

Abstract
Site-specific lysine acetylation and methylation on histones are critical post-translational modifications (PTMs) that govern ordered gene transcription in chromatin. Mis-regulation of these histone PTM-mediated processes has been shown to be associated with human diseases. Since the 2010 landmark reports of small molecules (+)-JQ1 and I-BET762 that target the acetyl-lysine ‘reader’ Bromodomain and Extra Terminal domain (BET) proteins, there have been relentless efforts to develop epigenetic therapy with small molecules to modulate molecular interactions of epigenome reader domain proteins with PTMs. In addition to BET, the other emerging targets include non-BET acetyl- and methyl-lysine reader domains. This review covers the key chemical modulators of the aforementioned epigenome reader proteins.
Graphical abstract
1. Introduction
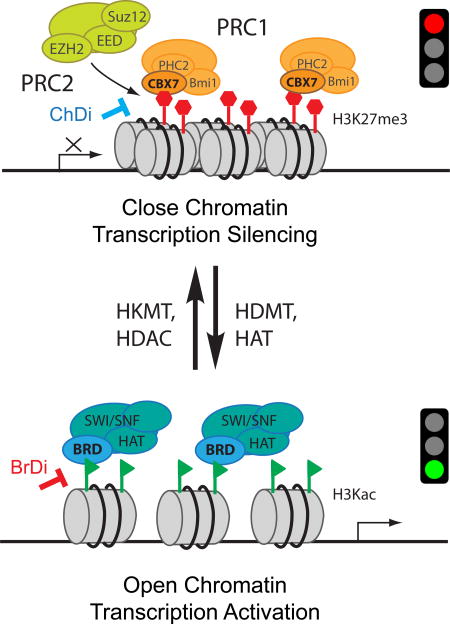
Epigenetic regulation of gene transcription plays a crucial role in normal cellular homeostasis. Mis-regulation of this process via abnormal expression or mutations of key proteins can lead to the onset and progression of a broad range of diseases including cancer and inflammation.[1] These key proteins typically involved in epigenetic control of gene transcription in chromatin are referred to as ‘writers’ [such as histone acetyltransferase (HAT), histone lysine methyltransferase (HKMT)], ‘erasers’ [i.e. histone deacetylase (HDAC), histone demethyltransferase (HDMT)] and ‘readers’ [i.e. bromodomains (BrDs), chromatin organization modifier domains (chromodomains, ChDs), malignant brain tumor (MBT) domains, plant homeodomain (PHD) and Tudor domains].[1–3] While writers and erasers deposit or remove post-translational modifications (PTMs), respectively, epigenome readers recognize these marks and in turn direct PTM-mediated protein-protein interactions (PPIs) in regulation of gene transcription in the context of chromatin. These three types of proteins work in close collaboration with other chromatin associated proteins to furnish either a closed chromatin state responsible for gene transcriptional silencing or an open chromatin state that dictates transcriptional activation (Figure 1). Such tightly regulated molecular activities provide opportunities for therapeutic intervention of PPIs with small molecule chemical inhibitors designed to target acetyl-lysine binding bromodomains (BrDis) or methyl-lysine binding chromodomains (ChDis).[4, 5] Common PTMs include histone lysine acetylation (KAc) and histone lysine mono-, di- or tri-methylation (Kme, Kme2, or Kme3). For the purposes of transcription, acetylation PTM is implicated in activation, while methylation PTMs are associated with either gene activation or silencing.[6]
Figure 1. Scheme illustrating modulation of chromatin structure changes and gene transcription by histone modifications.

Chromatin exists in a condensed or closed state, (known as heterochromatin) (top), or in an open state (euchromatin) (bottom), which correlates to gene transcriptional silencing or activation, respectively. Change of post-translational modifications (PTMs) such as removal of histone H3 lysine 27 tri-methylation (H3K27me3, red hexagons) by histone demethyltransferase (HDMT) followed by lysine acetylation (green triangles) by histone acetyltransferase (HAT) can covert heterochromatin to euchromatin. The reverse process, i.e. conversion of euchromatin to heterochromatin, can be catalyzed by action of histone deacetylase (HDAC) followed by histone lysine methyltransferase (HKMT). Top: The H3K27me3 PTM is added by a lysine methyltransferase EZH2 (Enhancer of zeste homolog 2) that is part of the Polycomb Repressive Complex 2 (PRC2). Following this, the methylated PTM is bound by PRC1, via the chromodomain (ChD) of the chromobox protein CBX7. This results in chromatin condensation and target gene repression. The protein-protein interaction (PPI) between CBX7 ChD and H3K27me3 can be disrupted by an inhibitor (ChDi) to result in gene transcriptional de-repression. Bottom: The H3Kac PTM is added by a HAT that is part of the SWI/SNF (SWItch/Sucrose Non-Fermentable) nucleosome remodeling complex. Following this, the acetylated PTM is bound via the bromodomain (BrD) of the complex. This dislodges DNA from its core histones due to an increase in steric hindrance and a decrease in positive charge, making it more accessible to transcription factors and other cofactors, allowing for target gene activation. The PPI between BrD and H3K27me3 can be disrupted by an inhibitor (BrDi) that results in gene transcriptional deactivation.
Small molecule chemical modulators of epigenome readers have established a new paradigm in epigenetic therapy highlighted by several small molecule modulators of BrDs currently in clinical trials (Supplementary Table 1) (https://clinicaltrials.gov/). As the field has evolved, it is important to classify chemical modulators of epigenome readers as ‘chemical probes’ or ‘inhibitors’.[7] A ‘chemical probe’ has been defined by the Structural Genomics Consortium (SGC) as an entity that exhibits binding to the target of interest with an in vitro potency <100 nM (KD or IC50), selectivity >30-fold against other protein families, and cellular target engagement <1 μM (http://www.thesgc.org/chemical-probes). Additional criteria for a small molecule to be deemed as a chemical probe may include availability of a negative control compound, acceptable toxicity profile, and even higher selectivity. One of the principal goals of developing chemical probes is to increase the reliability of published literature along with the robustness of target validation.[8] The term ‘inhibitor’ in this arena is now frequently used to identify a chemical entity that cannot meet the stringent requirements of a chemical probe or has missing data. The focus of this review is to highlight the representative chemical modulators of epigenome reader domains that read acetylated and methylated lysine PTMs in histones and transcription proteins.
Chemical modulators for acetyl-lysine binding domains
Bromodomains are evolutionarily conserved modular domains in chromatin and transcription-associated proteins and represent the first group of epigenome reader domains that function to recognize KAc’s on histones and other protein.[9] Brds are ~110 residues in length. The human genome comprises of 61 Brds that can be categorized into eight subfamilies based on their sequence and structures (Supplementary Figure 1). Zhou and colleagues [9] reported the first three-dimensional structure of the BrD of PCAF (p300/CBP-associated factor) in both free form and in complex with a KAc mimic: acetylhistamine, and discovered the bromodomain as the bona fide histone acetyl-lysine binding domain. The BrD is composed of a left-handed bundle of four α- helices (αZ, αA, αB, and αC), and this structure is conserved across the majority of BrDs (Figure 2A–B). The acetyl-lysine binding site is a hydrophobic pocket formed by a large loop between αZ and αA (ZA loop) and a shorter loop between αB and αC (BC loop). Most of the BrDs (48 cases) have a conserved Asn in the BC loop, and all reported potent inhibitors have exhibited interactions with this residue. There are few cases where in place of the Asn, a Tyr, Thr, or Asp (8, 4, or 1 example(s) respectively) is present. A second highly conserved structural feature in BrDs is an arrangement of five water molecules stably bound at the bottom of the KAc recognition site through hydrogen bond interactions with the protein residues. Representative chemical modulators of the BrD subfamilies are depicted in Figure 3 and are discussed below in detail.
Figure 2. Molecular basis of ligand recognition by the bromodomains.

A. Crystal Structure of human BRD4-BD1 in complex with a diacetylated histone 4 peptide (H4K12acK16ac) (PDB: 3UVX). The left-handed bundle of four α- helices (αZ, αA, αB, and αC) along with the ZA and BC loop are displayed. In the binding pocket, the right KAc forms a hydrogen bond with Asn 140 and a water-mediated hydrogen bond with Tyr 97. The interactions shown here along with the array of bound water molecules are conserved in majority of human BrDs. Note that KAc also forms a water-mediated hydrogen bond in the ZA loop with the hydrophobic “WPF” shelf. B. Crystal Structure of human BRD4-BD1 in complex with a small molecule inhibitor MS402 (PDB: 5ULA). C. Crystal structure of L3MBTL1 in complex with H4K20Me2 peptide (PDB: 2RJF). Interactions of K20Me2 in the aromatic cage are displayed. D. Crystal structure of L3MBTL1 in complex with UNC669 (PDB: 3P8H), an example of a ‘cavity insertion’ type binding mode. E. Crystal structure of chromobox homolog 7 (CBX7) chromodomain (ChD) with H3K27me3 peptide (PDB: 4X3K). Interactions of K27Me3 in the aromatic cage are displayed. F. Crystal structure of CBX7 ChD with a small molecule inhibitor MS37452 (PDB: 4X3T), an example of a ‘surface groove’ type binding mode.
Figure 3. Representative chemical modulators of bromodomains categorized by the target sub-family.

Sub-family I
PCAF (p300/CBP Associated Factor) is a multi-domain protein bearing a single BrD and is predicted to be highly druggable.[10] Its deregulation has been linked to cancer, HIV infection, and neuroinflammation.[11–13] The Zhou group reported the first ever BrD inhibitor called NP1 (Supplementary Figure 2) bound to PCAF.[14] The aniline scaffold bearing NP1 interacted with a conserved asparagine (Asn 798) and showed a disruption of the PPI between PCAF and HIV-1 Tat, providing a direct proof of concept that small molecules can bind to BrDs and disrupt PPIs. Of the PCAF inhibitors reported so far compound 1 (IC50 = 13 nM AlphaLISA) (Figure 3) and its analogs developed by Constellation/Genentech were shown to be very potent but lacked selectivity over other BrDs.[11] CERC2 (Cat Eye Syndrome Chromosome Region, candidate 2), is another highly druggable target.[10] Chemical probe NVS-CERC2-1 displayed high affinity (IC50 = 47 nM, KD = 80 nM) and robust in-cell target engagement (0.1 μM, FRAP assay) [http://www.thesgc.org/chemical-probes/NVS-1]. BPTF (FALZ), another highly druggable target,[10] has been linked to bladder and colorectal cancers, melanoma, and leukemia. The 19F assay was used to discover AU1 (KD = 2.8 μM), which is a weakly active inhibitor of BPTF.[15]
Sub-family II
This group is also called the BrD and Extra Terminal (BET) and comprises four proteins (BRD2, BRD3, BRD4, and BRDT) each containing two N-terminal BrD modules (BD1 and BD2). This is the most intensely studied family with 17 BET BrD blocking molecules (BETi’s) currently in clinical trials (Supplementary Table 1; as of April 7, 2017). The field of epigenetic drug discovery gained significant momentum after the 2010 reports of two triazolodiazepine BET BrDis: (+)-JQ1[16] and I-BET762.[17] Quinazolinone RVX-208 is the most advanced BETi that has reached phase III clinical trails for type II diabetes mellitus, coronary artery disease, and cardiovascular disease. Four diazepines (OTX-015, CPI-0610, I-BET762, TEN-010) and pyrrolo[2,3-c]pyridine derivative ABBV-075 are at various stages of clinical trials, mostly for cancer therapy. Recent reviews[2–4, 18–20] have covered the extensive work done on BETi’s and have underscored their potential.
All BET BrDs display 95% sequence identity in the KAc binding site, 75% sequence identity in the N-terminal tandem domains (BD1 and BD2), and 38% cross-domain sequence identity. Chemical probes that are highly selective between different BET family members (e.g. BRD2 vs BRD4), different bromodomains of the same protein (e.g. BRD4-BD1 vs. BRD4-BD2), or similar bromodomains of different BET family members (e.g. BRD4-BD1 vs. BRD2-BD1) are key in understanding the individual role of these proteins. RVX-208 is 15–30 fold selective for BD2s of BET family members over their BD1s. Three compounds reported by the Zhou lab [21, 22] – MS436, MS611 (both diazobenzenes), and Olinone (tetrahydro-5H-pyrido[4,3-b]indolone) – show preference for BET BD1s over their BD2s. The same group recently reported a BD1-selective vinylogous amide MS402 that prevents and ameliorates T-cell transfer-induced colitis in mice.[23] The ‘bump-and-hole’ approach was utilized by Ciulli et al.[24] where-by the ethyl derivative of I-BET762 showed that it binds leucine/alanine mutant BrDs with nanomolar affinity and achieves up to 540-fold selectivity relative to wild-type BrDs.
Sub-family III
CBP (CREBBP/KAT3A) and p300 (EP300/KAT3B) have a high degree of sequence identity and are linked to Rubinstein-Taybi syndrome, cancer, inflammation, and neuropsychiatric disorders.[25–28] The Zhou group reported tetrahydrocarbazolone MS7972 (CBP KD = 19.6 μM)[29] and diazobenzene Ischemin (CBP KD = 19.0 μM)[30] confirming that the CBP active site could tolerate diverse scaffolds. Starting from a fragment hit, Hay et al.[31] developed a series of 5-isoxazolyl-benzimidazoles, of which SGC-CBP30 emerged as a potent (CBP KD = 21 nM, p300 KD = 32 nM) and selective (40-fold selectivity for CBP over BRD4-BD1) analog. Oxazepine I-CBP112 was another potent (CBP KD = 208 nM, p300 KD = 343 nM) and selective (26-fold selectivity for CBP over BRD4-BD1) inhibitor that also sensitizes leukemic cells to JQ1 treatment.[32] The PHIP(2) BrD has been predicted to be highly druggable[10] and is implicated in metastatic melanoma. Mildly active, compound 2 (IC50 = 128 μM) is the best hit reported so far.[33]
Sub-family IV
Understanding the biological function of BRD7 and BRD9 from this group is of interest to multiple research groups. Although their overall sequence similarity is low (36%), their BrDs share 72% sequence identity.[34] Fragment based screening lead to quinolone LP99 (BRD9 IC50 = 99 nM, BRD7 IC50 = 909 nM) that elucidated the role of BRD7/9 in inflammatory pathways. Structure-based drug discovery afforded thienopyridone I-BRD9 (BRD9 KD = 50 nM) with 700-fold selectivity over the BET family and 200-fold over BRD7, and was used to identify genes regulated by BRD9 in Kasumi-1 cells involved in oncology and immune response pathways.[35] Two potent and selective pyridinones, BI-7273 (BRD9 IC50 = 19 nM, BRD7 IC50 = 117 nM) and BI-9564 (BRD9 IC50 = 75 nM, BRD7 IC50 = 3410 nM) were shown to modulate BRD9 bromodomain cellular function and display antitumor activity in an AML xenograft model.[36]
The BRPF (Bromodomain and PHD Finger-containing) family of BRPF1, BRPF2/BRD1, and BRPF3 functions in assembly of MYST-family HAT complexes. A benzimidazolone GSK6853 was reported as a potent (pIC50 = 8.1), soluble, cell active, and highly selective (>1600-fold in BROMOscan) inhibitor of the BRPF1 BrD, and revealed properties suitable for in vivo studies.[37]
ATAD2 overexpression is linked to a variety of cancers, however its BrD is predicted to be among the least druggable.[10] Naphthyridone 3 exhibits excellent potency (ATAD2 KD = 8 nM) and selectivity (>2.8 log units over BRD4 BrDs).[38] Its enantiomer is significantly weaker against ATAD2; therefore serves as a useful negative control. The design of this compound was the first instance wherein CF2 was validated as a polar hydrophobic isostere of SO2.
Sub-family V
BAZ2A overexpression has been linked to prostate cancer. Mutations in BAZ2B have been linked to sudden cardiac death, and its overexpression negatively affects pediatric B cell acute lymphoblastic leukemia (B-ALL) outcome. However these proteins have been considered the least druggable.[10] Structure-based discovery produced a potent, selective and cell active inhibitor called BAZ2-ICR (BAZ2A IC50 = 0.13 μM, BAZ2B IC50 = 0.18 μM).[39] BrDs from TRIM24 have also been considered interesting targets and inhibitors have been reported against these.
No Inhibitors have been reported for Sub-family VI.
Sub-family VII
TAF1(2) inhibitor, compound 4 bears a pyrrolopyridone core and displayed excellent binding affinity (IC50 = 46 nM) and selectivity (30-fold over BRD9).[40].
