

Abstract
Purpose
Evaluation of the clinician’s role in optimal interpretation of clinical exome sequencing (ES) results.
Methods
Retrospective chart review of the first 155 patients who underwent clinical ES in our Exome Clinic and direct interaction with the ordering geneticist to evaluate the process of interpretation of results.
Results
The most common primary indication was neurodevelopmental problems (~66%), followed by multiple congenital anomalies (~10%). The overall diagnostic yield was 36% based on sequencing data. After assessment by the medical geneticist, incorporation of detailed phenotypic and molecular data, and utilization of additional diagnostic modalities, the final diagnostic yield was increased to 43%. Seven patients of our cohort were included in initial case series that described novel genetic syndromes, and 23% of patients were involved in subsequent research studies directly related to their results or involved in efforts to move beyond clinical ES for diagnosis. The clinical management was directly altered due to the ES findings in 12% of definitively diagnosed cases.
Conclusions
Our results emphasize the usefulness of ES, demonstrate the significant role of the medical geneticist in the diagnostic process of patients undergoing ES, and illustrate the benefits of post-analytical diagnostic work-up in solving the “diagnostic odyssey.”
Keywords: Exome Clinic, Medical Geneticist, Genetic Counseling, Diagnostic Yield, Exome Sequencing
INTRODUCTION
Clinical exome sequencing (ES) has revolutionized the diagnostic work-up in patients with genetic disease and has changed the diagnostic process in medical genetics practice1. The increasing utilization of ES has rapidly identified new genetic syndromes and contributed to solving many diagnostic odysseys2. Reports of the yield of exome sequencing through diagnostic laboratories have ranged from 25% to 30%3–5. Trio sequencing and focusing on specific disease subgroups can raise the diagnostic rate5,6. Many (23–30%) of these diagnosed patients were found to have mutations in genes that had been reported in association with the respective phenotype within the prior 2 to 3 years3,5.
Exome sequencing has provided insights into the genetic and phenotypic heterogeneity (e.g., atypical and milder presentations) of Mendelian disorders, and highlighted the importance of de novo mutations and “blended phenotypes” (co-existing diagnoses that combine the clinical features of each) in rare genetic disorders3–5. The application of this unbiased whole genome technology has led to shifting of the diagnostic skills of the medical geneticist from focusing on detailed phenotypic characterization to identify the genetic etiology to “next-generation phenotyping”: the interpretation and validation of molecular test results in clinical practice by analyzing observed clinical features7.
To date, there have only been a few attempts to study the role played by the medical geneticist in interpretation of results as part of the diagnostic process of ES, the concordance rate between the laboratory exome results and the geneticist’s interpretation, and the ability of ES to alter a patient’s or family’s medical management. Duke recently reported that the medical geneticists and the laboratories were 90% concordant in their interpretation of the exome results, and discordance occurred when the medical geneticist reconsidered additional clinical information and/or additional laboratory tests and genotyping of family members8. Another study showed that establishing a diagnosis through ES can lead to discontinuation of additional planned studies, screening patients for additional manifestations, altering management, identification of disease in other at risk family members, and reproductive planning9. The potential cost-effectiveness of ES has also been evaluated by calculating the cost of previous diagnostic workups, concluding that in some cases it may be most cost-effective to perform ES as a first test10.
In this study, we present our experience with the “Exome Clinic” with special emphasis on the diagnostic course after ES has been completed by the laboratory. We evaluate the role of the medical geneticist in interpretation of results, auxiliary studies performed to determine pathogenicity of genetic variants, follow up clinical tests, and post-exome enrollment in research studies. We discuss the diagnostic yield of ES in our cohort as a function of different phenotypic features. The utility of exome reanalysis 1–2 years after the original report is also presented. Finally, we recorded details of the social and financial implications of our exome results, such as determinations of misattributed paternity and the patient’s out-of-pocket cost.
MATERIALS AND METHODS
Chart Review and Clinical Evaluation
The Washington University School of Medicine Institutional Review Board approved this study. Clinical data were obtained by retrospective chart review and interview with the ordering medical geneticists and genetic counselors (Supplementary Material 1).
ES Laboratory Results
Exomes for 155 probands were ordered between March 2012 and January 2015. Exomes were performed in 3 different laboratories: 127 were analyzed through GeneDx (Gaithersburg, Maryland), 20 through Ambry Genetics (Aliso Viejo, California) and 8 through Baylor Genetics (Houston, Texas). Laboratories reported genetic variants as pathogenic, likely pathogenic, or variants of uncertain significance (VUS) but did not report benign or likely benign variants. We refer to this classification as variant-level assertion. GeneDx also classified the variants in relation to the patient’s phenotype as either definitively or possibly related and reported potential candidate genes for new genetic syndromes, which had not previously been associated with a human phenotype. Ambry Genetics classified variants as either likely positive, which we interpreted as possible, or positive, which we considered as definitively associated with the phenotype. Baylor Genetics classified the variants under “disease genes related to clinical phenotype” as either “deleterious” or “VUS.” We considered “deleterious” and “VUS” as definitive and possible, respectively. All three laboratories also reported incidental variants. Definitions of these terms were adapted from Retterer et al. 20156. We refer to these definitive, possible, candidate, and incidental classifications as case-level assertion, which is a synthesis of all the molecular data in a single subject specifying whether the test results provide a molecular diagnosis, according to the testing laboratory.
Clinical Assessment of ES Findings
Results of ES were discussed individually with the ordering medical geneticist and exome findings were confirmed or reclassified as needed as definitively, likely, possibly, or unlikely causative of the patient’s symptoms based on the molecular data (variant and case-level classifications) and the geneticist’s clinical assessment (Supplementary Material 1). We refer to this classification as clinical-level assertion. This clinical impression was then categorized as concordant or discordant with the laboratory’s case-level assertion to allow us to analyze how the geneticist’s interpretation influenced the final diagnosis (Supplementary Material 1). The statistical tools used for data analysis are presented in Supplementary Material 1.
RESULTS
Characteristics of the Cohort
Detailed descriptions of the clinical characteristics and molecular findings of the patients are documented in Supplementary Table 1. Demographic and phenotypic characteristics of our cohort are recorded in Table 1 and Supplementary Material 1. Sequencing cost for Medicaid patients was not covered by their insurance plans, and was either paid for by philanthropic support or absorbed by the hospital that sent the testing. Out-of-pocket costs to families with private insurance and for whom ES was sent as outpatients were available for 82 cases (Figure 1A). 54 of these cases had an out-of-pocket cost of $0, and the average cost was $386.31 with a maximum cost of $4,012.
Table 1.
