

Abstract
Mutational signatures in cancer genomes have implicated the APOBEC3 cytosine deaminases in oncogenesis, possibly offering a therapeutic vulnerability. Elevated APOBEC3B expression has been detected in solid tumors, but expression of APOBEC3A (A3A) in cancer has not been described to date. Here we report that A3A is highly expressed in subsets of pediatric and adult acute myeloid leukemia (AML). We modeled A3A expression in the THP1 AML cell line by introducing an inducible A3A gene. A3A expression caused ATR-dependent phosphorylation of Chk1 and cell cycle arrest, consistent with replication checkpoint activation. Further, replication checkpoint blockade via small molecule inhibition of ATR kinase in cells expressing A3A led to apoptosis and cell death. Although DNA damage checkpoints are broadly activated in response to A3A activity, synthetic lethality was specific to ATR signaling via Chk1 and did not occur with ATM inhibition. Our findings identify elevation of A3A in AML cells, enabling apoptotic sensitivity to inhibitors of the DNA replication checkpoint and suggesting it as a candidate biomarker for ATR inhibitor therapy.
Keywords: APOBEC3, ATR kinase, Chk1 kinase, acute myeloid leukemia, replication checkpoint
INTRODUCTION
Mutational signatures in cancer genomes have elucidated potential etiologies of DNA damage and oncogenesis (1,2). Possible mutagens include both endogenous and exogenous sources. One mutational signature, which consists of clustered cytosine mutations and predominantly C→T transitions within a TC context, has been attributed to the APOBEC3 (A3) family of DNA cytosine deaminases. A3 enzymes are best defined by their ability to mutate viral DNA and restrict infection, although the full scope of their endogenous function is unknown (3). Two A3 family members, A3A and A3B, are localized in the nucleus (4) and their activity results in cellular DNA damage (5–7). A3 enzymes are postulated to be under tight regulation to prevent detrimental genotoxicity.
The association between A3 family members and cancer mutagenesis has emerged through identification of the A3 mutational signature in cancer genomes, as well as increased A3 expression in human tumors. Most notably, several studies have detected elevated A3B expression correlated with mutational hallmarks in breast cancer (7,8). The related A3A has also been implicated in mutational signatures identified in cancer genomes (9,10). The mutational signatures of A3A and A3B are similar but distinguishable and that of A3A is the more prominent signature in cancer genomes (9). A germline polymorphism that deletes the A3B coding sequence and fuses A3A to the 3’UTR of A3B has been associated with increased stability of A3A transcripts, a higher burden of A3 mutational signatures, and cancer susceptibility (11–13). Despite these strong associations between A3A and mutational burden in cancer genomes, A3A expression in human cancers has not been established. Endogenous expression of A3 enzymes is largely restricted to immune tissues, although variations exist across A3 family members (14). A3A is highly expressed in myeloid lineage cells and is interferon-inducible in lymphocytes (15–17). Although A3A expression in peripheral blood mononuclear cells represents the highest level of endogenous expression of any A3 family member in human tissue (14), A3A expression in hematologic malignancies has not been investigated.
A3A expression results in robust activation of cellular DNA damage responses (5,6). Ectopic A3A expression has been shown to cause phosphorylation of histone variant H2AX and ATM (Ataxia-Telangiectasia Mutated) kinase, indicative of a cellular response to double-stranded DNA breaks (DSBs) (6). We recently showed that A3A expression also causes replication stress, as evidenced by activation of ATR (ATM- and Rad3-related) kinase, phosphorylation of downstream Chk1 (Checkpoint Kinase 1), and cell cycle arrest (5). The ATR-Chk1 pathway is activated in response to replication stress when Replication Protein A (RPA) binds exposed ssDNA and recruits ATR localization and activation (18). ATR signaling promotes replication fork stability and enforces the replication checkpoint which arrests cellular division to allow for repair of damaged DNA prior to mitosis (19). Inhibition of the replication checkpoint promotes replication fork collapse into DSBs and genotoxicity, particularly in the context of DNA damage that impedes DNA polymerase progression. These conditions can be cancer-associated, including the effects of oncogene expression (20,21). Thus, the ATR-Chk1 pathway has been investigated as a target for cancer therapy (22,23).
Effective cancer therapy relies on differences between healthy cells and cancer cells that present opportunities for selective targeting. Traditional chemotherapeutics target fundamental cellular processes, such as DNA replication, which results in death of rapidly dividing cancer cells but have a high level of toxicity in proliferating healthy cells. One potential for more refined therapies occurs in the context of synthetic lethality, in which two abnormal gene products co-operate to impair cell fitness more than either one alone (24). Synthetic lethality can be prompted by drug-induced inhibition of essential pathways in genetically altered cancer cells. Here, we investigate the hypothesis that inhibition of genome-protective DNA damage responses enables A3A-induced genotoxicity and can result in synthetic lethality.
In this study, we show that A3A expression is elevated in subsets of human acute myeloid leukemia (AML). Our findings present the first demonstration of elevated A3A expression in human cancer, and the first association of A3 enzymes with hematologic malignancies. To model the effects of high A3A expression, we developed an AML cell line with inducible A3A expression. We demonstrate that the DNA replication checkpoint is robustly activated by A3A in leukemia cells and that A3A sensitizes AML cells to treatment with small molecule inhibitors of ATR and Chk1 kinases. We determined that A3A deamination renders cancer cells dependent on the replication checkpoint for genome protection and that this can be exploited to induce synthetic lethality. Taken together, our data contribute to a new, targeted treatment paradigm for human cancers with elevated A3A expression.
