

Abstract
Technologies that induce antigen-specific immune tolerance by mimicking naturally occurring mechanisms have the potential to revolutionize the treatment of many immune-mediated pathologies such as autoimmunity, allograft rejection, and allergy. The immune system intrinsically has central and peripheral tolerance pathways for eliminating or modulating antigen-specific responses, which are being exploited through emerging technologies. Antigen-specific tolerogenic responses have been achieved through the functional reprogramming of antigen-presenting cells or lymphocytes. Alternatively, immune privileged sites have been mimicked using biomaterial scaffolds to locally suppress immune responses and promote long-term allograft survival. This review describes natural mechanisms of peripheral tolerance induction and the various technologies being developed to achieve antigen-specific immune tolerance in vivo. As currently approved therapies are non-specific and carry significant associated risks, these therapies offer significant progress towards replacing systemic immune suppression with antigen-specific therapies to curb aberrant immune responses.
Keywords: Immune tolerance, nanoparticle, autoimmune disease, transplantation, allergy, drug delivery, regulatory T cells
Graphical abstract
1. Introduction
Induction of antigen (Ag)-specific immune tolerance is a complex process that requires the collaboration of multiple immunological pathways. Aberrant activation of T cells in vivo results in cellular damage against specific tissues and is responsible for the development of autoimmune diseases. To minimize the occurrence of undesirable immune responses to self-Ags, most self-reactive lymphocytes are eliminated in the thymus and bone marrow by a mechanism known as central tolerance. Unfortunately, this process is only 60-75% effective and potentially harmful Ag-specific cells with possible effector activity are released into the blood and tissues [1, 2]. To suppress potentially autoreactive cells that have avoided elimination by central tolerance, peripheral tolerance mechanisms exist. Intrinsic peripheral tolerance mechanisms are sometimes insufficient to curb inappropriate immune activation, necessitating therapeutic intervention to enable the body to limit responses to “self.” Common therapies used to subdue abnormal immune activation are not Ag-specific and involve systemic immune suppression or immunodepletion therapies that target the T cell receptor (TCR), co-signaling molecules, cytokines, or inhibit leukocyte trafficking, among other mechanisms [3, 4]. However, administration of these non-specific treatments over a prolonged period of time is associated with numerous adverse effects, including increased patient susceptibility to opportunistic infections [5], viral reactivation [6], and neoplasia [7].
Ag-specific tolerance approaches are needed to restore immune homeostasis in the cases of autoimmune disease as indicated above, and can be extended to establish selective Ag tolerance in the cases of allogeneic transplant and allergy. In Ag-specific tolerance, undesired immune activation is suppressed while the activity of the remaining immune system is maintained. Thus, the desirability of therapies to address these conditions has gained significant traction over several decades as the incidence of immune-mediated diseases has steadily risen [8, 9]. T cell-mediated autoimmune diseases are driven by the continued presentation of self-Ag by Ag-presenting cells (APCs) to autoreactive T cells. Conversely, allograft rejection involves a combination of allorecognition by T cells and alloantibody production by B cells [10]. Allergic reactions involve the activation of granulocytes such as mast cells, basophils, and eosinophils by allergen binding to antibodies [11]. Important immune elements of these diseases are the development of Ag-specific effector T-helper type 1 (Th1) and Th17, or Th2 responses that are associated with the clinical features of disease progression [12]. The acquired phenotype of a T cell that differentiates from a naïve T cell is determined by its type of interaction with an APC as well as other factors that include the microenvironment, co-signaling molecule expression, type and load of Ag, and the intramolecular signals transduced [12]. A thorough discussion of the molecular mechanisms of these conditions is beyond the scope of this review and readers are directed towards several excellent reviews [10, 13-18].
Peripheral tolerance can be induced in vivo using a variety of technologies (Figure 1). For Ag-specific tolerance, the Ag is presented by APCs in the presence of low levels of co-stimulatory molecule expression and in the absence of other activating stimuli (i.e. absence of inflammation, infectious agents, and other pathologies) [3, 19]. These specific interactions aid in driving Ag-specific effector T cells towards an unreactive state (anergy or deletion) or induce regulatory T cells (Tregs) that can modify the activity of other T cells [4]. To drive immune responses towards tolerance, the Ag must be delivered to the appropriate cell types and initiate a cascade of tolerogenic signaling pathways. Other technologies, such as biomaterial scaffolds, mimic immune privileged sites in the body and can bolster tolerogenic responses through modulation of the local microenvironment. In this review, we will briefly introduce natural mechanisms of peripheral tolerance that will serve as a backdrop for an in-depth discussion of the state-of-the-art technologies available to reprogram immune cells to induce Ag-specific immune tolerance. Systematically, we will discuss technologies that promote tolerogenic responses by acting on APCs, lymphocytes, and by the creation of immune privileged sites using examples for the treatment autoimmune disease, allograft transplantation, and allergy as each of these therapies has unique immunological characteristics that motivate/influence the design of new technologies.
Figure 1.
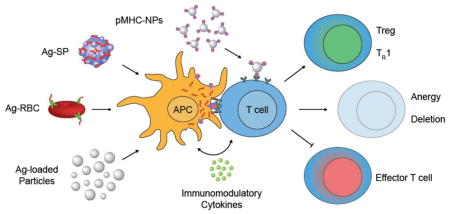
Highlighted approaches of technologies implemented for antigen-specific tolerance induction. Most antigen-specific tolerance strategies result in reprogramming lymphocytes through antigen presenting cells (APCs), however, there are platforms that target T cells and specifically recognize their autoreactive T cell receptors. Inspired by the natural clearance of apoptotic cells which results in peripheral tolerance maintenance, antigen has been delivered by various platforms including antigen-coupled splenocytes (Ag-SP), erythrocyte-targeted peptides (Ag-RBC), and antigen-loaded synthetic particles. These carriers are internalized, processed by APCs, and induce tolerogenic costimulation and soluble signaling pathways that direct T cell phenotypes away from immunogenic effector T cell activation and toward regulatory T cells (Tregs), anergy, or deletion. Direct interaction of particle-bound peptide-major histocompatibility complexes (pMHC-NPs) with antigen-experienced T cells can induce a tolerogenic regulatory-like TR1 phenotype that can mitigate immune-mediated disease progression.
2. Peripheral tolerance mechanisms
The development of T cell tolerance to self-Ags begins in the thymus and is refined in the periphery. As mentioned earlier, negative selection is not completely effective and autoreactive T cells that escape to the periphery are potentially harmful since they may provoke immune responses towards common Ags such as those in food or organ tissues [20]. T cells evade negative selection through multiple mechanisms that include low TCR avidity, TCR crossreactivity, and incomplete self-Ag representation by thymocytes [21]. When autoreactive T cells reach the periphery, their activity is modulated by anergy and deletion mechanisms. However, if either of these mechanisms fails, T cells can become activated due to the presence of activating stimuli such as inflammation, infection, or other pathologies that lead to the enhanced expression of co-signaling molecules that aid in the development of effector T cell responses. This autoreactivity can propagate the development of autoimmunity, however, the body has acquired mechanisms to restrain self-Ag-reactive T cells.
Activation of naïve T helper (Th) cells has been generally assumed to be the result of 2 signals: (1) TCR stimulation (signal 1) and (2) co-signaling molecules (signal 2). However, data that has been recently accumulated provided evidence for a 3 signal model for T cell activation, where signal 3 is provided by inflammatory cytokines such as interferon-gamma and interleukin-12 [22]. To enable TCR stimulation, proteolytically processed antigenic peptides are presented on the surface of APCs on major histocompatibility (MHC) complexes and recognized by cognate TCRs on T cells [3, 18]. A diverse repertoire of co-signaling molecules exists that function as either co-stimulatory or co-inhibitory receptors that influence the outcome of TCR signaling [23, 24]. The effects of co-signaling molecules are Ag-independent and act to modify signal 1. T cell co-signaling pathways have diverse immune regulation functions that can control effector, memory, and Treg functions [25]. Furthermore, the presence of inflammatory cytokines in the microenvironment aid in Th1 polarization. This complex interplay between co-stimulatory molecules, co-inhibitory molecules, and cytokines directs downstream immunity or tolerogenic signaling pathways and is critical to understand the type and strength of immune responses generated. These findings have given rise to the concept of the tidal model of co-signaling, where changing environmental conditions can lead to dynamic cell-surface interactions and intracellular signaling [23]. Self-tolerance and immune homeostasis are naturally maintained as self-Ags are presented to T cells in the absence of inflammation through steady-state processes such as clearance of apoptotic cells, exosomes, and by other cell death mechanisms involved in peripheral T cell tolerance.
2.1. Apoptotic cells
The clearance of apoptotic cells is a natural process that acts to maintain homeostasis and promotes the maintenance of peripheral tolerance through noninflammatory pathways. This process is unlike another cell death process, pyroptosis, which results in the activation and release of pro-inflammatory cytokines such as IL-1p and IL-18 that may hinder tolerance induction [26, 27]. For apoptosis, a combination of signals regulate dead cell clearance (efferocytosis) and aids to ensure that immunity is not generated towards self-Ags [28]. Dysfunction of these natural clearance mechanisms has been shown to result in autoimmunity [29]. When a cell dies, phosphatidylserine normally present on the inner leaflet of the plasma membrane is externalized and serves as an apoptosis indicator [30, 31]. In one pathway, the interaction of apoptotic membrane-associated ligands (Growth arrest specific-6 and Protein S) with Tyro-3, Axl, and Mer (TAM) receptor kinases on dendritic cells (DCs) interferes with Toll-like Receptor (TLR) signaling and prevents DC maturation [32]. Evidence supports that the TAM family interactions affect other phagocytic pathways which do not necessarily depend on phosphatidylserine binding. Integrin-based systems and scavenger receptors, for example, are implicated in TAM interaction and have been targeted by tolerogenic technologies but may not necessarily involve phosphatidylserine [33].
An important co-stimulatory pathway involved in apoptosis and maintenance of peripheral tolerance involves the programmed death-1 (PD-1) receptor and the corresponding ligands PD-L1 (known also as B7-H1 or CD274) and PD-L2 (known also as B7-DC or CD273) [34]. PD-1 is part of the cluster of differentiation 28 (CD28)/cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) superfamily and is expressed on a variety of cells, including T cells, B cells, monocytes, natural killer cells, and myeloid cells. PD-L1 is commonly expressed on non-activated T cells, B cells, dendritic cells, and macrophages while PD-L2 is primarily expressed by macrophages and dendritic cells [35]. The PD-1PD-L1/2 pathway has been implicated in immune suppression in models of autoimmunity, transplantation, and allergy through a variety of mechanisms, including promotion of T cell apoptosis [34]. The complex mechanisms of PD-1-dependent immune suppression are discussed more fully in other reviews, but binding of PD-1 by PD-L1/2 typically results in cross-linkage between PD-1 and the TCR, leading to tyrosine phosphorylation of the tyrosine-based inhibitory or switch motifs (ITIM and ITSM, respectively) of PD-1 [36, 37]. The binding of Sarcoma Homology 2 domain Phosphatase 1 or 2 (SHP-1 or SHP-2) to these phosphorylated regions limits activation of the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathway by dephosphorylation of relevant signaling molecules, eventually resulting in increased apoptosis, decreased cell proliferation, and decreased IL-2 production, among other suppressive outcomes [36, 37].
2.2. Naturally occurring exosomes
Exosomes are 30-100 nm cell-derived extracellular vesicles that are produced during intraluminal vesicle formation of endosomal membranes [38, 39]. The mechanism of exosome formation is not entirely clear, yet likely involves the endosomal sorting complex or ceramide-dependent lipid raft formation [40]. Exosomes are frequent carriers of micro/messengerRNA (mi/mRNA), proteins/lipids, and other cellular material. Evidence suggests that Tregs can also produce exosomes and frequently utilize this mechanism of gene transfer to direct T cells towards tolerogenic phenotypes [41]. Additionally, recent studies support that Treg exosomes may target and influence cells other than T cells towards stable tolerance induction through a variety of pathways, including antibody inhibition, cytokine modulation, or presentation of exosome-delivered Ags [40, 42].
2.3. Non-specific deletion
In peripheral tolerance, T cell deletion occurs through apoptotic cell death via apoptosis stimulating fragment (Fas)- and Bcl-2-interacting mediator of cell death (Bim)-regulated pathways. T cells express Fas (CD95) and, following Ag and IL-2 signaling, FasL (CD178). Activation of Fas by FasL or certain antibodies initiates activation-induced cell death (AICD), involving the formation of the death-inducing signaling complex (DISC) and downstream activation of apoptotic caspases. Alternatively, Bim may act as antagonist survival protein Bcl-2 to induce apoptosis through mitochondrial membrane permeabilization [20].
3. APC Reprogramming
APCs are skilled phagocytes that are well-adapted to process and present Ag derived from native and foreign sources. This makes them a natural target for immunotherapies. APCs efficiently scavenge and traffic to T cell compartments, so carrier-based tolerance strategies rely less on targeted delivery, and more on Ag (signal 1) and co-signaling (signal 2), and cytokine (signal 3) modulation. This section discusses strategies for the delivery of Ag to APCs to promote tolerogenic phenotypes in the context of autoimmunity, allograft transplantation, and allergy.
3.1. Cell-based therapeutics
Cell-based technologies have been developed to induce Ag-specific tolerance by exploiting naturally-occurring peripheral tolerance processes responsible for maintaining immune system homeostasis. Skewing APCs toward tolerogenic phenotypes ex vivo has demonstrated Ag-specific tolerance and has been extensively reviewed elsewhere [43-45]. Using cells as tolerogenic agents typically utilizes ex vivo biochemical modification, however in situ modification has been successfully demonstrated [46-48]. Cell-based carriers have the advantage of closely recapitulating the body's natural clearance mechanisms such as apoptotic cell clearance resulting in secretion of inhibitory cytokines, increased expression of co-inhibitory molecules, and expansion of Ag-specific Tregs [49]. However, ex vivo manipulation of autologous patient or donor cells requires extreme care and must be conducted using good laboratory practices [50, 51]. The high costs and standards for procedural care may limit the translational potential for cell-based therapies that require ex vivo handling. Cell-based therapies that utilize in situ targeting strategies (e.g. Ag targeting to red blood cells) represent an “off-the-shelf” option for harnessing autologous cells destined for clearance by APCs [52]. Together, technologies that recapitulate the natural clearance of apoptotic cells represent promising cell-based strategies for inducing Ag-specific tolerance.
3.2. Particle-based therapeutics
Nanotechnology has demonstrated great potential to revolutionize the treatment of disorders with an underlying immunological pathogenesis. Nano/microparticles comprised of safe and widely-available biocompatible materials have gained traction as relevant surrogates for cell-based carriers to induce Ag-specific tolerance [53]. Synthetic materials offer the ability to achieve fine control over the structure, surface properties, and various interactions between particles and biological systems (i.e. nanobio interactions) [54], that is not always possible using cell-based platforms. Modulation of various physicochemical properties such as composition, size, and surface charge can direct biological responses [53, 55-58].
Tolerogenic responses have been achieved in vivo using Ag-associated particles or particles engineered to deliver non-specific immune modulators such as small molecule immune suppressants, anti-sense oligonucleotides, short-interfering RNA, and others [59-62]. The carrier itself has also been shown to modulate immune responses for some applications [63]. Particles are combined with disease-relevant peptides or proteins, either through surface-coupling or encapsulation-based approaches, to obtain Ag-specificity. Importantly, some particle platforms induce tolerance by delivering Ag alone [64], while others require the co-incorporation of immune modifiers such as rapamycin, (2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester) (IDO), IL-10, or others [65-67]. This requirement for an immune modifier is attributed to the many differences in particle physicochemical properties and differences in biological mechanisms that have not been directly compared.
Particles have been fabricated with a variety of natural and synthetic materials, yet the most common composition of particles developed to modulate immune responses have been comprised of biocompatible and biodegradable polymers such as poly(lactide-co-glycolide) (PLGA), polylactide (PLA), and co-polymers thereof such as PLA-poly(ethylene glycol) (PLA-PEG) [53, 65]. Particles comprised of inorganic materials such as gold [66, 68], or iron oxide and quantum dots [69] have also shown success to induce tolerance. Regardless of the particle composition, to be effective, the particle must deliver its therapeutic payload at a sufficient level and distribute to the appropriate site of action in the body.
For tolerance induction, delivery of tolerogenic Ags using particles provides numerous advantages over delivery of soluble Ags. A summary of particles that have been demonstrated to promote Ag-specific immune regulation are detailed in Table 1. Through fine-tuning of the particle size (approximately 500 nm), the biodistribution can be tailored to target tolerogenic organs such as the liver and spleen [64, 70-72]. Furthermore, in contrast to soluble Ag, particles deliver a bolus of Ag to APCs which may result in lower therapeutic doses and safer Ag profiles. Directing the Ag distribution in the body is especially important for antibody-mediated immune reactions such as allergy, where encapsulation of Ags within particles alleviates potentially deadly side effects such as IgE-mediated anaphylaxis (Figure 2) [73]. Lastly, the surface properties of particles such as charge can be modulated to mimic naturally occurring apoptotic bodies (i.e. highly negative) by coating the particle surface with synthetic (poly(ethylenealt-maleic anhydride)) or natural materials (phosphatidylserine) to mediate uptake by cell debris-clearance pathways [74, 75].
Table 1. Nanoparticles investigated for antigen-specific immune regulation delivering protein or peptide antigens.
| Indication | Particle type | Antigen | Important finding | Refere nce |
|---|---|---|---|---|
| EAE | Ag-coupled PLGA and PS | PLP139-151; PLP178-191; MBP35-55 | MARCO scavenger receptor mediated tolerance induction | [64] |
| EAE | Ag-coupled PLGA | PLP139-151; PLP178-191 | Lower negative charge on particles resulted in improved efficacy | [74] |
| EAE | Ag-coupled PLGA | OVA323-339; PLP139-151 | Increased Ag conjugation and particle concentration enhanced Ag presentation and reduced co-stimulatory expression | [154] |
| EAE | Ag-encapsulated PLGA | OVA323-339; PLP139-151; PLP178-191 | Tolerance induction was not completely dependent on the spleen | [70] |
| EAE | Ag-polymer conjugate PLGA | OVA323-339; PLP139-151; PLP178-191 | Modular Ag loading, negligible burst release, tolerance induction to multiple epitopes | [94] |
| EAE | Ag-encapsulated PLGA and IL-10 encapsulated PLGA | MOG35-55 | Subcutaneous prophylactic administration reduced clinical disease. Co-administration of IL-10 PLGA was necessary to suppress disease | [67] |
| EAE | Ag and rapamycin co-encapsulated PLGA/PLA-PEG | PLP139-151 | Significantly reduced clinical disease score when Ag co-encapsulated with rapamycin. | [65] |
| EAE | Poly(maleic anhydride-alt-1-octadecene)-coated superparamagnet ic iron oxide nanocrystals | MBPAC-1-9 (4Tyr); MOG35-55 | Ag delivery to LSECs by particles induced Ag-specific Tregs and suppressed clinical disease | [69] |
| EAE | Ag and ITE-loaded | MOG35-55; PLP139-151; | Co-encapsulation of ITE with particles expanded Tregs | [66] |
| PEGylated gold | PLP178-191 | and suppressed clinical disease | ||
| EAE | Peptide-MHCII complex-conjugated iron oxide | N/A | Tolerance mediated by expansion of Ag-specific TR1-like cells and suppressive regulatory B cells | [133] |
| Diabetes | Peptide-MHCI complex-conjugated iron oxide | N/A | Expanded CD8+ T cells with regulatory potential but conventional memory-like phenotype | [149] |
| Diabetes | Peptide-MHCII complex-conjugated iron oxide | N/A | Tolerance mediated by expansion of Ag-specific TR1-like cells and suppressive regulatory B cells | [133] |
| Glomerular | Ag-coupled latex | OVA | Tolerance in the liver is | [97] |
| nephritis | dependent on KCs in a noninflammatory microenvironment | |||
| Collagen | Ag-encapsulated | CII | Oral administration of | [155] |
| induced | PLGA | particles suppressed arthritis | ||
| arthritis | symptoms | |||
| Collagen | Peptide-MHCII | N/A | Tolerance mediated by | [133] |
| induced | complex- | expansion of Ag-specific | ||
| arthritis | conjugated iron oxide | TR1-like cells and suppressive regulatory B cells | ||
| Proteoglyc | Ag-encapsulated | Hsp 70- | Intranasal delivery of | [156] |
| an induced | PLGA and | peptide | particles suppressed arthritis | |
| arthritis | PLGA-TMC | mB29a | symptoms | |
| Islet | Ag-coupled | Donor cell | Full MHC-mismatched | [71] |
| transplant | PLGA | lysate | murine allogeneic transplantation was achieved in 20% of recipients and improved to 60% with short course rapamycin | |
| Bone | Ag-encapsulated | Dby, Uty | Delivery of CD4 Dby epitope | [49] |
| marrow | PLGA | prevented transplant | ||
| transplant | rejection and delivery of CD8 epitope Uty did not induce tolerance | |||
| Hemophilia | Ag-encapsulated PLGA/PLA-PEG | Factor VIII | Significantly reduced antibody formation when Ag co-encapsulated with rapamycin. | [65] |
| Hemophilia | Ag-conjugated liposomes | Factor VIII | Suppression of antibody responses and prevented bleeding when delivered with CD22 ligand | [119] |
| Anti-drug antibody | Ag-conjugated liposomes | OVA; MOG1-120 | Suppression of antibody responses when delivered with CD22 ligand | [119] |
| Anti-drug antibody | Soluble Ag and rapamycin-encapsulated PLGA/PLA-PEG | OVA; OVA323-339; adalimumab; pegsiticase | Delivery of rapamycin in particles and Ag within 1 day of particle administration was necessary to suppress antibody responses | [118] |
| Allergy | Ag-encapsulated PLGA/PLA-PEG | OVA; OVA323-339 | Significantly reduced antibody formation when Ag co-encapsulated with rapamycin. | [65] |
| Allergy | Liposomes | OVA | Suppression of antibody responses when delivered with CD22 ligand | [119] |
| Allergy | Ag-encapsulated PLGA | Bet v 1 | Subcutaneous administration of particles modulated Th2 response | [157] |
| Allergy | Ag-encapsulated PLGA | OE109-130 | Intranasal administration of particles suppressed IgE and IgG1 production but increased IgG2a | [158] |
| Allergy | Ag-encapsulated PLGA | OVA | Inhibited Th2 responses in models of allergic airway inflammation | [73] |
PLGA (poly(lactide-co-glycolide)); PS (polystyrene); PLA-PEG (polylactide-poly(ethylene glycol)); TMC (trimethyl chitosan); LSEC (liver sinusoidal endothelial cell); KC (Kupffer cell); OVA (Ovalbumin); PLP (proteolipid protein); MOG (myelin oligodendrocyte protein); MBP (Myelin basic protein); CII (type II collagen); Bet v 1 (Birch pollen allergen); OE (Olive allergen); ITE (2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester)
Figure 2.
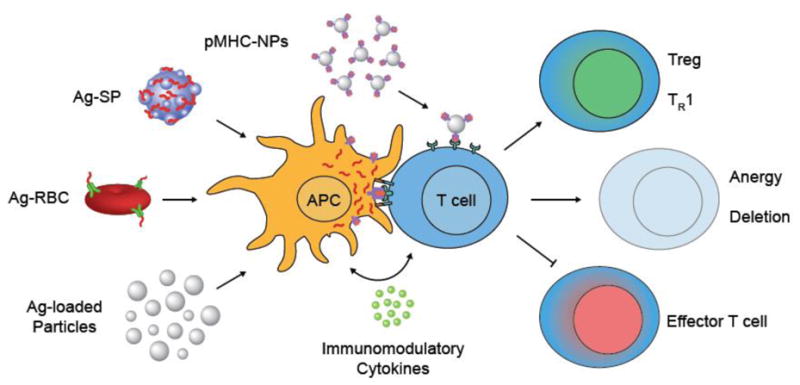
Mode of antigen-association with particles affects the risk of anaphylaxis following intravenous administration in individuals with prior antigen sensitization. Antigen is associated with particles by surface-coupling (NP-Ag) or by encapsulation (NP(Ag)) methods. In vivo, granulocyte activation occurs when NP-Ag or soluble antigen is recognized by circulating IgE antibodies or binds to pre-bound IgE on granulocytes. Cross-linking of IgE on granulocytes triggers degranulation and subsequent release of histamine and other inflammatory mediators that cause increased permeability, distension of blood capillaries, and anaphylaxis. (i) Binding of antigen-specific IgE to NP-Ag can trigger granulocyte activation. (ii) Binding of antigen-specific IgG to NP-Ag reduces potential granulocyte activation but can result in off-targeted biodistribution and reduce tolerance induction. (iii) Pre-mature antigen release from NP(Ag) can result in granulocyte activation but to a lesser extent than NP-Ag. (iv) NP(Ag) with negligible rate of antigen release reduces the risk of granulocyte activation and enables unaffected distribution to the liver and spleen to induce tolerogenic responses.
3.3. Autoantigen delivery
There are at least 81 types of autoimmune diseases that are heterogeneously observed in humans and their estimated worldwide prevalence is 4.5% [76] .These diseases are associated with complex pathologies in which the immune cells of the body attack healthy cells and tissues resulting in a state of persistent inflammation and chronic tissue destruction. Many immunotherapies are not Ag-specific and target signaling pathways that curb pathogenic T cells in active autoimmune disease, however they are not always effective [77]. Notably, the difficulty in developing therapies for autoimmune diseases, such as multiple sclerosis (MS), is that the primary disease-specific Ags involved in occurrence and progression are numerous and not always well-defined. However, proteins found in the tissues targeted by autoimmune diseases are good starting targets when designing Ag-specific therapies. For example, in MS, known autoAgs are associated with the myelin sheath proteolipid protein (PLP), myelin basic protein (MBP), and myelin oligodendrocyte protein (MOG). Delivery of disease-relevant Ags using cell- and particle-based carriers to APCs results in the initiation of tolerogenic signaling cascades that can reprogram autoreactive responses. In this section, we describe the various cell- and particle-based approaches to induce Ag-specific immune tolerance to treat autoimmunity.
3.3.1. Cell-based approaches
For the past three decades, autoAg-coupled syngeneic splenocytes (Ag-SPs) have been investigated for their ability to induce tolerance in models of autoimmunity (Figure 1) based on direct and indirect T cell interaction pathways [78, 79]. These Ag carriers have demonstrated tolerance to autoAg in Th1/17-mediated autoimmune models of experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, and the non-obese diabetic (NOD) model of type 1 diabetes (T1D) [80, 81]. After intravenous infusion, Ag-SPs accumulate in the marginal zone of the spleen where they are phagocytosed by marginal zone macrophages. Ag-SP uptake is followed by IL-10 production and upregulation of PD-L1 by macrophages [82]. Tolerance was not attained in IL-10 knockout mice, or mice treated with anti-PD-L1 antibody, suggesting that IL-10 and PD-L1 are necessary for Ag-SP-mediated tolerance. Direct presentation of Ag-SPs to T cells has been demonstrated to result in unresponsiveness, but studies coupling whole protein to splenocytes or using MHC-deficient or MHC-mismatched splenocytes have shown that the indirect presentation pathway is sufficient for tolerance [80]. Furthermore, the accumulation of Ag-SPs in the spleen was necessary, as autoAg tolerance was not achieved in models of splenectomy or subcutaneous administration.
The reality of human MS is that treatment needs to be effective in individuals with pre-existing disease and that the variety of autoAgs is not well-characterized. Due to epitope spreading during chronic autoimmunity, a person may have several Ags causing the pathogenesis of their disease [83]. A study by Smith et al. evaluated the feasibility of preventing EAE using Ag-SP coupled with an array of autoAgs (i.e. PLP139-151, PLP178–191, MBP84–104, and MOG92–106) [84]. EAE clinical scores and delayed type-hypersensitivity (DTH) responses indicated that disease suppression in the therapeutic tolerance model was different than in the prophylactic model where an increase in anti-inflammatory cytokine production (IL-10 and TGF-β) was observed. This study, in addition to the years of experimental success by Ag-SPs, provided justification for a first-in-man clinical trial using Ag-coupled cells to treat MS.
In 2013, a clinical trial was published that built on the work of Miller et al. using Ag-coupled cells for the treatment of autoimmune disease. In this study, autologous peripheral blood mononuclear cells (PBMCs) were coupled with seven known myelin peptides described to be potentially antigenic in MS. This trial demonstrated that the treatment was safe, and patients that received doses of Ag-PBMCs higher than 1 × 109 cells showed a decrease in Ag-specific T cell responses following therapy [51]. This study provided evidence that autologous cells could be used to induce tolerogenic responses in humans, although further studies using larger cohorts of patients will be required to demonstrate its wide-scale applicability and versatility.
Another tolerance therapy that takes advantage of the body's natural apoptotic clearance mechanisms targets autoAgs to circulating erythrocytes (Ag-RBC) (Figure 1). Kontos et al. have utilized Ag-conjugates that targeted murine glycophorin A-binding (TER119) on erythrocytes [48]. After intravenous infusion, the Ags become associated with erythrocytes and are cleared by naturally tolerogenic pathways. This treatment resulted in cross-presentation of Ag, and proliferation of CD8+ T cells with upregulated PD-1 and annexin-V indicating potential exhaustion and deletion outcomes. In the NOD T1D model, complete prevention of hyperglycemia was demonstrated with injections of erythrocyte targeting fusion peptides containing the diabetogenic mimetope peptide p31 (Figure 3). By utilizing in situ association to cells bound for apoptotic clearance, ex vivo manipulation of cells has been avoided, making this cell-based strategy a strong candidate for clinical translation.
Figure 3.
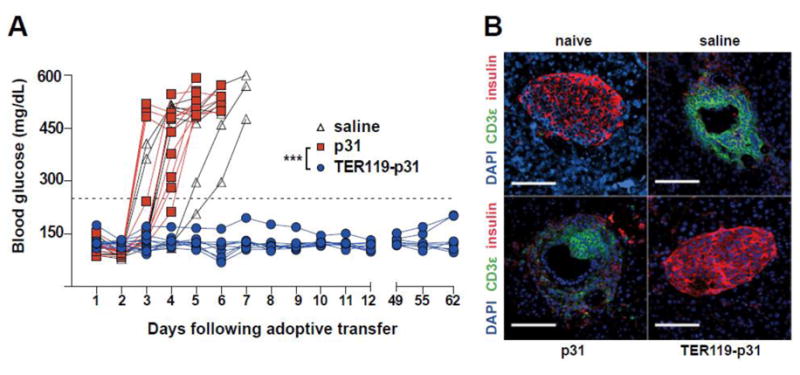
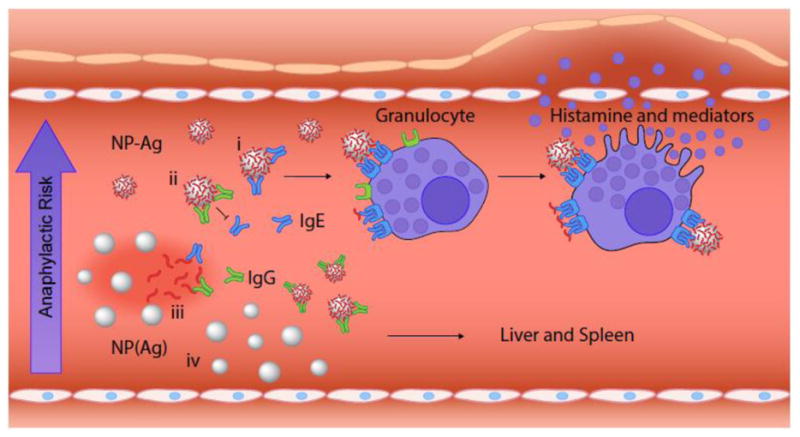
Erythrocyte-binding TER119 scFv antibodies fused with autoantigen (p31) specifically target red blood cells in situ after intravenous administration and induce antigen-specific tolerance in a type 1 diabetes model. (A) TER119-p31 induces tolerance and results in normoglycemia. Normoglycemic NOD/ShiLtJ mice received adoptive transfer of diabetogenic BDC2.5 CD4+ T cells and 3 intravenous treatments of saline, p31, or TER119-p31 (n =8, n = 9, and n = 9, respectively) over the first week. ***P < 0.0001. (B) Immunohistochemistry of pancreatic islets excised 4 days after treatment and stained for CD3ε T cells (green), insulin (red), and nuclei (blue). Saline-treated and untargeted autoantigen mimetope p31 resulted in T cell infiltration and islet destruction in contrast to mice treated with red blood cell-binding autoantigen (TER119-p31) which prevented T cell infiltration and preserved insulin production. (Scale bar = 100 μm). Reproduced from [48] with permission.
Expanding on the work of immunoglobulin G-mediated tolerance, B cells have been retrovirally transduced to produce conjugates of the IgG heavy chain and Ags of interest (peptide-IgG) [85]. In one example of this technology, the HIV TAT protein was fused with a peptide and IgG to form TAT-Ag-IgG, with the TAT protein functioning to mediate cell entry of the relevant Ag, as shown in previous studies [86, 87]. B cells activated with LPS and reprogrammed by TAT-MOG35-55-IgG displayed the ability to reduce EAE disease score when injected 10 days after immunization with MOG35-55, but interestingly did not reduce EAE disease score when injected one week prior to disease induction. In NOD T1D, B cells incubated with a fusion protein incorporating islet Ag B9-23 (TAT-B9-23-IgG) displayed a delayed onset of diabetes when administered prophylactically [85]. In each of these studies, delivery of irrelevant peptide-transduced B cells were unable to reduce or delay disease progression, suggesting Ag-specificity of the platform [85]. Preliminary mechanistic studies indicate that the tolerance induction by transduced B cells required CD4+CD25+ Tregs as well as CTLA-4/B7-dependent interaction between B and T cells [88]. Similar successes were observed using this platform in murine models of hemophilia, experimental autoimmune uveitis, and arthritis, and represent a promising method of tolerance induction [89-93].
3.3.2. Particle-based approaches
Peripheral tolerance induction by recapitulating natural tolerance maintenance in the hematopoietic compartment has offered insight into potential avenues to develop particulate delivery systems that follow a similar mechanism of action. Intravenously injected 500 nm Ag-coupled carboxylated polystyrene particles or PLGA particles were found to localize in similar regions of the splenic marginal zone and liver as Ag-SPs and target specific scavenger receptors on APCs that play an active role in tolerance induction (Figure 1) [64]. These particles efficiently prevented the onset of EAE, prevented epitope spreading, and reversed the progression of EAE in a therapeutic disease model. Importantly, tolerance was dependent on the highly-negative charge (less than -30 mV) and size (500 nm) of the particles, which targeted their distribution to macrophages expressing macrophage receptor with collagenous structures (MARCO) in the spleen. In a follow up study, Ag-coupled PLGA particles induced tolerance in EAE as demonstrated by significantly reduced mean clinical scores, reduced DTH responses, and reduced central nervous system (CNS) immune infiltration of Th1/17 cells [74].
The previous examples have demonstrated that tolerance can be induced by particles with surface-coupled Ag. However, the covalent modification of the particle surface with Ags affects physicochemical properties of particles such as their size, charge, and solution stability [73, 74]. Therefore, encapsulation of Ag within particles represents a more advantageous method to deliver Ag in vivo. McCarthy et al. recently described the encapsulation of peptide Ags (OVA323-339, PLP139-151, and PLP178-191) into PLGA particles (PLGA(Ag)) to treat EAE [70]. PLGA(Ag) particles significantly abrogated EAE induction in vivo and inhibited Th1/17 Ag recall responses (proliferation, IFN-y, and IL-17a) in vitro. Corroborating previous studies using peptide-coupled PLGA particles, the biodistribution of intravenously injected PLGA(Ag) particles was found primarily in liver and to a lesser extent in the spleen and lungs. Previous studies using Ag-SP demonstrated the dependence of the spleen for tolerance induction [82], however, prophylactic tolerance induction by PLGA(Ag) particles was not solely dependent on the spleen [70]. Pearson et al. developed Ag-polymer conjugate PLGA (acNP) particles that displayed modular Ag loading (up to 150 μg peptide per mg particle), low burst release, and minimally exposed surface Ag [94]. In vitro, acNPs were effective at inducing Tregs in a co-culture model of BMDCs, naïve OTII T cells, and TGF-p1. Treg induction was dependent on Ag loading and particle concentration, where the highest Ag loading in acNPs enabled a 10-fold lower concentration to be used. Furthermore, acNPs were effective at treating EAE that was induced by single of multiple peptides (Figure 4). This approach holds promise to deliver several therapeutic antigens that can tolerize multiple disease relevant epitopes.
Figure 4.
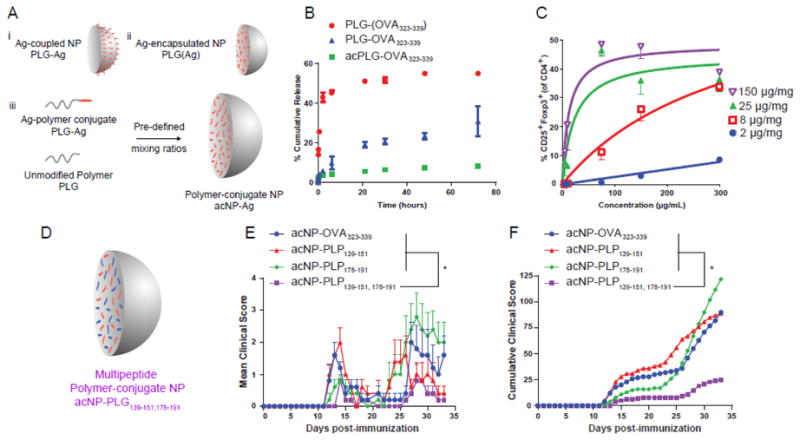
Antigen-polymer conjugate nanoparticles display favorable physicochemical and biological properties for tolerance induction. (A) Schematic representation of Ag-coupled, Ag-encapsulated, and polymer-conjugate nanoparticles. (B) Release profile of NP(OVA323-339), NP-OVA323-339, and acNP-OVA323-339. (C) Regulatory T cell induction is dependent on nanoparticle concentration. BMDCs were treated for 3 hr with various concentrations of acNP-OVA323-339 (2, 8, 25, 150 μg/mg loading). Excess acNP-OVA323-339 particles were subsequently washed from the cell surface prior to addition of OT-II T cells and 2 ng/mL of TGF-β1. (D) Schematic representation of antigen-polymer conjugate nanoparticles delivering multiple Ags. (E) Clinical scores of SJL/J mice treated with 1.25 mg of acNP-OVA323-339 (8 μg/mg OVA323-339), acNP-PLP139-151 (8 μg/mg PLP139-151), acNP-PLP178-191 (8 μg/mg PLP178-191), or acNP-PLP139-151,178-191 (8 μg/mg PLP139-151 and 8 μg/mg PLP178-191) and immunized with PLP139-151 and PLP178-191 in CFA to induce R-EAE 7 days later. (F) Corresponding cumulative clinical score for mice treated with particles (n = 5). Differences between disease courses of different treatment groups were analyzed for statistical significance using the Kruskal-Wallis test (one-way ANOVA non-parametric test) with Dunn's multiple comparisons test (p < 0.05) [94].
Additional in vivo tolerance mechanisms have implicated the liver as critical for tolerance induction since it is exposed to numerous self and non-self Ags regularly and therefore must balance immunity and tolerance [95]. Many APCs in the liver have a low abundance of MHC and co-stimulatory molecule expression, which can reduce immune cell activation. Kupffer cells (KCs) and liver sinusoidal endothelial cells (LSECs) internalize a majority of particles administered intravenously [96]. Heymann et al. utilized carboxylated latex particles to study the mechanism of action of tolerance induction using particle-delivered Ags. The liver was specifically identified as important for tolerance induction where Kupffer cells were demonstrated to be associated with Ag-presentation that induced CD4+ T cell arrest and expansion of naturally occurring Tregs [97]. In a model of Ag-specific glomerular nephritis, particle delivery protected against kidney damage by reducing T cell infiltration, reduced glomerular damage, and reduced periglomerular infiltration. However, in models of liver injury, Kupffer cells lost expression of their tolerogenic phenotype, and tolerance was abrogated due to the redistribution of Ag to infiltrating monocyte-derived macrophages away from the hepatic phagocyte compartment. Similarly, the liver was determined to play an important role in tolerance induction by Carambia et al. [69]. Small poly(maleic anhydride-alt-1-octadecene)-coated particles induced tolerance though LSECs. Mice treated with particles displayed higher frequencies of Tregs and tolerance induction was abrogated when Tregs were inactivated using anti-CD25 antibody. These studies provided support that the liver plays an important role in tolerance induction by particles, however, it should be noted that due to distinct differences in particle physicochemical properties (size, charge, composition), the mechanism of action for each particle platform must be determined experimentally and cannot be assumed.
Co-delivery of immune modulating agents such as immune suppressants, cytokines, or other immune modifiers with Ag by particles is sometimes necessary to induce Ag-specific tolerance. One of the most commonly delivered agents for immune modulation is rapamycin, a mammalian target of rapamycin (mTOR) inhibitor. Encapsulation of rapamycin within PLGA nanoparticles induced a tolerizing phenotype in vitro where particles decreased the expression of maturation markers MHCII, CD86, and CD40 expression and increased levels of TGF-β using bone-marrow derived dendritic cells (BMDCs) [98]. Maldonado et al. developed tolerizing nanoparticles (tNPs) comprised of a mixture of PLGA and polyethylene glycol)-polylactide (PEG-PLA) that co-encapsulated Ag and rapamycin. These tNPs delivered antigenic peptides or whole proteins and could tolerize against both cellular and humoral immune responses. The co-encapsulation of rapamycin with Ag into tNPs was necessary to induce tolerance, whereas co-delivery of soluble rapamycin with Ag-encapsulated particles did not elicit tolerogenic effects but rather propagated humoral immunity [65]. In another example, immune modifying agent 2-(1-indole-3-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE), an activator of the aryl hydrocarbon receptor transcription factor (AhR), was loaded into 60 nm gold particles with MOG35-55 peptide [66]. In the EAE model, particle treated mice displayed expanded populations of Tregs and abrogated EAE clinical disease symptoms. In a subsequent study, the same gold particles delivering (3 cell proinsulin and ITE were found to induce a tolerogenic phenotype in dendritic cells by inducing Socs2-mediated inhibition of NF-κB and suppression of inflammatory cytokines as well as the promotion of Treg generation in vivo [68]. MOG35-55 peptide and IL-10 were separately encapsulated into 200 nm PLGA particles for the treatment of EAE by the subcutaneous route of administration. Co-administration of IL-10 containing NPs was required to significantly mitigate EAE clinical scores, while IFN-γ and IL-17 were significantly decreased [67].
Immune polyelectrolyte multilayers (iPEMs) have been built on calcium carbonate templates to promote immunological tolerance [99]. Delivery of a regulatory antagonist ligand of TLR9, GpG oligonucleotide, along with a MOG-triarginine peptide was hypothesized as able to restrain the pro-inflammatory signaling and redirect T cell differentiation from inflammatory populations and towards regulatory phenotypes such as Tregs. Interestingly, iPEMs reduced TLR9 signaling, reduced dendritic cell activation, and polarized myelin-specific T cells towards a tolerogenic phenotype and function. In an EAE model, iPEMs abrogated disease, which coincided with Treg expansion and reduced IL-17, IL-6, and IFN-γ production, with no effect on T cell proliferation.
3.4. Alloantigen Delivery
Achieving allogeneic tolerance to multiple foreign Ags represents a significant challenge to the field of transplantation. Transplantation of allogeneic or ‘non-self’ tissues between genetically different individuals of the same species leads to a T cell-mediated immune response resulting in rejection and graft destruction. Current methodologies for improving transplant tolerance are similar to those of treating autoimmunity (such as the chronic use of immunosuppressive agents) that are associated with numerous risks [18]. The severity of the immune response against the transplanted tissue depends on the differences in Ag between the donor and recipient as well as the intragraft expression of inflammatory cytokines following transplantation [100]. There are three pathways through which allograft recognition and rejection can occur: 1) direct, 2) indirect, and 3) semi-direct. In the direct pathway, recipient T cells recognize MHC molecules present on the surface of donor APCs. In the indirect pathway, recipient APCs internalize allogeneic proteins and present it to recipient T cells on recipient MHC molecules. It has been observed that CD4+ T cells with indirect specificity can provide “unlinked” T cell help to CD8+ T cells with direct specificity, which does not agree with the commonly-accepted “linked” model of immune response in which the same APCs activate both CD4+ and CD8+ T cells. An explanation for this phenomenon is the acquisition of MHC molecules from donor APCs by recipient APCs and subsequent direct presentation to CD8+ T cells. This is known as the semi-direct pathway and is under investigation [101].
The wide breadth of Ags necessary to tolerize against in allogeneic transplants and the multiplicity of allorejection pathways pose significant challenges to the development of new tolerance therapies. The primary Ags that influence rejection in mice are MHCs, and in humans are called human leukocyte Ags (HLAs). These HLA proteins are of two types, Class I and Class II, and are involved in Ag presentation and recognition, such as between APCs and T cells. Both major and minor mismatches in HLAs between the donor and recipient are highly deterministic of the success of a transplant. In addition to the major Ags, mismatches in numerous other proteins of the cell, which are termed minor Ags, can also mediate rejection. Tolerance induction strategies have aimed to affect the direct or indirect pathways of allograft rejection and are the focus of many Ag carrier therapies.
3.4.1. Cell-based approaches
Immunosuppression-free allogeneic transplant tolerance has been achieved in mice by pretreatment with ethylene carbodiimide (ECDI)-fixed splenocytes (ECDI-SP) that mimicked donor apoptotic cells. Long-term tolerance to allogeneic islets was achieved by two infusions of ECDI-SP from the donor mouse strain, 1-week prior to, and 1-day post, kidney-capsule transplant [47]. The effect was shown to be strain-specific and dependent on ECDI treatment. The durability of this tolerance was challenged with a second islet transplant 60 days after the initial treatment. Donor strain-matched islets were protected by the tolerance, whereas third party islets led to rejection as indicated by hyperglycemia. The authors attributed the allograft tolerance induction, but not maintenance, to an increase in Tregs in the spleen as Treg ablation early (day -9), but not late (day 15), prevented engraftment. Furthermore, graft rejection dominated in PD-L1 deficient mouse suggesting a potential role for the PD-1 pathway in mediating ECDI-SP allotolerance. Since the kidney capsule is not considered a translatable transplantation site, this tolerance protocol was combined with islet transplantation on a PLGA microporous scaffolds into the mouse peritoneal fat, an analogue to human omentum [102]. Tolerance for islets transplanted on the PLGA scaffold was as effective as the kidney capsule, with both providing a site for successful long-term allogeneic islet engraftment. Combined with tolerance directed toward islet Ag InsB9-23, allotolerance by ECDI-SP offers a potential solution for islet replacement therapy for T1D patients.
Further investigation of ECDI-SP induced allotolerance revealed contribution of both direct and indirect tolerance mechanisms [46]. In vivo selective depletion experiments showed that DCs, but not B cells or macrophages, were necessary for ECDI-SP induced allotolerance. Furthermore, there were T cell responses to APC-processed EDCI-SPs (indirect) as well as presentation of un-phagocytosed ECDI-SP alloAg to T cells (direct). After uptake of ECDI-SPs, CD11c+ DCs upregulated co-inhibitory molecules PD-L1 and PD-L2 and caused the rapid expansion of T cells, followed by clonal deletion and hindered migration to the site of engraftment. The directly stimulated T cells experienced weak proliferation and became unresponsive to subsequent stimulatory signals. Thus, in addition to the expansion of Tregs in the spleen, graft, and graft draining lymph nodes, ECDI-SPs induced tolerance by causing clonal deletion and anergy of alloreactive T cells. In other disease models, ECDI-SPs have been used to prolong cardiac allografts (indefinitely if combined with a short course of rapamycin) [103], and have induced long-lasting xenograft tolerance to rat islets transplanted into mouse kidney capsules when combined with transient B cell depletion [104].
3.4.2. Particle-based approaches
The induction of donor-specific tolerance to transplanted cells and organs remains of utmost importance to mitigate allograft rejection. The first uses of particles to improve allotransplant acceptance involved the delivery of small molecule immune suppressants such as rapamycin or calcineurin inhibitors [60]. Achieving Ag-specific suppression of anti-donor immune responses is complicated due to the wide array of major and minor Ags. PLGA particles have been used to induce donor-specific tolerance and mediate long-term acceptance of full MHC-mismatched allografts using an allogeneic islet transplant model (BALB/c to C57BL/6). Donor Ags were obtained from the lysate of BALB/c splenocytes, with the lysate Ag conjugated to the surface of 500 nm PLGA particles using ECDI chemistry. Delivery of PLGA particles with surface-coupled donor Ags to transplanted C57BL/6 mice led to tolerance in 20% of recipients. In combination with a short course of low-dose rapamycin at the time of transplant, tolerance was greatly improved to 60% (Figure 5) [71]. In another study, PLGA particles were used to induce tolerance in minor histocompatibility Ag sex-mismatched C57BL/6 model of bone marrow transplantation. Peptide Ags Dby and Uty are respective CD4+ and CD8+ T cell Ags that mediate male bone marrow transplant rejection in females. Interestingly, delivery of Dby peptide either by conjugation to the surface or encapsulation promoted transplant tolerance. However, delivery of Uty peptide using particles did not induce tolerance. In this model, depletion of Tregs using anti-CD25 antibody did not alter tolerance induction, suggesting other potential tolerance induction mechanisms [49].
Figure 5.
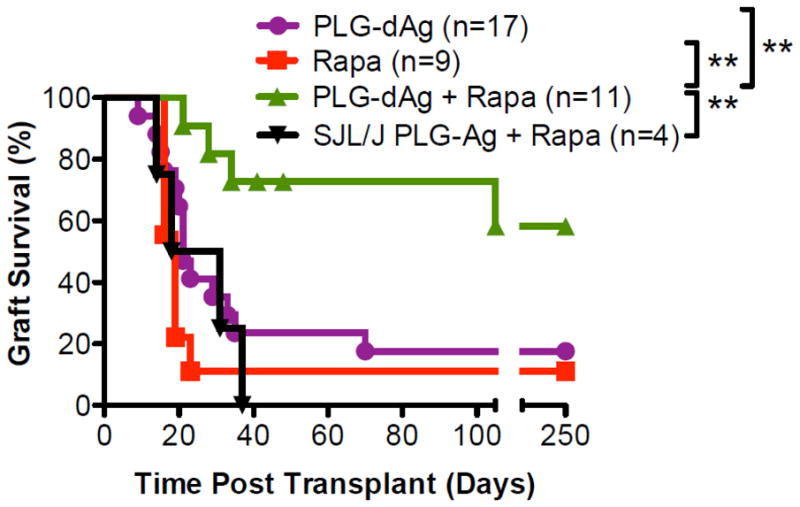
A short course of low dose rapamycin synergizes with PLG-donorAg (dAg) to enhance tolerance efficacy. Recipient mice (C57BL/6) receiving donor (BALB/c) PLG-dAg injections at days 7 and +1 in combination with a 4-day course (days 1, 0. +1, and +2) of low dose (0.1 mg/kg) rapamycin demonstrated significantly greater islet allograft survival (n=11) compared with mice treated with rapamycin alone (n=9), BALB/c PLG-dAg alone (n=17), or PLG particles coupled with lysate proteins from a third party donor SJL/J (n=4). **p < 0.01. Reproduced from [71] with permission from Elsevier.
Exosomes have also been used to induce tolerance in allogeneic transplant models. Exosomes derived from PBMCs have been used to prolong cardiac transplants in a C57BL/6 to BALB/c model [105]. Song et al. could extend cardiac allografts 40 days using 2 doses of exosomes from donor C57BL/6 mice. The authors attributed the tolerance to a skewing of Th2 T cells toward a regulatory phenotype. While they ruled out the possibility of direct induction of Tregs by exosomes, they concluded that MMP1 carried by the donor exosomes was necessary for Treg induction in a Th2:DC co-culture system.
In the same C57BL/6 to BALB/c cardiac transplant scheme, another group of investigators used immature dendritic cell exosomes (imDex) to prolong cardiac graft survival [106]. Cardiac allografts were extended 25 days using 3 doses of imDex from C57BL/6 donor mice. Interestingly, infusion of more or less than 10 μg resulted in minimal graft protection. When rapamycin was administered for 11 days, half of the grafts were maintained indefinitely. The tolerogenic effects were donor-specific and dependent on rapamycin. The same group also demonstrated liver transplant tolerance using imDex [107]. In a rat model, imDex from donor rats (Brown Norway) prolonged liver graft survival about 25 days in recipient Lewis rats. Instead of administering rapamycin to extend graft survival, recipients were infused with donor-specific recipient Tregs, which resulted in an indefinite survival of 70% of liver grafts. The mechanism by which DC-derived exosomes induce tolerance is unknown. Like ECDI-SP and PLGA NPs mentioned earlier, intravenously delivered DC-derived exosomes have accumulated in liver Kupffer cells as well as marginal zone macrophages and DCs [108]. Like their immature DC source, these exosomes express low levels of MHC class I and II as well as subimmunogenic levels of co-stimulatory molecules. In the cardiac allograft model, the data suggested that splenic T cells become hyporesponsive to alloAg challenge, and there was an observed increase in Foxp3+ splenic T cells following a co-treatment with imDex and rapamycin [106]. Co-administration and in vivo proliferation of Tregs seems to support their role in exosome induced allotolerance [107]. It is unknown whether the T cell interactions result from direct presentation of exosome Ags by fusion with host APCs, or indirect presentation of exosome Ags following APC phagocytosis [107].
3.5. Allergen Delivery
Allergic diseases such as asthma and food allergies are becoming increasingly common in developed nations. Immediate allergic reactions (Type 1), involve an overreaction of the immune system and the formation of IgE antibodies. The development of allergic responses is reliant on CD4+ Th2 cells as they produce cytokines that induce immunoglobulin class switching to IgE. IgE binds with high affinity to mast cells and basophils that release pro-inflammatory mediators once they encounter the allergen [11]. At current, the most effective treatment regimen consists of avoidance or other specific immune tolerance (SIT) strategies that deliver soluble Ag at increasing dosages to mitigate symptoms. SIT is usually carried out by repetitive subcutaneous injections or sublingual delivery of increasing doses of the allergen [109]. However, SITs feature numerous potential issues especially for treating food allergies that pose a risk of developing adverse reactions including life-threatening anaphylaxis. Additionally, SIT by subcutaneous injections often requires 3-5 years of treatment, involving multiple sessions per week in the build-up phase [110]. Recent work has focused the delivery of antigen to the lymphatic system by ultrasound guided intranodal injections which show promise in decreasing the injection frequency and duration of SIT regimes. A study comparing a 3 year subcutaneous SIT regime (54 injections) to a 2 month intralymphatic regime (3 injections) demonstrated equivalent tolerance to pollen as measured by hay fever symptoms, skin reactivity, and decreased allergen-specific serum IgE [111]. A similar study of 3 intralymphatic inguinal resulted in a decrease in seasonal allergic symptoms and nasal inflammatory leukocytes compared to adjuvant delivery without pollen (aluminum hydroxide only) [112]. Other allergen-specific tolerance strategies include using Tregs, anti-IgE, or by blocking Th2 cytokines. SIT to allergens leads to a shift in Th2 and Th17 towards a Th1 response and Treg induction [113]. This results in reduced production of IgE production, IL-4, and IL-13 and an increased production of IFN-γ and IgG subtypes that can act as blocking antibodies and capture the allergen before activating effector cells [114].
B cells express a variety of B cell receptor (BCR) inhibitory co-receptors that aid in setting a threshold for B cell activation. Among them are CD22 and SIGLEC-G (SIGLEC-10 in humans), and members of the SIGLEC (sialic-acid binding Ig-like lectin) immunoglobulin family that recognize sialic acid-containing glycans of glycoproteins and glycolipids and ligands [115]. Targeting Ag-specific B cells is a mechanism to induce systemic humoral tolerance. Favorable approaches to induce B cell tolerance involve taking advantage of mechanisms to suppress B cell activation.
3.5.1. Cell-based approaches
One of the methods currently available to induce Ag-specific immune tolerance is the use of Ag-SP. As described earlier in this review, Ag-SP have been prophylactically and therapeutically tolerogenic in models of Th1/17-mediated autoimmune disease. An early demonstration of Ag-SP to treat Th2-associated models was performed by Smarr et al. [85]. This study utilized two models of allergy: peanut hypersensitivity as well as ovalbumin (OVA)-induced allergic airway inflammation. Whole peanut extract (WPE) or whole OVA protein was coupled to splenocytes using ECDI, and the Ag-SPs were intravenously infused resulting in decreased local and systemic Th2-related disease. In the peanut hypersensitivity model, characterized by mast cell-mediated anaphylaxis, prophylactic WPE-SP administration diminished the levels of Ag-specific IgE (but not IgG), eosinophil numbers, and IL-3, IL-4, and IL-13 in recall assays that resulted in a prevention of anaphylactic symptoms in an oral WPE challenge. The authors observed anaphylactic symptoms when Tregs were inactivated by anti-CD25 antibody indicating a partial role for Tregs in the tolerance mechanism. In the OVA-induced allergic airway model, prophylactic OVA-SP treatment similarly reduced IgE, eosinophil numbers, and Th2-associated cytokines (in vivo and ex vivo). IgG levels were also reduced and the tolerogenic effects were Treg independent. Regardless of allergy model and Treg activity, treating with Ag-specific Ag-SPs decreased IgE levels indicating inactivation of B cell class switching which the authors posit is a result of previously observed decreased CD40L levels on helper T cells (previously observed) [85, 116]. Though IgE levels were decreased compared to controls, the levels were increased compared to Ag-inexperienced animals, however, this did not result in anaphylaxis.
The increased use of biologics in medicine has led to the development of antidrug antibodies (ADAs) in patients. This undesired immune recognition has hindered the effective use of biologics and has created a demand for drug-specific tolerance. The usage of erythrocyte-targeted Ags has recently been applied to prevent development of ADAs against acute lymphoblastic leukemia (ALL) therapeutic enzyme Escherichia coli L-asparaginase (ASNase) [117]. An infusion of ASNase conjugated to glycophorin A-binding peptide (ERY1-ASNase) significantly reduced anti-ASNase IgG titers against 6 weekly challenges, compared to infusion of untargeted ASNase. This reduction in anti-ASNase titer was observed in all IgG subclasses. The treatment showed no effect on the development of humoral responses to OVA indicating that this therapy is both effective and Ag-specific in mice.
3.5.2. Particle-based approaches
PLG particles have been used to tolerize against OVA in a Th2-mediated allergic airway inflammation model both pre- and post-sensitization [73]. Since activation of mast cells is APC independent, avoiding Ag recognition by antibodies is desirable. Encapsulation of allergens into particles shielded their detection by IgE auto-antibodies on mast cells and basophils thus preventing any undesired side effects associated with SIT and Ag-coupled particles (Figure 2). Encapsulation of OVA protein within PLGA particles (PLGA(OVA)) eliminated the presence of particle surface-associated protein that could lead to deleterious biological effects such as anaphylaxis. In a prophylactic disease model, PLGA(OVA) particles inhibited the production of anti-OVA IgE and suppressed the production of Th2-mediated cytokines IL-4, IL-5, IL-13, IL-10, and IL-17. In a therapeutic model of allergic airway inflammation, PLGA(OVA) particles inhibited the Th2 responses and airway inflammation but led to increases in OVA-specific IgE (Figure 6) [73]. Importantly, the encapsulation of OVA into PLG particles eliminated OVA-specific IgG and IgE binding to the surface of the particle preventing anaphylaxis.
Figure 6.
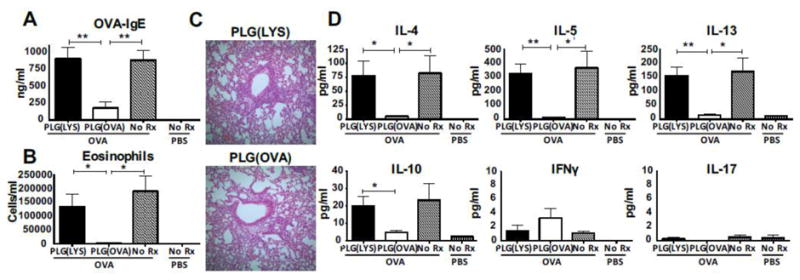
Prophylactic treatment with PLG(OVA) inhibits Th2-induced airway inflammation. Naive female BALB/c mice (n = 5) were treated i.v. with 2.5 mg PLG(OVA) or control PLG(LYS) on days −7 and +7 relative to i.p. immunization with 10 μg of OVA in 3 mg of alum or alum alone on days 0 and +14 before aerosol challenge with 10 mg/mL OVA for 20 min on days +28–30 and sample collection on day +31. LYS, lysozyme. (A) Concentration of serum OVA-IgE was determined by sandwich ELISA. (B) Lungs were flushed with BALF, total cell counts were determined, and samples were cytospun onto slides before DiffQuik staining for differential cell counts of bronchoalveolar lavage eosinophils. (C) Lungs were fixed in formalin and stained with H&E. (D) Cytokines from BALF supernatant were analyzed by Milliplex. Results are mean ± SEM and are representative of three separate experiments. *P < 0.05; **P < 0.01. Reproduced with permission from [73].
Particles loaded with rapamycin that co-delivered PEGylated uricase or adalimumab demonstrated suppression of ADAs. Interestingly, the ability to suppress ADA production was dependent on the timing of rapamycin-encapsulating particle administration. Significant suppression of anti-keyhole limpet hemocyanin (KLH) antibodies was only achieved if the protein was administered within 1 day of particle administration [118]. Macauley et al. demonstrated that SIGLEC-engaging tolerance-inducing antigenic liposomes (STALs) (liposomes that displayed Ag as well as glycan ligands for CD22) could induce Ag-specific B cell apoptosis. CD22-dependent tolerance induction was demonstrated for both T cell-independent (nitrophenol) and T cell-dependent (hen egg lysozyme) Ags as Ag-specific antibody titers were significantly reduced. Furthermore, STALs induced tolerance to FVIII using a hemophilia mouse model. STALs significantly reduced anti-FVIII titers and prevented bleeding [119].
3.6. Carrier-free therapeutics
Although much recent focus has been on cell-based and particle-based technologies for bulk delivery of Ag, molecular-scale carrier-free platforms (less than 200 kDa) offer distinct advantages. The unique properties of carrier-free technologies such as their small size enable enhanced lymphatic drainage and bioavailability from the subcutaneous route of administration. For example, subcutaneously administered Ag-graft polymers (less than 100 kDa) showed improved tolerogenic properties compared to similarly functionalized particles (500 nm) due to their decreased size and increased solubility that improved interstitial drainage and bioavailability of the Ag-grafted polymer [120]. Furthermore, soluble carrier-free platforms often incorporate specific binding motifs that target specific cell types or interrupt inflammatory signaling pathways associated with cell activation. Fusion peptides comprised of Ag-antibody bioconjugates have demonstrated improved outcomes by targeting delivery of autoAg to tolerogenic APC subtypes [121]. Alternatively, polymer platforms co-grafted with autoAg and peptide inhibitors of co-stimulation have demonstrated Ag-specific tolerance that is dependent on interrupting APC:T cell interactions [120]. Soluble carrier-free systems deliver similar payloads as particle and cell-based systems, but their small scale, solubility, and engineered specificity make these systems unique.
3.6.1. Fusion proteins
Fusion proteins are a class of biologics that have seen remarkable success in research and clinical applications. Many design variations exist, but most are composed of a protein/peptide of interest linked to an antibody (often an IgG subclass) at the fragment crystallizable (Fc) region. Conjugation to IgG provides several benefits to therapeutics, including increased serum half-life (through increased size and, consequently, reduced clearance) and recycling (through increased interaction with the neonatal Fc receptor, which protects IgG from degradation) [122, 123].
Multiple tolerogenic fusion proteins have demonstrated the ability to induce Ag-specific tolerance. Recently, blood clotting protein FVIII conjugated to human IgG1 (rFVIIIFc) demonstrated the ability to diminish anti-FVIII antibody responses associated with FVIII protein replacement therapy. In mice, this treatment was accompanied by development of Tregs and a general shift towards a tolerogenic phenotype, as indicated by up-regulation of IL-10, TGF-β, IL-35, and IDO-1, as well as down-regulation of IL-17 measured by real time polymerase chain reaction [124]. Importantly, mice pretreated with rFVIIIFc retained the ability to mount an antibody response to both dinitrophenol and OVA, demonstrating the Ag-specificity of the platform. The mechanisms behind these observations are being investigated, and studies support a role of the Fcy class of Fc receptors, which are particularly important in phagocytosis, and the neonatal Fc receptor, which the authors suggests implicate B cells and dendritic cells [124].
Antibody-Ag fusion proteins combine solubility and specificity to achieve efficient delivery of autoAg to specific cell types. Coupling of autoAg to anti-DEC205 antibody resulted in in vivo targeting of the endocytic receptor DEC205 on tolerogenic dendritic cells and demonstrated tolerance in models of EAE, NOD T1D, and experimental autoimmune arthritis (EAA) [125-127]. In the EAE model, intraperitoneal injection of anti-DEC205 antibody linked with PLP, but not isotype fusion protein or anti-DEC205 with irrelevant peptide, ameliorated disease onset. The protective effects in this context were attributed to hindered proliferation of IL-17-producing T cells and anergy in the remaining Ag-specific T cell population [125]. In the NOD T1D mouse model, it was shown that delivery of β cell peptide mimotope on anti-DEC205 antibody results in cross-presentation of peptide to CD8+ T cells and subsequent clonal deletion [126]. In EAA, decreased disease scores were associated with lower B cell counts and IgG1 and IgG2a serum levels resulting from insufficient T follicular helper cell differentiation. Mechanistic studies showed that targeting autoAg to migratory DC subsets using anti-DEC205-Ag and anti-Langerin-Ag prevented EAE onset and caused an increase in Foxp3+ T cells, whereas targeting lymphoid-resident DCs with anti-DCIR2-Ag and anti-Treml4-Ag only mitigated symptoms [121]. These outcomes demonstrate the specificity and utility of using antibodies to deliver autoAg to tolerogenic APC subsets.
3.6.2. Soluble Ag arrays
AutoAg are being coupled to targeting proteins and peptides to deliver Ags to specific cells and internalization pathways to dictate tolerogenic Ag presentation. Soluble Ag arrays (SAgAs) are composed of hyaluronic acid polymers with grafted Ag (e.g., PLP139-151) and a co-stimulatory molecule inhibitor (B7 inhibiting peptide). Hyaluronic acid is employed as it is hydrophilic, and its relatively large molecular weight (≈106 Da) provides the opportunity to conjugate many Ags/inhibitor molecules. SAgAs that co-delivered B7-inhibiting peptides with PLP139-151 decreased clinical disease scores in the EAE model with three subcutaneous injections. The resulting decrease in co-stimulation in the context of TCR stimulation is thought to lead to the observed tolerance, as disease was only alleviated when the inhibitor and Ag were presented on the same polymer and not when any other combination of components was administered. Specifically, it is suggested that the coincident binding of signals 1 and 2 interferes with APC interaction and dampens T and B cell clonal expansion [120, 128].
4. Lymphocyte Reprogramming
T cells play a central role in orchestrating adaptive immune responses and their phenotypes dictate the pathophysiology of immunity and tolerance [129]. As such, they have been the target of many tolerogenic therapies. Specifically, strategies for treating autoimmunity have explored Ag-specific Tregs, and in the case of allogeneic transplant, polyclonal populations [130]. Tregs have been extensively studied for their ability to suppress Ag-specific effector T cells, and even dampen the inflammatory effects associated with inflammatory environments that lead to epitope spreading [83]. Reprogrammed T cells with chimeric Ag receptors (CAR T cells) have been studied extensively in the field of cancer immunotherapy, but have recently been applied to immune tolerance. These engineered cells have been used as effector cells to delete autoreactive cells, and more commonly as regulatory cells that specifically target autoAg and alloAg [107, 131, 132]. Particle-based systems, though common for implementing tolerance through APCs, have also been designed to target T cells with specific Ag-reactivity resulting in a regulatory TR1-like phenotype [133]. These T cell-targeted approaches for Ag-specific tolerance are discussed in the following section.
4.1. Regulatory T cell induction
CD4+CD25+Foxp3+ Tregs are a critical component of peripheral tolerance mechanisms, and deficiency of Tregs is associated with severe autoimmune diseases and chronic inflammation [134]. Regulatory T cells suppress immune responses to a broad range Ags and indirectly limit immune inflammation-mediated tissue damage through multiple mechanisms [135]. Tregs actively suppress activated T cells through the production of anti-inflammatory cytokines (IL-10, IL-35, and TGF-β), cytolysis, metabolic disruption, and by targeting the maturation or function of DCs [134]. There are three main subclasses of Foxp3+ Tregs that are classified by the location of their differentiation: thymus-derived Tregs (tTregs), peripherally-derived Tregs (pTregs), and in vitro-induced Tregs (iTregs) [136]. tTregs are effectors of central tolerance and have been shown to permit differentiation of Th1 and Th17 cells in the lymphatic system, but prevent circulation of these effector T cells into the tissues containing cognate antigens [137, 138]. In contrast, iTregs, which are generated in vitro by stimulating T cells in the presence of TGF-β, are thought to suppress APCs by local release of anti-inflammatory cytokines such as IL-10 [137, 139, 140]. As a result, the APCs become less potent primers of effector T cells. iTregs have shown adept regulatory behavior, but have a phenotype that is considered incomplete compared to in vivo derived tTregs and pTregs. The role of pTregs is thought to be complementary to tTregs in maintaining tolerance especially by transiently bolster tolerance in the peripheral compartment [141]. Strategies to increase Tregs and restore a healthy T cell balance have been extensively researched and reached clinical trials.
A method to generate Ag-specific Tregs in vivo has been developed that was effective at treating EAE and NOD diabetes in mice. Mice were treated with a systemic sublethal irradiation or depletion of B and CD8+ T cells followed by administration of autoAg peptides. The irradiation of cells induced apoptosis which triggered professional phagocytes to produce TGF-β, under which the autoAg peptides directed naïve CD4+ T cells to differentiate into Foxp3+ Treg cells instead of effector T cells [142].
4.2. CAR T cells
The development of chimeric Ag receptor (CAR) technology more than 25 years ago for enhanced immunity by Eshher et al., has also been employed to reverse autoimmunity [132, 143]. In vitro expanded CD4+CD25+ Tregs were engineered with CARs directed toward the carcinoembryonic Ag (CEA), which is overexpressed in inflamed colon tissue and colon cancer. In colitis-induced by adoptive transfer of CEA-specific effector CAR T cells, adoptive transfer of CEA-specific CAR Tregs (1:1 TregTeff) increased the 4-wk survival to 75% compared to 25% by non-specific CAR Tregs. Tolerance was also tested in the azoxymethane-dextran sodium sulfate (AOM-DSS) induced model that combines the pathogenesis of colitis and colon cancer. Here, treatment by CEA-specific CAR Tregs halved the average colitis score compared to non-specific CAR Tregs. Interestingly and paradoxically, treatment with CEA-specific Tregs also significantly decreased tumor burden. Whereas the anti-inflammatory effects of Tregs are often implicated in the unchecked progression of tumors, here they acted to reduce the tumor burden. The authors postulated that the CAR Tregs acted to reduce the inflammatory mediators associated with intestinal polyps resulting in a reduction in neoplastic exacerbation.
The usage of CD19+ B cell depleting CAR T cells has demonstrated effective treatment of acute lymphoblastic leukemia (ALL), and has been recently adapted to target and eliminate autoreactive B cells responsible for causing pemphigus vulgaris (PV) [131, 144-146]. PV is currently treated with systemic corticosteroids, but the off-label use of rituximab (combined with IVIG therapy) has shown effectiveness in creating long-term remission. Unfortunately, the resulting B cell depletion by anti-CD20 treatment resulted in cases of patient infection and septicemia [147]. These chimeric autoantibody receptor (CAAR) T cells were used to selectively deplete B lymphocytes that produce antibodies against keratinocyte desmosome adhesion protein desmoglein 3 (Dsg3). The CAAR-T cell strategy utilizes human T cells transduced with lentiviral vectors to express transmembrane receptors with extracellular chimeric autoAg Dsg3 with an intracellular signaling CD173-CD3ζ domain [131]. The investigators demonstrated in vitro that these CAAR-T cells induce lysis of hybridomas producing antibodies against the extracellular cadherin domains on Dsg3, but not control hybridomas producing irrelevant antibodies. In vivo, the CAAR-T cells were evaluated in NSG mice infused with B cell hybridomas secreting antibodies against Dsg3 domains or with hybridomas cloned to express antibodies that target Dsg3 in human PV. Compared to control CAR treatment, Dsg3 CAARs depleted circulating autoAg-secreting B cells, reduced the anti-Dsg3 IgG levels in serum, and prevented mucosal blistering. Interestingly, the presence of soluble anti-Dsg3 IgG did not prohibit effectiveness of the CAAR-T cells in vitro or in vivo. This successful experimental treatment of PV shows the promise of using CAAR T cells to treat B cell-mediated autoimmune disorders in an Ag-specific manner.
In contrast to CAR T cells expressing autoAgs, others have used the chimeric receptor to home toward sites containing autoAg. Loskog et al. used a lentiviral vector system to express both a CAR single-chain variable fragment (scFv) for MOG as well as to stably express Foxp3 in CD4+ T cells [148]. After intranasal administration, the MOG-specific CAR Tregs, also transduced with GFP, were histologically observed in several regions of the brain and cerebellum. In the MOG-induced EAE model, MOG-specific CAR Tregs delivered at peak of disease resulted in complete remission of disease symptoms in ten days. Importantly, this treatment resulted in astrogliosis and remyelination. However, administration of mock-transduced T cells also resulted in a decrease in symptoms (but to a lesser extent). The therapeutic effect of these non-CAR T cells may have been affected by the naturally occurring Treg population, yet questions about specificity remain.
While most uses of CAR T cells for tolerance have focused on targeting specific autoAg epitopes, Levings et al. have developed a regulatory CAR T cell for treating alloreactivity by targeting HLA-A2 (A2-CAR Tregs), a major histocompatibility complex often implicated in transplant rejection [107]. The human CD25hiCD45RA+ A2-CAR Tregs expressed CD25, Foxp3, LAP, GARP, CTLA-4 expression. Compared to irrelevant control CAR T cells, injection of A2-CAR T cells with HLA-A2+ xenogeneic PMBCs resulted in a prolongation of survival and a 2-fold increase in the time to graft versus host disease (GVHD) onset. Additionally, the A2-CAR T cells maintained their Foxp3 expression for twice as long. When A2-CAR Tregs were infused into HLA-A2+ mice with HLA-A2- PBMCs, the mice experienced no observed tissue cytotoxicity (in contrast to infusion with CD25-CD45RA+ A2-CAR T cells).
4.3. Peptide-MHC complexes on particles
Peptide-MHC (pMHC) complexes attached to the surface of crosslinked dextran-coated or PEGylated iron oxide particles (pMHC-NPs) offer a new therapeutic strategy, targeted directly at specific T cells, to treat autoimmune diseases in a disease- and organ-specific manner. To date, pMHC-NPs have demonstrated efficacy in multiple mouse models of autoimmunity (diabetes, MS, collagen-induced arthritis) using MHC class I or II complexes (Figure 1) [133, 149]. Importantly, as the Ag diversity in autoimmune diseases is highly complex, it has been demonstrated that pMHC-NPs that carry subdominant disease-relevant Ags show similar biological effects towards inducing tolerogenic responses as dominant Ags. In the first study, it was initially hypothesized that the delivery of multiple Ags within MHC molecules was necessary to abrogate disease in a mouse model of diabetes [149]. However, it was found that functionalization of NPs with monospecific pMHC complexes suppressed T1D progression in pre-diabetic mice as well as restored normoglycemia in recently diagnosed diabetic mice. pMHC-NPs expanded autoAg-experienced CD8+ T cells that suppressed the activation and recruitment of cells with non-cognate specificities to islets. Furthermore, in a humanized mouse model of diabetes, particles coated with disease-relevant pHLA complexes restored normoglycemia. Studies showed that cognate T cells internalize these NPs without accumulating in CD11b+, CD11c+, or B cells. The ability to blunt disease progression was demonstrated to be due to the expansion of subsets of CD8+ T cells with regulatory potential but a conventional memory-like phenotype [133, 149].
More recently [133], Clemente-Casares et al. demonstrated that pMHCII-NPs could induce Ag-specific regulatory CD4+ T cell type 1 (TR1)-like cells in various mouse models of autoimmunity as well as in humanized mice (Figure 7). By using ten human or mouse autoimmune-disease-relevant pMHC-NPs, the reversion of pathology was shown to be independent of genetic background or type of disease. Note that peptide-functionalized particles in NP-Ag discussed earlier did not induce TR1 responses suggesting significantly different mechanisms of action. To induce the Ag-specific suppressive effects, pMHC-NPs rely on previous autoAg experience in the T cell repertoire whereas without previous experience, favorable outcomes were not observed. Disease suppression was dependent on IFN-γ and IL-10 as transgenic mice deficient in IFN-γ or IL-10 abrogated disease suppression. Taken together, the data from both studies support that the attachment of a single disease-related pMHC to NP could be used to suppress autoimmunity.
Figure 7.
pMHC-NPs suppress MOG-induced EAE in vivo. C57BL/6 mice were immunized with pMOG35-55. (A) EAE scores of mice treated from day 14 (n=4 each). (B) EAE scores of mice treated from day 21 (n=10, 7 and 3 from top). (C) Representative microglial IBA1 stainings and relative rank scores in the cerebellum of mice from B (n=4–5). Reproduced with permission from [133]. ©2016 Macmillan Publishers Ltd.
5. Immune privileged sites
Biomaterial scaffolds have been employed for several regenerative medicine and cell transplantation applications, and more recently, these scaffolds have been designed to modulate the local environment [4, 150]. The strategy aims to mimic natural sites in the body, such as testes or eye, that intrinsically have local immunomodulation [151]. Scaffolds have been implanted that release cytokines, co-transplant cells, or locally present T-cell apoptosis inducing factor FasL [152]. While these strategies have had some efficacy in delaying rejection or promoting long-term engraftment, these strategies are not typically Ag specific, which represents an opportunity for the field.
Microporous PLG scaffolds have been used to co-transplant islets along with Tregs specific for islet Ag to enhance graft survival [153]. Islet Ag (BDC2.5)-specific Tregs were cultured in vitro and were co-incorporated with 300 NOD scid gamma (NSG) islets before implantation within the abdominal cavity. In control scaffolds lacking Tregs, no insulin-producing islet cells were detected at day 25 of implant, while islets from scaffolds containing Tregs survived significantly longer which was associated with a significant survival benefit. Interestingly, a second implant possessing islets but lacking Tregs into the kidney capsule, 97 days after the scaffold implant, did not result in rejection of those islet cells, even after removal of the original scaffold. This protection of the second islet-only transplant demonstrated a systemic tolerance. Following removal of the scaffold and kidney capsule transplant, mice became hyperglycemic, confirming that the transplanted islets were responsible for euglycemia.
6. Conclusion and future directions
Ag-specific therapies offer numerous advantages to improve the treatment of diseases with immune-mediated pathology. However, significant challenges remain to be addressed regarding the heterogeneity and breadth of disease-relevant Ags, understanding the molecular mechanisms of tolerance induction, the refinement of the production and characterization of therapeutics, and identifying the most favorable applications to apply Ag-specific tolerance therapies.
The wide-scale application of Ag-specific immune tolerance technologies is complex, as disease progression can be mediated by a single or many Ags and multiple immune pathways are often involved. The wide-breadth of Ags implicated in clinical disease makes it difficult to produce Ag-specific therapies especially for autoimmunity, allograft transplantation, and allergy. In diseases such as MS, additional layers of complexity exist due to processes such as epitope spreading and the relative reactivity of a patient's cells to particular autoAgs [3]. In a small cohort of MS patients, PBMCs coupled with a library of seven relevant antigenic peptides (Ag-PBMCs) demonstrated promising results [51]. It is possible that the use of cell lysates or protein extracts containing a plethora of immunogenic epitopes may be more effective in these disorders, however, as the complexity of these Ag mixtures increases, the ease of formulation and characterization of the Ag carriers is likely to decrease. Furthermore, the low concentrations of individual Ags present in the extracts may not yield significant tolerogenic responses in vivo as it is likely that there is an Ag loading requirement necessary for tolerance induction. Future research targeted towards the discovery of additional epitopes will aid in the development of Ag-specific tolerogenic therapies.
Molecular mechanisms described for Ag-specific therapies are complex and not completely understood. The majority of efforts for the development of therapeutics have focused on inducing tolerogenic responses through APC or lymphocyte reprogramming (Figure 1). Modulation of signal 1 and signal 2 of T cell activation through APCs represents an important pathway to alter T cell activation and induce tolerance. Other mechanisms such as direct interactions of therapeutics and CAR T cells with lymphocytes to induce tolerogenic phenotypes such as Tregs and deletion represent additional methods to curb aberrant immune activation. However, as each technology is different, individual mechanisms of action need to be identified and should not be extrapolated from one to another.
Developing improved strategies to produce therapeutics that deliver peptide and protein Ags will be required to enable the large-scale progression of these technologies to the clinic. Cell-based technologies have shown promise in clinical trials, but may have limited translatability due to limited sources of cells, complicated ex vivo manipulations, and poor shelf-life. Particle-based therapeutics may address these issues and are expected to gain significant traction in these applications, however, difficulties regarding scale up and controlling physicochemical properties such as size, charge, and Ag release as well as methods to characterize the presence of relevant epitopes within the particle will need to be overcome.
There is an excellent opportunity to develop novel Ag-specific therapies to improve the efficacy and long-term usability of therapeutic proteins and antibodies for which the formation of anti-drug antibodies limits their clinical applicability. ADA-specific tolerance strategies may revitalize the use of previously discovered therapies. Since the Ags are well described when tolerizing against therapeutic proteins and limiting ADAs, the use of Ag-specific tolerance therapies for this purpose is highly promising.
Acknowledgments
This work was supported by the National Institutes of Health (grant number EB-013198). Conflict of interest disclosure: RMP, SDM, and LDS have financial interests in Cour Pharmaceuticals Development Co.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parish IA, Heath WR. Too dangerous to ignore: self-tolerance and the control of ignorant autoreactive T cells. Immunology and Cell Biology. 2008;86:146–152. doi: 10.1038/sj.icb.7100161. [DOI] [PubMed] [Google Scholar]
- 2.Richards DM, Kyewski B, Feuerer M. Re-examining the Nature and Function of Self-Reactive T cells. Trends Immunol. 2016;37:114–125. doi: 10.1016/j.it.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol. 2007;7:665–677. doi: 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
- 4.Luo X, Miller SD, Shea LD. Immune Tolerance for Autoimmune Disease and Cell Transplantation. Annu Rev Biomed Eng. 2016;18:181–205. doi: 10.1146/annurev-bioeng-110315-020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peleg AY, Husain S, Kwak EJ, Silveira FP, Ndirangu M, Tran J, Shutt KA, Shapiro R, Thai N, Abu-Elmagd K. Opportunistic infections in 547 organ transplant recipients receiving alemtuzumab, a humanized monoclonal CD-52 antibody. Clin Infect Dis. 2007;44:204–212. doi: 10.1086/510388. [DOI] [PubMed] [Google Scholar]
- 6.Calabrese LH, Zein N, Vassilopoulos D. Hepatitis B virus (HBV) reactivation with immunosuppressive therapy in rheumatic diseases: assessment and preventive strategies. Ann Rheum Dis. 2006;65:983–989. doi: 10.1136/ard.2005.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adami J, Gäbel H, Lindelöf B, Ekström K, Rydh B, Glimelius B, Ekbom A, Adami HO, Granath F. Cancer risk following organ transplantation: a nationwide cohort study in Sweden. Br J Cancer, BJC. 2003;89:1221–1227. doi: 10.1038/sj.bjc.6601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lerner A, Jeremias P, Matthias T. The World Incidence and Prevalence of Autoimmune Diseases is Increasing. Int J Celiac Dis. 2016;3:151–155. [Google Scholar]
- 9.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 10.Ingulli E. Mechanism of cellular rejection in transplantation. Pediatr Nephrol. 2010;25:61–74. doi: 10.1007/s00467-008-1020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2010;125:S73–80. doi: 10.1016/j.jaci.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaiko GE, Horvat JC, Beagley KW, Hansbro PM. Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology. 2008;123:326–338. doi: 10.1111/j.1365-2567.2007.02719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atassi MZ, Casali P. Molecular mechanisms of autoimmunity. Autoimmunity. 2008;41:123–132. doi: 10.1080/08916930801929021. [DOI] [PubMed] [Google Scholar]
- 14.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3(+)CD25(+)CD4(+) natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 15.Bolon B. Cellular and molecular mechanisms of autoimmune disease. Toxicol Pathol. 2012;40:216–229. doi: 10.1177/0192623311428481. [DOI] [PubMed] [Google Scholar]
- 16.Broide DH. Molecular and cellular mechanisms of allergic disease. J Allergy Clin Immunol. 2001;108:S65–S71. doi: 10.1067/mai.2001.116436. [DOI] [PubMed] [Google Scholar]
- 17.Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8:205–217. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 18.Priyadharshini B, Greiner DL, Brehm MA. T-cell activation and transplantation tolerance. Transpl Rev. 2012;26:212–222. doi: 10.1016/j.trre.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Podojil JR, Miller SD. Molecular mechanisms of T-cell receptor and costimulatory molecule ligation/blockade in autoimmune disease therapy. Immunol Rev. 2009;229:337–355. doi: 10.1111/j.1600-065X.2009.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957. doi: 10.1101/cshperspect.a006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderton SM, Wraith DC. Selection and fine-tuning of the autoimmune T-cell repertoire. Nat Rev Immunol. 2002;2:487–498. doi: 10.1038/nri842. [DOI] [PubMed] [Google Scholar]
- 22.Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. 2010;22:333–340. doi: 10.1016/j.coi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity. 2016;44:955–972. doi: 10.1016/j.immuni.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharpe AH. Mechanisms of costimulation. Immunol Rev. 2009;229:5–11. doi: 10.1111/j.1600-065X.2009.00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Y, Xu S, Chen H, He M, Deng Y, Cao Z, Pi H, Chen C, Li M, Ma Q, Gao P, Ji Y, Zhang L, Yu Z, Zhou Z. CdSe/ZnS quantum dots induce hepatocyte pyroptosis and liver inflammation via NLRP3 inflammasome activation. Biomaterials. 2016;90:27–39. doi: 10.1016/j.biomaterials.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. 2013;5:a008748. doi: 10.1101/cshperspect.a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 30.Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annual review of biophysics. 2010;39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 31.Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, Barcinski M, Brekken RA, Huang X, Hutchins JT, Freimark B, Empig C, Mercer J, Schroit AJ, Schett G, Herrmann M. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23:962–978. doi: 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 33.Lemke G, Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci. 2010;1209:23–29. doi: 10.1111/j.1749-6632.2010.05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Sci Rep. 2015;5:13110. doi: 10.1038/srep13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14:1212–1218. doi: 10.1038/ni.2762. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat Rev Immunol. 2015;15:45–56. doi: 10.1038/nri3790. [DOI] [PubMed] [Google Scholar]
- 38.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ribeiro MF, Zhu H, Millard RW, Fan GC. Exosomes Function in Pro- and Anti-Angiogenesis. Curr Angiogenes. 2013;2:54–59. doi: 10.2174/22115528113020020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chatila TA, Williams CB. Regulatory T cells: exosomes deliver tolerance. Immunity. 2014;41:3–5. doi: 10.1016/j.immuni.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T, Seabra MC, Wilson MS. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity. 2014;41:89–103. doi: 10.1016/j.immuni.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agarwal A, Fanelli G, Letizia M, Tung SL, Boardman D, Lechler R, Lombardi G, Smyth LA. Regulatory T cell-derived exosomes: possible therapeutic and diagnostic tools in transplantation. Front Immunol. 2014;5:555. doi: 10.3389/fimmu.2014.00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maldonado RA, von Andrian UH. Chapter 4 - How Tolerogenic Dendritic Cells Induce Regulatory T Cells. In: Frederick KFATHFMJWU, Alt W, Emil RU, editors. Advances in Immunology. Academic Press; 2010. pp. 111–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ten Brinke A, Hilkens CMU, Cools N, Geissler EK, Hutchinson JA, Lombardi G, Lord P, Sawitzki B, Trzonkowski P, Van Ham SM, Martinez-Caceres EM. Clinical Use of Tolerogenic Dendritic Cells-Harmonization Approach in European Collaborative Effort. Mediators Inflamm. 2015;2015:8. doi: 10.1155/2015/471719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hilkens CMU, Isaacs JD. Tolerogenic dendritic cell therapy for rheumatoid arthritis: where are we now? Clin Exp Immunol. 2013;172:148–157. doi: 10.1111/cei.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kheradmand T, Wang S, Bryant J, Tasch JJ, Lerret N, Pothoven KL, Houlihan JL, Miller SD, Zhang ZJ, Luo X. Ethylenecarbodiimide-fixed donor splenocyte infusions differentially target direct and indirect pathways of allorecognition for induction of transplant tolerance. J Immunol. 2012;189:804–812. doi: 10.4049/jimmunol.1103705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo X, Pothoven KL, McCarthy D, DeGutes M, Martin A, Getts DR, Xia G, He J, Zhang X, Kaufman DB. ECDI-fixed allogeneic splenocytes induce donor-specific tolerance for long-term survival of islet transplants via two distinct mechanisms. Proc Natl Acad Sci U S A. 2008;105:14527–14532. doi: 10.1073/pnas.0805204105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kontos S, Kourtis IC, Dane KY, Hubbell JA. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc Natl Acad Sci U S A. 2013;110:E60–E68. doi: 10.1073/pnas.1216353110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hlavaty KA, McCarthy DP, Saito E, Yap WT, Miller SD, Shea LD. Tolerance induction using nanoparticles bearing HY peptides in bone marrow transplantation. Biomaterials. 2016;76:1–10. doi: 10.1016/j.biomaterials.2015.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, Fouweather T, MacDonald C, Chadwick T, McColl E, Dunn J, Dickinson AM, Hilkens CMU, Isaacs JD. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis. 2017;76:227–234. doi: 10.1136/annrheumdis-2015-208456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, Schippling S, Miller SD, Sospedra M, Martin R. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med. 2013;5:188ra175. doi: 10.1126/scitranslmed.3006168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grimm AJ, Kontos S, Diaceri G, Quaglia-Thermes X, Hubbell JA. Memory of tolerance and induction of regulatory T cells by erythrocyte-targeted antigens. Sci Rep. 2015;5:15907. doi: 10.1038/srep15907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Getts DR, Shea LD, Miller SD, King NJ. Harnessing nanoparticles for immune modulation. Trends Immunol. 2015;36:419–427. doi: 10.1016/j.it.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pearson RM, Hsu Hj, Bugno J, Hong S. Understanding nano-bio interactions to improve nanocarriers for drug delivery. MRS Bull. 2014;39:227–237. [Google Scholar]
- 55.Jiao Q, Li L, Mu Q, Zhang Q. Immunomodulation of nanoparticles in nanomedicine applications. BioMed research international. 2014;2014:426028. doi: 10.1155/2014/426028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 57.Smith DM, Simon JK, Baker JR., Jr Applications of nanotechnology for immunology. Nat Rev Immunol. 2013;13:592–605. doi: 10.1038/nri3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pearson RM, Juettner V, Hong S. Biomolecular Corona on Nanoparticles: A Survey of Recent Literature and its Implications in Targeted Drug Delivery. Front Chem. 2014;2 doi: 10.3389/fchem.2014.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein-Barash H, Cantley W, Wong J, Cortez-Retamozo V, Newton A, Love K, Libby P, Pittet MJ, Swirski FK, Koteliansky V, Langer R, Weissleder R, Anderson DG, Nahrendorf M. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–1010. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fisher JD, Acharya AP, Little SR. Micro and nanoparticle drug delivery systems for preventing allotransplant rejection. Clin Immunol. 2015;160:24–35. doi: 10.1016/j.clim.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hlavaty KA, Luo X, Shea LD, Miller SD. Cellular and molecular targeting for nanotherapeutics in transplantation tolerance. Clin Immunol. 2015;160:14–23. doi: 10.1016/j.clim.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Serra P, Santamaria P. Nanoparticle-based autoimmune disease therapy. Clin Immunol. 2015;160:3–13. doi: 10.1016/j.clim.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 63.Getts DR, Terry RL, Getts MT, Deffrasnes C, Muller M, van Vreden C, Ashhurst TM, Chami B, McCarthy D, Wu H, Ma J, Martin A, Shae LD, Witting P, Kansas GS, Kuhn J, Hafezi W, Campbell IL, Reilly D, Say J, Brown L, White MY, Cordwell SJ, Chadban SJ, Thorp EB, Bao S, Miller SD, King NJ. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med. 2014;6:219ra217. doi: 10.1126/scitranslmed.3007563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, Miller SD. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol. 2012;30:1217–1224. doi: 10.1038/nbt.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maldonado RA, LaMothe RA, Ferrari JD, Zhang A, Rossi RJ, Kolte PN, Griset AP, O'Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH, Kishimoto TK. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci U S A. 2014;112:E156–165. doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109:11270–11275. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, Gigliotti CL, Boggio E, Clemente N, Comi C, Dianzani C, Boldorini R, Chiocchetti A, Reno F, Dianzani U. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine. 2014;32:5681–5689. doi: 10.1016/j.vaccine.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 68.Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, Tukpah AM, Babon JAB, DeNicola M, Kent SC, Pozo D, Quintana FJ. Tolerogenic nanoparticles inhibit T cell–mediated autoimmunity through SOCS2. Sci Signal. 2016;9:ra61–ra61. doi: 10.1126/scisignal.aad0612. [DOI] [PubMed] [Google Scholar]
- 69.Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, Reimer R, Heine M, Huber S, Waurisch C, Eychmuller A, Wraith DC, Korn T, Nielsen P, Weller H, Schramm C, Luth S, Lohse AW, Heeren J, Herkel J. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J Hepatol. 2015;6:1349–1356. doi: 10.1016/j.jhep.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 70.McCarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, Miller SD, Shea LD. An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy. Nanomedicine: NBM. 2017;13:191–200. doi: 10.1016/j.nano.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bryant J, Hlavaty KA, Zhang X, Yap WT, Zhang L, Shea LD, Luo X. Nanoparticle delivery of donor antigens for transplant tolerance in allogeneic islet transplantation. Biomaterials. 2014;35:8887–8894. doi: 10.1016/j.biomaterials.2014.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater. 2014;13:125–138. doi: 10.1038/nmat3780. [DOI] [PubMed] [Google Scholar]
- 73.Smarr CB, Yap WT, Neef TP, Pearson RM, Hunter ZN, Ifergan I, Getts DR, Bryce PJ, Shea LD, Miller SD. Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre- and postsensitization. Proc Natl Acad Sci U S A. 2016;113:5059–5064. doi: 10.1073/pnas.1505782113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, Miller SD. A Biodegradable Nanoparticle Platform for the Induction of Antigen-Specific Immune Tolerance for Treatment of Autoimmune Disease. ACS Nano. 2014;8:2148–2160. doi: 10.1021/nn405033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roberts RA, Eitas TK, Byrne JD, Johnson BM, Short PJ, McKinnon KP, Reisdorf S, Luft JC, DeSimone JM, Ting JP. Towards programming immune tolerance through geometric manipulation of phosphatidylserine. Biomaterials. 2015;72:1–10. doi: 10.1016/j.biomaterials.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hayter SM, Cook MC. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev. 2012;11:754–765. doi: 10.1016/j.autrev.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 77.Bluestone JA, Bour-Jordan H. Current and future immunomodulation strategies to restore tolerance in autoimmune diseases. Cold Spring Harb Perspect Biol. 2012;4:a007542. doi: 10.1101/cshperspect.a007542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miller S, Wetzig RP, Claman HN. The induction of cell-mediated immunity and tolerance with protein antigens coupled to syngeneic lymphoid cells. J Expt Med. 1979;149:758–773. doi: 10.1084/jem.149.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Expt Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Turley DM, Miller SD. Peripheral tolerance induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2212–2220. doi: 10.4049/jimmunol.178.4.2212. [DOI] [PubMed] [Google Scholar]
- 81.Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9-23 epitope and involves functional epitope spreading. J Autoimmun. 2012;39:347–353. doi: 10.1016/j.jaut.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo X, Terry RL, King NJ, Miller SD. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol. 2011;187:2405–2417. doi: 10.4049/jimmunol.1004175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 84.Smith CE, Miller SD. Multi-peptide coupled-cell tolerance ameliorates ongoing relapsing EAE associated with multiple pathogenic autoreactivities. J Autoimmun. 2006;27:218–231. doi: 10.1016/j.jaut.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smarr CB, Hsu CL, Byrne AJ, Miller SD, Bryce PJ. Antigen-fixed leukocytes tolerize Th2 responses in mouse models of allergy. J Immunol. 2011;187:5090–5098. doi: 10.4049/jimmunol.1100608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Becker-Hapak M, McAllister SS, Dowdy SF. TAT-mediated protein transduction into mammalian cells. Methods. 2001;24:247–256. doi: 10.1006/meth.2001.1186. [DOI] [PubMed] [Google Scholar]
- 87.Gump JM, Dowdy SF. TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol Med. 2007;13:443–448. doi: 10.1016/j.molmed.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 88.Lei TC, Scott DW. Induction of tolerance to factor VIII inhibitors by gene therapy with immunodominant A2 and C2 domains presented by B cells as Ig fusion proteins. Blood. 2005;105:4865–4870. doi: 10.1182/blood-2004-11-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Skupsky J, Zhang AH, Su Y, Scott DW. B-cell-delivered gene therapy induces functional T regulatory cells and leads to a loss of antigen-specific effector cells. Mol Ther. 2010;18:1527–1535. doi: 10.1038/mt.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matheu MP, Su Y, Greenberg ML, Blanc Ca, Parker I, Scott DW, Cahalan MD. Toll-like receptor 4-activated B cells out-compete Toll-like receptor 9-activated B cells to establish peripheral immunological tolerance. Proc Natl Acad Sci U S A. 2012;109:E1258–1266. doi: 10.1073/pnas.1205150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Agarwal RK, Kang Y, Zambidis E, Scott DW, Chan CC, Caspi RR. Retroviral gene therapy with an immunoglobulin-antigen fusion construct protects from experimental autoimmune uveitis. J Clin Invest. 2000;106:245–252. doi: 10.1172/JCI9168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Melo MEF, Qian J, El-Amine M, Agarwal RK, Soukhareva N, Kang Y, Scott DW. Gene Transfer of Ig-Fusion Proteins Into B Cells Prevents and Treats Autoimmune Diseases. J Immunol. 2002;168:4788–4795. doi: 10.4049/jimmunol.168.9.4788. [DOI] [PubMed] [Google Scholar]
- 93.Xu B, Scott DW. A novel retroviral gene therapy approach to inhibit specific antibody production and suppress experimental autoimmune encephalomyelitis induced by MOG and MBP. Clin Immunol. 2004;111:47–52. doi: 10.1016/j.clim.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 94.Pearson RM, Casey LM, Hughes KR, Wang LZ, North MG, Getts DR, Miller SD, Shea LD. Controlled Delivery of Single or Multiple Antigens in Tolerogenic Nanoparticles using Peptide-polymer Bioconjugates. Mol Ther. 2017 doi: 10.1016/j.ymthe.2017.04.015. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14:996–1006. doi: 10.1038/ni.2691. [DOI] [PubMed] [Google Scholar]
- 96.Park JK, Utsumi T, Seo YE, Deng Y, Satoh A, Saltzman WM, Iwakiri Y. Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomedicine: NBM. 2016;12:1365–1374. doi: 10.1016/j.nano.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, Martin C, van Rooijen N, Ochando JC, Randolph GJ, Luedde T, Ginhoux F, Kurts C, Trautwein C, Tacke F. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62:279–291. doi: 10.1002/hep.27793. [DOI] [PubMed] [Google Scholar]
- 98.Haddadi A, Elamanchili P, Lavasanifar A, Das S, Shapiro J, Samuel J. Delivery of rapamycin by PLGA nanoparticles enhances its suppressive activity on dendritic cells. J Biomed Res Part A. 2008;84:885–898. doi: 10.1002/jbm.a.31373. [DOI] [PubMed] [Google Scholar]
- 99.Tostanoski LH, Chiu YC, Andorko JI, Guo M, Zeng X, Zhang P, Royal W, 3rd, Jewell CM. Design of Polyelectrolyte Multilayers to Promote Immunological Tolerance. ACS Nano. 2016;10:9334–9345. doi: 10.1021/acsnano.6b04001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sanchez-Fueyo A, Strom TB. Immunologic basis of graft rejection and tolerance following transplantation of liver or other solid organs. Gastroenterology. 2011;140:51–64. doi: 10.1053/j.gastro.2010.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Afzali B, Lombardi G, Lechler RI. Pathways of major histocompatibility complex allorecognition. Curr Opin Organ Transplant. 2008;13:438–444. doi: 10.1097/MOT.0b013e328309ee31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kheradmand T, Wang S, Gibly RF, Zhang X, Holland S, Tasch J, Graham JG, Kaufman DB, Miller SD, Shea LD. Permanent protection of PLG scaffold transplanted allogeneic islet grafts in diabetic mice treated with ECDI-fixed donor splenocyte infusions. Biomaterials. 2011;32:4517–4524. doi: 10.1016/j.biomaterials.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen G, Kheradmand T, Bryant J, Wang S, Tasch J, Wang Jj, Zhang Z, Luo X. Intragraft CD11b+ IDO+ Cells Mediate Cardiac Allograft Tolerance by ECDI-Fixed Donor Splenocyte Infusions. Am J Transpl. 2012;12:2920–2929. doi: 10.1111/j.1600-6143.2012.04203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang S, Tasch J, Kheradmand T, Ulaszek J, Ely S, