

Abstract
Background & Aims
Intestinal epithelial homeostasis is maintained by complex interactions among epithelial cells, commensal gut microorganisms, and immune cells. Disruption of this homeostasis is associated with disorders such as inflammatory bowel disease, but the mechanisms of this process are not clear. We investigated how Sirtuin 1 (SIRT1), a conserved mammalian NAD+-dependent protein deacetylase, senses environmental stress to alter intestinal integrity.
Methods
We performed studies of mice with disruption of Sirt1 specifically in the intestinal epithelium (SIRT1 iKO, villin-Cre+, Sirt1flox/flox mice) and control mice (villinCre-, Sirt1flox/flox) on a C57BL/6 background. Acute colitis was induced in some mice by addition of 2.5% dextran sodium sulfate to drinking water for 5–9 consecutive days. Some mice were given antibiotics via their drinking water for 4 weeks to deplete their microbiota. Some mice were fed with a cholestyramine containing diet for 7 days to sequester their bile acids. Feces were collected and proportions of microbiota were analyzed by 16S rRNA amplicon sequencing and quantitative PCR. Intestines were collected from mice and gene expression profiles were compared by microarray and quantitative PCR analyses. We compared levels of specific mRNAs between colon tissues from age-matched patients with ulcerative colitis (n=10) vs without inflammatory bowel disease (n=8, controls).
Results
Mice with intestinal deletion of SIRT1 (SIRT1 iKO) had abnormal activation of Paneth cells starting at the age of 5–8 months, with increased activation of NF-κB, stress pathways, and spontaneous inflammation at 22–24 months of age, compared with control mice. SIRT1 iKO mice also had altered fecal microbiota starting at 4–6 months of age compared with control mice, in part due to altered bile acid metabolism. Moreover, SIRT1 iKO mice with defective gut microbiota developed more severe colitis than control mice. Intestinal tissues from patients with ulcerative colitis expressed significantly lower levels of SIRT1 mRNA than controls. Intestinal tissues from SIRT1 iKO mice given antibiotics, however, did not have signs of inflammation at 22–24 months of age, and did not develop more severe colitis than control mice at 4–6 months.
Conclusions
In analyses of intestinal tissues, colitis induction, and gut microbiota in mice with intestinal disruption of SIRT1, we found this protein to prevent intestinal inflammation by regulating the gut microbiota. SIRT1 might therefore be an important mediator of host–microbiome interactions. Agents designed to activate SIRT1 might be developed as treatments for inflammatory bowel diseases.
Keywords: IBD, mouse model, microbiome, bacteria
Introduction
The intestinal epithelium is a major metabolic tissue with a monolayer of columnar epithelial cells organized into crypts and villi. It contains absorptive enterocytes and secretory Paneth, goblet, and enteroendocrine cells, which are replenished continuously from stem cells residing at the bottom of the crypts 1. With colonizing microbiota and immune cells, the intestinal epithelium is enriched in environment–host interactions and is constantly flooded with foreign agents, including diet, drugs, pathogens and environmental toxins. Disruption of the intestinal epithelial homeostasis due to abnormal gene-environment interactions leads to various intestinal disorders, including IBD and small intestinal or colonic cancers. However, the underlying molecular mechanisms of these diseases remain ambiguous.
Sirtuins are highly conserved NAD+-dependent protein deacetylases and/or ADP ribosyltransferases that are important for regulation of metabolism, development, tumorigenesis, as well as aging and longevity 2, 3. The mammalian genome encodes seven sirtuins, SIRT1 to SIRT7 4, among which, SIRT1 is the most evolutionally conserved NAD+-dependent protein deacetylase. As a predominant nuclear sirtuin, SIRT1 is a critical nuclear metabolic sensor that regulates a variety of cellular biological processes including stress resistance, metabolism, apoptosis, and inflammation in response to environmental signals 5, 6. In particular, SIRT1 has been identified as a key repressor of inflammation in multiple tissues/cells over the past few years 7, 8. A number of recent studies have suggested a critical role of SIRT1 in regulation of intestinal inflammation and tissue homeostasis 9-13. However, whether SIRT1 stimulates or represses intestinal inflammation remains controversial 9-13, and how intestinal epithelial SIRT1 modulates complex environment–host interactions to regulate intestinal epithelial integrity is still unclear.
To investigate the possible role of SIRT1 in intestinal physiology, we recently generated an intestinal epithelium-specific SIRT1 knockout mouse model (SIRT1 iKO) 14. In the present study, we show that intestinal epithelial SIRT1 is critically involved in regulation of intestinal tissue homeostasis through modulation of the gut microbiota. SIRT1 deficiency in the intestinal epithelium results in increased fecal bile acid concentrations, which in turn lead to an altered gut microbial composition, enhanced intestinal inflammation, and increased susceptibility to colitis.
Materials and Methods
Animal experiments
Intestinal epithelial specific SIRT1 knockout mice (SIRT1 iKO, Villin-Cre+, Sirt1flox/flox) and their age- and gender- matched littermate Flox controls (Villin-Cre-, Sirt1flox/flox) on a C57BL/6 background were generated as described 14. SIRT1 Flox/Lgr5-EGFP and iKO/Lgr5-EGFP mice were generated by breeding SIRT1 Flox and iKO mice with Lgr5-EGFP-IRES-creERT2 mice (Jackson Laboratory, 008875). Age and gender-matched mice were used for all experiments. All mice at the NIEHS animal facility are maintained under strict SPF conditions. All of our mice were housed in micro-isolator static cages (Techniplast, Exton, PA) with autoclaved nesting material (Nestlet, Ancare Corp., Bellmore, NY) and housed on hardwood bedding (Sani-chips, PJ Murphy, Montville, NJ). Mice were provided ad libitum irradiated or autoclaved rodent diet (NIH31, Harlan Laboratories, Madison, WI) and deionized water treated by reverse osmosis. Mice were negative for mouse hepatitis virus, Sendai virus, pneumonia virus of mice, mouse parvovirus 1 and 2, epizootic diarrhea of infant mice, mouse norovirus, Mycoplasma pulmonis, Helicobacter spp., and endo- and ectoparasites, and no pathogens were detected in sentinel mice during our study.
Acute colitis was induced by adding 2.5% DSS in the drinking water ad libitum for five to nine consecutive days. Body weights and rectal bleeding were monitored daily, and the stool blood scores were measured at the end of treatment. According to the animal protocol, mice with more than 20% loss of their initial body mass were sacrificed during the experiments.
To deplete gut microbiota, mice were treated with an antibiotic cocktail in their drinking water containing 1 g/L Ampicillin, 500 mg/L Vancomycin, 1 g/L Neomycin, and 1 g/L Metronidazole for 4 weeks, and the depletion of gut microbiota were analyzed at the end of treatment with high throughput sequencing of bacterial 16S rRNA genes in stool DNA samples. The successful depletion of gut microbiota was evident by the observation that no sufficient bacterial 16S rRNA genes amplicons were obtained from treated mice for the library preparation.
To determine the fecal bile acid contents, feces were freshly collected and fecal bile acids were extracted with 75% ethanol at 50 °C for 2 hours. Bile acids in the resulting supernatants were measured using a total bile acid kit (Diazyme Laboratories, Poway, CA).
To test the impact of bile acids on intestinal SIRT1 deficiency induced gut dysbiosis and intestinal inflammation, 10-12 month old age- and gender-matched Flox and SIRT1 iKO mice were fed with a chow diet containing 2% cholestyramine (custom made from Research Diets) for 7 days, followed by 2.5% DSS water feeding for additional 6 days.
All animal procedures were reviewed and approved by National Institute of Environmental Health Sciences Animal Care and Use Committee. All animals were housed, cared for, and used in compliance with the Guide for the Care and Use of Laboratory Animals and housed and used in an Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALAC) Program.
Microarray study and data analysis
To analyze the transcriptomes of the intestine, total RNA isolated from the colon and ileum was analyzed using Agilent Whole Mouse Genome 4×44 multiplex format oligo arrays (014850) (Agilent Technologies, Santa Clara, CA), following the Agilent 1-color microarray-based gene expression analysis protocol. Data was obtained using the Agilent Feature Extraction software (v9.5), using the 1-color defaults for all parameters. The Agilent Feature Extraction Software performed error modeling, adjusting for additive and multiplicative noise. The microarray data (GSE86475) can be accessed in the Gene Expression Omnibus repository at the National Center for Biotechnology Information.
Meta-Analysis was conducted by NEXTBIO (www.nextbio.com). The gene list from aged colons was used as input to query a collection of individual biosets in Nextbio database to derive a consensus gene signature and/or discover sets of commonly regulated biogroups. The most consistently and highly regulated genes across multiple biosets were identified. GO and Canonical pathway in biogroups were filtered.
16S rRNA amplicon sequencing
Stool DNA samples were analyzed for microbiome at the University of North Carolina Chapel Hill Microbiome Core Facility using Ion Torrent PGM sequencing technology. Analysis of sequencing data was carried out using the QIIME pipeline as described 15, 16. For analysis of Ion Torrent fastq files the 400-bp reads were first truncated at any site if more than three sequential bases receiving a quality score of <20, and any read containing ambiguous base calls or barcode/primer errors was discarded, as were truncated reads. After the Operational Taxonomic Unit (OTU) picking step, chimeras and singletons were removed using ChimeraSlayer 17. Sequences were grouped into OTUs at a 97% level using UCLUST. After taxonomic assignation of OTUs, sequences were aligned and phylogenetic trees were built. QIIME was also used to calculate alpha diversity on rarefied OTU tables to assess sampling depth coverage using observed species, Shannon and phylogenetic diversity (PD) metrics.
Quantitative PCR for microbiota analysis
Total DNA from the feces or from the stool and intestinal contents was extracted using the QIAamp DNA Stool Mini kit (Qiagen, Germantown, MD). The abundance of different intestinal bacterial groups was measured by real-time PCR using group specific 16S rDNA primers. The primers used are Total bacteria (forward ACTCCTACGGGAGGCAGCAGT and reverse, ATTACCGCGGCTGCTGGC), Eubacterium rectal-/Clostridium coccoides (Erec) (forward ACTCCTACGGGAGGCAGC and reverse, GCTTCTTAGTCAGGTACCGTCAT), Clostridium leptum (Clept) (forward GTTGACAAAACGGAGGAAGG and reverse, GACGGGCGGTGTGTACAA), Lactobacillus sp. (Lact) (forward AGCAGTAGGGAATCTTCCA and reverse, CACCGCTACACATGGAG), Bacteroides sp. (Bact) (forward GGTTCTGAGAGGAGGTCCC and reverse, GCTGCCTCCCGTAGGAGT), Mouse Intestinal Bacteroides (MIB) (forward CCAGCAGCCGCGGTAATA and reverse, CGCATTCCGCATACTTCTC), and segmented filamentous bacteria (SFB) (forward GACGCTGAGGCATGAGAGCAT and reverse, GACGGCACGGATTGTTATTCA). The results were quantified by calculating abundance of bacterial group specific 16S rRNA genes relative to total bacterial rRNA genes.
Gut microbiota bile tolerance assays
Four common commensal gut bacterial strains, including two Lactobacillus strains, Lactobacillus johnsonii and Lactobacillus murinus, as well as two control Gram-negative strains, Escherichia coli and Enterobacter cloacae, were cultured in either Elliker Broth (for Lactobacillus strains) or Lysogeny Broth (for control strains). To test their sensitivities to bile, each of above strains were inoculated into their respective broth containing 0, 0.001, 0.01, 0.1, 0.3, 0.5, 1, 5, 10, or 20% ox-bile (Sigma-Aldrich, St. Louis, MO) at the optical density at 620 nm (OD620) of 0.05. Broth without inoculation was used as a negative control. Changes in OD620 were then measured after 16 hours of incubation at 37 °C. Survival of each strain in broth containing 0 or 0.3% ox-bile was tested by plating onto Blood Agar plates after 4 hours of incubation at 37°C.
Human subjects
Age matched human colonic RNAs from 8 normal and 10 Ulcerative colitis subjects (between 22 to 45; both males and females) were obtained from OriGene, Rockville, MD. Total RNA was purified from pathologist verified, frozen human colon tissues collected under strict consent and IRB protocols by OriGene technologies, Inc. OriGene has de-identified these human colon RNAs in accordance with 45 CFR 164.514(b)(2). Human colons were provided by NIEHS-Duke collaborative initiative and are approved by IRB protocols.
Statistical analysis
Values are expressed as mean + standard error of mean (s.e.m.). Significant differences between means were analyzed by the two-tailed, unpaired, non-parametric Mann-Whitney test, and differences were considered significant at P < .05.
Results
Deletion of intestinal epithelial SIRT1 activates intestinal secretory cells and induces inflammation in the small intestine of aged mice
SIRT1 is widely expressed in all intestinal epithelial cells (IECs) in both the colon and small intestine, with relatively high levels in the crypts (Supplementary Figure 1A-D), suggesting an important role for this protein factor in maintenance of intestinal tissue homeostasis. To dissect the possible functions of SIRT1 in the intestinal epithelium, we have previously generated an intestinal epithelium-specific SIRT1 knockout mouse strain (SIRT1 iKO) in which the exon 4 of the mouse Sirt1 gene was deleted throughout the length of the intestinal epithelium 14, including intestine-originated innate immune cells Paneth cells (Supplementary Figure 1E), and Lgr5 positive intestinal stem cells (iSCs, Supplementary Figure 1F).