Demographic Details of Cohort.
| Gender | |
| Male | 87 (56%) |
| Female | 68 (44%) |
| Ethnicity | |
| Caucasian | 130 (84%) |
| Mixed | 14 (9%) |
| African-American | 8 (5%) |
| Hispanic | 3 (2%) |
| Patient location | |
| Outpatient | 133 (86%) |
| Inpatient | 22 (14%) |
| Insurance (133 cases) | |
| Private | 90 (68%) |
| Medicaid | 43 (32%) |
| Dysmorphism (154 cases) | |
| Yes | 73 (47%) |
| Mild | 17 (11%) |
| No | 64 (42%) |
| OFC | |
| Normal | 93 (61%) |
| < −1.88 SD | 42 (28%) |
| > +1.88 SD | 17 (11%) |
| Height | |
| Normal | 99 (64%) |
| < 5th centile | 50 (32%) |
| > 95th centile | 6 (4%) |
| Weight | |
| Normal | 106 (68%) |
| < 5th centile | 36 (23%) |
| > 95th centile | 13 (8%) |
| Consanguinity | 6 (3.9%) |
| Average age at ES (range) | 6 years (3 days–33 years) |
| Average turnaround time in months (range) | 4.7 (1.3–7.9) |
Figure 1. Cost and phenotypic characterization of the cohort.
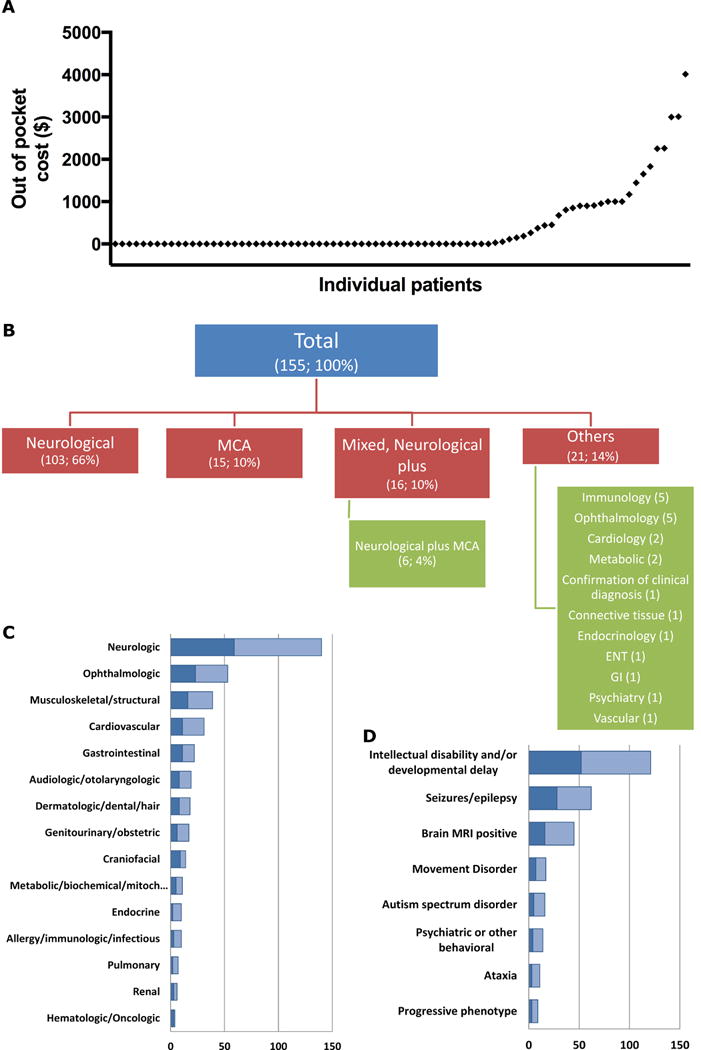
A) Scatter plot of the out-of-pocket cost in ascending order. B) Each case was assigned a phenotype-based, single primary indication for performing ES. The number and percentage of cases are shown in parenthesis. MCA: Multiple congenital anomalies. C) Each phenotypic feature of the probands was assigned to an organ system, and the total count of cases is displayed. D) The frequency and distribution of the neurodevelopmental phenotypes in the cohort. The darker portion of the bar in C and D indicates the proportion of cases that achieved a definitive diagnosis.
The average age at which symptoms in patients began was 11 months, with a median of 7 weeks, ranging from birth to 22 years. Of note, 63 patients (41%) had onset of symptoms at birth. Patients were first seen by a medical geneticist at an average age of 3 years, with a median of 14 months and a range from birth to 31 years old.
The primary indications for ES, the most common affected organ systems, and the most common neurodevelopmental findings are presented in Figure 1B, 1C, and 1D, respectively. The average number of organ systems affected in our cohort was 2.6 (median 2 and range 1 to 7 out of 15 possible organ systems). The average number of services (other than genetics) involved in the care of the patients in our cohort was 3.3 (median 3 and range 0 to 10 out of 19 possible services).
Variant Classification and Interpretation
The diagnostic laboratory reported 237 genetic variants, with an average of 1.5 variants reported per patient and a range from 0 to 6. The distribution of genetic variants based on variant-level assertion was as follows: 79 pathogenic, 37 likely pathogenic, and 107 VUS as well as 14 incidental findings (Figure S2, Tables S1, S2) that were classified by the laboratory as known pathogenic (12) or expected pathogenic (2). Among the 155 cases, 56 cases (36%) have definitive diagnosis based on case-level assertion by the laboratory, 60 cases were reported as possible, 10 cases as candidate, and 29 cases as negative (Figures 2A, S1, Tables S3). Due to the presence of autosomal recessive (AR) conditions and blended phenotypes among the 56 definitive cases, the number of variants was 71. Definitive diagnoses in 4 genes were identified in more than one unrelated case: ARID1B (2), GABRB2 (3), NGLY1 (2), and PTPN11 (2). Eleven cases had mitochondrial genome sequencing completed as part of the ES order but none of these yielded abnormal results. Misattributed or non-paternity was found in two families as a result of ES testing.
Figure 2. Characterization of case-level and clinical-level assertions.