Sub-family VIII
PB1 contains six BrDs, while SMARCA2 and SMARCA4 each have a single BrD. All three have been associated with multiple cancer types. PFI-3 demonstrated excellent potency [PB1(5) KD = 54 nM, SMARCA2/SMARCA4 KD < 0.1 μM] and selectivity against >40 BRDs from other families.[41]
Chemical modulators for methyl-lysine binding domains
Some of the key methyl-lysine PTM reading families include the malignant brain tumor (MBT) domains (9 members), chromodomains (29 members), plant homeodomain (PHD) fingers (99 members), Tudor domains (41 members), and proline-tryptophan-tryptophan-proline (PWWP) domains (24 members).[5] Methyl-lysine reader domains are emerging as an important class of targets with antagonists reported for members of the former four families. The latter four families along with another family, the plant Agenet domains, collectively embody the Royal family domains.[42] A WD40 repeat-containing protein, the embryonic ectoderm development protein (EED) is also known to recognize methyl-lysine PTMs.[43]
A methyl-lysine binding site in a reader protein usually comprises two to four aromatic residues (typically Tryptophan, Phenylalanine, and Tyrosine) and is referred to as an ‘aromatic-cage’ (Figure 2C–F). Depending on their size, shape and charge, the Kme, Kme2, and Kme3 residues engage in cation-π and hydrophobic interactions in the aromatic cage. Based on their binding mode, Patel et al.[44] classified methyl-lysine binding readers into ‘cavity-insertion’ and ‘surface-groove’ binders. In the cavity-insertion binding mode, the methylammonium is buried deep within a protein cavity. In the surface-groove binding mode, the methyl-lysine lies along a protein surface groove.
MBT family
MBT domains bind to mono- or di-methylated lysine causing transcriptional repression. Their role in oncogenesis, and their high-predicted druggability makes them promising targets.[45] Frye and co-workers reported the first ever inhibitors targeting a methyl-lysine reader protein, in form of UNC669 (KD = 5 μM, MBT domain of L3MBTL1).[46] UNC669 was equipotent against the highly similar L3MBTL3. Additional analog design lead to UNC926 (IC50 = 3 μM, L3MBTL1).[47] The same group targeted the L3MBTL3 domain leading to potent inhibitor UNC1215 (KD = 120 nM, L3MBTL3).[48] This inhibitor binds to L3MBTL3 at its two MBT domains in a 2:2 complex as indicated by its co-crystal structure. Compound UNC2533 (KD = 370 nM, L3MBTL3) displayed a similar 2:2 binding mode.[49] UNC1215 was at least 50-fold selective over other MBT proteins in the assay, and served as a chemical probe for further studies in which L3MBTL3 function was studied.[50, 51] Optimization of UNC1215 lead to L3MBTL3 inhibitors UNC1679 (KD = 470 nM) and 5 (KD = 350 nM), which were 150- and 380- fold selective over L3MBTL1.
Chromodomain family
The HP1 (CBX -1, -3, -5) and CBX (CBX -2, -4, -6, -7, -8) sub-families have been the focus of research efforts. Both sub-families are associated with gene repression by binding to H3K9me3 and H3K27me3 marks, and facilitate a surface groove-binding mode of inhibitors. CBX7 has been predicted to be the most druggable member and is implicated in several cancers.[45] The Hof lab[52] reported the first CBX7 antagonists, in form of several 5-mer peptides (KD = 0.2–4.1 μM). The best peptide 6 had a KD of ~200 nM for CBX7 and a 10/400- fold selectivity over CBX8/CBX1. The Frye group [53] reported a cell permeable peptide UNC3866 (KD ~100 nM CBX4 and CBX7) that is 6- to 18- fold selective over seven CBX and CDY chromodomains. This peptide demonstrates target engagement via a biotinylated derivative, inhibits PC3 prostate cancer cells, and also has a negative control called UNC4219.
The first small molecule inhibitor of CBX7 was reported by the Zhou group in form of MS37452 (KD = 29 μM) which displayed 3-fold selectivity over CBX4 and 10-fold selectivity over CBX2/6/8.[54] MS37452 decreased the occupancy of CBX7 on the INK4A/ARF locus in human prostate cancer cells, as revealed by chromatin immunoprecipitation. In addition, de-repression of gene products p14ARF and p16INK4a was also demonstrated. Further optimization lead to upto 7- fold increase (Ki = 4.8 μM) in potency but showed decreased cell permeability.[55] The same report[55] described another class of small molecule inhibitors of CBX7 that act by an allosteric mechanism and is represented by MS351 (KD = 23.8 μM CBX7). The binding of this substituted benzimidazole to CBX7, induces binding of ANRIL RNA to a neighboring binding pocket on CBX7. MS351 was also shown to induce p16INK4a de-repression.
A CBX6 inhibitory peptide 7 (KD = 0.9 μM) was identified by the Hof group that displayed 7–90 fold selectivity over CBX1/2/4/7/8.[56]
PHD family
PHD fingers can recognize methylated, unmethylated, and acetylated lysine residues. They are linked to cancer and immune disorders. The PHD finger of JARID1A was the first member of the PHD family to be targeted.[57] WAG-003 bound JARID1A with an IC50 of 30 μM, but exhibits lack of selectivity as it has similar affinity for the PHD domains of JMJD2A and ING2. The PHD finger of Pygo protein was the second PHD finger to be targeted. An NMR fragment screen identified weakly active inhibitors CF4 and CF16 with weak binding affinities of 2.5 and 7.3 mM respectively.