SIRT1 iKO mice were phenotypically normal on the chow diet at the age of 2-4 months 14 (Supplementary Figure 1 and 2). They only exhibited altered expression levels of one Paneth cell maker (Lysozyme) and one iSC marker (telomerase reverse transcriptase, mTert) in the purified Paneth cell and iSC fractions from the ileum (Supplementary Figure 1E and 1F). However, during the process of aging, the expression levels of intestinal SIRT1 were gradually reduced (Figure 1A), and SIRT1 iKO mice began to exhibit abnormal activation of Paneth cells from the age of 5-8 months, as indicated by increased number and size of lysozyme positive Paneth cells (Figure 1B-1D) and elevated expression of a number of Paneth cell anti-microbial peptides (AMPs) in the ileum (Figure 1E). In addition, the number of goblet cells, cells that are specific for mucus secretion, was also significantly elevated in aged SIRT1 iKO mice (Figure 1F). No additional defects were observed in other cell types (Supplementary Figure 3) or in the proliferation rate of IECs (Supplementary Figure 4), suggesting that SIRT1 deficiency in the intestine specifically activates secretory cells in response to age-induced stress.
Figure 1. Deletion of SIRT1 in the intestinal epithelium results in hyperactivation of Paneth cells and goblet cells in aged mice.

(A) The expression of Sirt1 is decreased with increasing age in the jejunum (n=6, *P < .05). (B) Increased expression of Paneth cell markers in the jejunum of 8-month old SIRT1 iKO mice. Paneth cells were immuno-stained with anti-lysozyme antibodies. (C) Increased Paneth cell number in the jejunum of 14-month old SIRT1 iKO mice. Paneth cell number was determined by the lysozyme-based FACS method (n=6, *P < .05). (D) Increased expression of lysozyme and Paneth cell number in the jejunum of 24-month old SIRT1 iKO mice (n=6, *P < .05). Bars, 200 μm. (E) Elevated mRNA levels of Paneth cell products in the ileum of SIRT1 iKO mice (n=6, *P < .05). (F) Increased number of Goblet cells in the jejunum of 24-month old SIRT1 iKO mice. Goblet cells were immuno-stained with Alcian blue (n=6, *P < .05). Bars, 200 μm.
Aged SIRT1 iKO mice also experienced enhanced inflammation in the small intestine. As shown in Figure 2A-2C, 12 to 24-month old SIRT1 iKO mice had elevated expression levels of pro-inflammatory cytokines (Figure 2A), increased accumulation of F4/80 positive macrophages (Figure 2B), along with an enhanced transcriptional activity of NF-κB (Figure 2C), a master transcription regulator of inflammation and a well-established SIRT1 deacetylation substrate 18, in the ileum.
Figure 2. Deletion of intestinal epithelial SIRT1 enhances age-induced inflammation and oxidative stress in the ileum.

(A) SIRT1 iKO mice have increased expression levels of several proinflammatory cytokines in the ileum (n=6, .05 < #P < 0.1, *P < .05). (B) Aged SIRT1 iKO mice have increased accumulation of F4/80 positive macrophages in the ileum. Bars, 100 μm. (C) Aged SIRT1 iKO ileum have increased NF-κB activity (n=8, *P < .05). (D) Flox and SIRT1 iKO mice have distinct age-induced alterations in gene expression profiles in the ileum. (Left) The numbers of differentially expressed gene probes between young (2-month old) and aged (24-month old) Flox controls and SIRT1 iKO mice. (Right) Venn-diagram representation of age-induced common gene probes (2976), as well as unique gene probes (2535 unique in Flox mice, and 3037 unique in SIRT1 iKO mice). (E) Aged Flox and SIRT1 iKO ileum display differential expression patterns of a subset of genes involved in mitochondrial functions, inflammation, and stress response. (n=3-4, P < .05) (F) Flox and SIRT1 iKO ileum have distinct age-dependent alterations in cell death, mitochondrial functions, NF-κB signaling, and NRF2-mediated oxidative stress response pathways. Unique gene lists from Flox and SIRT1 iKO ileum were analyzed by the IPA software, and the top 5 toxicity lists were listed (n=3-4).
To elucidate the molecular mechanisms underlying SIRT1 deficiency-induced age-dependent secretory cell hyperactivation and intestinal inflammation, we determined the transcriptomes of young (6 to 8-week old) and aged (24-month old) Flox and SIRT1 iKO ilea by microarrays. Consistent with the observation that young SIRT1 iKO mice were relatively normal, only 960 out of the 41,175 tested gene probes were differentially expressed between Flox and SIRT1 iKO ilea in young mice (Figure 2D, left). Aging altered the expression of 8,548 gene probes in the ileum, among which only 2,976 were common to both Flox and SIRT1 iKO mice (Figure 2D, right), suggesting that Flox and SIRT1 iKO mice respond to aging differently. Heatmap analyses of these total 8,548 gene probes confirmed that the ileum of young SIRT1 iKO mice did not significantly differ from that of Flox control mice, whereas aged Flox and SIRT1 iKO mice displayed markedly distinct gene expression profiles (Figure 2E). Ingenuity Pathway Analysis (IPA) further showed that in response to aging, ilea from Flox mice were enriched in genes involved in mitochondrial function, cell death, DNA damage and cell cycle, whereas ilea from SIRT1 iKO mice were primarily impaired in mitochondrial functions, NF-κB-induced inflammatory signaling, and NRF2-mediated oxidative stress response pathways (Figure 2F). Disruption in mitochondrial functions, activation of NF-κB- signaling and NRF2-mediated oxidative stress could all contribute to the symptoms of enhanced inflammation and secretory cell hyperactivation in aged SIRT1 iKO ileum.
Intestinal epithelial SIRT1 deficiency induces spontaneous inflammation in the colon of aged mice
SIRT1 deficiency-induced activation of Paneth cells and goblet cells, increase in NF-κB activity, and elevation of pro-inflammatory genes and antimicrobial proteins in the small intestine (Figure 1 and 2) suggest that SIRT1 deficient colons may also display symptoms of spontaneous inflammation and other impairments. In line with this possibility, aged SIRT1 iKO mice had slightly but significantly decreased colon lengths (Figure 3A) along with increased damage of intestinal barrier function, as indicated by elevated lipopolysaccharide (LPS) in mesenteric lymph nodes (Figure 3B). Morphological analyses of colon H&E sections revealed that aged SIRT1 iKO mice had marked increases in leukocyte infiltration (Figure 3C and 3D). Consistently, aged SIRT1 deficient colons had increased expression of pro-inflammatory cytokines, angiogenins (Angs), as well as massive elevation of immunoglobulin genes yet significantly reduced levels of several nutrient transporters, as revealed by microarray (Figure 3D and 3E). The increased expression of immunoglobulin genes suggests an infiltration of B-lymphocytes or increased activation of resident B-lymphocytes in the aged colons.
Figure 3. Deletion of intestinal epithelial SIRT1 promotes age-induced spontaneous inflammation and tissue damage in the colon.