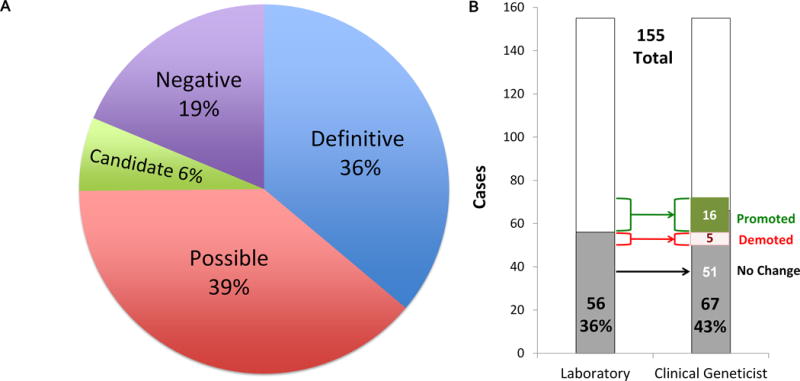
A) The relative percentages of each case-level classification as reported by the testing laboratory. B) The diagnostic rates according to case-level and clinical-level assertions are shown as the proportion of cases, in gray. The change in classification of cases is indicated, with 16 cases promoted and 5 demoted.
Based on the assessment of the ordering medical geneticist, the final diagnosis was changed in 21 subjects (14%) (Figures 2B,S1, S2, Tables S1, S2, S3, 2, 3). The diagnosis in 16 subjects was promoted such that the clinical geneticist determined that the variant was more definitively related to the phenotype, and in 5 subjects it was demoted. Consequently, there was a net gain of 11 additional definitive diagnoses, for a total of 67 cases (43%) definitively diagnosed (Table 2). There were multiple reasons for changing the case-level classification (Table 3). First, the clinical geneticist has direct and detailed knowledge of the patient’s phenotype and the opportunity to order follow up studies including biochemical and radiological studies, segregation analysis in relatives, and/or single-gene re-sequencing or deletion/duplication studies to search for a mutation in the second allele. Furthermore, there were variants in candidate genes that were promoted because of subsequent publication of new syndromes, either in other similarly affected patients or contribution of these patients to syndrome discovery themselves11–16. Thirty-two (48%) out of the 67 definitive cases had mutations in genes described in 2011 or later. This includes 7 (10%) being described as new genetic syndromes12–16 (WES038, WES052, WES057, WES062, WES079, WES105, WES121), 3 of which are in the process of being published. Five cases (7.5%) had definitive variants in two genes resulting in “blended phenotypes” (WES02817, WES030, WES060, WES070, WES128). Reanalysis of the exome data was performed in 14 cases by the molecular laboratory, usually 12 to 18 months after the initial report was generated. In 7 cases the reanalysis resulted in no change, in 4 cases it resulted in a new definitive diagnosis (WES013, WES019, WES039, WES13118) due to subsequently published new syndromes or functional analysis of variants, and in one case a previously reported variant was demoted (WES002). The remaining two cases (WES099, WES112) involved efforts by the laboratory to identify candidate disease genes for which there have not yet been human phenotypes associated.
Table 2.
Description of Definitively Diagnosed Cases.
| Case Number |
Gene | Variant(s) | De Novo | Inherited | Unknown Inheritance |
Sequencing Company Classification(s) |
Medical Geneticist’s Interpretation |
Testing Laboratorya |
Disease Mode of Inheritanceb |
Phenotype MIM Number |
Final clinical diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WES152 | ADAR | c.577C>G, p.P193A; c.3020-3C>G, IVS11-3C>G | X, X | Definitive | Concordant | G | AR | 615010 | Aicardi-Goutieres syndrome | ||
| WES111 | AHDC1 | c.2373_2374delTG, p.C791WfsX57 | X | Definitive | Concordant | G | AD | 615829 | Xia-Gibbs syndrome | ||
| WES056 | ANKRD11 | c.6159_6162delGGCT, p.A2054PfsX32 | X | Definitive | Concordant | G | AD | 148050 | KBG syndrome | ||
| WES066 | ARID1B | c.3644delC, p.1215QfsX9 | X | Definitive | Concordant | G | AD | 135900 | Coffin-Siris syndrome 1 | ||
| WES030 | ARID1B / FGFR3 | c.2281G>A, p.G761S / c.445+(2_5)delTAGG, IVS4+(2_5)delTAGG | X (FGFR3) | X (ARID1B) | Possible / Possible | Promoted / Promoted | G | AD / AD | 135900 / 149730 | Coffin-Siris syndrome 1/ LADD syndrome | |
| WES126 | ATM | c.3993+1G>A, IVS26+1G>A; c.5763-1050A>G, IVS39-1050A>G | X, X | Possible | Promoted | G | AR | 208900 | Ataxia telangectasia | ||
| WES028 | ATP2B3 / LAMA1 | c.1445G>A, p.R482H / c.6074C>T, p.T2025M; c.1741C>T, p.R2381C | X / X, X | Possible / Possible | Promoted / Promoted | G | XL / AR | 302500 / 615960 | ATP2B3-related disorder / LAMA1-related disorder | ||
| WES076 | CHD7 | c.8279delA, p.N2760IfsX39 | X | Definitive | Concordant | G | AD | 214800 | CHARGE syndrome | ||
| WES086 | COL1A1 | c.652G>T, p.G218C | X | Definitive | Concordant | G | AD | 166200 | Osteogenesis imperfecta type 1 | ||
| WES085 | COL4A1 | c.2291G>A, p.G764D | X | Definitive | Concordant | G | AD | 607595 | COL4A1-related disorders | ||
| WES121 | COQ4 | c.245T>A, p.L82Q; c.473G>A, p.R158Q | X, X | Candidate | Promoted | G | AR | 616276 | COQ4-related disorder | ||
| WES038 | CTBP1 | c.991C>T, p.R331W | X | Candidate | Promoted | G | AD | None yet | CTBP1-related disorder | ||
| WES050 | CYB5R3 | c.250C>T, p.R84X | Xc | Possible | Promoted | G | AR | 250800 | Methemoglobinemia type II | ||
| WES109 | CYP11A1 | c.1078C>T, p.R360W | Xc | Definitive | Concordant | G | AR | 613743 | CYP11A1-related adrenal insufficiency with sex reversal | ||
| WES131 | DNMT3A | c.2645G>A, p.R882H | X | Possible | Promoted | G | AD | 615879 | Tatton-Brown-Rahman syndrome | ||
| WES128 | DYRK1A / STK11 | c.889_893dupAGGTT, p.F298LfsX40; c.665-9_665-5delCTCTT, IVS5-9_IVS5-5delCTCTT | X, X | Definitive | Concordant | G | AD / AD | 614104 / 175200 | DYRK1A-related intellectual disability / Peutz-Jeghers syndrome | ||
| WES047 | EIF2B5 | c.318A>T, p.L106F; c.799C>T, p.Q267X | X | X | Definitive | Concordant | G | AR | 603896 | Vanishing white matter disease | |
| WES117 | FGD1 | c.563_570delTGCCTGCC, p.L188RfsX26 | X | Definitive | Concordant | G | XL | 305400 | Aarskog syndrome | ||
| WES134 | FGFR1 | c.2152C>G, p.R718G | X | Definitive | Concordant | G | AD | 615465 | Hartsfield syndrome | ||
| WES020 | FHL1 | c.799delC, p.H267Tfs*23 | X | Definitive | Concordant | A | XL | 300696 | Emery-Dreifuss muscular dystophy type 6 | ||
