

Abstract
The SinI and EcoRII DNA methyltransferases recognize sequences (GGA/TCC and CCA/TGG, respectively), which are characterized by an A/T ambiguity. Recognition of the A·T and T·A base pair was studied by in vitro methyltransferase assays using oligonucleotide substrates containing a hypoxanthine·C base pair in the central position of the recognition sequence. Both enzymes methylated the substituted oligonucleotide with an efficiency that was comparable to methylation of the canonical substrate. These observations indicate that M.SinI and M.EcoRII discriminate between their canonical recognition site and the site containing a G·C or a C·G base pair in the center of the recognition sequence (GGG/CCC and CCG/CGG, respectively) by interaction(s) in the DNA minor groove. M.SinI mutants displaying a decreased capacity to discriminate between the GGA/TCC and GGG/CCC sequences were isolated by random mutagenesis and selection for the relaxed specificity phenotype. These mutations led to amino acid substitutions outside the variable region, previously thought to be the sole determinant of sequence specificity. These observations indicate that A/T versus G/C discrimination is mediated by interactions between the large domain of the methyltransferase and the minor groove surface of the DNA.
INTRODUCTION
DNA (cytosine-5) methyltransferases (C5-MTases) catalyze the transfer of a methyl group from S-adenosyl-l-methionine (AdoMet) to the C5 carbon of cytosine in specific DNA sequences. These enzymes play an important role in several biological phenomena, e.g. restriction–modification in bacteria, differentiation, regulation of gene expression in eukaryotes and carcinogenesis (1–5).
C5-MTases consist of a single polypeptide chain. A key feature of the catalytic mechanism of C5 methylation is the formation of a transient covalent bond between the C6 carbon of the substrate cytosine and the sulfur of the active site cysteine, which is conserved in all C5-MTases (6–9). Most of our knowledge about C5-MTases is based on studies with bacterial enzymes. Eukaryotic C5-MTases are larger proteins, but the sequence homology they share with bacterial C5-MTases and the available experimental data suggest that they act using the same catalytic mechanism (4,10). Bacterial C5-MTases share a common architecture, they contain 10 conserved sequence motifs and a so-called variable region located between conserved motifs VIII and IX (8,9). The variable region is thought to be responsible for sequence-specific DNA recognition. The hypothesis that DNA recognition by C5-Mtases is mediated by the variable region was originally based on mutational analysis of multispecific C5-MTases (11) and domain swap experiments (12,13). The available X-ray structures of enzyme–DNA co-crystals of two methyltransferases (M.HhaI, recognition sequence GCGC, target cytosine underlined; M.HaeIII, GGCC; 14,15) are consistent with this view. The X-ray structures revealed that both enzymes fold in two domains. The large domain encompasses most of the conserved motifs, whereas the small domain contains the variable region. The two domains form a cleft where the DNA substrate fits with the major groove facing the small domain and the minor groove facing the large domain. In the M.HhaI and M.HaeIII co-crystals all protein–DNA interactions mediating sequence specificity were at the small domain–major groove interface (14,15).
In this study we have investigated methylation of oligonucleotide substrates by two C5-MTases recognizing an A·T or a T·A base pair (commonly represented as W but indicated as A/T in this paper) in the center of their target sequence. We present data showing that M.SinI (GGA/TCC) and M.EcoRII (CCA/TGG) discriminate between their canonical recognition site and the site containing a G·C or C·G base pair in the center of the recognition sequence (GGG/CCC and CCG/CGG, respectively) by interaction(s) in the DNA minor groove. Moreover, we could isolate M.SinI mutants displaying a relaxed specificity phenotype, i.e. a decreased capacity to discriminate between the GGA/TCC and GGG/CCC sequences. These mutations lead to amino acid substitutions outside the variable region, adding support to the interpretation that A/T versus G/C discrimination is mediated by interactions between the large domain of the MTase and the minor groove surface of DNA.
MATERIALS AND METHODS
Strains and media
The Escherichia coli strains SURE e14–(McrA–) Δ(mcrCB-hsdMR-mrr)171 endA1 supE44 thi-1 gyrA96 relA1 lac recB recJ sbcC umuC::Tn5 (Kanr) uvrC [F′ proAB lacIqZΔM15 Tn10 (Tetr)] and XL-1 Blue MRF′ Kan ΔmcrA183 Δ(mcrCB-hsdMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac [F′ proAB lacIqZΔM15 Tn5 (Kanr)] were purchased from Stratagene. Strain ER1398 hsdR2 mcrB1 (16) was used to overproduce M.SinI. The M.EcoRII overproducer strain BL21(DE3, pT71-Cys) (17) was a gift of A. Bhagwat. Bacteria were grown in LB medium (18) at 30 or 37°C. Ampicillin (Amp) and kanamycin (Kan) were used at 100 and 50 µg/ml concentrations, respectively.