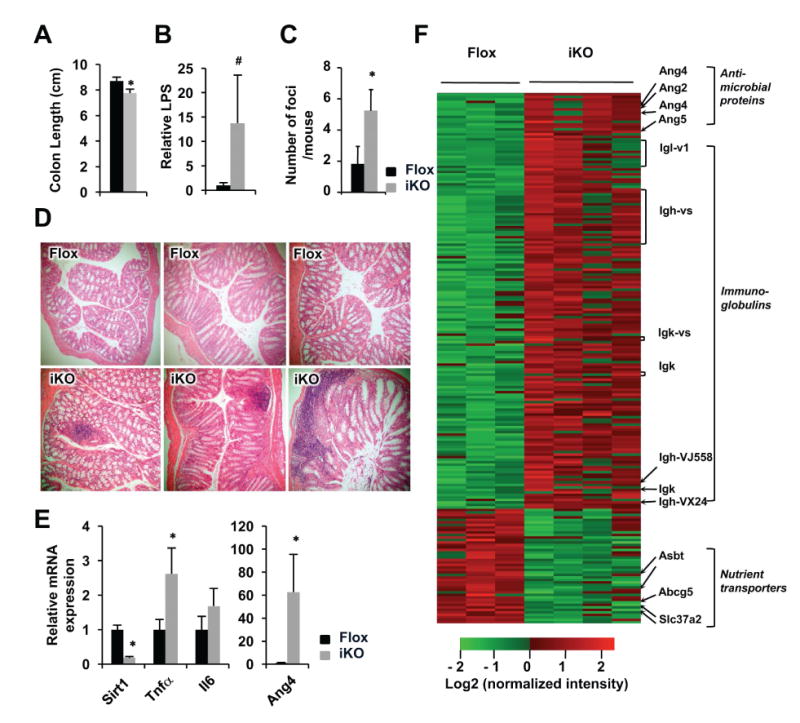
(A) Aged (24-month old) SIRT1 iKO mice have reduced colon length (n=8, *P < .05). (B) Aged SIRT1 iKO mice have elevated LPS levels in the mesenteric lymph node (n=5, .05 < # P < .1). (C-D) Aged SIRT1 iKO display elevated infiltration of leukocytes in the colon (n=6, *P < .05). (E) SIRT1 iKO colons have increased mRNA levels of inflammatory genes and AMPs. The colonic mRNA were analyzed by qPCR (n=6-8, *P < .05). (F) Aged SIRT1 iKO mice have increased expression levels of anti-microbial proteins, enhanced leukocyte gene expression signature, but reduced levels of nutrient transporters in the colon. The colonic total RNA were analyzed by mouse whole genome microarray (n=3-4, fold changes >2, P < .05).
Deletion of intestinal epithelial SIRT1 increases susceptibility to colitis
Intestinal epithelial SIRT1 deficiency induced spontaneous inflammation raises the possibility that SIRT1 iKO mice may be sensitive to IBD, including Crohn's disease and ulcerative colitis (UC). In support of this possibility, meta-analyses of colon gene expression profiles of SIRT1 iKO vs Flox controls against 70,297 biosets using the NextBio software indicated that aged SIRT1 iKO colons shared the highest similarity with various mouse models of colitis, colon inflammation/tumors, and systemic inflammation (Figure 4A). Importantly, the gene expression profiles of aged SIRT1 iKO colons significantly overlapped with those from colons of UC patients in two independent studies 19, 20 (Figure 4B). In particular, the most significantly correlated GO functional categories between SIRT1 iKO colons and UC patient colons included inflammatory response and mitochondrial functions (Supplementary Figure 5A). Specifically, the vast majority of overlapped genes in the mitochondrial function category were down regulated in both SIRT1 deficient colons and UC patient colons (Supplementary Figure 5B), indicating the importance of mitochondria and oxidative stress in the pathogenesis of clinical symptoms of SIRT1 deficient colons and UC patient colons. These observations suggest that SIRT1 may be a critical genetic factor that regulates the susceptibility to environment-induced inflammation and colitis. In line with this idea, the mRNA levels of human SIRT1 were significantly reduced in colons of human UC patients in two published study cohorts 19, 20 (Figure 4C and Supplementary Figure 5C), which were confirmed by qPCR in an independent UC patient cohort (Figure 4D). Intriguingly, the mRNA levels of SIRT6, another nuclear sirtuin that is critically involved in regulation of genome stability, inflammation, and metabolism 21, as well as SIRT5, a mitochondrial sirtuin, were also reduced in the UC patients (Figure 4D and Supplementary Figure 5C), indicating a general role of sirtuins in regulating intestinal epithelial homeostasis. Collectively, our observations suggest that SIRT1 may be critical in modulating susceptibility to colitis in both humans and mice.
Figure 4. Intestinal epithelial SIRT1 deficiency is associated with colitis in both humans and mice.

(A) The mRNA expression profile of SIRT1 iKO colon strongly correlates with those of colitis and inflammatory mouse models. The gene expression profiles from aged SIRT1 iKO vs old Flox control colons were analyzed against 70,297 biosets using the NextBio meta-analysis program. The top six correlated biosets are shown with the corresponding significant scores. (B) The mRNA expression profile of SIRT1 iKO colon significantly overlaps with mRNA profiles from the colons of human UC patients. P = 3.4×10-11 with Olsen et al dataset 19, and P = 6.0×10-11 with Planell et al dataset 20. (C) The mRNA levels of SIRT1 are reduced in colons from UC patients (based on microarray data from Olsen et al 19 and Planell et al 20). (D) SIRT1, SIRT5, and SIRT6 mRNA levels are reduced in human UC patients. mRNA from colons of human UC patients and controls were analyzed by qPCR (n=8-10, *P < .05).
To test this possibility and to further assess the significance of intestinal SIRT1 in regulating intestinal epithelium functions in response to environmental stress, we challenged Flox and SIRT1 iKO mice at the age of 3-6 months with 2.5% Dextran Sodium Sulphate (DSS) in drinking water for 5-7 days. SIRT1 iKO mice within this age range did not display spontaneous colonic inflammation. However, after DSS challenge, they experienced significantly enhanced rectal bleeding (Figure 5A), bloody diarrhea (Figure 5B), increased colonic shortening (Figure 5C), and elevated colonic crypt erosion (Figure 5D) compared to Flox control mice. These pathological changes were accompanied with elevated expression of a number of pro-inflammatory markers, such as Tnfα, Cd68, and Ang4 (Figure 5E and Supplementary Figure 5D), indicating that intestinal epithelial SIRT1 deficiency enhances the sensitivity to chemical-induced colitis.
Figure 5. Intestinal epithelial SIRT1 deficiency is associated with increased susceptibility to DSS-induced colitis in mice.

(A-D) SIRT1 iKO mice are more sensitive to DSS-induced colitis than Flox controls. Flox and SIRT1 iKO mice were treated with 2.5% DSS in drinking water for 6 days. Their rectal bleeding (A), stool blood (B), colon length (C), and colon histology (D) were analyzed (n=23-27, *P < .05). Bar in (C), 1 cm; Bar in (D), 200 μm. (E) SIRT1 deficient colons have increased expression levels of pro-inflammatory genes and antibacterial proteins after DSS administration (n=6-8, *P < .05).