| WES104 | FLG | c.1501C>T, p.R501X | X | Definitive | Concordant | G | AD | 146700 | Ichthyosis vulgaris | ||
| WES051 | FOXG1 | c.700T>C, p.S234P | X | Definitive | Concordant | G | AD | 613454 | FOXG1-related disorder, Rett-like | ||
| WES049 | GABRA1 | c.643C>G, p.L215V | X | Definitive | Concordant | G | AD | 615744 | GABRA1-related disorder | ||
| WES052 | GABRB2 | c.909G>T, p.K303N | X | Candidate | Promoted | G | AD | None yet | GABRB2-related disorder | ||
| WES062 | GABRB2 | c.863T>G, p.I288S | X | Definitive | Concordant | G | AD | None yet | GABRB2-related disorder | ||
| WES105 | GABRB2 | c.845T>C, p.V282A | X | Definitive | Concordant | G | AD | None yet | GABRB2-related disorder | ||
| WES070 | GALNS / SUFU | c.1485C>G, p.N495K; c.539T>C, p.V180A / c.794_808del15, p.N265_V269del | X / X | X | Possible / Possible | Promoted / Promoted | B | AR / AD | 253000 / 109400 | Morquio syndrome / Gorlin syndrome | |
| WES140 | GNAO1 | c.833_835delAGA, p.K278del | X | Definitive | Concordant | G | AD | 615473 | GNAO1-related disorder (Early infantile epileptic encephalopathy 17) | ||
| WES019 | GRIN2B | c.1916C>T, p.A639V | X | Possible | Promoted | A | AD | 616139 | GRIN2B-related disorder (Early infantile epileptic encephalopathy 27) | ||
| WES039 | HNRNPK | c.1008+1G>A, IVS12+1G>A | X | Definitive | Concordant | G | AD | 616580 | Au-Kline syndrome | ||
| WES120 | KAT6B | c.2184T>G, p.Y728X | X | Definitive | Concordant | G | AD | 603736 | KAT6B-related disorder | ||
| WES095 | KCNB1 | c.629C>T, p.T210M | X | Definitive | Concordant | G | AD | 616056 | KCNB1-related disorder (Early infantile epileptic encephalopathy 26) | ||
| WES071 | KCNQ2 | c.740C>T, p.S247L | X | Definitive | Concordant | G | AD | 613720 | KCNQ2-related disorder (Early infantile epileptic encephalopathy 7) | ||
| WES107 | KCNT1 | c.1193G>A, p.R398Q | X | Definitive | Concordant | G | AD | 614959 | KCNT1-related disorder (Early infantile epileptic encephalopathy 14) | ||
| WES114 | KMT2A | c.7419delT, p.P2474LfsX35 | X | Definitive | Concordant | G | AD | 605130 | Wiedemann-Steiner syndrome | ||
| WES029 | KMT2D | c.12039_12046delAGCCCTGG, p.A4014SfsX23 | X | Definitive | Concordant | G | AD | 147920 | Kabuki syndrome | ||
| WES153 | NBAS | c.688dupT, p.S230QfsX4; c.2524G>T, p.V842F | X, X | Definitive | Concordant | G | AR | 616483 | Infantile liver failure syndrome 2 | ||
| WES037 | NGLY1 | c.347C>G, p.S116X; c.881+5G>T, IVS5+5G>T | X, X | Definitive | Concordant | G | AR | 615273 | NGLY1-related congenital disorder of deglycosylation | ||
| WES096 | NGLY1 | c.953T>C, p.L318P; c.1169G>C, p.R390P | X, X | Definitive | Concordant | G | AR | 615273 | NGLY1-related congenital disorder of deglycosylation | ||
| WES007 | OCA2 | c.1327G>A, p.V443I | X | Definitive | Concordant | B | AR | 203200 | Oculocutaneous Albinism, type II | ||
| WES155 | OPHN1 | c.155-2A>C, IVS2-2A>C | X | Definitive | Concordant | G | XL | 300486 | OPHN1-related disorder | ||
| WES065 | PANK2 | c.1561G>A, p.G521R; c.1264T>C, p.C422R | X | X | Definitive | Concordant | G | AR | 234200 | PANK2-related disorder (Neurodegeneration with Brain Iron Accumulation) | |
| WES129 | PGAP1 | c.1546_1549delGTCA, p.V516KfsX4; c.1077T>G, p.Y359X | X, X | Possible | Promoted | G | AR | 615802 | PGAP-related disorder | ||
| WES025 | PHF6 | c.915_916delTGinsAA, p.C305X | X | Definitive | Concordant | A | XL | 301900 | Borjeson-Forssman-Lehmann syndrome | ||
| WES059 | PHGDH | c.1538C>T, p.S513F; c.1078+1G>A, IVS9+1G>A | X, X | Definitive | Concordant | G | AR | 601815 | Phosphoglycerate dehydrogenase deficiency | ||
| WES089 | PIK3CD | c.3061G>A, p.E1021K | X | Definitive | Concordant | G | AD | 615513 | Primary immunodeficiency 14 | ||
| WES040 | PKHD1 | c.930delC, p.T311LfsX8; c.5134G>A, p.G1712R | X, X | Definitive | Concordant | G | AR | 263200 | Autosomal recessive polycystic kidney disease | ||
| WES087 | PLA2G6 | c.1613G>A, p.R538H; c.319dupC, p.L107PfsX10 | X, X | Definitive | Concordant | G | AR | 256600 | Infantile neuronal axonal dystrophy type 1 | ||
| WES148 | POLR3B | c.2570+5G>A, IVS22+5G>A; c.3317T>C, p.I1106T | X, X | Possible | Promoted | G | AR | 614381 | Hypomyelinating leukodystrophy type 8 | ||
| WES077 | PTPN11 | c.922A>G, p.N308D | X | Definitive | Concordant | G | AD | 163950 | Noonan syndrome | ||
| WES154 | PTPN11 | c.836A>G, p.Y279C | X | Definitive | Concordant | G | AD | 151100 | Noonan syndrome with multiple lentigines | ||
| WES074 | SCN1A | c.677C>T, p.T226M | X | Definitive | Concordant | G | AD | 604403 | SCN1A-related epilepsy disorder | ||
| WES013 | SCYL1 | c.1039C>T, p.Q347* | Xc | Possible | Promoted | A | AR | 616719 | SCYL1-related disorder | ||
| WES113 | SLC16A2 | c.623_624delGCinsAA, p.G208E | X | Definitive | Concordant | G | XL | 300523 | Allan-Herndon-Dudley syndrome | ||
| WES122 | SNX27 | c.510C>G, p.Y170X; c.1295G>A, p.C432Y | X, X | Candidate | Promoted | G | AR | None yet | SNX27-related disorder | ||
| WES009 | STXBP1 | c.875G>T, p.R292L | X | Definitive | Concordant | A | AD | 612164 | STXBP1-related disorder (Early infantile epileptic encephalopathy 4) | ||
| WES147 | TBC1D24 | c.1008delT, p.H336QfsX12; c.680G>T, p.R227L | X, X | Definitive | Concordant | G | AR | 220500 | DOORS syndrome (deafness, onychodystrophy, osteodystrophy, mental retardation and seizures) | ||
| WES082 | TBX1 | c.1392_1403del12, p.A473_A476del | X | Definitive | Concordant | G | AD | 188400 | TBX-1-related DiGeorge syndrome | ||
| WES118 | TBX3 | c.1090G>T, p.E364X | X | Definitive | Concordant | G | AD | 181450 | Ulnar-mammary syndrome | ||
