Notch ternary complex. Taken together, these data reveal the existence of a stable transcriptional repressor complex comprising EZH2, LSD1 and the Notch ternary complex.
Table 1.
MS analysis showed significant enrichment of PRC2 and LSD1 along with the Notch ternary complex from 4084 lysates in comparison to 6780 derived lysates.
| Complex/Protein names | Gene names | MS/MS Count 4084A250 | MS/MS Count 6780A250 | Intensity Fold Change |
|---|---|---|---|---|
| Notch ternary complex | Notch1 | 10 | 0 | 2.38E+08 |
| Maml1 | 9 | 0 | 1.77E+08 | |
| Rbpj | 12 | 0 | 3.84E+08 | |
| PRC2 | Ezh2 | 24 | 1 | 4.50E+02 |
| Suz12 | 24 | 2 | 3.40E+02 | |
| Eed | 17 | 3 | 3.41E+01 | |
| Rbbp4 | 37 | 17 | 1.38E+01 | |
| LSD1 | Kdm1a | 34 | 4 | 5.90E+01 |
Discussion
Many expression analysis studies have reported that induction of Notch signaling results in both the activation and repression of gene expression. It has long been thought that Notch functions as a transcriptional activator that initiates a transcriptional cascade. In such a model, Notch would repress gene expression by first activating the expression of transcriptional repressors such as the Hes/Hey family (canonical Notch target genes), which in turn would repress transcription of their target genes. For example, induction of Notch represses the expression of PTEN and Fbw7. In this case, repression is directly mediated by Hes/Hey, which is a secondary effect of Notch (17,19,22). In other cases, such as the regulation of Sox9 in chondrogenesis and miR-155 during inflammation, Notch does not invoke Hes/Hey repressors to turn these genes off but rather it appears to be a direct effect of Notch as evidenced by ChIP analysis (16,18). However, to date, this “direct” effect is only supported by a circumstantial evidence, as no mechanism for repression by Notch has been reported. Herein, we present evidence that reveals a novel mechanism by which Notch directly represses gene transcription through binding to target promoters. We demonstrate that the down-regulation of Arf and Klf4 by Notch is a direct effect and is accompanied by changes in the epigenetic landscape to favor repression. Furthermore, we demonstrate that PRC2, the histone methyl transferase complex responsible for H3K27me3, is recruited by Notch to these repressed promoters. Data also indicate that recruitment of PRC2 and the subsequent enrichment of H3K27me3 require the activity of LSD1. Furthermore, using a two-step chromatography purification scheme we have identified a stable Notch repressor complex comprising EZH2, LSD1 and the Notch ternary complex. Based on these data, we propose the following model for the assembly of a Notch directed repressor complex on target promoters: in that, Notch binding to the target promoters recruits co-factors Maml and PRC2, together with LSD1, to drive the reduction of H3K4me3 and the accumulation of H3K27me3 resulting in repression of transcription (Figure 6G).
PRC2 is a component of the Notch Transcriptional cascade
It has been established that several epigenetic modifiers including EZH2 are aberrantly expressed in multiple human malignancies (34,39). EZH2 is known as a core enzymatic component of PRC2 which mediates the methylation of H3K27 and functions as a transcriptional repressor complex (33). Our data demonstrates that by recruiting PRC2 to target promoters, Notch directly mediates target gene repression. Is PRC2 a direct transcriptional target of Notch? Preliminary data suggests that Notch activation leads to direct transcriptional induction of both Ezh2 and Suz12. Although there is no evidence indicating that Ezh2 and Suz12 are direct Notch target genes, our results suggests that they are indeed part of the transcriptional cascade, as are Hes and Hey genes. We have demonstrated that NACK is a direct transcriptional target of Notch and acts as a critical co-activator in Notch-mediated transcription (26). Considering this, a model emerges that suggests that upon activation of Notch there is a series of target genes that are activated and in turn function as co-transcriptional regulators of Notch-mediated transcription. This mechanism serves to enforce a Notch transcriptional profile of activated and repressed genes. Therefore, an interesting question is: what determines how Notch becomes an activator or a repressor of transcription. The answer likely involves the local environment and specific modifications to the Notch ternary complex in a context-dependent manner.
LSD1 is required for both Notch transcriptional activation and repression
LSD1 is a histone demethylase which has been reported to be involved in Notch-mediated transcriptional activation. However, a mechanism of action for this role of LSD1 has not been resolved. Mulligan et al. have also suggested that LSD1 serves as a repressor for Notch target genes by forming a complex with CSL and that induction of Notch replaces LSD1 from the target promoters (40). However, data from Benkirane’s group indicates that upon Notch activation, LSD1 switches its function from a co-repressor to a co-activator by modifying different histone marks (3,41). In this study, we sought to investigate the role of LSD1 in Notch mediated gene repression. Our data have revealed that LSD1 is recruited by Notch to the Arf promoter and its demethylase activity is required for the assembly of the Notch repressor complex. Inhibiting LSD1 activity with TCP dissociates the repressor complex from the Arf promoter and rescues Arf transcription from Notch repression. However, we have also detected the binding of LSD1 to the Hey2 promoter, which is a gene activated by Notch. Inhibition of LSD1 activity with TCP also blocks activation of Hey2 transcription by Notch and therefore is not specific to either of these activities. Taken together, our study indicates that LSD1 is required for both Notch-mediated activation and repression, perhaps through influencing assembly of the ternary complex on DNA.
Assembly of the Notch repressor complex on target promoters
In this study, we demonstrate a link between Notch, LSD1 and PRC2 in Notch-mediated gene repression. Moreover, protein purification analysis has identified a stable Notch repressor complex comprising EZH2, LSD1 and the Notch ternary complex. The mechanistic details describing how the complex assembles, however, remain to be fully understood. Maml, a core factor in Notch transcriptional complexes, is acetylated by p300 during Notch-mediated transcriptional activation (42). Unpublished data from our lab demonstrate that this modification is important for recruiting the co-activator NACK and activating transcription (Ke at al, submitted). Evidence derived from this study reveals that Maml is also involved in the repressor complex. Therefore, one possibility is that post-translational modifications on Maml by distinct epigenetic enzymes dictate whether Notch activates or represses transcription by the subsequent recruitment of secondary complexes. How is PRC2 recruited to the Notch ternary complex? Mechanisms for the recruitment of PRC2 to transcription factors remain elusive, but several models have been proposed (43–47). Jaird2 has been shown to link PRC2 to chromatin in ES cells (48). Histone deacetylases (HDAC) 1 and 2 associate with PRC2 core complex to drive gene repression (49). The long non-coding RNA HOTAIR has been shown to serve as a bridge between LSD1 and PRC2 (50). Whether or not any of these factors contribute to the Notch repressor complex assembly and activity is yet to be explored.
Based on data presented herein, we propose a novel regulatory mechanism of Notch. That is, in addition to activating gene expression by forming a transcriptional activation complex. Notch directs the assembly of a repressor complex and acts as a transcriptional repressor. Since ARF is an important tumor suppressor that triggers the induction of p53 activity, repression of ARF transcription by Notch is fundamental to its role in driving tumorigenesis. Therefore, this study reveals potential new targets in the attack on Notch-dependent tumors. Specifically, chemical attack on the PRC2 enzyme EZH2 in combination with inhibition of Notch might provide an effective therapeutic approach.
Supplementary Material
Implications.
This study provides rationale for the targeting of epigenetic enzymes to inhibit Notch activity or use in combinatorial therapy to provide a more profound therapeutic response.
Acknowledgments
The authors thank members of the Capobianco and Robbins laboratory for support and technical assistance.
Funding: NCI RO1 CA 83736, RO1 CA 169805-01 to A.J.C.
Grant Support
This work was supported by the NCI (NCI R01CA083736-12A1, NCIR01CA125044-02 to A.J. Capobianco). This project was also generously supported by funding from the Dewitt Daughtry Family Department of Surgery and the Sylvester Comprehensive Cancer Center (A.J. Capobianco).
Footnotes
No potential conflicts of interest were disclosed.
References
- 1.Davidson EH. Emerging properties of animal gene regulatory networks. Nature. 2010;468:911–20. doi: 10.1038/nature09645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peter IS, Davidson EH. Evolution of gene regulatory networks controlling body plan development. Cell. 2011;144:970–85. doi: 10.1016/j.cell.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer cell. 2014;25:318–34. doi: 10.1016/j.ccr.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–7. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 5.Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. The Journal of biological chemistry. 2007;282:33994–4002. doi: 10.1074/jbc.M701847200. [DOI] [PubMed] [Google Scholar]
- 6.Koch U, Radtke F. Notch and cancer: a double-edged sword. Cellular and molecular life sciences: CMLS. 2007;64:2746–62. doi: 10.1007/s00018-007-7164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demarest RM, Ratti F, Capobianco AJ. It’s T-ALL about Notch. Oncogene. 2008;27:5082–91. doi: 10.1038/onc.2008.222. [DOI] [PubMed] [Google Scholar]
- 8.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nature reviews Cancer. 2011;11:338–51. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 9.Kovall RA. More complicated than it looks: assembly of Notch pathway transcription complexes. Oncogene. 2008;27:5099–109. doi: 10.1038/onc.2008.223. [DOI] [PubMed] [Google Scholar]
- 10.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–33. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bray SJ. Notch signalling: a simple pathway becomes complex. Nature reviews Molecular cell biology. 2006;7:678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 12.Gordon WR, Arnett KL, Blacklow SC. The molecular logic of Notch signaling–a structural and biochemical perspective. Journal of cell science. 2008;121:3109–19. doi: 10.1242/jcs.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becam I, Rafel N, Hong X, Cohen SM, Milan M. Notch-mediated repression of bantam miRNA contributes to boundary formation in the Drosophila wing. Development. 2011;138:3781–9. doi: 10.1242/dev.064774. [DOI] [PubMed] [Google Scholar]
- 14.Kapuria S, Karpac J, Biteau B, Hwangbo D, Jasper H. Notch-mediated suppression of TSC2 expression regulates cell differentiation in the Drosophila intestinal stem cell lineage. PLoS genetics. 2012;8:e1003045. doi: 10.1371/journal.pgen.1003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lei T, Bi Y, Gao MJ, Gao SM, Zhou LL, Zheng HL, et al. HES1 inhibits adipogenesis of porcine mesenchymal stem cells via transcriptional repression of FAD24. Domestic animal endocrinology. 2013;45:28–32. doi: 10.1016/j.domaniend.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, Zhang H, Rodriguez S, Cao L, Parish J, Mumaw C, et al. Notch-Dependent Repression of miR-155 in the Bone Marrow Niche Regulates Hematopoiesis in an NF-kappaB-Dependent Manner. Cell stem cell. 2014;15:51–65. doi: 10.1016/j.stem.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sancho R, Blake SM, Tendeng C, Clurman BE, Lewis J, Behrens A. Fbw7 repression by hes5 creates a feedback loop that modulates Notch-mediated intestinal and neural stem cell fate decisions. PLoS biology. 2013;11:e1001586. doi: 10.1371/journal.pbio.1001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S, Tao J, Bae Y, Jiang MM, Bertin T, Chen Y, et al. Notch gain of function inhibits chondrocyte differentiation via Rbpj-dependent suppression of Sox9. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2013;28:649–59. doi: 10.1002/jbmr.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whelan JT, Kellogg A, Shewchuk BM, Hewan-Lowe K, Bertrand FE. Notch-1 signaling is lost in prostate adenocarcinoma and promotes PTEN gene expression. Journal of cellular biochemistry. 2009;107:992–1001. doi: 10.1002/jcb.22199. [DOI] [PubMed] [Google Scholar]
- 20.Zheng H, Pritchard DM, Yang X, Bennett E, Liu G, Liu C, et al. KLF4 gene expression is inhibited by the notch signaling pathway that controls goblet cell differentiation in mouse gastrointestinal tract. American journal of physiology Gastrointestinal and liver physiology. 2009;296:G490–8. doi: 10.1152/ajpgi.90393.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghaleb AM, Aggarwal G, Bialkowska AB, Nandan MO, Yang VW. Notch inhibits expression of the Kruppel-like factor 4 tumor suppressor in the intestinal epithelium. Molecular cancer research: MCR. 2008;6:1920–7. doi: 10.1158/1541-7786.MCR-08-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. Journal of cellular physiology. 2003;194:237–55. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 23.Kageyama R, Ohtsuka T, Kobayashi T. The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development. 2007;134:1243–51. doi: 10.1242/dev.000786. [DOI] [PubMed] [Google Scholar]
- 24.Beverly LJ, Felsher DW, Capobianco AJ. Suppression of p53 by Notch in lymphomagenesis: implications for initiation and regression. Cancer research. 2005;65:7159–68. doi: 10.1158/0008-5472.CAN-05-1664. [DOI] [PubMed] [Google Scholar]
- 25.Jozefczuk J, Drews K, Adjaye J. Preparation of mouse embryonic fibroblast cells suitable for culturing human embryonic and induced pluripotent stem cells. Journal of visualized experiments: JoVE. 2012 doi: 10.3791/3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weaver KL, Alves-Guerra MC, Jin K, Wang Z, Han X, Ranganathan P, et al. NACK is an integral component of the Notch transcriptional activation complex and is critical for development and tumorigenesis. Cancer research. 2014 doi: 10.1158/0008-5472.CAN-14-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeffries S, Robbins DJ, Capobianco AJ. Characterization of a high-molecular-weight Notch complex in the nucleus of Notch(ic)-transformed RKE cells and in a human T-cell leukemia cell line. Molecular and cellular biology. 2002;22:3927–41. doi: 10.1128/MCB.22.11.3927-3941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Da Silva TG, Jin K, Han X, Ranganathan P, Zhu X, et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer research. 2014;74:6364–74. doi: 10.1158/0008-5472.CAN-14-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13028–33. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Garcia E, Licht JD. Deregulation of H3K27 methylation in cancer. Nature genetics. 2010;42:100–1. doi: 10.1038/ng0210-100. [DOI] [PubMed] [Google Scholar]
- 32.Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nature reviews Molecular cell biology. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 33.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simon C, Chagraoui J, Krosl J, Gendron P, Wilhelm B, Lemieux S, et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes & development. 2012;26:651–6. doi: 10.1101/gad.186411.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nature medicine. 2016;22:128–34. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morera L, Lubbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clinical epigenetics. 2016;8:57. doi: 10.1186/s13148-016-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–53. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Hayward D, Cole PA. LSD1 Histone Demethylase Assays and Inhibition. Methods in enzymology. 2016;573:261–78. doi: 10.1016/bs.mie.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melnick A. Epigenetic therapy leaps ahead with specific targeting of EZH2. Cancer cell. 2012;22:569–70. doi: 10.1016/j.ccr.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mulligan P, Yang F, Di Stefano L, Ji JY, Ouyang J, Nishikawa JL, et al. A SIRT1-LSD1 corepressor complex regulates Notch target gene expression and development. Molecular cell. 2011;42:689–99. doi: 10.1016/j.molcel.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yatim A, Benne C, Sobhian B, Laurent-Chabalier S, Deas O, Judde JG, et al. NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Molecular cell. 2012;48:445–58. doi: 10.1016/j.molcel.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saint Just Ribeiro M, Hansson ML, Wallberg AE. A proline repeat domain in the Notch co-activator MAML1 is important for the p300-mediated acetylation of MAML1. The Biochemical journal. 2007;404:289–98. doi: 10.1042/BJ20061900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seelk S, Adrian-Kalchhauser I, Hargitai B, Hajduskova M, Gutnik S, Tursun B, et al. Increasing Notch signaling antagonizes RC2-mediated silencing to promote reprograming of germ cells into neurons. eLife. 2016;5 doi: 10.7554/eLife.15477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127:42–52. doi: 10.1182/blood-2015-07-604512. [DOI] [PubMed] [Google Scholar]
- 45.Koppens MA, Bounova G, Gargiulo G, Tanger E, Janssen H, Cornelissen-Steijger P, et al. Deletion of Polycomb Repressive Complex 2 From Mouse Intestine Causes Loss of Stem Cells. Gastroenterology. 2016;151:684–97 e12. doi: 10.1053/j.gastro.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 46.Tolhuis B, de Wit E, Muijrers I, Teunissen H, Talhout W, van Steensel B, et al. Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nature genetics. 2006;38:694–9. doi: 10.1038/ng1792. [DOI] [PubMed] [Google Scholar]
- 47.Oravecz A, Apostolov A, Polak K, Jost B, Le Gras S, Chan S, et al. Ikaros mediates gene silencing in T cells through Polycomb repressive complex 2. Nature communications. 2015;6:8823. doi: 10.1038/ncomms9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, et al. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature. 2010;464:306–10. doi: 10.1038/nature08788. [DOI] [PubMed] [Google Scholar]
- 49.van der Vlag J, Otte AP. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nature genetics. 1999;23:474–8. doi: 10.1038/70602. [DOI] [PubMed] [Google Scholar]
- 50.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.