MATERIALS AND METHODS
Cell lines
Authenticated THP1, K562, CEM, Ramos, and U2OS cells were purchased from the American Type Culture Collection (ATCC). U937 cells were a kind gift from Sarah Tasian. Cell lines were not further authenticated. All cells were obtained within 5 years prior to use in experiments described. Cells were tested and confirmed to be mycoplasma free. All cell lines were maintained in Roswell Park Memorial Institute medium (RPMI) containing 100 U/mL of penicillin and 100 μg/mL of streptomycin, and supplemented with 10% tetracycline free fetal bovine serum (FBS). Cells were used within 8 weeks of thawing for experiments described. To construct inducible A3A cell lines, HA-tagged A3A cDNAs were inserted into an entry vector with the CMV promoter and upstream Tetracycline Response Element (pEN_Tmcs). Cloning primer sequences are available upon request. A pSLIK-A3A plasmid was generated by Gateway cloning of the pEN_Tmcs-A3A plasmid into a pSLIK lentivirus vector with a constitutively expressed neomycin resistance gene and tetracycline transactivator (rTA) (25) to generate a single vector system of doxycycline-inducible expression of A3A-HA. Production of lentiviral particles was achieved by transfection of 293T cells as previously described (26). Following transduction with the pSLIK-A3A lentivirus, cells were selected in 1 mg/mL neomycin. After selection, cells were maintained in 0.5 mg/mL neomycin. Knockdown of endogenous A3A in U937 cells was achieved by stable expression of short hairpin RNA against A3A (shA3A). Lentivirus vectors (pLKO) containing shA3A and shControl were purchased from Sigma. Production of lentivirus was achieved as above, and following transduction with the pLKO-shRNA lentivirus, cells were selected in 1 μg/mL puromycin. All cells were grown at 37°C in a humidified atmosphere containing 5% CO2.
Small molecule inhibitors
The ATR kinase inhibitor VE-822 (Vertex), Chk1 kinase inhibitor PF-477736 (Pfizer), and ATM kinase inhibitor KU55933 (Abcam) were each dissolved in dimethyl sulfoxide (DMSO) to make a stock solution and stored at −20°C. Solutions were further diluted in DMSO for treatment of cells in culture. Controls were treated with an equal volume of vehicle (DMSO).
Cell-cycle analysis
THP1, K562, CEM, and Ramos cells were grown in the presence or absence of doxycycline (1 μg/ml, Clontech), fixed in 70% ice-cold ethanol, washed in PBS, and resuspended in staining solution containing 20 mg/ml propidium iodide (Sigma) and 200 mg/ml RNAse A (Roche). Data were collected using an Accuri C6 Flow Cytometer (BD Bioscience) and analyzed by FlowJo software (Version 10.2). Experiments were each performed three times, and at least 20,000 cells were analyzed per sample.
Immunoblotting and immunofluorescence
For immunoblotting, lysates were prepared by harvesting cells in 10% SDS sample buffer (Novex) and 5% b-Mercaptoethanol (Sigma) and boiling for 10 minutes, then run on bis-tris gels and transferred to a polyvinylidene fluoride membrane (PVDF, Amersham). For immunofluorescence, cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 for 10 minutes. Nuclei were visualized by staining with 4,6-diamidino-2-phenylindole (DAPI, Sigma). Images were acquired using a Zeiss LSM 710 confocal microscope. Quantification of cell staining was performed on at least 50 cells per condition. Representative images are shown.
Antibodies
Commercially available antibodies used in this study were obtained from Cell Signaling (Chk2-p-T68, Chk1-p-S345, Chk1-p-S317), Millipore (Chk2), Epitomics (ATM), Abcam (ATM-p-S1981), Santa Cruz (Chk1, Actin), Bethyl (RPA32), GeneTex (GAPDH), Biolegend (HA), and the NIH AIDS Reagent Program (A3A/A3G C-terminal polyclonal antibody). Secondary antibodies for immunoblotting included goat anti-rabbit IgG and goat anti-mouse IgG (Jackson ImmunoResearch).
Proliferation, viability and apoptosis assays
Proliferation assay: Leukemia cells were plated at density of 1,000–5,000 cells/well in a 96 well plate. Cells were pre-cultured for 24 hours, then treated with doxycycline (1 ug/ml) and a range of small molecule inhibitor doses or vehicle control (DMSO). Cells were then cultured for 48 hours. Water-soluble tetrazolium salt reagent (CCK-8, Dijoundo) was added 2–4h prior to analysis. Data were collected using an Infinity M1000 Pro plate reader (Tecan). Viability assay: Leukemia cells were plated at density of 300,000 cells/well in a 6 well plate. Cells were treated with doxycycline (1 ug/ml) and small molecule inhibitor or vehicle control for 48 hours. Cells were stained using the Live/Dead Kit (Invitrogen) according to the manufacturer’s instructions. Data were collected using an Accuri C6 Flow Cytometer (BD Bioscience). Apoptosis assay: Leukemia cells were plated at density of 300,000 cells/well in a 6 well plate. Cells were treated with doxycycline (0.1 ug/ml) and small molecule inhibitor or vehicle control. Cells were cultured for 48hr following treatment and then stained using FITC Annexin-V Kit (BD Bioscience) according to the manufacturer’s instructions.
RNA isolation, cDNA synthesis and qPCR
Primary acute myeloid leukemia samples were obtained from the University of Pennsylvania Stem Cell and Xenograft Core. RNA was extracted from primary samples and cell lines using the RNeasy Minikit (Qiagen). cDNA was made using the High Capacity RNA to cDNA kit (Applied Biosystems). Quantitative PCR (qPCR) was performed with SYBR Green PCR Master Mix (Applied Biosystems) on a ViiA 7 real-time PCR instrument (Applied Biosystems). Primers were designed to distinguish A3A from A3B and are as follows: A3A forward CACAACCAGGCTAAGAATCTTCTC, A3A reverse CAGTGCTTAAATTCATCGTAGGTC, A3B forward GAATCCACAGATCAGAAATCCGA, and A3B reverse TTTCACTTCATAGCACAGCCA. The housekeeping gene cyclophillin was used for normalization. For comparison of primary AML samples, a pool of all primary samples was used for delta delta CT analysis. For comparison of THP1-A3A to primary AML samples, all samples were normalized to THP1-A3A cells induced with dox for delta delta CT analysis. For comparison of AML cell lines, a pool of all cell lines was used for delta delta CT analysis.
Bioinformatic analysis
Primary tumor RNA sequencing data were obtained from public sources. Raw count data for genes expressed in TCGA-LAML (n=151) and TARGET-AML (n=282) were downloaded from the GDC Data Portal. Data were normalized using the bioconducter package DESeq2 (27). The normalized gene expression for A3A was separated into two groups, using the 1.5 times the IQR to determine high-expression outliers (28). The results shown here are based upon data generated by the TCGA Research Network (http://cancergenome.nih.gov), and by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative managed by the NCI (http://ocg.cancer.gov/programs/target). The data used for this analysis are available at GDC Data portal (https://gdc-portal.nci.nih.gov/).