| WES060 | TCF12 / EFTUD2 | c.1319delA, p.N440TfsX79 / c.270A>G, p.T90T | X / X | Definitive / Definitive | Concordant / Concordant | G | AD / AD | 615314 / 610536 | Craniosnostosis type 3 / Mandibulofacial dysostosis Guion-Almeida type | ||
| WES079 | TELO2 | c.1100G>T, p.C367F; c.2296G>A, p.V766M | X, X | Candidate | Promoted | G | AR | 616954 | TELO2-related disorder | ||
| WES132 | TNNT2 | c.833G>A, p.R278H | X | Definitive | Concordant | G | AD | 115195 | TNNT2-related disorder | ||
| WES017 | TRMU | c.718C>T, p.R240X | Xc | Definitive | Concordant | G | AR | 613070 | Combined Respiratory Chain Deficiency (Infantile Liver Failure) | ||
| WES130 | TUBA1A | c.1168C>T, p.R390C | X | Definitive | Concordant | G | AD | 611603 | Lissencephaly type 3 | ||
| WES084 | UBE3A | c.2563_2566dupCTTA, p.K856TfsX2 | X | Definitive | Concordant | G | AD | 105830 | UBE3A-related disorder | ||
| WES015 | UBE3B | c.2990G>C, p.R997P | Xc | Possible | Promoted | A | AR | 244450 | Blepharophimosis-Ptosis-Intellectual disability syndrome (Kaufman oculocerebrofacial syndrome) | ||
| WES057 | WAC | c.1721G>A, p.W574X | X | Definitive | Concordant | G | AD | 616708 | DeSanto-Shinawi syndrome |
G: GeneDx, B: Baylor Genetics, C: Ambry Genetics.
AR: autosomal recessive; AD: autosomal dominant; XL: X-linked.
Homozygous variant.
Table 3.
Reasons for Changing the Diagnosis.
| Case Number | Gene(s) | Variant(s) | Testing Laboratory | Laboratory Case-Level Classification | Clinical Geneticist Clinical-Level Classification | Reason |
|---|---|---|---|---|---|---|
| Cases That Were Demoted by the Clinical Geneticist | ||||||
| WES002 | HEXA / VPS13B | c.1073+1G>AIVS9+1G>A / c.11256_11290+10del, IVS58+10delC | B | Definitive | Unlikely | Hexosaminidase A activity was normal and clinical phenotype is not consistent with Tay Sachs / Lack of a second mutation in VSP13B and phenotype is not consistent with Cohen syndrome |
| WES003 | PANK2 | c.1561G>A, p.G521R | B | Definitive | Unlikely | Brain MRI and clinical course are not consistent with PANK2-related phenotype |
| WES069 | UPB1 / GAMT | c.917-1G>A, IVS8-1G>A / c.327G>A, p.K109K | B | Definitive | Unlikely | Negative biochemical studies for creatine deficiency syndromes and pyrimidine metabolism defects |
| WES090 | DPYD | c.1905+1G>A, IVS14+1G>A; c.1679T>G, p.I560S | G | Definitive | Possible | Biochemical studies were consistent but clinical phenotype did not fit with the phenotype of dihydropyrimidine dehydrogenase deficiency |
| WES091 | DMD | Deletion of exons 45-51 | G | Definitive | Possible | The neurological and cardiac phenotypes, normal muscle histopathological findings, and normal CK are not consistent with the expected clinical findings of this in-frame DMD deletion |
| Cases That Were Promoted by the Clinical Geneticist | ||||||
| WES013 | SCYL1 | c.1039C>T, p.Q347* | A | Possible | Definitive | Clinical phenotype of the patient matched a newly described syndrome 2 years after initial analysis |
| WES015 | UBE3B | c.2990G>C, p.R997P | A | Possible | Definitive | Facial features and clinical phenotype of the patient matched published syndrome |
| WES019 | GRIN2B | c.1916C>T, p.A639V | A | Possible | Definitive | Clinical phenotype of the patient matched neurological findings reported in patients with GRIN2B mutations |
| WES028 | ATP2B3 / LAMA1 | c.1445G>A, p.R482H / c.6074C>T, p.T2025M; c.1741C>T, p.R2381C | G | Possible | Definitive | In vitro functional studies showed impaired PMCA3 pump function and data supported a synergistic effect with LAMA1 mutations17 |
| WES030 | ARID1B / FGFR3 | c.2281G>A, p.G761S / c.445+(2_5)delTAGG, IVS4+(2_5)delTAGG | G | Possible | Definitive | The blended phenotype in the patient matched published syndromes related to these genes |
| WES038 | CTBP1 | c.991C>T, p.R331W | G | Candidate | Definitive | The patient was one of 4 patients described with a new genetic syndrome15 |
| WES050 | CYB5R3 | c.250C>T, p.R84X | G | Possible | Definitive | Follow up measurement of NADH to cytochrome b5 activity and methemoglobin level in blood were consistent with CYB5R3 deficiency |
| WES052 | GABRB2 | c.909G>T, p.K303N | G | Candidate | Definitive | Subsequent publication of new syndrome in other patients12; the patient is part of an ongoing study on a series of patients to define the phenotype |
| WES070 | GALNS / SUFU | c.1485C>G, p.N495K; c.539T>C, p.V180A / c.794_808del15, p.N265_V269del | G | Possible | Definitive | Clinical phenotype of the patient matched the two published syndromes |
| WES079 | TELO2 | c.1100G>T, p.C367F; c.2296G>A, p.V766M | G | Candidate | Definitive | The patient was 1 of 6 patients described with a new genetic syndrome16 |
| WES121 | COQ4 | c.245T>A, p.L82Q; c.473G>A, p.R158Q | G | Candidate | Definitive | The patient was 1 of 4 patients described with a new CoQ10 deficiency syndrome13 |
| WES122 | SNX27 | c.510C>G, p.Y170X; c.1295G>A, p.C432Y | G | Candidate | Definitive | Brain MRI and neurological phenotype were consistent with newly described syndrome11 |
| WES126 | ATM | c.3993+1G>A, IVS26+1G>A; c.5763-1050A>G, IVS39-1050A>G | G | Possible | Definitive | Re-sequencing of ATM detected a second mutation; elevated AFP and neurological findings matched the diagnosis |
| WES129 | PGAP1 | c.1546_1549delGTCA, p.V516KfsX4; c.1077T>G, p.Y359X | G | Possible | Definitive | Clinical and neurological phenotype of the patient matched published syndrome |
| WES131 | DNMT3A | c.2645G>A, p.R882H | G | Possible | Definitive | Clinical phenotype of the patient was consistent with a newly described syndrome18 |
| WES148 | POLR3B | c.2570+5G>A, IVS22+5G>A; c.3317T>C, p.I1106T | G | Possible | Definitive | Brain MRI and clinical phenotype of the patient matched published syndrome |
G: GeneDx, B: Baylor Genetics, A: Ambry Genetics.