Enzymes, oligonucleotides and chemicals
Sau96I endonuclease was purified by a published procedure (19). Other restriction enzymes, T4 DNA ligase and DNA polymerase large (Klenow) fragment were either from Fermentas or from New England Biolabs, Taq DNA polymerase from Pharmacia and deoxyoligonucleotides from Integrated DNA Technologies (IDT). The following double-stranded oligonucleotides were used as substrates for M.SinI and M.EcoRII:
M.SinI 5′-GACGTCAGGXCCACTCCTC-3′
3′-CTGCAGTCCYGGTGAGGAG-5′
M.EcoRII 5′-GACGTCACCXGGACTCCTC-3′
3′-CTGCAGTGGYCCTGAGGAG-5′
[recognition sites shown in bold; X = A, G or I (hypoxanthine); Y = T or C]. Complementary oligonucleotides were annealed at 100 µM concentration in 10 mM Tris–HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA by heating to 65°C and then slow cooling to room temperature. Tm values of the substrate oligonucleotides were 48.66°C or higher in 50 mM NaCl as specified by the supplier (IDT). Unlabeled AdoMet was from Sigma.
Plasmids
Plasmid pSI4 (m+ r+; from C. Karreman) carries the complete SinI restriction–modification system (GenBank accession no. J03391; 20). Plasmid pSin5 (m+ r–), which is identical to pΔHH2 (20), was obtained by deleting the small HindIII fragment carrying the 3′-end of the sinIR gene in pSI4. Plasmid pSin5-1 differs from pSin5 in that the NdeI site of the pUC19 vector has been eliminated. All relevant restriction sites used in further plasmid constructions are shown in Figure 1. Plasmid pSin17 was constructed by cloning the SacI–HindIII fragment of pSin5 between the SacI and HindIII sites of pBluescriptSKII(+). Plasmid pSin10-19 was obtained from pSin5-1 by random mutagenesis (see below). It carries the 10-19 (Asn172Ser) allele of the sinIM gene. Plasmid pSin26-19, which is similar to pSin17 but encodes the Asn172Ser mutant M.SinI, was constructed by replacing the XbaI–HindIII fragment of pSin17 by the XbaI–HindIII fragment of pSin10-19.
Figure 1.

Schematic map of the sinIM gene cloned in pSin5 and amino acid sequence of a part of the SinI methyltransferase. The coding sequence of the sinIM gene is indicated by a filled bar, the variable region by an empty bar, conserved sequence elements by roman numerals I–X and pUC19 vector sequence by a broken line. Amino acid substitutions in two M.SinI mutants are shown below the protein sequence.
Plasmid pSin7 was prepared by cloning the sinIM gene in the expression plasmid vector pER23S(–ATG). pER23S(–ATG) is a derivative of pER23(–ATG) (21), which differs from the parental plasmid by a SalI linker inserted into the PvuII cloning site (T.Lukacsovich, unpublished). Genes cloned in this plasmid are transcribed from the E.coli rrnB P2 promoter and expression is controlled by the lac repressor. To construct pSin7, pSI4 was digested with NruI, then partially digested with NsiI. The sinIM gene contains two NsiI sites. One site overlaps the translational initiator codon, while the other is in the coding region (20). The partially digested 1567 bp NsiI–NruI fragment encompassing the sinIM gene was purified from an agarose gel, the 3′-overhang of the NsiI end was removed with Klenow polymerase, then the fragment was ligated to pER23S(–ATG) that had been digested with SalI and subsequently treated with Klenow polymerase to fill in the SalI ends. The ligated DNA was transformed into ER1398 (pVH1). Plasmid pVH1 (Kanr, lacIQ; 22) served to repress transcription from the rrnB P2 promoter. To obtain an overproducer for the M.SinI(Asn172Ser) mutant, the XbaI–HindIII fragment of pSin26-19 was substituted for the XbaI–HindIII fragment of pSin7. Although the HindIII site downstream of the sinIM gene was lost when pSin7 was constructed, this exchange of fragments was possible because pSin7 contains a HindIII site in the vector part of the plasmid, 94 bp downstream of the SalI cloning site (21). The resulting plasmid, pSin27-19, was maintained in ER1398 containing pVH1.
Enzyme purification
ER1398(pVH1+pSin7) was grown in 1 l LB/Kan/Amp medium at 37°C to an OD550 of 0.6. M.SinI production was induced by addition of 0.2% lactose and shaking was continued at 30°C for 4 h. Cells (6 g) were harvested by centrifugation at 4°C, resuspended in 50 ml of TEM buffer (20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 7 mM β-mercaptoethanol), then disrupted by sonication. Cell debris was removed by centrifugation (20 000 r.p.m., 45 min). Nucleic acid concentration was estimated by measuring A260 of the supernatant, then 1 ml of a 10% streptomycin solution was added for every 900 A260 units. The precipitate was collected by centrifugation, then dissolved in 50 ml of TEM buffer. The solution was dialyzed against PC buffer (10 mM potassium phosphate, pH 7.4, 10 mM β-mercaptoethanol, 0.1 mM EDTA, 10% glycerol), then loaded onto a 110 ml phosphocellulose column equilibrated with PC buffer. Proteins were eluted with an 800 ml gradient of 0–1 M NaCl in PC buffer. M.SinI eluted between 0.3 and 0.4 M NaCl. In the peak fractions M.SinI was at least 95% pure as judged by SDS–PAGE. Protein concentration was estimated by the Bradford reaction (23) using bovine serum albumin as the standard.