Statistical analysis
To distinguish significant differences between high and low A3A expression groups we applied the Mann–Whitney U-test. Boxplot statistics were computed with the function “boxplot” of R programming language. P values and standard error of the mean (SEM) for cell cycle analysis, Annexin V staining, and Live/Dead staining were obtained by paired two-tailed t-tests. P values and SEM for proliferation assays were determined by sum-of-squares F-test. Results were considered significant at p < 0.05. GraphPad Prism 7 software was used for statistical analysis.
RESULTS
A3A is highly expressed in a subset of pediatric and adult AML
To determine which types of human leukemia are most impacted by A3A activity, we examined RNA-sequencing (RNA-seq) data from two major databases of primary leukemias: The Cancer Genome Atlas (TCGA) which comprises expression data from adult-onset tumors, and Therapeutically Applicable Research to Generate Effective Treatments (TARGET), which includes expression profiles of childhood cancers. We limited our evaluation of A3A expression to samples with RNA-seq data, since microarray probes are insufficient to distinguish specific A3 transcripts given their high degree of homology (7). Analysis of RNA-seq from available data showed that high A3A expression occurs in subsets of both pediatric and adult AML (Fig 1a–b, Table S1). A3A expression levels 1.5 times greater than the interquartile range (IQR) were considered high expression outliers (28). The remaining values were assigned to the low expression group. The high A3A expression group comprised 14% of the TCGA samples and 11% of the TARGET samples. Since A3B is reported to be overexpressed in many human cancers (29), we evaluated expression levels of A3B in the same patient samples (Fig 1a–b, Fig S1a–b). When the high A3A and low A3A groups were analyzed in aggregate, we found no difference in average A3B expression between the groups (Fig 1a–b). An additional analysis of samples with outlier A3B expression showed that this group did not overlap with samples in the outlier A3A expression group. There were fewer samples with A3B outlier expression (6% of TCGA and 4% of TARGET), and A3B outlier expression was several log-fold lower than the expression of A3A outliers (Fig S1a–b).
Figure 1. APOBEC3A is highly expressed in acute myeloid leukemia.
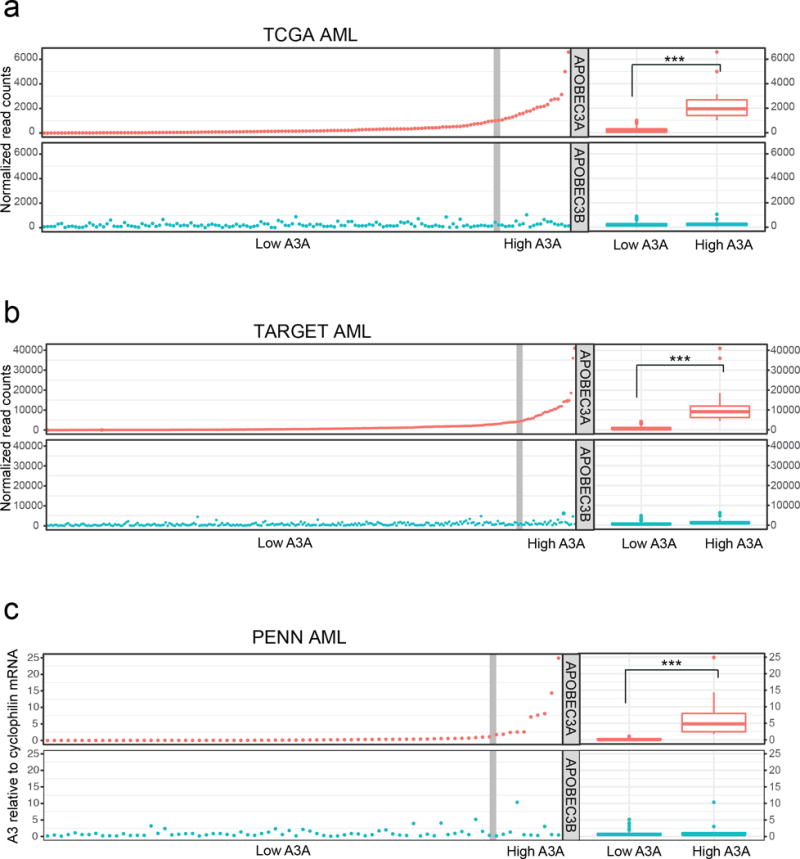
APOBEC3A (red) and APOBEC3B (blue) expression was evaluated in three large cohorts of primary AML. A3 expression in AML samples from the TCGA (a) and TARGET (b) databases was determined by RNA sequencing analysis. Evaluation of A3 mRNA levels in primary AML samples from the University of Pennsylvania (PENN) biological repository (c) was performed by quantitative PCR; A3 mRNA level is displayed as fold change relative to a pooled control. Scatter plots (left) show normalized expression of A3A and A3B in individual samples. Vertical gray line denotes a subset of high A3A expression outliers (right) determined by 1.5× interquartile range. Box plots (far right) display aggregate expression of A3A and A3B in high A3A and low A3A groups. Statistical analysis was performed using the Mann-Whitney U-test.
We obtained primary AML samples from a biorepository at the University of Pennsylvania (PENN) and evaluated A3A mRNA expression by quantitative real-time PCR (qPCR). Consistent with results from TCGA and TARGET datasets, 13.3% of PENN patient samples exhibited high outlier A3A expression (Fig 1c, Table S1). Analysis of high A3A and low A3A groups indicated no difference in average A3B mRNA levels in either group (Fig 1c). Additionally, in primary AML samples, outlier A3B expression did not correlate with elevated A3A expression (Fig S1c). Together, these data indicate that A3A is highly expressed in a subset of pediatric and adult AML.