We then assessed the relationship between the diagnostic yield, as determined by the medical geneticist, and various demographic and phenotypic characteristics (Table S4). Our results indicated a higher diagnostic yield for females (47%), patients with a craniofacial anomaly (64%), and patients with an abnormal head circumference, specifically microcephaly (50%), but none of these effects were statistically significant. Caucasians had a statistically significant higher rate of diagnosis compared to all other racial groups (46% vs. 24%, p=0.04), which persisted after adjusting for craniofacial anomaly in the multivariable logistic regression model, demonstrating the disproportionately low diagnosis rate for non-Caucasians. The following additional categories were tested for effect on diagnostic rate and were found to be not significant: inpatient versus outpatient status, all other phenotypic categories, death, abnormal height or weight, dysmorphism, and positive family history.
The inheritance patterns in the 72 conditions (67 subjects; 5 with two conditions caused by variants in different genes) that were determined to be definitive are as follows: 42 (58%) autosomal dominant (AD), 24 (33%) AR, and 6 (8%) X-linked. Of the 89 variants that are associated with these 72 conditions, 34 (38%) were de novo, including one variant in each of two cases with AR conditions (Table 2). The average paternal age at delivery of the 42 patients with de novo mutations was 32 years with a median of 32 years and a range of 22 to 49 years. For the inherited variants, 25 were passed from the mother, 18 from the father, 4 from both (homozygous for recessive condition), and 8 had unknown inheritance due to at least one parent not being sequenced. We observed reduced penetrance of 5 variants that were associated with AD conditions and inherited from seemingly unaffected parents, although parental cardiac evaluations are pending in 2 of these cases.
In 9 cases, ES was sent prior to the implementation of the 2013 ACMG guidelines for reporting incidental findings19. Of the remaining 146 cases, 5 (3%) families opted out and 141 (97%) families elected to receive the findings. 14 patients (10%) had one incidental finding each. Incidental findings were found in the following genes from the ACMG-recommended list of 56 genes: BRCA2 (2), FBN1, LDLR (2), MYBPC3 (4), MYH7, RET, SCN5A, TTN (2) (Table S5). Although the laboratories’ reports indicate that these incidental variants are known pathogenic in 12 cases, only 5 of these 12 are uniformly classified as pathogenic in ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) and the remainder have conflicting interpretations of pathogenicity, with some submitters even identifying two of these variants as likely benign (Table S5). Follow up assessment or evaluation was done based on established guidelines and protocols for these cases and their carrier relatives (Table S5).
The Effect of Exome Results on Auxiliary Tests, Management and Research Studies
We examined whether the exome results affected subsequent diagnostic work-up or changed patient management. Additional diagnostic studies were performed in 84 subjects (54%), including molecular studies (proband or family members) in 37 (24%), imaging studies in 29 (19%), and biochemical and/or chemistry tests in 22 (14%). The distribution of the 84 cases based on clinical-level assertion was the following: 48 were definitive, 4 were likely, 8 were possible, 20 were unlikely, 1 was incidental only, and 3 had completely negative results, but had follow up genetic testing performed due to concerns regarding poor coverage of the exome data at particular genes of interest (Supplementary Material 1, Table S6). In 12 out of the 84 cases, these follow up studies were due to the discovery of an ACMG-designated incidental finding. An echocardiogram was performed in 19 (12%) probands or family members, 7 of which were due to incidental findings. In addition, cancer surveillance protocols were initiated in 7 probands or related family members due to variants found by ES, 2 of which were incidental. Three families used the ES information for prenatal or pre-implantation genetic diagnosis.
In 8 out of the 67 definitive cases (12%), clinical care was directly altered due to primary ES findings, as follows: 1) discontinuation of levothyroxine (WES113, SLC16A2); 2) cardiac ablation in an asymptomatic patient (WES118, TBX3) found to have Wolff-Parkinson-White syndrome on the EKG that was ordered based on ES results; 3) prophylactic thyroidectomy and Hirschprung’s diagnosis (WES018, RET); 4) neuropsychology evaluation because of known deficits associated with this condition, although not obviously present in this case, which showed ADHD and anxiety disorder and resulted in an atomoxetine prescription (WES057, WAC); 5) orthopedics referral of a patient (WES025, PHF6) with a condition known to cause musculoskeletal phenotypes which led to diagnosis and surgical repair of her scoliosis; 6) amantadine trial initiated for ataxia telangiectasia (WES126, ATM); 7) a trial of methylene blue and vitamin C in a patient (WES050, CYB5R3) with methemoglobinemia; and, lastly, 8) serine prescription for serine-responsive seizures (WES059, PHGDH). Thirty-six patients were enrolled in research studies related to their ES results. These include efforts to characterize the potential functional effect of a particular variant, as well as reanalysis of otherwise negative clinical exome data for research purposes.