Intestinal epithelial SIRT1 deficiency age-dependently alters gut microbiota in part through bile acids
Dysregulation of intestinal tissue homeostasis and increased intestinal inflammation are closely associated with alterations in intestinal microbiota 22-24. To evaluate whether intestinal epithelial SIRT1 deficiency-induced alterations in epithelial cell homeostasis and intestinal inflammation are associated with changes of the gut microbial ecosystem, we analyzed total fecal microbiota profiles from co-housed paired Flox and SIRT1 iKO littermates by 16S rRNA amplicon sequencing. While minimal changes in relative abundance of bacterial taxa were observed between 2-month old young Flox and SIRT1 iKO mice, 24-month old aged SIRT1 iKO mice showed a trend of reduction in the total number of observed bacterial species richness and phylogenetic diversity values compared to aged Flox mice (Supplementary Figure 6A). Most notably, 24-month old SIRT1 iKO mice consistently exhibited reduced abundance of the class Bacilli (Figure 6A, 6B, and Supplementary Figure 6B), including all genera such as Lactobacillus (Figure 6C). The significant reduction of fecal Lactobacillus abundance in SIRT1 iKO mice was independently confirmed in whole intestinal samples (tissues together with feces) from 22 to 24-month old SIRT1 iKO mice by q-PCR (Supplementary Figure 6C). Further time course analyses revealed that the abundance of fecal Lactobacillus was increased with age in control mice, but this age-dependent increase was blunted in SIRT1 iKO mice (Figure 6D), and the significant inhibition of fecal Lactobacillus in SIRT1 iKO mice was detectable as early as 4-month old (Figure 6D). Lactobacillus is an important genus of bacteria that is reduced in IBD patients 25, and administration of probiotic Lactobacillus strains can protect against intestinal inflammation in humans and mice 26, 27. Therefore, our observations suggest that SIRT1 may regulate intestinal inflammation and susceptibility to IBD through modulation of gut microbiota.
Figure 6. Deletion of SIRT1 in the intestinal epithelium age-dependently alters gut microbial composition through bile acids.

(A-B) Aged SIRT1 iKO mice have reduced abundance of the Bacilli class in feces. Total stool DNA from co-housed and paired 24-month old Flox and SIRT1 iKO littermates were analyzed as described in Materials and Methods (n=7-8, *P < .05). (C) Aged SIRT1 iKO have reduced levels of all detectable genera of the Bacilli class in feces (n=7, *P < .05). (D) Intestinal epithelial SIRT1 deficiency age-dependently inhibits Lactobacillus in feces. Total stool DNA from co-housed Flox and SIRT1 iKO mice at different ages were extracted and analyzed by qPCR using specific primers targeting total or different bacterial 16S rRNA genes (n=15-30 for the 1.5 to 3-month group, n=14-21 for the 4 to 10-month group, n=13 for the 22 to 24-month group, *P < .05). (E) Lactobacillus strains have increased sensitivity to bile salts compared to gram-negative gut microbiota. L. johnsonii and L. murinus, as well as two gram-negative gut microbiota strains, E. coli and E. cloacae, were plated on Blood Agar plates after culturing in medium containing 0 or 0.3% ox-bile for 4 hours. (F) Ten to twelve-month old Flox and SIRT1 iKO mice have comparable abundance of fecal Lactobacillus after feeding with a bile acid sequestrant. 10-12 month old Flox and SIRT1 iKO mice were fed with a chow diet containing 2% cholestyramine for 7 days. Their fecal bile acid contents, total serum cholesterol levels, and relative abundance of indicated fecal bacteria before (Before) and after (After) feeding were analyzed and compared (n=8, *P < .05, ** P < .01, and *** P < .001).
The homeostasis of intestinal microbiota is regulated by a number of genetic and environmental factors 22-24. For instance, Paneth cells have been shown to be crucial in regulating the gut microbiota and its associated (patho)physiologies through antimicrobial peptides 22-24, 28. The observation that SIRT1 deficiency leads to hyperactivation of Paneth cells (Figure 1) suggests that Paneth cell dysfunction may contribute to the observed gut dysbiosis in aged SIRT1 iKO mice. However, our preliminary data indicated that deletion of SIRT1 specifically in Paneth cells failed to inhibit Lactobacillus, moreover, these mice were protected from DSS-induced colitis and inflammation (not shown), suggesting that SIRT1 modulates gut microbiota through alternative mechanisms.
In addition to Paneth cells, the homeostasis of gut microbiota is also under the influence of bile acids, end products of hepatic cholesterol catabolism. Bile acids have direct antimicrobial effects on gut microbes and indirect effects through FXR induced antimicrobial peptides 29, 30. Intriguingly, different bacteria strains have distinct tolerance to bile salts, and the Gram-negative bacteria are inherently more resistant to bile than Gram-positive bacteria (e. g. Lactobacillus) 29, 31, 32. For instance, it has been reported that the majority of Lactobacillus strains exhibit growth delay or fail to grow at all in broth containing a low concentration of bile salts 32. We have previously shown that deletion of intestinal epithelial SIRT1 decreases the HNF1α/FXR signaling pathways, reducing ileal bile acid absorption and increasing fecal contents of bile acids 14. Consistent with this previous observation, SIRT1 iKO mice had significantly higher fecal bile acid contents at different ages (Supplementary Figure 6D). Therefore, it is possible that the increased concentration of fecal bile acids drives our observed Lactobacillus depletion phenotype in SIRT1 iKO mice.
To test this possibility, we first examined the growth of four common commensal gut bacterial strains, including two Lactobacillus strains, Lactobacillus johnsonii and Lactobacillus murinus, as well as two control Gram-negative strains, Escherichia coli and Enterobacter cloacae, in response to increased concentrations of bile salt (ox-bile) in culture medium. Both Lactobacillus strains started to display reduced growth below 0.5% ox-bile, whereas two control Gram-negative strains did not show any delay in growth until ox-bile concentrations reached to 5-10% (Supplementary Figure 6E). Moreover, in contrast to the strong resistance of E. coli and E. cloacae to 0.3% ox-bile, very few bacteria from L. johnsonii and L. murinus survived after 4-hour treatment of 0.3% ox-bile (Figure 6F), indicating that many Lactobacillus strains are indeed more sensitive to bile salts than Gram-negative strains. We then fed 10-12 month old Flox and SIRT1 iKO mice with a chow diet containing 2% cholestyramine, a bile acid sequestrant (Figure 6F). Cholestyramine is a strong ion exchange resin that binds bile acids strongly in the resin matrix. In the intestine, they form insoluble complexes with bile acids (thereby sequestering them) and are then excreted in the feces. Therefore, treatment with cholestyramine containing diet has been shown to increase fecal bile acids and lower plasma cholesterol levels as a result of increased hepatic conversion of cholesterol to bile acids 33. As shown in Figure 6F, 7-day feeding of the cholestyramine diet significantly increased fecal bile acid contents in Flox mice but not further in SIRT1 iKO mice (Figure 6F, Fecal bile acids), although the serum cholesterol levels were reduced in both Flox and SIRT1 iKO mice (Figure 6F, Serum cholesterol). As a result, SIRT1 iKO mice had higher fecal bile acid contents than Flox mice before but not after cholestyramine diet feeding (Figure 6F, Fecal bile acids). Remarkably, sequestration of bile acids increased the fecal levels of Lactobacillus (Before vs After), and elimination of the fecal bile acid difference by cholestyramine also rescued the Lactobacillus depletion phenotype of SIRT1 iKO mice (Figure 6F, Lactobacillus). Together, our finding indicates that intestinal epithelial SIRT1 regulates gut microbiota, at least in part, through regulation of bile acid metabolism.