Tudor domain family
Tudor domains play a role in RNA metabolism, histone modification, DNA damage responses, and development. The tandem Tudor domain of a protein 53BP1 recognizes H4K20me2 and is associated with DNA damage. 53BP1 is implicated in immune responses and oncogenesis as indicated by knockdown studies. UNC2170 (KD = 22 μM) was >17-fold selective for 53BP1 over other proteins in the screen.[58] Another protein SPIN1 contains three Tudor-like domains, of which domain-2 displayed strong affinity for H3K4me3. SPIN1 was shown to be highly expressed in several tumor types. Inhibitor A366 interacted with SPIN1 with an IC50 of 200 nM.[59]
WD40 repeat-containing proteins
With 349 human WD40 repeat-containing proteins, the WD40 domain is one of the most abundant domains, and it functions as a protein-protein or protein-DNA interaction platform to regulate several different cellular processes.[60] The WD40 repeat-containing protein EED is one of the core subunits of PRC2 complex (Figure 1).[43] Dysregulation of PRC2 is observed in multiple cancers. A report from Novartis[61] demonstrated that inhibition of PRC2 by pharmacologically targeting EED is a viable strategy to accomplish anticancer effect. Prudent modifications of a high-throughput screen (HTS) derived hit, afforded potent (Kd = 82 nM) and selective allosteric inhibitor EED226 that was shown to directly bind the H3K27me3 pocket of EED, and induce regression of tumor xenograft in vivo. This inhibitor is active against cancer cells with acquired resistance to SAM-competitive EZH2 inhibitors, and also shows a synergistic effect against cancer cell growth in combination with EZH2 inhibitors. An account from AbbVie,[62] identified A-395, a highly potent (Kd = 1.5 nM) and selective allosteric antagonist of EED that also binds in the H3K27me3 pocket, shows strong efficacy against a human tumor xenograft model, and can potently inhibit proliferation of EZH2 inhibitor resistant cell lines. A-395 was derived by optimization of a lead obtained from a high throughput thermal shift assay (TSA). It also has a closely related negative control A-395N. Both EED226 and A-395 modulate H3K27 methylation in cells. The Frye lab[63] used weakly active peptide Jarid2 against EED (IC50 ~ 15 μM) as a starting point, and devised a paired combinatorial and structure-based optimization approach to identify UNC5114 (IC50 ~ 2 μM) and UNC5115 (IC50 ~ 4 μM) as small peptidomimetic ligands of EED with improved physicochemical properties. These ligands also function as allosteric modulators to abrogate PRC2 activity. It is worth noting that the aforementioned ligands of EED induce a rearrangement of the aromatic cage in the H3K27me3 binding pocket of EED; such an outcome is not trivial to design or predict using a traditional structure based drug design program. It is positive to see industrial interest in methyl-lysine readers evident by their efforts in developing robust high-quality HTS assays that are required productive drug discovery. This practice is bound to open up new chemical space for this class of targets. Inhibitors have been reported for another WD40 repeat containing protein, WDR5.[64, 65] As this protein binds methylated arginine PTMs, it is not discussed in detail here.
Conclusions
Epigenome readers are promising targets for development of therapies for a broad range of human diseases. Of the eight sub-families of acetyl-lysine reader domains, remarkable advances have been made for the BET family proteins, with several BET antagonists being evaluated in clinical trials and several reports of chemically diverse BET family antagonists. Selectivity between BrDs and BD1/2 of BET proteins remains a formidable challenge. Promising inroads for six other BrD sub-families have recently been made with the discovery of potent chemical modulators for even difficult to target BrDs including those of ATAD2, BAZ2A and BAZ2B. Structure based and fragment based drug design have been at the forefront of chemical probe and inhibitor development. Chemical modulators for methyl-lysine reader domains have been discovered for the MBT, Chromodomain, PHD, and Tudor domain families. Although this area is lagging behind relative to BrDs, potent and selective peptides like UNC1215 and UNC3866, and moderately active small molecules are leading the way for the development of future analogs. Altogether, these epigenome reader chemical modulators seem to be paving the way to the emergence of new epigenetic therapies for a wide array of human diseases.
Supplementary Material
Figure 4. Representative chemical modulators of methyl-lysine binding domains categorized by the target family.

Highlights.
Acetyl- and methyl-lysine reader domains are promising drug targets
Major accomplishments made in chemical modulation of BET family bromodomains
What is the progress made in non-BET chemical modulators?
What are the advances in chemical modulators of methyl-lysine reader domains?
Acknowledgments
We thank Dr. Jamel Meslamani and the other members of the Zhou Lab for helpful discussion. This work was supported in part by the research grants from the Department of Defense Breast Cancer Program and the National Institutes of Health (to M.-M.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: A new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 2.Ferri E, Petosa C, McKenna CE. Bromodomains: Structure, function and pharmacology of inhibition. Biochem Pharmacol. 2016;106:1–18. doi: 10.1016/j.bcp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Greschik H, Schule R, Gunther T. Selective targeting of epigenetic reader domains. Expert Opin Drug Discov. 2017;12(5):449–463. doi: 10.1080/17460441.2017.1303474. [DOI] [PubMed] [Google Scholar]