DISCUSSION
Although there have been several studies reporting clinical ES results, most of these reports are from diagnostic laboratories and do not focus on the medical geneticists’ interpretation of the findings. The main purpose of this study was to evaluate the medical geneticist’s role in the optimal interpretation of the exome results, and how this might alter the final diagnostic yield. The overall definitive diagnosis rate of clinical ES in our cohort was 36% based on laboratory sequencing data but this increased to 43% after the integration of the molecular and phenotypic data by the medical geneticist as well as the incorporation of additional diagnostic modalities. Fifty-four percent of patients in our cohort underwent “post-analytical” auxiliary diagnostic studies, including biochemical analyses, imaging studies, complementary molecular tests (e.g., deletion and duplication analysis of a specific gene or Sanger sequencing of a gene with low exome coverage), and/or genotyping affected and unaffected family members for segregation analysis. Furthermore, each genetic variant was evaluated by a thorough literature review and searching databases such as ExAC and ClinVar. This extensive post-exome assessment by the clinician is time consuming and illustrates that ES results as reported by the molecular laboratory require clinical context. The laboratory identifies sequence changes and provides information about suspected pathogenicity, but the medical geneticist must compare the expected phenotype associated with the molecular finding to the patient’s phenotype to determine if they align, and whether the molecular finding may account for the patient’s clinical presentation. In 5 cases, we determined that the molecular finding was not consistent with the patient’s phenotype, and the genetic variant was considered to be either benign or not completely explanatory. In 16 other cases, the classification was promoted to a more definitive category and ultimately the final diagnosis was modified (Table 3). However, in other patients the final diagnosis is still uncertain and pathogenicity of the variants is difficult to establish due to lack of functional data, inability to perform segregation analysis, incomplete explanation of the phenotype by the variant, or candidate gene status. These limitations pose challenges to the clinician and demonstrate that receiving the exome results can be the beginning of a continuing exploration process rather than the end of the “diagnostic odyssey.”
As evidenced by large-scale research studies that use ES as a tool for discovery such as the Deciphering Developmental Disorders20, the rate of discovery of new genetic syndromes is rapidly increasing. Therefore, reanalysis of previously reported clinical ES data has the potential to increase the sensitivity of the test. In fact, 48% of definitive cases in our study had mutations in genes with associated syndromes described in 2011 or later. Subsequent reanalysis of the exome data, either at the request of the medical geneticist or at the prompting of internal reanalysis by the diagnostic laboratory, directly resulted in 7 additional definitive diagnoses than would have otherwise been obtained, illustrating the need to perform ongoing data mining for previously submitted cases with negative exome results.
The increased diagnostic yield in our cohort relative to previously reported clinical series3–6,8–10 can be in part attributed to the selection process we apply for subspecialty referrals for the Exome Clinic, including an ES-specific referral form (Supplementary Material 2) and review of the suitability of the case by a medical geneticist. It is also possible that there was a selection bias toward the most severely affected patients referred to a tertiary medical center, reflected by a relatively high number of organ systems, services involved, as well as highly skewed growth parameters and high rate of dysmorphism in the probands when the test was initially implemented in our institution. We cannot exclude the contribution of other factors such as a high trio rate (83%), different categories of indications, or differences in sample size.
This study has a number of important limitations. For example, ES was ordered through 3 different laboratories that used different terminology to classify the variants in relation to patient’s phenotype, which limits cross-case comparison. In addition, the laboratories’ data analysis processes changed over time as algorithms have improved and ACMG guidelines have been implemented. However, there were no statistically significant differences among the three different diagnostic laboratories regarding the number of cases with incidental findings, the proportion of cases that received a definitive diagnosis at the case-level, and whether the case-level classification was revised by the clinician (Supplementary Material 1). Another factor limiting the generalizability of our findings is that these patients were all part of a highly selected population that was evaluated at a tertiary medical center.
Our study shows that clinical ES is a powerful diagnostic tool especially for atypical and mild presentations of well-established genetic syndromes. For example, none of the patients who received diagnoses of CHARGE, Noonan, ataxia telangiectasia and LADD syndromes met clinical diagnostic criteria, but rather exhibited partial phenotypes. Furthermore, the discovery of five patients in our cohort having “blended phenotypes”, as similarly described in other cohorts3,21,22, should change our traditional diagnostic approach. ES is a valuable gene discovery tool as illustrated in 7 patients who were included in initial case series that described novel genetic syndromes. Other unexpected exome results were related to potential germline mosaicism in one case (WES057) and uniparental disomy in another case (WES050). This information about non-Mendelian modes of inheritance was very important for providing accurate recurrence risks in future pregnancies. ES also uncovered non-paternity in two cases, which required a consultation with our institutional ethics committee and ultimately led to altered strategies for pretest counseling regarding this complicated issue.
Incidental findings present in 9% of our cohort patients often resulted in additional interventions in both the probands and their carrier relatives. This number is higher than we would have expected by comparison to previous cohorts.3–5,23 However, based on the conflicting assertions in ClinVar (Table S5), it is clear that the performing laboratories over called incidental findings and the actual rate is 3.8% (6/14). These data illustrate the challenges in variant classification and the need for simple and consistent criteria for classification based on variant-specific databases and knowledge bases23. We speculate that this lack of uniformity may be due to changes in how variants are classified over time, especially after the release of the 2015 ACMG guidelines24. The role of the medical geneticist in following up these incidental results is as important as it is for following up primary results because subsequent monitoring, such as cancer screening and cardiac monitoring, can have life-saving consequences for the patients and their relatives. However, the conflicting interpretations of the data as presented here and the workup performed for patients with uncertain incidental findings (Table S5) illustrate the challenges the medical geneticist faces and reveal one of the significant drawbacks of ES related to false positive incidental findings, which could lead to substantial harmful consequences including performing unnecessary and potentially harmful tests and procedures, increased healthcare costs due to performing unnecessary follow-up evaluations, and causing anxiety among a percentage of patients undergoing ES23,25,26. These are important points that should be carefully considered prior to ordering ES and during pretesting counseling.
For many patients, ending a diagnostic odyssey limits additional expensive, time-consuming, and potentially invasive diagnostic procedures. It also allows precise determination of recurrence risk and prognosis. ES results were used by 3 families from our cohort for prenatal diagnosis testing. Although the discovery of a treatable condition can dramatically change the clinical outcome, the exome resulted in specific treatments in only a limited number of our patients. Nevertheless, clinical management was directly altered due to primary ES findings in 8 patients, which is 5.2% of all patients who underwent ES. It is also possible that careful clinical assessment for part of these cases would detect clinical findings that might ultimately change the management even without the molecular data.