The replication checkpoint is activated by A3A in leukemia cells
We previously showed that replicating cells are more susceptible to DNA damage by A3A than quiescent cells (5), likely due to deamination of single-stranded DNA (ssDNA) substrate at replication forks (30,31). We sought to evaluate whether this process occurs in leukemia cells in which A3A is active. We constructed a model AML cell line (THP1) with a doxycycline-inducible HA-tagged A3A gene, in which titration of doxycycline dosing enables control of A3A expression levels. In our inducible THP1-A3A cell line, treatment with 1 ug/mL doxycycline resulted in A3A expression similar to that of primary AML samples from PENN categorized as having high A3A expression (Fig 2a). We examined checkpoint activation by immunoblotting with phospho-specific antibodies that recognize sites attributable to ATR kinase activity. We observed a dose-response relationship between A3A protein level and Chk1 phosphorylation at serines 317 and 345 (32) (Fig 2b). To evaluate whether deaminase activity is required for ATR activation, we generated a THP1 cell line with an inducible HA-tagged A3A mutant (C106S) lacking enzymatic activity (15). Immunoblotting showed that when A3A-C106S was induced there was no significant change in phosphorylation of Chk1 at serines 317 and 345 (Fig S2). Thus, the enzymatic activity of A3A appears to be required for replication checkpoint activation. We also show that RPA foci accumulate upon A3A expression, supporting the conclusion that A3A expression results in replication stress and accumulated ssDNA (Fig S3). We then evaluated THP1 cell cycle profiles upon A3A induction by propidium iodide staining. A3A induction resulted in G2 arrest, consistent with replication checkpoint activation (Fig 2c–d). Similar results were observed in a panel of A3A-inducible leukemia cell lines representative of chronic myeloid leukemia (K562), acute T-lymphoblastic leukemia (CEM), and B-lymphoblastic leukemia (Burkitt, Ramos) (Fig S4a–c). These data indicate that A3A deamination results in replication checkpoint activation in hematopoietic cells.
Figure 2. APOBEC3A expression results in ATR activation and checkpoint arrest in AML cells.
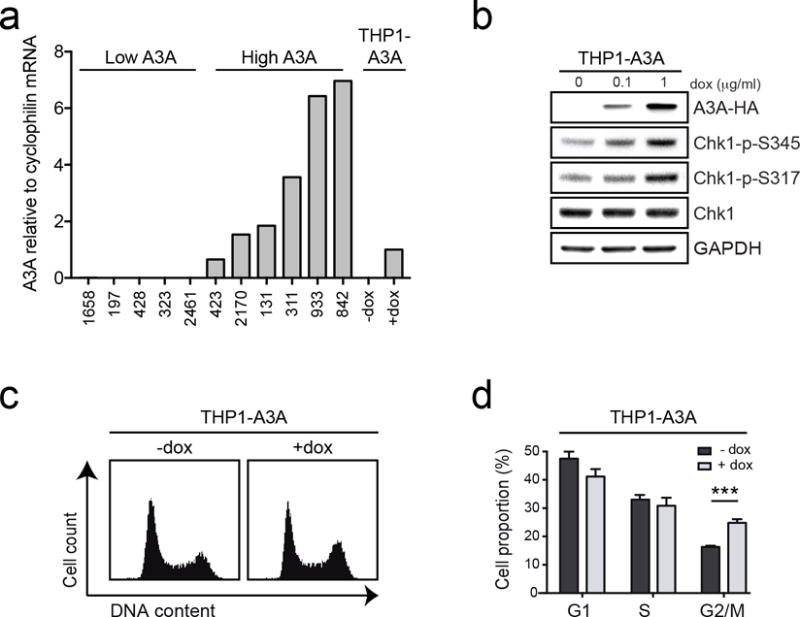
The acute myeloid leukemia cell line, THP1, was generated to express A3A upon treatment with doxycycline (dox). (a) Expression levels of A3A in inducible THP1 cells are similar to those of primary AML samples with high A3A expression. Evaluation of A3A mRNA levels was performed by qPCR in THP1-A3A cells treated with dox (1 μg/mL) alongside primary AML samples from the PENN dataset. Primary AML samples included a selection of those with high A3A and low A3A expression. A3A expression level is displayed as fold change relative to dox-treated THP1-A3A cells. (b) ATR signaling is activated by A3A expression. Cells were treated with indicated concentrations of dox and analyzed by immunoblotting using antibodies to HA, Chk1, and phosphorylated Chk1 (S345 and S317). GAPDH was used as a loading control. (c) A3A expression results in G2 arrest. Inducible cells were treated with dox (1 μg/mL) for 48 hours and analyzed for cell cycle progression by propidium iodide staining. (d) Accompanying chart shows fraction of cells in G1, S, and G2 phase pre- and post-induction. Error bars indicate SEM. Statistical analysis was performed using a paired two-tail t test. Results are representative of three independent replicates.
A3A sensitizes AML cells to ATR inhibition
Since A3A activates the replication checkpoint, we hypothesized that inhibition of checkpoint signaling would result in genotoxicity in AML cells with high A3A expression. We therefore treated the THP1-A3A AML cell line with the ATR kinase inhibitor VE-822 (ATRi), a potent and specific drug that is currently being evaluated in clinical trials in combination with chemotherapy for refractory solid tumors (33–35). We examined the impact of ATR inhibition in our THP1-A3A cells by inducing A3A and then treating with a range of doses of ATRi. THP1 cells that were induced to express A3A had significantly decreased viability after treatment with ATRi when compared to uninduced cells (Fig 3a, left). Notably, A3A induction resulted in an impressive shift of the ATRi half maximal effective concentration (EC50) from 5700 nM to 150 nM. Immunoblotting showed decreased phosphorylation of Chk1 in cells treated with ATRi, confirming inhibition of ATR activity (Fig 3a, right).
Figure 3. APOBEC3A sensitizes AML cells to ATR inhibition.
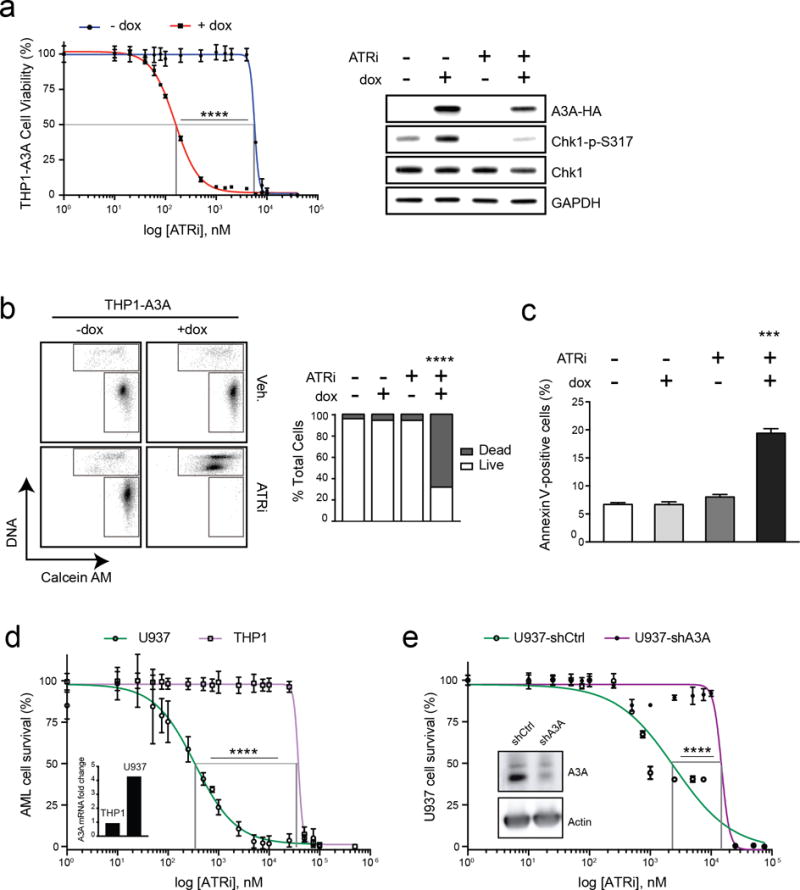
(a) A3A expression was induced in THP1-A3A cells by addition of dox (1 μg/mL), and then treated with indicated doses of a small molecule inhibitor of ATR kinase (ATRi, VE-822) 48 hours prior to analysis. Viability was determined by colorimetric change after addition of a water-soluble tetrazolium salt. Statistical analysis of EC50 was performed using a sum-of-squares F-test. Error bars are SEM. Accompanying immunoblot shows impact of ATRi treatment on THP1-A3A cells treated with dox, ATRi (80 nM), or combinations. Cell lysates were probed with antibodies to HA, Chk1, and phosphorylated Chk1. GAPDH was used as loading control. (b) THP1-A3A cells were treated with dox (1 μg/mL), ATRi (200 nM), or combinations, incubated with fluorescent-labeled calcein AM (live) and DNA (dead) stains, then evaluated by FACS. The upper gate includes dead cells and the lower gate includes live cells. Accompanying bar chart shows quantitation of results averaged over three replicates. Statistical analysis was performed using a paired two-tail t-test. Error bars indicate SEM. (c) THP1-A3A cells were treated with 0.1 μg/mL dox, ATRi (80 nM), or combinations. Apoptosis was evaluated by FACS analysis after Annexin V staining and results are displayed as quantitation of three replicate experiments. Statistical analysis was performed using a paired two-tail t test. Error bars indicate SEM. (d) Endogenous A3A expression was evaluated by qPCR in the U937 and THP1 AML cell lines (inset). A3A level is shown as fold-change relative to a pooled control sample. AML cell lines were treated with indicated doses of ATRi for 48 hours prior to analysis of cell viability. (e) Short hairpin RNA (shRNA) was used to deplete endogenous A3A in the U937 AML cell line. Cells transduced with lentivirus containing control (U937-shCtrl) and A3A (U937-shA3A) shRNAs were evaluated by immunoblot using an antibody to endogenous A3A (inset). Actin was used as a loading control. U937 cells were treated with indicated doses of ATRi for 48 hours prior to analysis of cell viability. Statistical analysis of EC50 was performed using a sum-of-squares F-test. Error bars are SEM.
We further assessed cytotoxicity of ATRi treatment in THP1-A3A cells using a DNA stain and a fluorescent-labeled cell membrane stain (calcein AM) that permeates intact cells. Flow cytometric analysis showed a significant increase in the fraction of dead cells when A3A expression was combined with low-dose ATRi as compared to controls (Fig 3b). Additionally, we found increased Annexin V staining in cells treated with both dox and ATRi, suggesting that cell death occurs by apoptosis (Fig 3c). Together, these findings indicate that A3A expression sensitizes AML cells to treatment with a selective ATR inhibitor.
To determine the contribution of endogenous A3A to sensitivity of AML cells by ATR inhibition, we used two approaches. First, we evaluated expression of A3A in AML cell lines and found that the U937 cell line had relatively high A3A expression compared to THP1 (Fig 3d, inset). We assessed viability of these cell lines after treatment with ATRi and found that U937 cells were more susceptible to ATRi treatment than THP1 (Fig 3d). Second, we knocked down endogenous A3A in U937 cells using short hairpin RNA against A3A (shA3A). By immunoblotting, we show that A3A expression is decreased in cells with stable expression of shA3A compared to a control shRNA (Fig 3e, inset). We treated U937-shA3A and U937-shCtrl cells with ATRi and observed decreased viability in cells with higher A3A expression (Fig 3e). Thus, high endogenous A3A expression correlates with increased sensitivity of AML cell lines to ATRi treatment.
Synthetic lethality with high A3A expression is specific to the ATR-Chk1 pathway
We previously showed that A3A activates parallel arms of the DNA damage response via both ATM and ATR kinases (5,6). Based on the finding that A3A sensitizes leukemia cells to ATRi, we hypothesized that an ATM inhibitor (ATMi) would also result in synthetic lethality. We tested this hypothesis by treating the THP1-A3A cell line with a specific ATMi (KU55933) (36) but found no difference in viability of induced and uninduced cells (Fig 4a). Immunoblotting confirmed that ATMi efficiently decreased Chk2 phosphorylation, and therefore blocked ATM activity (Fig 4b). To strengthen this finding we used two cell models of inducible A3A expression with genetic abrogation of ATM. First, we knocked down ATM expression by siRNA in a U2OS-A3A cell line. A3A expression resulted in phosphorylation of ATM and Chk2, which was effectively inhibited by siRNA knock-down of ATM (Fig S5a, right). However, we observed that ATM knockdown had no effect on cell viability (Fig S5a, left). Second, we examined immortalized human fibroblasts derived from a patient with Ataxia-Telangiectasia that express non-functional ATM (-ATM), and compared to isogenic cells complemented with wild type ATM (+ATM) (37). When ectopic A3A was expressed in both cell lines, no difference in viability was observed (Fig S5b). Together, these data show that A3A activity does not sensitize cells to ATM inhibition.
Figure 4. APOBEC3A does not sensitize leukemia to ATM inhibition.
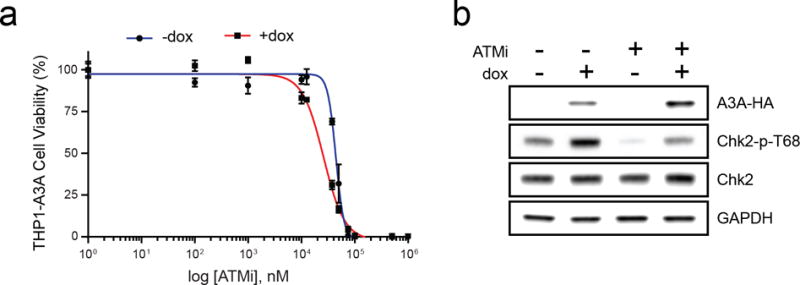
THP1-A3A cells were treated with dox (1 μg/mL) to induce A3A expression and a small molecule inhibitor of ATM kinase (ATMi, KU55933). (a) Viability was determined by colorimetric change after incubation with a water-soluble tetrazolium salt. Statistical analysis was performed using a sum-of-squares F-test. Error bars indicate SEM. (b) Inhibition of ATM activity was analyzed by immunoblot of THP1-A3A cell lysates treated with dox (1 μg/mL), ATMi (10 μM), and combination. Antibodies to HA, Chk2, and phosphorylated Chk2 (T68) were used. GAPDH was used as a loading control.
We hypothesized that the ATR-Chk1 function in cell cycle checkpoint activation is central to susceptibility of cells with high A3A expression. Checkpoint activation occurs through ATR phosphorylation of the downstream effector Chk1 (38). We examined cell cycle profiles in THP1-A3A cells expressing A3A in the presence of either ATRi or a small molecule Chk1 inhibitor (Chk1i, PF477736) (39,40). Upon A3A induction, cell cycle profiles of THP1 cells showed an arrest in G2, which returned to baseline after treatment with either ATRi or Chk1i (Fig 5a, Fig S6a–b). Thus, inhibition of the ATR-Chk1 pathway abrogates A3A-induced cell cycle arrest. We then examined the susceptibility of THP1-A3A cells to Chk1i. Treatment with Chk1i led to decreased viability and cell death when A3A was expressed (Fig 5b–c). Chk1i exhibited a very large therapeutic index with a shift in EC50 from 250,000 nM to 66 nM upon A3A induction. Inhibition of Chk1 activity by PF477736 was confirmed by diminished phosphorylation of Chk1 at serine 296 (41) (Fig 5b, right). These data show that the ATR-Chk1 checkpoint function is activated by A3A-induced replication stress and that abrogation of checkpoint activation leads to death of leukemia cells.
Figure 5. Sensitization to ATR inhibition by A3A is dependent on the ATR checkpoint function.
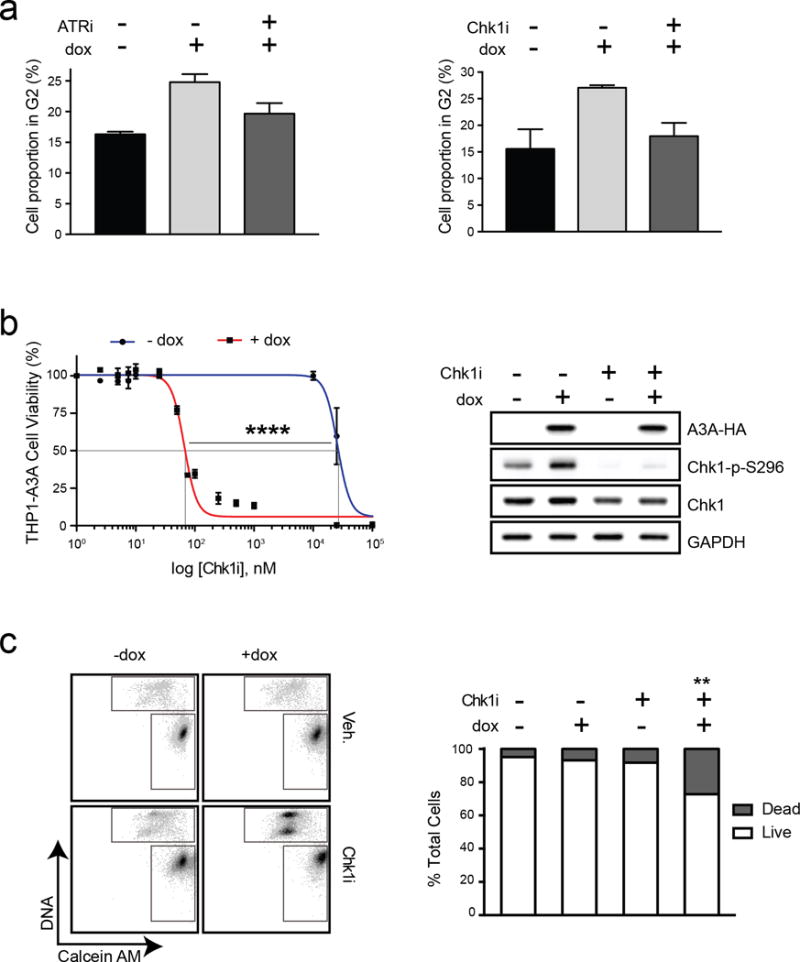
THP1-A3A cells were treated with 1 μg/mL dox to induce A3A expression, and a small molecule inhibitor of ATR or Chk1 kinase prior to analysis. (a) Checkpoint arrest is abrogated by small molecule inhibition of ATR or Chk1. THP1-A3A cells were treated with dox for 48 hours and analyzed for cell cycle progression by propidium iodide staining. Accompanying chart shows fraction of cells in G2 phase before dox induction, after dox induction, and after dox induction combined with kinase inhibitor (80 nM ATRi or 30 nM Chk1i) treatment. Statistical analysis was performed using a paired two-tailed t-test. Error bars indicate SEM. (b) Viability of THP1-A3A cells was measured after treatment with indicated doses of Chk1i. Cell viability was determined by metabolism of a water-soluble tetrazolium salt. Statistical analysis of EC50 was performed using a sum-of-squares F-test. Error bars are SEM. Immunoblotting shows inhibition of Chk1 kinase activity upon treatment with Chk1i (30 nM) indicated by decreased phosphorylation of Chk1 at serine 296. Antibodies to HA, Chk1, and phosphorylated Chk1 (S296) were used. GAPDH was used as a loading control. (c) Cell death was measured by staining THP1-A3A cells with fluorescent-labeled calcein AM (live) and DNA (dead) stains after treatment with dox (1 μg/mL), Chk1i (100 nM), or combinations. The upper gate on the FACS plot includes dead cells and the lower gate includes live cells. Accompanying bar chart shows quantitation of FACS results averaged over three replicates. Statistical analysis was performed using a paired two-tailed t-test. Error bars indicate SEM.