Figure 7. Intestinal epithelial SIRT1 deficiency induced enhancement in intestinal inflammation is largely dependent on gut microbiota.
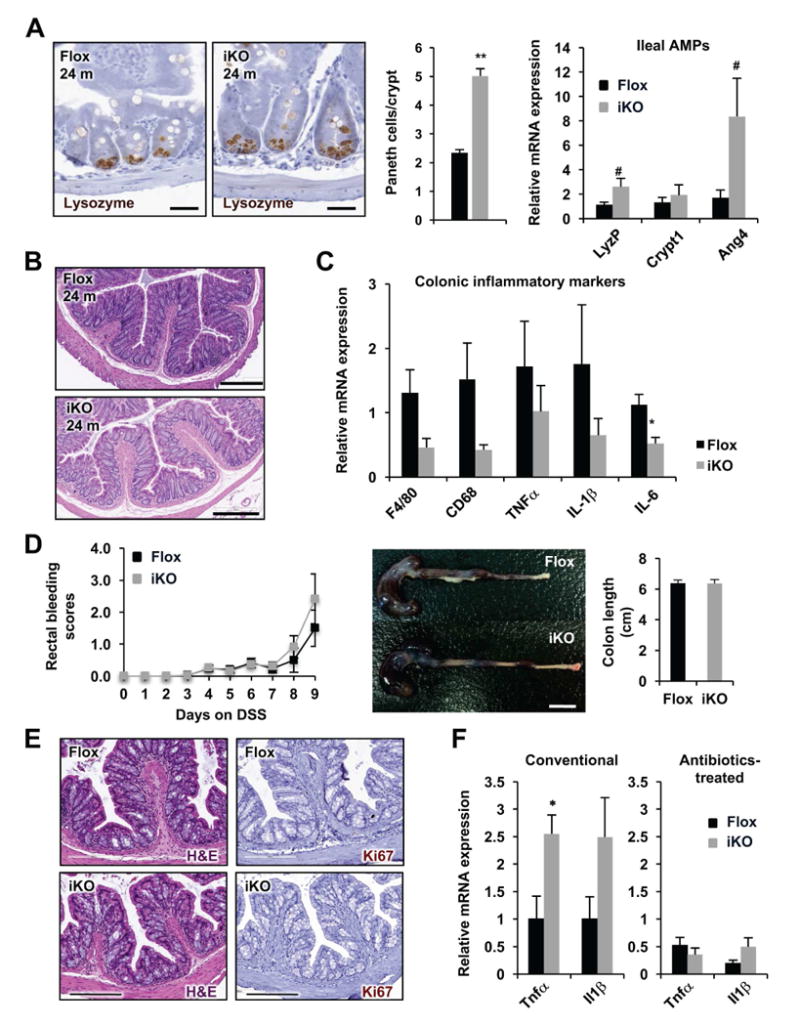
(A) Antibiotics-treated aged SIRT1 iKO mice have increased Paneth cells and increased expression of AMPs in the ileum (n=6-8, ** P < .01, .05 < # P < .1). The number of lysozyme positive Paneth cells were counted in 100 crypts/mouse. Bars, 20 μm. (B and C) Antibiotics-treated aged SIRT1 iKO mice have normal colon histology (B) and a reduced trend of expression of proinflammatory genes in colon (C) (n=6-8, *P < .05). Bars in B, 200 μm. (D-F) Antibiotic-treated aged SIRT1 iKO mice have a comparable sensitivity to DSS-induced colitis compared to Flox controls. Flox and SIRT1 iKO mice pre-treated with antibiotic cocktail water for 4 weeks were administrated with 2.5% DSS in drinking water for 9 days. Their rectal bleeding and colon length (A), as well as colon histology (E) were analyzed (n=9-11). Bar in (D), 1 cm; Bar in (E), 200 μm. (F) SIRT1 deficient colons have normal expression levels of pro-inflammatory genes after antibiotics/DSS treatment (Conventional conditions, n=6-8; Antibiotics-treated, n=9-11; *P < .05)
SIRT1 deficiency-induced colonic inflammation and hypersensitivity to DSS-colitis are largely dependent on gut microbiota
Intestinal epithelial homeostasis is maintained through complex interplays between epithelial cells, commensal gut microorganisms, and immune cells. For example, intestinal secretory cells have been shown to closely interact with gut microbiota to regulate intestinal epithelial homeostasis and inflammation 22-24. On the other hand, alterations in gut microbial ecosystem (gut dysbiosis) have also been shown to modify intestinal inflammation and epithelial cell activity, playing a role in initiating and sustaining IBD 34, 35. Specifically, the intestinal microbiota in patients with IBD is characterized by a reduced bacterial diversity, a decrease of Firmicutes (including Lactobacillus) and an increase of Proteobacteria, and therapeutic strategies aimed to correct dysbiosis have been shown to be promising in IBD 34, 35.
To dissect the primary defects caused by intestinal epithelial SIRT1 deletion and to further understand the cause-effect relationship among hyperactivated Paneth cells, increased inflammation, and altered gut microbiota observed in aged SIRT1 iKO mice, we generated gut microbiota-depleted mice by treating 22-month old aged Flox and SIRT1 iKO mice with drinking water containing an antibiotic cocktail for four weeks (Figure 7A-C). Interestingly, antibiotic-treated aged SIRT1 iKO mice still exhibited hyperactivation of Paneth cells in the small intestine (Figure 7A), although the number and morphology of their goblet cells appeared to be normal (Supplementary Figure 7A). However, remarkably, depletion of gut microbiota by antibiotics diminished the spontaneous inflammation in the colon of aged SIRT1 iKO mice, as indicated by a normal colon length (Supplementary Figure 7B), minimal leukocyte infiltration (Figure 7B), and normal to reduced expression of proinflammatory markers (Figure 7C). These observations demonstrate that alteration of gut microbiota is the primary cause for enhanced colonic inflammation in aged SIRT1 iKO mice.