The correlation of diagnostic yield in our cohort with various demographic and phenotypic characteristics showed a higher yield for Caucasians, females, patients with craniofacial anomalies, and patients with abnormal head circumference, but none of these reached statistical significance except for ethnic background (Supplementary Material 1, Table S4). It is important to note that patients from minority populations are under-represented in our cohort, suggesting a need for increased access to ES for individuals from these backgrounds. While the average out-of-pocket cost for ES was $386 per family and although we do not have detailed socioeconomic data for our cohort, we speculate that economic factors may play a role in this discrepancy. Publicly funded insurance plans do not routinely provide coverage for ES and families with high out-of-pocket costs sometimes self-selected not to pursue this testing. Compounding this situation, non-Caucasians achieved a significantly lower diagnostic rate of only 24%. This finding may be due in part to an underrepresentation of minority populations in variant databases, causing challenges in interpreting the clinical significance of variants found in these populations.
Taking into account the work involved in interpreting and following up both primary and incidental exome findings, the complex phenotype of patients referred for ES, as well as the constantly evolving nature of these results due to re-analysis and publication of new genetic syndromes, medical geneticists serve an essential role in this complex diagnostic process. This study shows that the partnership of the clinician with the molecular laboratory can increase the diagnostic yield by 7%. An accurate molecular diagnosis ends a diagnostic odyssey, allows for precise genetic counseling, and has the potential to change clinical management. It is also the launching point for the development of targeted pharmacologic therapies, which can hopefully translate these discoveries into efficacious novel treatments to achieve the promise of personalized genomic medicine.
Supplementary Material
Acknowledgments
D.B. is supported by the National Institute of Mental Health of the National Institutes of Health (NIH) under award number T32MH014677 and the National Heart, Lung, and Blood Institute of the NIH under award number K12HL120002. M.V. is partially supported by the Eunice Kennedy Shriver National Institute Of Child Health & Human Development of the NIH under award number U54HD087011, the Intellectual and Developmental Disabilities Research Center at Washington University.
References
- 1.Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. The New England journal of medicine. 2014;370(25):2418–2425. doi: 10.1056/NEJMra1312543. [DOI] [PubMed] [Google Scholar]
- 2.Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nature reviews Genetics. 2011;12(11):745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 3.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. Jama. 2014;312(18):1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. Jama. 2014;312(18):1880–1887. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farwell KD, Shahmirzadi L, El-Khechen D, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(7):578–586. doi: 10.1038/gim.2014.154. [DOI] [PubMed] [Google Scholar]
- 6.Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genetics in medicine : official journal of the American College of Medical Genetics. 2015 doi: 10.1038/gim.2015.148. [DOI] [PubMed] [Google Scholar]
- 7.Hennekam RC, Biesecker LG. Next-generation sequencing demands next-generation phenotyping. Hum Mutat. 2012;33(5):884–886. doi: 10.1002/humu.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shashi V, McConkie-Rosell A, Schoch K, et al. Practical considerations in the clinical application of whole-exome sequencing. Clinical genetics. 2016;89(2):173–181. doi: 10.1111/cge.12569. [DOI] [PubMed] [Google Scholar]
- 9.Iglesias A, Anyane-Yeboa K, Wynn J, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genetics in medicine : official journal of the American College of Medical Genetics. 2014;16(12):922–931. doi: 10.1038/gim.2014.58. [DOI] [PubMed] [Google Scholar]
- 10.Valencia CA, Husami A, Holle J, et al. Clinical Impact and Cost-Effectiveness of Whole Exome Sequencing as a Diagnostic Tool: A Pediatric Center’s Experience. Frontiers in pediatrics. 2015;3:67. doi: 10.3389/fped.2015.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damseh N, Danson CM, Al-Ashhab M, et al. A defect in the retromer accessory protein, SNX27, manifests by infantile myoclonic epilepsy and neurodegeneration. Neurogenetics. 2015;16(3):215–221. doi: 10.1007/s10048-015-0446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava S, Cohen J, Pevsner J, et al. A novel variant in GABRB2 associated with intellectual disability and epilepsy. American journal of medical genetics Part A. 2014;164A(11):2914–2921. doi: 10.1002/ajmg.a.36714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung WK, Martin K, Jalas C, et al. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. Journal of medical genetics. 2015;52(9):627–635. doi: 10.1136/jmedgenet-2015-103140. [DOI] [PubMed] [Google Scholar]
- 14.DeSanto C, D’Aco K, Araujo GC, et al. WAC loss-of-function mutations cause a recognisable syndrome characterised by dysmorphic features, developmental delay and hypotonia and recapitulate 10p11.23 microdeletion syndrome. Journal of medical genetics. 2015;52(11):754–761. doi: 10.1136/jmedgenet-2015-103069. [DOI] [PubMed] [Google Scholar]
- 15.Beck DB, Cho MT, Millan F, et al. A recurrent de novo CTBP1 mutation is associated with developmental delay, hypotonia, ataxia, and tooth enamel defects. Neurogenetics. 2016 doi: 10.1007/s10048-016-0482-4. [DOI] [PubMed] [Google Scholar]
- 16.You J, Sobreira NL, Gable DL, et al. A Syndromic Intellectual Disability Disorder Caused by Variants in TELO2, a Gene Encoding a Component of the TTT Complex. American journal of human genetics. 2016;98(5):909–918. doi: 10.1016/j.ajhg.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cali T, Lopreiato R, Shimony J, et al. A Novel Mutation in Isoform 3 of the Plasma Membrane Ca2+ Pump Impairs Cellular Ca2+ Homeostasis in a Patient with Cerebellar Ataxia and Laminin Subunit 1alpha Mutations. J Biol Chem. 2015;290(26):16132–16141. doi: 10.1074/jbc.M115.656496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tatton-Brown K, Seal S, Ruark E, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46(4):385–388. doi: 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2013;15(7):565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akawi N, McRae J, Ansari M, et al. Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4,125 families. Nat Genet. 2015;47(11):1363–1369. doi: 10.1038/ng.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Posey JE, Rosenfeld JA, James RA, et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genetics in medicine : official journal of the American College of Medical Genetics. 2015 doi: 10.1038/gim.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Salfelder KO, Schwab A, et al. Against all odds: blended phenotypes of three single-gene defects. European journal of human genetics : EJHG. 2016 doi: 10.1038/ejhg.2015.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amendola LM, Dorschner MO, Robertson PD, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome research. 2015;25(3):305–315. doi: 10.1101/gr.183483.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biesecker LG. Overcalling secondary findings. Genetics in medicine : official journal of the American College of Medical Genetics. 2016;18(4):416. doi: 10.1038/gim.2016.19. [DOI] [PubMed] [Google Scholar]
- 26.Manrai AK, Funke BH, Rehm HL, et al. Genetic Misdiagnoses and the Potential for Health Disparities. The New England journal of medicine. 2016;375(7):655–665. doi: 10.1056/NEJMsa1507092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.