- 4*.Galdeano C, Ciulli A. Selectivity on-target of bromodomain chemical probes by structure-guided medicinal chemistry and chemical biology. Future Med Chem. 2016;8(13):1655–1680. doi: 10.4155/fmc-2016-0059. Recent review on bromodomain chemical probe discovery by structure-guided medicinal chemistry and chemical biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5*.Milosevich N, Warmerdam Z, Hof F. Structural aspects of small-molecule inhibition of methyllysine reader proteins. Future Medicinal Chemistry. 2016;8(13):1681–1702. doi: 10.4155/fmc-2016-0082. Current and comprehensive review on chemical modulators of methyl-Lysine reader domains. [DOI] [PubMed] [Google Scholar]
- 6.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 7*.Moustakim M, Clark PGK, Hay DA, Dixon DJ, Brennan PE. Chemical probes and inhibitors of bromodomains outside the bet family. Medchemcomm. 2016;7(12):2246–2264. doi: 10.1039/c6md00373g. Recent review on chemical modulators of non-BET Bromodomains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, et al. The promise and peril of chemical probes. Nat Chem Biol. 2015;11(8):536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399(6735):491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 10.Vidler LR, Brown N, Knapp S, Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J Med Chem. 2012;55(17):7346–7359. doi: 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albrecht BK, Burdick DJ, Cote A, Duplessis M, Nasveschuk CG, Taylor AM. Pyridazinone derivatives and their use in the treatment of cancer. 2016 In: [Google Scholar]
- 12.Mujtaba S, He Y, Zeng L, Farooq A, Carlson JE, Ott M, Verdin E, Zhou MM. Structural basis of lysine-acetylated hiv-1 tat recognition by pcaf bromodomain. Mol Cell. 2002;9(3):575–586. doi: 10.1016/s1097-2765(02)00483-5. [DOI] [PubMed] [Google Scholar]
- 13.Zhou M-M, Gerona-Navarro G, Rodriguez-Fernandez Y, Casaccia P. Small molecule transcription modulators of bromodomains. 2015 In: [Google Scholar]
- 14.Zeng L, Li J, Muller M, Yan S, Mujtaba S, Pan C, Wang Z, Zhou M-M. Selective small molecules blocking hiv-1 tat and coactivator pcaf association. J Am Chem Soc. 2005;127(8):2376–2377. doi: 10.1021/ja044885g. [DOI] [PubMed] [Google Scholar]
- 15.Urick AK, Hawk LM, Cassel MK, Mishra NK, Liu S, Adhikari N, Zhang W, dos Santos CO, Hall JL, Pomerantz WC. Dual screening of bptf and brd4 using protein-observed fluorine nmr uncovers new bromodomain probe molecules. ACS Chem Biol. 2015;10(10):2246–2256. doi: 10.1021/acschembio.5b00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, et al. Selective inhibition of bet bromodomains. Nature (London, U K) 2010;468(7327):1067–1073. doi: 10.1038/nature09504. Recent review on chemical modulators of BRD4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung C-w, Chandwani R, Marazzi I, Wilson P, Coste H, White J, et al. Suppression of inflammation by a synthetic histone mimic. Nature (London, U K) 2010;468(7327):1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18*.Liu Z, Wang P, Chen H, Wold EA, Tian B, Brasier AR, Zhou J. Drug discovery targeting bromodomain-containing protein 4. J Med Chem. 2017 doi: 10.1021/acs.jmedchem.6b01761. Comprehensive review on chemical modulators of Bromodomains. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith SG, Zhou MM. The bromodomain: A new target in emerging epigenetic medicine. ACS Chem Biol. 2016;11(3):598–608. doi: 10.1021/acschembio.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang G, Smith SG, Zhou MM. Discovery of chemical inhibitors of human bromodomains. Chem Rev. 2015;115(21):11625–11668. doi: 10.1021/acs.chemrev.5b00205. [DOI] [PubMed] [Google Scholar]
- 21.Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J, Zeng L, Mujtaba S, Ohlmeyer M, Zhou MM. Structure-guided design of potent diazobenzene inhibitors for the bet bromodomains. J Med Chem. 2013;56(22):9251–9264. doi: 10.1021/jm401334s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gacias M, Gerona-Navarro G, Plotnikov AN, Zhang G, Zeng L, Kaur J, Moy G, Rusinova E, Rodriguez Y, Matikainen B, Vincek A, et al. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem Biol. 2014;21(7):841–854. doi: 10.1016/j.chembiol.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheung K, Lu G, Sharma R, Vincek A, Zhang R, Plotnikov AN, Zhang F, Zhang Q, Ju Y, Hu Y, Zhao L, et al. Bet n-terminal bromodomain inhibition selectively blocks th17 cell differentiation and ameliorates colitis in mice. Proc Natl Acad Sci U S A. 2017;114(11):2952–2957. doi: 10.1073/pnas.1615601114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baud MG, Lin-Shiao E, Cardote T, Tallant C, Pschibul A, Chan KH, Zengerle M, Garcia JR, Kwan TT, Ferguson FM, Ciulli A. Chemical biology. A bump-and-hole approach to engineer controlled selectivity of bet bromodomain chemical probes. Science. 2014;346(6209):638–641. doi: 10.1126/science.1249830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hennekam RC. Rubinstein-taybi syndrome. Eur J Hum Genet. 2006;14(9):981–985. doi: 10.1038/sj.ejhg.5201594. [DOI] [PubMed] [Google Scholar]
- 26.Valor LM, Viosca J, Lopez-Atalaya JP, Barco A. Lysine acetyltransferases cbp and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr Pharm Des. 2013;19(28):5051–5064. doi: 10.2174/13816128113199990382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Revilla Y, Granja AG. Viral mechanisms involved in the transcriptional cbp/p300 regulation of inflammatory and immune responses. Crit Rev Immunol. 2009;29(2):131–154. doi: 10.1615/critrevimmunol.v29.i2.30. [DOI] [PubMed] [Google Scholar]