DISCUSSION
Emerging data provide compelling evidence that APOBEC3 enzymes act on the cellular genome. Increased expression of A3 family members has been detected in several types of human cancer (29) and APOBEC3 mutational signatures have been discovered in tumor genomes (1,2,8). Mutational burden associated with A3 activity has led to speculation about their oncogenic potential (7), influence on clonal evolution of cancer cells, and contribution to treatment-resistance and aggressive phenotypes (42). In this study, we investigate an alternative outcome of the interaction of A3 enzymes with cancer genomes in which A3 activity can be exploited for targeted cancer therapy.
APOBEC3 enzymes are proposed sources of both clustered and dispersed cytidine mutations in cancer genomes (1,9,31). Clustered mutations, termed kataegis, are postulated to occur due to A3 mutagenesis of ssDNA generated by resection of ends at DNA breakpoints. Discontinuous synthesis at the lagging strand during replication likely provides substrate for dispersed patterns of cytosine deamination by A3 (30,31,43). We recently showed that replicating cells incur more DNA damage caused by A3A when compared to non-replicating cells (5). These data point to a model of A3 deamination at replication forks, which suggests that frequently replicating cells are particularly susceptible to damage by A3 enzymes. Consistent with this model, the replication checkpoint, regulated by ATR-Chk1 signaling, is activated in response to A3A expression. Our data suggest that A3A expression results in increased ssDNA accumulation, likely due to stalling of replicative polymerases at uracil lesions or abasic sites, providing further substrate for A3A activity. If unchecked, A3A deamination may result in increasing levels of mutagenesis, irreversible genome damage, and cytotoxicity. In support of this model, we find that cells with high A3A expression are dependent on the ATR-Chk1 checkpoint for survival, and inhibition of this pathway results in cell death.
Previous studies have shown broad DNA damage responses to A3A expression including phosphorylation of histone variant H2AX and activation of ATM-Chk2 signaling, which is indicative of DSBs (6,44). In our model system, we also demonstrate ATM-Chk2 activation by A3A, yet in contrast to the ATR-Chk1 axis, inhibition of ATM does not induce cell death. These findings suggest that the essential genome protective mechanism during A3A mutagenesis is a response to replication stress, and not to DSBs. A possible explanation for this conclusion is that ATR is first activated by extensive deamination at replication forks resulting in overwhelming replication stress and fork collapse, which secondarily leads to DSBs and subsequent ATM signaling.
Rapidly dividing cancer cells, such as AML blasts, are dependent on the replication checkpoint to maintain genome integrity. Accordingly, replication checkpoint inhibitors (RCi) are being investigated in clinical trials for their activity as chemotherapy sensitizers (35,45,46). These RCi approaches are attractive therapeutic options due to limited toxicity in healthy tissues, wide therapeutic window, and efficacy in a broad array of cancers (47). Few studies have addressed the potential for checkpoint inhibition as a therapeutic option in hematologic malignancies. In vitro data have shown efficacy for ATRi in killing hematopoietic tumor cell lines with excessive oncogene-induced replication stress, ATM-deficiency, or in combination with oxidative stressors (21,48,49). ATRi is not frequently investigated as monotherapy in cancers without additional DDR defects, however the efficacy of ATRi was recently reported in a murine model of MLL-rearranged AML (50). Here we show that the replication checkpoint is activated by A3A in AML cells, and that A3A expression significantly increases the therapeutic index of ATR and Chk1 inhibitors. Thus, A3A could serve as a powerful biomarker for RCi treatment in AML. Further studies are warranted to examine the impact of cell cycle checkpoint inhibition in other tumors with elevated expression of the A3 family members. Our data provide a strong rationale for further preclinical investigation of replication checkpoint inhibition in AML with high A3A expression.
Supplementary Material
Acknowledgments
We thank members of the Weitzman Lab for insightful discussions and input. We thank Daphne Avgousti, Mateusz Koptyra, and Rahul Sood for technical assistance. We are grateful to Sarah Tasian and Kristina Cole for generously sharing reagents. We thank Rahul Kohli and Eric Brown for critical reading of the manuscript. A.M. Green was supported by a Young Investigator Award from the Alex’s Lemonade Stand Foundation, and by the National Institutes of Health (K12 CA076931 and K08 CA212299). Research on APOBEC enzymes in the Weitzman lab was supported by grants to M.D. Weitzman from the National Institutes of Health (CA181259 and CA185799), and funds from The Children’s Hospital of Philadelphia.
Footnotes
Disclosure statement: The authors declare no competing financial interests.