Preparation of M.SinI and M.EcoRII crude extracts
To measure M.SinI activity in crude extracts, XL-1 Blue MRF′ Kan cells harboring plasmid pSin17 or pSin26-19 were grown to saturation in 20 ml LB/Amp at 37°C. For extracts of M.SinI overproducer clones, ER1398(pVH1+pSin7) or ER1398(pVH1+pSin27-19) was grown in 20 ml LB/Kan/Amp at 37°C to a density of OD550 = 0.5. Enzyme production was induced by adding 1 mM isopropyl-β-d-galactoside (IPTG), then shaking was continued at 30°C for 4 h. For an M.EcoRII extract, BL21(DE3, pT71-Cys) was grown at 37°C, then IPTG was added and growth was continued for 3 h. Cells were centrifuged, resuspended in 2 ml of 20 mM Tris–HCl, pH 8.0, 10 mM 2-mercaptoethanol, 1 mM EDTA, then disrupted by sonication. Cell debris was removed by centrifugation and the supernatants were used to determine methyltransferase activity.
DNA methyltransferase assay
For steady-state kinetic analysis of M.SinI, the concentration of one of the two substrates (DNA or AdoMet) was varied. Reactions contained MTase assay buffer (50 mM Tris–HCl, pH 8.5, 50 mM NaCl, 10 mM dithiothreitol), 667 nM or varying amounts (7, 13, 27, 67, 133, 267, 533, 667, 1333 or 2667 nM) of annealed oligonucleotides, 5 µM or varying amounts (0.078, 0.156, 0.313, 0.625, 1.25, 2.5 or 5 µM) of [methyl-3H]AdoMet (111 GBq/mmol; New England Nuclear) and 7 nM purified M.SinI. Reactions were started by adding the enzyme. After a 10 or 20 min incubation at 30°C, methylation was stopped by adding 4 µl 10% SDS, then the reaction mixtures were pipetted onto DE81 (Whatman) paper disks. The disks were washed and the filter-bound radioactivity was determined (24). Counting efficiency of 3H was assessed as described (25). Data were analyzed by non-linear regression fitting to the Michaelis–Menten equation using the GraphPad PRISM v.3.02 program.
In experiments using crude enzyme extracts, reactions contained MTase assay buffer, 2.6 µM oligonucleotide duplex and 5 µM [methyl-3H]AdoMet (111 GBq/mmol) and 2 µl of enzyme extract in 30 µl. After incubation at 30°C for 20 min, the reaction mixtures were processed as described above.
Random mutagenesis and selection of relaxed specificity M.SinI mutants
The XbaI–NdeI fragment of the sinIM gene was mutagenized in vitro by error-prone PCR (26). Reactions contained 10 mM Tris–HCl, pH 8.8, 50 mM KCl, 0.08% Nonidet P-40, 1.5 mM MgCl2, 20 pmol primers S5 and S3, 250 µM dNTPs, 0.3 ng pSin5-1 linearized by HindIII digestion and 2 U Taq DNA polymerase in 100 µl. Primer S5 (5′-CGTTTAGGTCTAGAAGATGAGAG) corresponds to positions 1394–1416 of the coding strand and primer S3 (5′-CGATTTTCCTTCCATTCATATGATC) to positions 2219–2195 of the non-coding strand of the sinIM gene (the XbaI and NdeI sites are shown in bold). After 30 cycles of amplification (95°C for 1 min, 55°C for 1 min and 72°C for 2 min), the PCR product was purified using a Wizard PCR Preps DNA Purification System (Promega), then it was digested with XbaI and NdeI. The digested DNA was ligated to the isolated large XbaI–NdeI fragment of pSin5-1. The ligated DNA was introduced by electroporation into E.coli SURE cells. Transformants (∼280 000 clones) were grown as a mixed culture in LB/Amp medium to saturation at 37°C. Approximately 1 µg plasmid DNA isolated from the mixed culture was digested with an excess (60 U) of Sau96I endonuclease, then ∼20 ng digested DNA was introduced, by electroporation, into E.coli SURE cells. Around 40 000 transformants were obtained, which were used to inoculate a mixed culture. Approximately 0.5 µg plasmid DNA purified from the culture was digested with Sau96I endonuclease, then the digested DNA was used to transform E.coli SURE cells.
DNA sequencing
DNA sequence was determined either manually using a T7 sequencing kit (Pharmacia) or using an ABI automated sequencer.
RESULTS
Methylation of a recognition site containing hypoxanthine
Some type II restriction–modification systems (e.g. AvaII, GGA/TCC; EcoRII, CCA/TGG; for a complete list see 27) are characterized by an A/T degeneracy in their recognition sequence. These enzymes accept A·T or T·A but exclude G·C or C·G base pairs at a particular position of the recognition sequence. Molecular modeling indicated that such discrimination can be best accomplished in the minor groove and predicted that recognition would be based on the presence or absence of the guanine 2-amino group protruding into the center of the minor groove (28). This model contradicts the view that sequence specificity of C5-MTases is mediated solely by interactions between the small domain and the major groove. To address this question, we performed DNA methylation experiments in vitro with one of the enzymes of this class, the SinI methyltransferase. M.SinI recognizes the sequence GGA/TCC and methylates the inner cytosine to yield 5-methylcytosine (29).
A plasmid (pSin7) overproducing M.SinI was constructed by cloning the sinIM gene into an expression plasmid vector and M.SinI was purified to near homogeneity. The purified enzyme was used to in vitro methylate double-stranded deoxyoligonucleotide substrates prepared by annealing two complementary 19mer strands. Duplexes A/T, G/C and I/C contained GGA/TCC, GGG/CCC and GGI/CCC sites, respectively, embedded in identical flanking sequences (see Materials and Methods; I = hypoxanthine). Hypoxanthine is a guanine analog which lacks the 2-amino group but can form Watson–Crick base pairs with cytosine. In the major groove an I·C base pair is similar to a G·C base pair, whereas in the minor groove, because of the missing 2-amino group, it is similar to an A·T base pair (Fig. 2). The G/C duplex was included in this study because Sau96I digestion of plasmids isolated from cells expressing M.SinI indicated that M.SinI can methylate, at a low rate, GGG/CCC sites (see below).
Figure 2.

Structure of the A·T and G·C base pairs (adapted from 30).
Steady-state kinetic parameters of M.SinI were determined as described in Materials and Methods. First, the Michaelis constant for AdoMet (KmAdoMet) was determined using the canonical A/T duplex as DNA substrate and was found to be 0.5 ± 0.095 µM. DNA concentration dependence of the methylation reaction for the three oligonucleotide substrates was investigated at 5 µM AdoMet. The velocity versus substrate concentration plots show that the enzyme could be saturated by all three substrates (Fig. 3). Table 1 summarizes the Km, kcat and kcat/Km values derived for the A/T, G/C and I/C substrates. As expected, the A/T substrate was methylated much more efficiently than the G/C duplex: the catalytic efficiency (kcat/Km) for the latter substrate was <1% of the value characterizing the canonical substrate. Nevertheless, the G/C duplex could function as a substrate, confirming in vivo observations (see below), which suggested that M.SinI can methylate GGG/CCC sites, albeit at much lower rates than GGA/TCC sites. The I/C duplex was methylated efficiently by M.SinI, with a kcat/Km value only four times lower than the canonical substrate (Table 1). We interpret this finding to mean that the enzyme recognized the GGA/TCC and GGI/CCC sites as similar.
Figure 3.

Steady-state kinetic analysis of M.SinI methylation using three different DNA substrates. Circles, GGA/TCC; squares, GGI/CCC; triangles, GGG/CCC. (Inset) Plot of GGG/CCC data using a smaller velocity scale. The curves were generated from three experiments as described in Materials and Methods. For clarity, standard error of the mean bars are shown only above the data points.
Table 1. Steady-state kinetic parameters of M.SinI.
| GGACCa | GGGCCa | GGICCa | |
| |
CCTGG |
CCCGG |
GGCCC |
| KmDNA (nM) | 17 ± 3 | 116 ± 20 | 95 ± 13 |
| kcat (min–1) | 0.3 ± 0.054 | 0.014 ± 0.0035 | 0.436 ± 0.077 |
| kcat/KmDNA (×105 M–1 s–1) | 2.94 ± 0.68 | 0.02 ± 0.002 | 0.76 ± 0.23 |
| Relative kcat/KmDNA | 1 | 0.007 | 0.26 |
aSubstrate sequence.
It was interesting to test whether this mechanism of substrate recognition holds for other C5-MTases characterized by an A/T recognition ambiguity. M.EcoRII, another representative of this group, recognizes the sequence CCA/TGG. We prepared enzyme extract from the overproducer E.coli strain BL21(DE3, pT71-Cys) and used it, at three different dilutions, to methylate double-stranded oligonucleotides containing CCA/TGG, CCG/CGG or CCI/CGG sites. The same tendency was observed as in the case of M.SinI: duplexes with sites containing A/T or I/C were good substrates, whereas the duplex with the G/C base pair was methylated at a much lower rate (Table 2).
Table 2. Methylation of different substrate sequences by M.EcoRII.
| Extract (fold dilution) | CCAGG | CCGGG | CCIGG |
| |
GGTCC |
GGCCC |
GGCCC |
| pT71-Cys (1×) | 71 646 ± 3850 | 1940 ± 263 | 109 724 ± 6641 |
| pT71-Cys (10×) | 13 336 ± 1562 | 688 ± 166 | 45 932 ± 6157 |
| pT71-Cys (100×) | 1996 ± 301 | 252 ± 35 | 7854 ± 1690 |
| No plasmid (1×) | 189 ± 5 | 195 ± 4 | 200 ± 9 |
In vitro methylation reactions using cell-free extracts of BL21(DE3) carrying pT71-Cys or no plasmid. Values (average of four experiments) indicate incorporated 3H radioactivity in c.p.m.
Mutagenesis of the sinIM gene and selection of relaxed specificity mutants
With the aim of identifying amino acids that play a role in recognition of the central A/T base pair, we tried to isolate M.SinI mutants displaying an impaired capacity to distinguish between GGA/TCC and GGG/CCC sites. Because this approach was initiated before experiments with the modified oligonucleotide substrates revealed the importance of minor groove contacts, we anticipated such specificity mutations to occur in the variable region. Therefore, we randomly mutagenized the pSin5-1 XbaI–NdeI fragment, which encodes the variable region and conserved motifs V–X (Fig. 1). Plasmid pSin5-1, which was used for mutagenesis, expresses M.SinI, making the GGA/TCC sites of the cell DNA resistant to SinI endonuclease digestion. The plasmid contains two GGA/TCC and eight GGG/CCC sites. Digestion of pSin5-1 with the GGNCC-specific (19) endonuclease Sau96I gave a partial digestion pattern (Fig. 4, lane 2), suggesting that M.SinI can also methylate GGG/CCC sites, but much less efficiently than GGA/TCC sites.