- 28.Wang F, Marshall CB, Ikura M. Transcriptional/epigenetic regulator cbp/p300 in tumorigenesis: Structural and functional versatility in target recognition. Cell Mol Life Sci. 2013;70(21):3989–4008. doi: 10.1007/s00018-012-1254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sachchidanand, Resnick-Silverman L, Yan S, Mutjaba S, Liu WJ, Zeng L, Manfredi JJ, Zhou MM. Target structure-based discovery of small molecules that block human p53 and creb binding protein association. Chem Biol. 2006;13(1):81–90. doi: 10.1016/j.chembiol.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 30.Borah JC, Mujtaba S, Karakikes I, Zeng L, Muller M, Patel J, Moshkina N, Morohashi K, Zhang W, Gerona-Navarro G, Hajjar RJ, et al. A small molecule binding to the coactivator creb-binding protein blocks apoptosis in cardiomyocytes. Chem Biol. 2011;18(4):531–541. doi: 10.1016/j.chembiol.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hay DA, Fedorov O, Martin S, Singleton DC, Tallant C, Wells C, Picaud S, Philpott M, Monteiro OP, Rogers CM, Conway SJ, et al. Discovery and optimization of small-molecule ligands for the cbp/p300 bromodomains. J Am Chem Soc. 2014;136(26):9308–9319. doi: 10.1021/ja412434f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J, Olzscha H, Monteiro O, Martin S, Philpott M, Tumber A, et al. Generation of a selective small molecule inhibitor of the cbp/p300 bromodomain for leukemia therapy. Cancer Res. 2015;75(23):5106–5119. doi: 10.1158/0008-5472.CAN-15-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cox OB, Krojer T, Collins P, Monteiro O, Talon R, Bradley A, Fedorov O, Amin J, Marsden BD, Spencer J, von Delft F, et al. A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of phip(2), an atypical bromodomain. Chem Sci. 2016;7(3):2322–2330. doi: 10.1039/c5sc03115j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hay DA, Rogers CM, Fedorov O, Tallant C, Martin S, Monteiro OP, Muller S, Knapp S, Schofield CJ, Brennan PE. Design and synthesis of potent and selective inhibitors of brd7 and brd9 bromodomains. MedChemComm. 2015;6(7):1381–1386. [Google Scholar]
- 35.Theodoulou NH, Bamborough P, Bannister AJ, Becher I, Bit RA, Che KH, Chung CW, Dittmann A, Drewes G, Drewry DH, Gordon L, et al. Discovery of i-brd9, a selective cell active chemical probe for bromodomain containing protein 9 inhibition. J Med Chem. 2016;59(4):1425–1439. doi: 10.1021/acs.jmedchem.5b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin LJ, Koegl M, Bader G, Cockcroft XL, Fedorov O, Fiegen D, Gerstberger T, Hofmann MH, Hohmann AF, Kessler D, Knapp S, et al. Structure-based design of an in vivo active selective brd9 inhibitor. J Med Chem. 2016;59(10):4462–4475. doi: 10.1021/acs.jmedchem.5b01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bamborough P, Barnett HA, Becher I, Bird MJ, Chung CW, Craggs PD, Demont EH, Diallo H, Fallon DJ, Gordon LJ, Grandi P, et al. Gsk6853, a chemical probe for inhibition of the brpf1 bromodomain. ACS Med Chem Lett. 2016;7(6):552–557. doi: 10.1021/acsmedchemlett.6b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bamborough P, Chung CW, Demont EH, Furze RC, Bannister AJ, Che KH, Diallo H, Douault C, Grandi P, Kouzarides T, Michon AM, et al. A chemical probe for the atad2 bromodomain. Angew Chem Int Edit. 2016;55(38):11382–11386. doi: 10.1002/anie.201603928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drouin L, McGrath S, Vidler LR, Chaikuad A, Monteiro O, Tallant C, Philpott M, Rogers C, Fedorov O, Liu MJ, Akhtar W, et al. Structure enabled design of baz2-icr, a chemical probe targeting the bromodomains of baz2a and baz2b. Journal of Medicinal Chemistry. 2015;58(5):2553–2559. doi: 10.1021/jm501963e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crawford TD, Tsui V, Flynn EM, Wang S, Taylor AM, Cote A, Audia JE, Beresini MH, Burdick DJ, Cummings R, Dakin LA, et al. Diving into the water: Inducible binding conformations for brd4, taf1(2), brd9, and cecr2 bromodomains. J Med Chem. 2016;59(11):5391–5402. doi: 10.1021/acs.jmedchem.6b00264. [DOI] [PubMed] [Google Scholar]
- 41.Gerstenberger BS, Trzupek JD, Tallant C, Fedorov O, Filippakopoulos P, Brennan PE, Fedele V, Martin S, Picaud S, Rogers C, Parikh M, et al. Identification of a chemical probe for family viii bromodomains through optimization of a fragment hit. J Med Chem. 2016;59(10):4800–4811. doi: 10.1021/acs.jmedchem.6b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP. The tudor domain 'royal family': Tudor, plant agenet, chromo, pwwp and mbt domains. Trends Biochem Sci. 2003;28(2):69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- 43.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, et al. Role of the polycomb protein eed in the propagation of repressive histone marks. Nature. 2009;461(7265):762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14(11):1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santiago C, Nguyen K, Schapira M. Druggability of methyl-lysine binding sites. J Comput Aid Mol Des. 2011;25(12):1171–1178. doi: 10.1007/s10822-011-9505-2. [DOI] [PubMed] [Google Scholar]
- 46**.Herold JM, Wigle TJ, Norris JL, Lam R, Korboukh VK, Gao C, Ingerman LA, Kireev DB, Senisterra G, Vedadi M, Tripathy A, et al. Small-molecule ligands of methyl-lysine binding proteins. Journal of Medicinal Chemistry. 2011;54(7):2504–2511. doi: 10.1021/jm200045v. This paper describes the first cell permeable peptidic chemical probe for CBX7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47**.Herold JM, James LI, Korboukh VK, Gao C, Coil KE, Bua DJ, Norris JL, Kireev DB, Brown PJ, Jin J, Janzen WP, et al. Structure-activity relationships of methyl-lysine reader antagonists. Medchemcomm. 2012;3(1):45–51. This papers reports the first small molecule inhibitor of CBX7. [Google Scholar]
- 48.James LI, Barsyte-Lovejoy D, Zhong N, Krichevsky L, Korboukh VK, Herold JM, MacNevin CJ, Norris JL, Sagum CA, Tempel W, Marcon E, et al. Discovery of a chemical probe for the l3mbtl3 methyllysine reader domain. Nat Chem Biol. 2013;9(3):184–191. doi: 10.1038/nchembio.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49**.Camerino MA, Zhong N, Dong A, Dickson BM, James LI, Baughman BM, Norris JL, Kireev DB, Janzen WP, Arrowsmith CH, Frye SV. The structure-activity relationships of l3mbtl3 inhibitors: Flexibility of the dimer interface. Medchemcomm. 2013;4(11):1501–1507. doi: 10.1039/C3MD00197K. This papers reports the selective inhibition of CBX6 over its closely related family members. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baughman BM, Pattenden SG, Norris JL, James LI, Frye SV. The l3mbtl3 methyl-lysine reader domain functions as a dimer. Acs Chemical Biology. 2016;11(3):722–728. doi: 10.1021/acschembio.5b00632. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51*.Esteve PO, Terragni J, Deepti K, Chin HG, Dai N, Espejo A, Correa IR, Bedford MT, Pradhan S. Methyllysine reader plant homeodomain ( phd) finger protein 20-like 1 ( phf20l1) antagonizes DNA ( cytosine-5) methyltransferase 1 ( dnmt1) proteasomal degradation. J Biol Chem. 2014;289(12):8277–8287. doi: 10.1074/jbc.M113.525279. First report of an inhibitor for a methyl-Lysine Tudor domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simhadri C, Daze KD, Douglas SF, Quon TTH, Dev A, Gignac MC, Peng FN, Heller M, Boulanger MJ, Wulff JE, Hof F. Chromodomain antagonists that target the polycomb-group methyllysine reader protein chromobox homo log 7 (cbx7) Journal of Medicinal Chemistry. 2014;57(7):2874–2883. doi: 10.1021/jm401487x. [DOI] [PubMed] [Google Scholar]
- 53.Stuckey JI, Dickson BM, Cheng N, Liu YL, Norris JL, Cholensky SH, Tempel W, Qin S, Huber KG, Sagum C, Black K, et al. A cellular chemical probe targeting the chromodomains of polycomb repressive complex 1. Nat Chem Biol. 2016;12(3):180–187. doi: 10.1038/nchembio.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ren CY, Morohashi K, Plotnikov AN, Jakoncic J, Smith SG, Li JJ, Zeng L, Rodriguez Y, Stojanoff V, Walsh M, Zhou MM. Small-molecule modulators of methyl-lysine binding for the cbx7 chromodomain. Chemistry & Biology. 2015;22(2):161–168. doi: 10.1016/j.chembiol.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ren CY, Smith SG, Yap K, Li SD, Li JJ, Mezei M, Rodriguez Y, Vincek A, Aguilo F, Walsh MJ, Zhou MM. Structure-guided discovery of selective antagonists for the chromodomain of polycomb repressive protein cbx7. Acs Medicinal Chemistry Letters. 2016;7(6):601–605. doi: 10.1021/acsmedchemlett.6b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Milosevich N, Gignac MC, McFarlane J, Simhadri C, Horvath S, Daze KD, Croft CS, Dheri A, Quon TT, Douglas SF, Wulff JE, et al. Selective inhibition of cbx6: A methyllysine reader protein in the polycomb family. ACS Med Chem Lett. 2016;7(2):139–144. doi: 10.1021/acsmedchemlett.5b00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wagner EK, Nath N, Flemming R, Feltenberger JB, Denu JM. Identification and characterization of small molecule inhibitors of a plant homeodomain finger. Biochemistry. 2012;51(41):8293–8306. doi: 10.1021/bi3009278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perfetti MT, Baughman BM, Dickson BM, Mu YX, Cui GF, Mader P, Dong AP, Norris JL, Rothbart SB, Strahl BD, Brown PJ, et al. Identification of a fragment-like small molecule ligand for the methyl-lysine binding protein, 53bp1. ACS Chemical Biology. 2015;10(4):1072–1081. doi: 10.1021/cb500956g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wagner T, Greschik H, Burgahn T, Schmidtkunz K, Schott AK, McMillan J, Baranauskiene L, Xiong Y, Fedorov O, Jin J, Oppermann U, et al. Identification of a small-molecule ligand of the epigenetic reader protein spindlin1 via a versatile screening platform. Nucleic Acids Research. 2016;44(9) doi: 10.1093/nar/gkw089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu C, Min J. Structure and function of wd40 domain proteins. Protein Cell. 2011;2(3):202–214. doi: 10.1007/s13238-011-1018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, Zhang M, Zhang J, Yu Z, Li L, Teng L, et al. An allosteric prc2 inhibitor targeting the h3k27me3 binding pocket of eed. Nat Chem Biol. 2017;13(4):381–388. doi: 10.1038/nchembio.2304. [DOI] [PubMed] [Google Scholar]
- 62.He Y, Selvaraju S, Curtin ML, Jakob CG, Zhu H, Comess KM, Shaw B, The J, Lima-Fernandes E, Szewczyk MM, Cheng D, et al. The eed protein-protein interaction inhibitor a-395 inactivates the prc2 complex. Nat Chem Biol. 2017;13(4):389–395. doi: 10.1038/nchembio.2306. [DOI] [PubMed] [Google Scholar]
- 63.Barnash KD, The J, Norris-Drouin JL, Cholensky SH, Worley BM, Li F, Stuckey JI, Brown PJ, Vedadi M, Arrowsmith CH, Frye SV, et al. Discovery of peptidomimetic ligands of eed as allosteric inhibitors of prc2. ACS Comb Sci. 2017;19(3):161–172. doi: 10.1021/acscombsci.6b00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Senisterra G, Wu H, Allali-Hassani A, Wasney GA, Barsyte-Lovejoy D, Dombrovski L, Dong A, Nguyen KT, Smil D, Bolshan Y, Hajian T, et al. Small-molecule inhibition of mll activity by disruption of its interaction with wdr5. Biochem J. 2013;449(1):151–159. doi: 10.1042/BJ20121280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R, Skucha A, Vittori S, Kuznetsova E, Smil D, Barsyte-Lovejoy D, et al. Pharmacological targeting of the wdr5-mll interaction in c/ebpalpha n-terminal leukemia. Nat Chem Biol. 2015;11(8):571–578. doi: 10.1038/nchembio.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.