References
- 1.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris RS, Dudley JP. APOBECs and virus restriction. Virology. 2015;479–480:131–45. doi: 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lackey L, Law EK, Brown WL, Harris RS. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle. 2013;12:762–72. doi: 10.4161/cc.23713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green AM, Landry S, Budagyan K, Avgousti DC, Shalhout S, Bhagwat AS, et al. APOBEC3A damages the cellular genome during DNA replication. Cell Cycle. 2016;15:998–1008. doi: 10.1080/15384101.2016.1152426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011;12:444–50. doi: 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–70. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nature Genet. 2013;45:970–6. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nature Genet. 2015;45:1067–72. doi: 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, et al. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife. 2013;2:e00534. doi: 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xuan D, Li G, Cai Q, Deming-Halverson S, Shrubsole MJ, Shu XO, et al. APOBEC3 deletion polymorphism is associated with breast cancer risk among women of European ancestry. Carcinogenesis. 2013;34:2240–3. doi: 10.1093/carcin/bgt185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nik-Zainal S, Wedge DC, Alexandrov LB, Petljak M, Butler AP, Bolli N, et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nature Genet. 2014;46:487–91. doi: 10.1038/ng.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caval V, Suspene R, Shapira M, Vartanian JP, Wain-Hobson S. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3′UTR enhances chromosomal DNA damage. Nat Commun. 2014;5:5129. doi: 10.1038/ncomms6129. [DOI] [PubMed] [Google Scholar]
- 14.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38:4274–84. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, et al. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Current Biol. 2006;16:480–5. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 16.Peng G, Greenwell-Wild T, Nares S, Jin W, Lei KJ, Rangel ZG, et al. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood. 2007;110:393–400. doi: 10.1182/blood-2006-10-051763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. Journal of virology. 2009;83:9474–85. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci USA. 2003;100:13827–32. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–27. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilad O, Nabet BY, Ragland RL, Schoppy DW, Smith KD, Durham AC, et al. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010;70:9693–702. doi: 10.1158/0008-5472.CAN-10-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schoppy DW, Ragland RL, Gilad O, Shastri N, Peters AA, Murga M, et al. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J Clin Invest. 2012;122:241–52. doi: 10.1172/JCI58928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–94. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 23.Puigvert JC, Sanjiv K, Helleday T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 2016;283:232–45. doi: 10.1111/febs.13574. [DOI] [PubMed] [Google Scholar]
- 24.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nature reviews Cancer. 2005;5:689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 25.Shin KJ, Wall EA, Zavzavadjian JR, Santat LA, Liu J, Hwang JI, et al. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc Natl Acad Sci USA. 2006;103:13759–64. doi: 10.1073/pnas.0606179103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Everett RD, Parsy ML, Orr A. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J Virol. 2009;83:4963–77. doi: 10.1128/JVI.02593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a Shared Vision for Cancer Genomic Data. N Engl J Med. 2016;375:1109–12. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tukey JW. Exploratory Data Analysis. Addison-Wesley; 1977. pp. 43–44. [Google Scholar]
- 29.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nature Genet. 2013;45:977–83. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoopes JIC, L M, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA. APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication. Cell Rep. 2016;14:1–10. doi: 10.1016/j.celrep.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016;25:174–182. doi: 10.1101/gr.197046.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Josse R, Martin SE, Guha R, Ormanoglu P, Pfister TD, Reaper PM, et al. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014;74:6968–79. doi: 10.1158/0008-5472.CAN-13-3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y, et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. 2014;5:5674–85. doi: 10.18632/oncotarget.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Carrigan B, de Miguel Luken MJ, Papadatos-Pastos D, Brown J, Tunariu N, Perez Lopez R, et al. Phase I trial of a first-in-class ATR inhibitor VX-970 as monotherapy (mono) or in combination (combo) with carboplatin (CP) incorporating pharmacodynamics (PD) studies. ASCO Annual Meeting Proceedings. 2016;2504 [Google Scholar]
- 36.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 37.Ziv Y, Jaspers NG, Etkin S, Danieli T, Trakhtenbrot L, Amiel A, et al. Cellular and molecular characteristics of an immortalized ataxia-telangiectasia (group AB) cell line. Cancer Res. 1989;49:2495–501. [PubMed] [Google Scholar]
- 38.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes & Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 39.Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7:2394–404. doi: 10.1158/1535-7163.MCT-07-2391. [DOI] [PubMed] [Google Scholar]
- 40.Zhang C, Yan Z, Painter CL, Zhang Q, Chen E, Arango ME, et al. PF-00477736 mediates checkpoint kinase 1 signaling pathway and potentiates docetaxel-induced efficacy in xenografts. Clin Cancer Res. 2009;15:4630–40. doi: 10.1158/1078-0432.CCR-08-3272. [DOI] [PubMed] [Google Scholar]
- 41.Iacobucci I, Di Rora AG, Falzacappa MV, Agostinelli C, Derenzini E, Ferrari A, et al. In vitro and in vivo single-agent efficacy of checkpoint kinase inhibition in acute lymphoblastic leukemia. J Hematol Oncol. 2015;8:125. doi: 10.1186/s13045-015-0206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faltas BM, Prandi D, Tagawa ST, Molina AM, Nanus DM, Sternberg C, et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nature Genet. 2016 doi: 10.1038/ng.3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhagwat ASH, Hao W, Townes JP, Lee H, Tang H, Foster PL. Strand-biased Cytosine deamination at the Replication Fork causes Cytosine to Thymine Mutations in Escherichia coli. Proc Natl Acad Sci USA. 2016;113:2176–81. doi: 10.1073/pnas.1522325113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mussil B, Suspene R, Aynaud MM, Gauvrit A, Vartanian JP, Wain-Hobson S. Human APOBEC3A Isoforms Translocate to the Nucleus and Induce DNA Double Strand Breaks Leading to Cell Stress and Death. PloS One. 2013;8:e73641. doi: 10.1371/journal.pone.0073641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perez RP, Lewis LD, Beelen AP, Olszanski AJ, Johnston N, Rhodes CH, et al. Modulation of cell cycle progression in human tumors: a pharmacokinetic and tumor molecular pharmacodynamic study of cisplatin plus the Chk1 inhibitor UCN-01 (NSC 638850) Clin Cancer Res. 2006;12:7079–85. doi: 10.1158/1078-0432.CCR-06-0197. [DOI] [PubMed] [Google Scholar]
- 46.Brega N, McArthur GA, Britten C, Wong SG, Wang E, Wilner KD, et al. Phase I clinical trial of gemcitabine (GEM) in combination with PF-00477736 (PF-736), a selective inhibitor of CHK1 kinase. ASCO Annual Meeting Proceedings. 2010;28:3062. [Google Scholar]
- 47.Manic G, Obrist F, Sistigu A, Vitale I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol Cell Oncol. 2015;2(4):e1012976. doi: 10.1080/23723556.2015.1012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016;127:582–95. doi: 10.1182/blood-2015-05-644872. [DOI] [PubMed] [Google Scholar]
- 49.Cottini F, Hideshima T, Suzuki R, Tai YT, Bianchini G, Richardson PG, et al. Synthetic Lethal Approaches Exploiting DNA Damage in Aggressive Myeloma. Cancer Discov. 2015;5:972–87. doi: 10.1158/2159-8290.CD-14-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgado-Palacin I, Day A, Murga M, Lafarga V, Anton ME, Tubbs A, et al. Targeting the kinase activities of ATR and ATM exhibits antitumoral activity in mouse models of MLL-rearranged AML. Sci Signal. 2016;9:ra91. doi: 10.1126/scisignal.aad8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.