Figure 4.
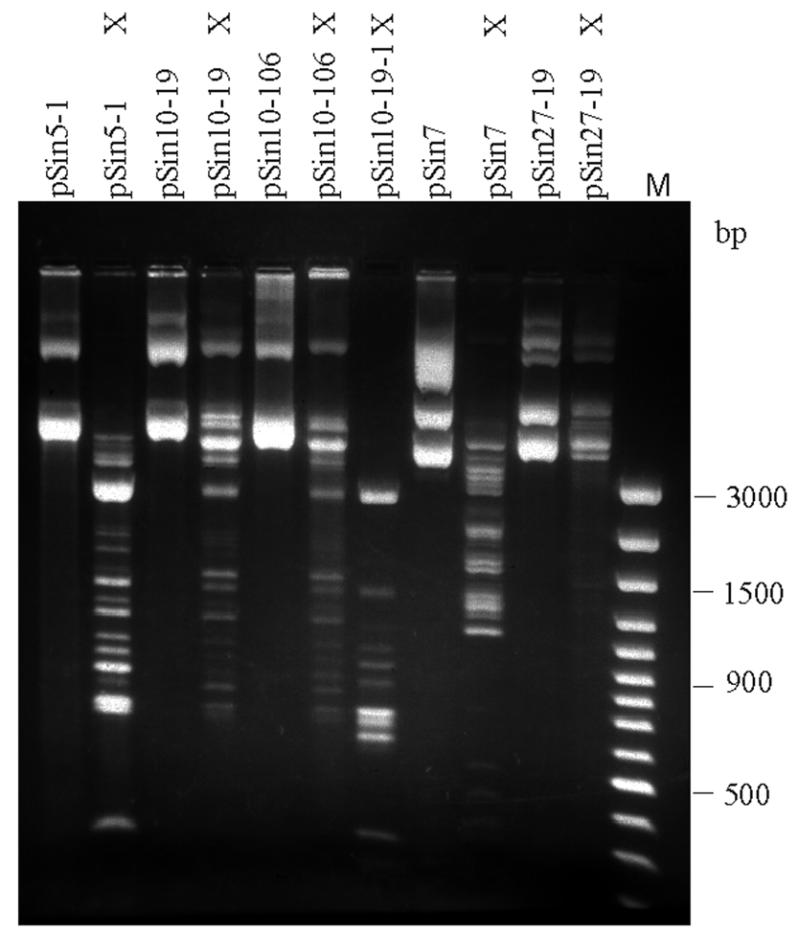
Protection of plasmid GGNCC sites by the Asn172Ser and Val173Leu SinI methyltranferase mutants in vivo. 1% agarose gel. X, Sau96I-digested samples; M, DNA Ladder Plus size marker (Fermentas).
Mutagenesis was performed in vitro by PCR taking advantage of the intrinsic error frequency of Taq DNA polymerase. The mutagenized fragment was reinserted into the plasmid backbone of pSin5-1 and a mutant plasmid library was established in E.coli. Clones encoding relaxed specificity M.SinI mutants were selected by digesting a sample of the mutagenized plasmid library with Sau96I endonuclease. Sau96I endonuclease does not cleave GGN5mCC sites (31). The selection was based on the idea that clones that have lost the ability to recognize the central A/T base pair would methylate both GGA/TCC and GGG/CCC sites, thus rendering the plasmid resistant to Sau96I digestion. Approximately half of the clones obtained after the second round of Sau96I digestion contained plasmids that showed higher resistance to Sau96I endonuclease than the parental pSin5-1. The Sau96I cleavage patterns of two mutant plasmids (pSin10-19 and pSin10-106) that displayed the highest resistance are shown in Figure 4. Although protection is not complete, some intact open circular form can be identified in the digestion patterns of both mutants, explaining why these plasmids could be recovered in this screen. Controls with unmodified λ phage DNA included in the reaction mixture showed that digestion of unmethylated sites went to completion (not shown). A frameshift introduced by cleaving the pSin10-19 plasmid with XbaI and filling in the ends with DNA polymerase large fragment abolished protection against Sau96I digestion (pSin10-19-1), indicating that protection was due to methylation rather than some other reason, e.g. loss of Sau96I sites (Fig. 4).
Characterization of the mutants
The entire gene for the most interesting mutants (pSin10-19 and pSin10-106) and the XbaI–NdeI fragment of some other mutants were sequenced. Plasmids pSin10-19 and pSin10-106 contained single point mutations (A1422G and G1424C) leading to amino acid substitutions Asn172Ser and Val173Leu, respectively (Fig. 1). Plasmid pSin10-123, which displayed weaker resistantance to Sau96I digestion than the former two plasmids, had a single base change within the mutagenized fragment resulting in an Arg232Gly substitution in the amino acid sequence. The XbaI–NdeI fragment of several other isolates displaying a phenotype similar to pSin10-19 and pSin10-106 was sequenced in its entirety or in part. All contained mutations that were of either the pSin10-19 or the pSin10-106 type.
We tested whether the mutant enzymes had a recognition specificity lower than GGNCC. The mutant plasmids were digested with the following restriction enzymes (the recognition sequences and the type of C5 cytosine methylation to which the enzyme is known to be sensitive are shown in parentheses): BspRI (GGCC), MspI (CCGG), HpaII (CCGG), BepI (CGCG) and HhaI (GCGC and GCGC). Digestion patterns characteristic for complete digestion were obtained, suggesting that the mutant enzymes methylate only GGNCC sites (not shown).
One of the mutants (Asn172Ser) was characterized in a more quantitative manner. In pSin5-1 and its mutant derivatives the sinIM gene has the opposite orientation relative to the vector lac promoter. To avoid potential interference by lac transcription, plasmids pSin17 and pSin26-19 were constructed as described in Materials and Methods. These plasmids carry the wild-type and the Asn172Ser mutant genes, respectively, in an orientation corresponding to transcription from the vector lac promoter. The in vitro methyltransferase assay performed with cell extracts showed that the Asn172Ser substitution reduced the GGA/TCC-specific activity substantially, however, it led to the appearance of a detectable level of methylation at GGG/CCC sites (Table 3). We also constructed a plasmid (pSin27-19) which, upon induction with IPTG, leads to overproduction of the Asn172Ser mutant protein. Plasmid pSin27-19 was compared with pSin7, the plasmid overproducing the wild-type enzyme. Methylation status of the two plasmids prepared from uninduced cultures was assessed by Sau96I digestion (Fig. 4). The mutant plasmid pSin27-19 (four GGA/TCC and eight GGG/CCC sites) showed a much higher protection than pSin7 (four GGA/TCC and nine GGG/CCC sites). These observations parallelled data from in vitro measurements of methyltransferase activity in crude extracts prepared from IPTG-induced cells. The GGG/CCC-specific methyltransferase activity was higher in cells carrying pSin27-19 than in cells with pSin7 (Table 3).
Table 3. Methylation of different substrate sequences by the wild-type and N172S mutant M.SinI.
| Clone | GGACC | GGGCC |
| |
CCTGG |
CCCGG |
| pSin17 (WT) | 23 067 ± 4269 | 63 ± 16 |
| pSin26-19 (Asn172Ser) | 1973 ± 714 | 913 ± 179 |
| pSin7 (WT) | 48 661 ± 4342 | 78 ± 14 |
| pSin27-19 (Asn172Ser) | 8713 ± 962 | 5020 ± 170 |
In vitro methylation reactions using cell-free extracts. Values (average of four experiments) indicate incorporated 3H radioactivity in c.p.m.
DISCUSSION
Base analogs are widely used in the analysis of sequence-specific DNA–protein interactions (32). Hypoxanthine is one of the purine analogs that is the least likely to cause a distortion of the double-helical structure, thus the observed effect can be, with great reliability, attributed to the structural difference between the normal base and the analog. The I·C base pair has a minor groove structure like that of an A·T base pair and a major groove structure like that of a G·C base pair (Fig. 2). Substitution of hypoxanthine for guanine has been used in numerous cases to assess the importance of the guanine exocyclic amino group in DNA recognition by proteins (33,34). In this study we used hypoxanthine-containing oligonucleotide substrates to test how the SinI and EcoRII methyltransferases recognize the central A/T base pair in their target sequence. For both enzymes the substituted oligonucleotide functioned with an efficiency comparable to that of the canonical (A/T) substrate, indicating that discrimination between the A/T and G/C sites is mediated by the guanine 2-amino group protruding into the minor groove.
This observation is fully consistent with results of the classic molecular modeling study by Rich and co-workers (28). Two of their conclusions are relevant to this paper. First, they have found that the minor groove was insensitive to base reversals, i.e. using minor groove contacts, a protein cannot distinguish between an A·T and T·A or a G·C and C·G base pair. (Such a type of ambiguity characterizes M.SinI and M.EcoRII.) Secondly, they have determined that the minor groove provides the best possibility of discriminating between A·T or T·A and G·C or C·G base pairs. The recognition was suggested to be based on the exocyclic 2-amino group of the guanine (28).
Our results modify the current view of substrate recognition by C5-MTases. We suggest that C5-MTases characterized by an A/T recognition degeneracy also employ, in addition to interactions between the major groove of the DNA and the small domain of the enzyme, minor groove contact(s) during substrate recognition and that this mechanism serves to exclude sites containing G/C base pairs. The available X-ray data (14,15) and the uniform architecture of C5-MTases (9,35) suggest that the structure responsible for this function resides in the large domain that is facing the minor groove. In this context it is interesting to note that the recently published X-ray structure of the ternary complex of an N6-adenine DNA methyltransferase (M.TaqI) revealed extensive interactions between the large domain of the enzyme and the minor groove of the DNA substrate (36).