Gut microbiota-depleted SIRT1 iKO mice not only had minimal colonic spontaneous inflammation, but also displayed a comparable sensitivity to DSS-induced colitis compared to age matched Flox controls. As shown in Figure 7D-F, antibiotics treated mice were relatively more resistant to DSS-induced colitis compared to mice maintained at the conventional condition (Figure 5). They required prolonged DSS administration to induce rectal bleeding and colon shortening (Figure 7D), and were able to preserve relatively intact crypt structures even after 9 days of DSS treatment (Figure 7E). Additionally, the expression levels of several proinflammatory cytokines were also reduced in antibiotic-treated DSS-colitis mice compared to conventional DSS-colitis mice (Figure 7F). Importantly, the hypersensitive phenotypes of SIRT1 iKO mice to DSS-colitis observed in the conventional condition (Figure 5) were almost completely eliminated in the gut microbiota-depleted condition (Figure 7D-F). Moreover, SIRT1 iKO mice that did not display alteration in their gut microbiota, including the 1.5 to 2-month old young SIRT1 iKO mice (Supplementary Figure 8) and the 10 to 12-month old SIRT1 iKO mice after feeding with 2% cholestyramine diet (Supplementary Figure 9), had a comparable sensitivity to DSS-colitis as control mice, highlighting the importance of gut dysbiosis in SIRT1 deficiency induced colitis. Taken together, these observations indicate that SIRT1 deficiency-induced colonic inflammation and hypersensitivity to DSS-colitis are largely dependent on gut microbiota.
Discussion
As one of the well-known genetic repressors of inflammation, SIRT1 exerts remarkable influences on a wide range of inflammation-associated diseases 8. However, the role of SIRT1 in intestinal tissue homeostasis and inflammation is still elusive. In the present study, using an intestinal epithelium-specific SIRT1 knockout mouse model together with DSS-colitis and gut-microbiota-depletion models, as well as tissues from human IBD patients, we show that intestinal epithelial SIRT1 is crucial in active modulation of intestinal epithelial cell homeostasis, intestinal inflammation, and gut microbial composition in response to age/chemical-induced stresses, thereby impacting intestinal epithelial function and affecting susceptibility to environment-induced intestinal inflammatory diseases. We further demonstrate that intestinal epithelial SIRT1 modulates gut microbiota in part through regulation of bile acid metabolism, and intestinal epithelial SIRT1 deficiency induced impairments in intestinal inflammation are largely dependent on gut microbiota. Our findings uncover a previously unknown role of SIRT1 in mediating host-microbiome interactions, and suggest that SIRT1-activating compounds may be therapeutically beneficial for the treatment of human IBD.
Gut microbiota exerts great influence on host health and disease. Maintenance of a normal homeostatic gut microbiota is critical for host nutrient metabolism and the immune system, whereas dysbiosis of the gut bacteria has been directly linked with the development of a number of human diseases, such as IBD, inflammation and infection, diabetes, and obesity 36, 37. In the present study, we discovered that SIRT1 modulates intestinal inflammation and susceptibility to IBD primarily through modulation of gut microbiota (Figure 6 and 7). Most notably, we found that aged SIRT1 iKO mice exhibited reduced abundance of Bacilli, particularly Lactobacillus (Figure 6), bacteria that are reduced in IBD patients 25 and have been shown to protect against intestinal inflammation in humans and mice 26, 27. Consistent with our observations, resveratrol, a well-known SIRT1 activator, has been shown to enhance the Lactobacillus population in a DSS colitis model 38. Therefore, SIRT1 may regulate intestinal inflammation and susceptibility to IBD through direct modulation of Lactobacillus. It will be of great interest to test whether administration of certain bile-resistant probiotic Lactobacillus strains will alleviate intestinal epithelial SIRT1 deficiency induced inflammation, and further investigate whether SIRT1 deficiency induced gut dysbiosis contributes to other SIRT1-mediated (patho)physiologies.
Several of the abnormalities we observed in the SIRT1 iKO mice, however, are in contrast to those reported by Lo Sasso et al. 13. Using a SIRT1 knockout mouse model in which exons 5, 6, and 7 of the mouse Sirt1 gene were deleted specifically in the intestinal epithelium, Lo Sasso et al. observed a similar hyperactivation of Paneth and goblet cells but at a much younger age. They further showed that the hyperactivated Paneth cells and goblet cells lead to rearrangement of the gut microbiota and protect SIRT1 KO animals from colitis 13. Based on our observations that the intestinal epithelial SIRT1 deficiency induced impairments in intestinal inflammation are largely dependent on gut dysbiosis, one possible factor contributing to the discrepancy between our observations and those of Lo Sasso et al. may be the difference in the gut microbial community between SIRT1 iKO mice raised in these two facilities. Notably, aged SIRT1 iKO mice in our facility were specifically depleted of the Bacilli class, including Lactobacillus (Figure 6), whereas a completely different set of microbes was altered in the intestinal epithelium-specific SIRT1 KO mice in Lo Sasso et al. study 13. The differential alterations of these gut microorganisms in these two studies may directly lead to distinct responses of SIRT1 iKO mice to DSS-induced colitis. Nevertheless, our observations in the present study are consistent with previous reports that reduced SIRT1 expression, inhibition of SIRT1 activity, or a SIRT1 mutation that decreases its activity, is associated with intestinal inflammation and colitis in both humans and rodents, whereas activation of SIRT1 by resveratrol, Cay10591, or SRT1720 prevents and cures experimental colitis 9-12. Our findings provide a direct link between SIRT1, gut microbiota, and intestinal inflammation, and highlight the therapeutic potential of SIRT1-activating compounds in the treatment of human IBD.
In summary, our study defines a new role for SIRT1 as a critical regulator of intestinal tissue homeostasis. In addition to providing a step in the understanding of secretory cell physiology especially during stress response, our study establishes SIRT1 as a critical molecular link in regulating gut microbiota, epithelial biology, and inflammation.
Supplementary Material
Acknowledgments
We thank Drs. Michael Fessler and Donald Cook, and members of the Li laboratory for critical reading of the manuscript; Dr. Frederic Alt at Harvard Medical School for providing SIRT1 exon 4 floxed allele; Dr. Arun Pandiri, Dr. Janardhan Kyathanahalli, Natasha Clayton, and histology core for the histology support. This research was supported by the Intramural Research Program of National Institute of Environmental Health Sciences of the NIH, to X.L. (Z01 ES102205), and a NIH grant to A. S. G. (K08 DK095917). The UNC Microbiome Core Facility is supported in part by NIH grant P30 DK34987.
Footnotes
Author Contributions: A. S. W. and M. R. M. designed experiments, carried out experiments, analyzed data, and wrote the manuscript. N. K., Q. X., A. C., M. T. S., and A. S. G. designed experiments, carried out experiments and analyzed data. X. X., and D. C. F. analyzed the microarray data. N. S. X. R., A. K. and W. C. carried out experiments. M. A. A-P did the bacterial 16S rDNA sequencing and data analysis. L. G. analyzed data and critically reviewed the manuscript. X. L. designed experiments, analyzed data, and wrote the manuscript.
The Gene Expression Omnibus (GEO) accession: GSE86475
Conflict of Interest: The authors declare that they have no conflict of interest.
All authors had access to the study data and reviewed and approved the final manuscript.
Author names in bold designate shared co-first authorship
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Noah TK, Shroyer NF. Notch in the Intestine: Regulation of Homeostasis and Pathogenesis. Annu Rev Physiol. 2013;75:24.1–24.26. doi: 10.1146/annurev-physiol-030212-183741. [DOI] [PubMed] [Google Scholar]
- 2.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–35. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 3.Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013;27:2072–85. doi: 10.1101/gad.227439.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–8. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol. 2007;21:1745–55. doi: 10.1210/me.2007-0079. [DOI] [PubMed] [Google Scholar]
- 6.Schug TT, Li X. Sirtuin 1 in lipid metabolism and obesity. Annals of Medicine. 2011;43:198–211. doi: 10.3109/07853890.2010.547211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guarente L, Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. 2011;364:2235–44. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- 8.Li X. SIRT1 and energy metabolism. Acta Biochim Biophys Sin (Shanghai) 2013;45:51–60. doi: 10.1093/abbs/gms108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biason-Lauber A, Boni-Schnetzler M, Hubbard BP, Bouzakri K, et al. Identification of a SIRT1 mutation in a family with type 1 diabetes. Cell Metab. 2013;17:448–55. doi: 10.1016/j.cmet.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh UP, Singh NP, Singh B, et al. Resveratrol (trans-3,5,4'-trihydroxystilbene) induces silent mating type information regulation-1 and down-regulates nuclear transcription factor-kappaB activation to abrogate dextran sulfate sodium-induced colitis. J Pharmacol Exp Ther. 2010;332:829–39. doi: 10.1124/jpet.109.160838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caruso R, Marafini I, Franze E, et al. Defective expression of SIRT1 contributes to sustain inflammatory pathways in the gut. Mucosal Immunol. 2014;7:1467–79. doi: 10.1038/mi.2014.35. [DOI] [PubMed] [Google Scholar]
- 12.Melhem H, Hansmannel F, Bressenot A, et al. Methyl-deficient diet promotes colitis and SIRT1-mediated endoplasmic reticulum stress. Gut. 2016;65:595–606. doi: 10.1136/gutjnl-2014-307030. [DOI] [PubMed] [Google Scholar]
- 13.Lo Sasso G, Ryu D, Mouchiroud L, et al. Loss of Sirt1 function improves intestinal anti-bacterial defense and protects from colitis-induced colorectal cancer. PLoS One. 2014;9:e102495. doi: 10.1371/journal.pone.0102495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kazgan N, Metukuri MR, Purushotham A, et al. Intestine-specific deletion of SIRT1 in mice impairs DCoH2-HNF-1alpha-FXR signaling and alters systemic bile acid homeostasis. Gastroenterology. 2014;146:1006–16. doi: 10.1053/j.gastro.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devine AA, Gonzalez A, Speck KE, et al. Impact of ileocecal resection and concomitant antibiotics on the microbiome of the murine jejunum and colon. PLoS One. 2013;8:e73140. doi: 10.1371/journal.pone.0073140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004;23:2369–80. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olsen J, Gerds TA, Seidelin JB, et al. Diagnosis of ulcerative colitis before onset of inflammation by multivariate modeling of genome-wide gene expression data. Inflamm Bowel Dis. 2009;15:1032–8. doi: 10.1002/ibd.20879. [DOI] [PubMed] [Google Scholar]
- 20.Planell N, Lozano JJ, Mora-Buch R, et al. Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut. 2012;62:967–76. doi: 10.1136/gutjnl-2012-303333. [DOI] [PubMed] [Google Scholar]
- 21.Kugel S, Mostoslavsky R. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem Sci. 2014;39:72–81. doi: 10.1016/j.tibs.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salzman NH. Paneth cell defensins and the regulation of the microbiome: detente at mucosal surfaces. Gut Microbes. 2010;1:401–6. doi: 10.4161/gmic.1.6.14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–6. doi: 10.1038/nature12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alenghat T, Osborne LC, Saenz SA, et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature. 2013;504:153–7. doi: 10.1038/nature12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ott SJ, Musfeldt M, Wenderoth DF, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–93. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borruel N, Carol M, Casellas F, et al. Increased mucosal tumour necrosis factor alpha production in Crohn's disease can be downregulated ex vivo by probiotic bacteria. Gut. 2002;51:659–64. doi: 10.1136/gut.51.5.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Llopis M, Antolin M, Carol M, et al. Lactobacillus casei downregulates commensals' inflammatory signals in Crohn's disease mucosa. Inflamm Bowel Dis. 2009;15:275–83. doi: 10.1002/ibd.20736. [DOI] [PubMed] [Google Scholar]
- 28.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356–68. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 29.Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29:625–51. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Ridlon JM, Kang DJ, Hylemon PB, et al. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30:332–8. doi: 10.1097/MOG.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noh DO, Gilliland SE. Influence of bile on cellular integrity and beta-galactosidase activity of Lactobacillus acidophilus. J Dairy Sci. 1993;76:1253–9. doi: 10.3168/jds.S0022-0302(93)77454-8. [DOI] [PubMed] [Google Scholar]
- 32.Jacobsen CN, Rosenfeldt Nielsen V, Hayford AE, et al. Screening of probiotic activities of forty-seven strains of Lactobacillus spp. by in vitro techniques and evaluation of the colonization ability of five selected strains in humans. Appl Environ Microbiol. 1999;65:4949–56. doi: 10.1128/aem.65.11.4949-4956.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jolley CD, Dietschy JM, Turley SD. Induction of bile acid synthesis by cholesterol and cholestyramine feeding is unimpaired in mice deficient in apolipoprotein AI. Hepatology. 2000;32:1309–16. doi: 10.1053/jhep.2000.19811. [DOI] [PubMed] [Google Scholar]
- 34.Dalal SR, Chang EB. The microbial basis of inflammatory bowel diseases. J Clin Invest. 2014;124:4190–6. doi: 10.1172/JCI72330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyoshi J, Chang EB. The gut microbiota and inflammatory bowel diseases. Transl Res. 2016 doi: 10.1016/j.trsl.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flint HJ, Scott KP, Louis P, et al. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9:577–89. doi: 10.1038/nrgastro.2012.156. [DOI] [PubMed] [Google Scholar]
- 37.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 38.Larrosa M, Yanez-Gascon MJ, Selma MV, et al. Effect of a low dose of dietary resveratrol on colon microbiota, inflammation and tissue damage in a DSS-induced colitis rat model. J Agric Food Chem. 2009;57:2211–20. doi: 10.1021/jf803638d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.