The conclusion about the role of the minor groove–large domain interaction is supported by the results of random mutagenesis. We mutagenized a segment of the sinIM gene encoding the variable region and selected for mutants that showed an increased capacity to methylate GGG/CCC sites. The selection was based on the same principle as the method that has been successfully used for the cloning of methyltransferase genes (37) and of complete restriction–modification systems (38,39). The mutations with the greatest change of phenotype affected two neighboring amino acids (Asn172Ser and Val173Leu) that are outside the variable region. Concomitantly, none of the single mutations identified in this screen was located in the variable region. Asn172 and Val173 are part of the weakly conserved motif V (8,9). The X-ray structures of M.HhaI and M.HaeIII (14,15) revealed that motif V is in the large domain. Leu100, the amino acid in M.HhaI that corresponds to Val173 in M.SinI, forms part of the AdoMet-binding pocket (35).
To our knowledge this is the first case when relaxed specificity mutants of a C5-MTase have been isolated. By stressing the importance of the M.SinI mutations we do not mean to imply that either Asn172 or Val173 contact the recognition site. They are more likely to play an indirect role in determining the sequence specificity of M.SinI. Their side chains might be important in maintaining the proper 3-D structure of a part of the large domain that approaches the DNA in the minor groove. Perturbation of this structure by the replacement of Asn172 and Val173 may have caused a decrease in recognition specificity.
Recognition of the A/T base pair by either M.SinI or M.EcoRII appears to be less accurate than that of the other four base pairs. This was suggested by the partial Sau96I digestion pattern of plasmids encoding wild-type M.SinI and by the detectable methylation of G/C sites in vitro. The reason might be that the structures recognizing the central A/T base pair and the structures recognizing the rest of the target sequence are located on separate domains. Lacking a 3-D structure for the M.SinI or M.EcoRII recognition complex, we can only speculate on the amino acid(s) that might play a role in the discrimination between A/T and G/C base pairs. We can postulate that an amino acid side chain could serve to exclude G/C base pairs by sterically clashing with the guanine amino group projecting into the minor groove.
Acknowledgments
ACKNOWLEDGEMENTS
We thank C. Karreman and A. Bhagwat for strains, R .J. Roberts for critical reading of the manuscript, C. Finta for discussions and Ibolya Anton for technical assistance. This project was supported by an International Research Scholar’s award from the Howard Hughes Medical Institute and grant T 029868 from the Hungarian Science Fund (OTKA).
References
- 1.Noyer-Weidner M. and Trautner,T.A. (1993) Methylation of DNA in prokaryotes. In Jost,J.P. and Saluz,H.P. (eds) DNA Methylation: Molecular Biology and Biological Significance. Birkhauser Verlag, Basel, Switzerland, pp. 39–108.
- 2.Walsh C.P. and Bestor,T.H. (1999) Cytosine methylation and mammalian development. Genes Dev., 13, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bird A.P. and Wolffe,A.P. (1999) Methylation-induced repression-belts, braces, and chromatin. Cell, 99, 451–454. [DOI] [PubMed] [Google Scholar]
- 4.Adams R.L.P. (1995) Eukaryotic DNA methyltransferases—structure and function. Bioessays, 17, 139–145. [DOI] [PubMed] [Google Scholar]
- 5.Jones P.A. and Laird,P.W. (1999) Cancer epigenetics comes of age. Nature Genet., 21, 163–167. [DOI] [PubMed] [Google Scholar]
- 6.Wu J.C. and Santi,D.V. (1987) Kinetic and catalytic mechanism of HhaI methyltransferase. J. Biol. Chem., 262, 4778–4786. [PubMed] [Google Scholar]
- 7.Chen L., MacMillan,A.M., Chang,W., Ezaz-Nikpay,K., Lane,W.S. and Verdine,G.L. (1991) Direct identification of the active-site nucleophile in a DNA (cytosine-5)-methyltransferase. Biochemistry, 30, 11018–11025. [DOI] [PubMed] [Google Scholar]
- 8.Pósfai J., Bhagwat,A., Pósfai,G. and Roberts,R. (1989) Predictive motifs derived from cytosine methyltransferases. Nucleic Acids Res., 17, 2421–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar S., Cheng,X., Klimasauskas,S., Mi,S., Pósfai,J., Roberts,R.J. and Wilson,G.G. (1994) The DNA (cytosine-5) methyltransferases. Nucleic Acids Res., 22, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pradhan S., Bacolla,A., Wells,R.D. and Roberts,R.J. (1999) Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem., 274, 33002–33010. [DOI] [PubMed] [Google Scholar]
- 11.Wilke K., Rauhut,E., Noyer-Weidner,M., Lauster,R., Pawlek,B., Behrens,B. and Trautner,T.A. (1988) Sequential order of target-recognizing domains in multispecific DNA-methyltransferases. EMBO J., 7, 2601–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balganesh T.S., Reiners,L., Lauster,R., Noyer-Weidner,M., Wilke,K. and Trautner,T.A. (1987) Construction and use of chimeric SPR/F3T DNA methyltransferases in the definition of sequence recognizing enzyme regions. EMBO J., 6, 3543–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klimasauskas S., Nelson,J.L. and Roberts,R.J. (1991) The sequence specificity domain of cytosine-C5 methylases. Nucleic Acids Res., 19, 6183–6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klimasauskas S., Kumar,S., Roberts,R.J. and Cheng,X. (1994) HhaI methyltransferase flips its target base out of the DNA helix. Cell, 76, 357–369. [DOI] [PubMed] [Google Scholar]
- 15.Reinisch K.M., Chen,L., Verdine,G.L. and Lipscomb,W.N. (1995) The crystal structure of HaeIII methyltransferase covalently complexed to DNA: an extrahelical cytosine and rearranged base pairing. Cell, 82, 143–153. [DOI] [PubMed] [Google Scholar]
- 16.Raleigh E.A. and Wilson,G. (1986) Escherichia coli K-12 restricts DNA containing 5-methylcytosine. Proc. Natl Acad. Sci. USA, 83, 9070–9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wyszynski M.W., Gabbara,S. and Bhagwat,A.S. (1992) Substitutions of a cysteine conserved among DNA cytosine methylases result in a variety of phenotypes. Nucleic Acids Res., 20, 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sambrook J., Fritsch,E. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 19.Sussenbach J.S., Steenbergh,P.H., Rost,J.A., van Leeuwen,W.J. and van Embden,J.D.A. (1978) A second site-specific restriction endonuclease from Staphylococcus aureus. Nucleic Acids Res., 5, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karreman C. and de Waard,A. (1988) Cloning and complete nucleotide sequences of the type II restriction-modification genes of Salmonella infantis. J. Bacteriol ., 170, 2527–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukacsovich T., Orosz,A., Balikó,G. and Venetianer,P. (1990) A family of expression vectors based on the rrnB P2 promoter of Escherichia coli. J. Biotechnol., 16, 49–56. [DOI] [PubMed] [Google Scholar]
- 22.Haring V., Scholz,P., Scherzinger,E., Frey,J., Derbyshire,K., Hatfull,G., Willetts,N.S. and Bagdasarian,M. (1985) Protein RepC is involved in copy number control of the broad host range plasmid RSF1010. Proc. Natl Acad. Sci. USA, 82, 6090–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradford M.M. (1976) A rapid and sensitive method for the quantition of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 24.Greene P.J., Poonian,M.S., Nussbaum,A.L., Tobias,L., Garfin,D.E., Boyer,H.W. and Goodman,H.M. (1975) Restriction and modification of a self-complementary octanucleotide containing the EcoRI substrate. J. Mol. Biol., 99, 237–261. [DOI] [PubMed] [Google Scholar]
- 25.Brennan C.A., Van Cleve,M.D. and Gumport,R.I. (1986) The effects of base analogue substitutions on the methylation by the EcoRI modification methylase of octadeoxyribonucleotides containing modified EcoRI recognition sequences. J. Biol. Chem., 261, 7279–7286. [PubMed] [Google Scholar]
- 26.Zhou Y., Zhang,X. and Ebright,R.H. (1991) Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase. Nucleic Acids Res., 19, 6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts R.J. and Macelis,D. (2001) REBASE—restriction enzymes and methylases. Nucleic Acids Res., 29, 268–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seeman N.C., Rosenberg,J.M. and Rich,A. (1976) Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl Acad. Sci. USA, 73, 804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karreman C. and de Waard,A. (1988) Isolation and characterization of the modification methylase M.SinI. J. Bacteriol ., 170, 2533–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stryer L. (1995) Biochemistry, 4th Edn. W.H. Freeman and Co., New York, NY. [Google Scholar]
- 31.Szilák L., Venetianer,P. and Kiss,A. (1990) Cloning and nucleotide sequence of the genes coding for the Sau96I restriction and modification enzymes. Nucleic Acids Res., 18, 4659–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aiken C.R. and Gumport,R.I. (1991) Base analogs in study of restriction enzyme-DNA interactions. Methods Enzymol., 208, 433–457. [DOI] [PubMed] [Google Scholar]
- 33.Starr D.B. and Hawley,D.K. (1991) TFIID binds in the minor groove of the TATA box. Cell, 67, 1231–1240. [DOI] [PubMed] [Google Scholar]
- 34.Wang S., Cosstick,R., Gardner,J.F. and Gumport,R.I. (1995) The specific binding of Escherichia coli integration host factor involves both major and minor grooves of DNA. Biochemistry, 34, 13082–13090. [DOI] [PubMed] [Google Scholar]
- 35.Cheng X. (1995) Structure and function of DNA methyltransferases. Annu. Rev. Biophys. Biomol. Struct., 24, 293–318. [DOI] [PubMed] [Google Scholar]
- 36.Goedecke K., Pignot,M., Goody,R.S., Scheidig,A.J. and Weinhold,E. (2001) Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nature Struct. Biol., 8, 121–125. [DOI] [PubMed] [Google Scholar]
- 37.Szomolányi ,É., Kiss,A. and Venetianer,P. (1980) Cloning the modification methylase gene of Bacillus sphaericus R in Escherichia coli. Gene, 10, 219–225. [DOI] [PubMed] [Google Scholar]
- 38.Kiss A., Pósfai,G., Keller,C.C., Venetianer,P. and Roberts,R.J. (1985) Nucleotide sequence of the BsuRI restriction-modification system. Nucleic Acids Res., 13, 6403–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lunnen K.D., Barsomian,J.M., Camp,R.R., Card,C.O., Chen,S.Z., Croft,R., Looney,M.C., Meda,M.M., Moran,L.S., Nwankwo,D.O., Slatko,B.E., Van Cott,E.M. and Wilson,G.G. (1988) Cloning type-II restriction and modification genes. Gene, 74, 25–32. [DOI] [PubMed] [Google Scholar]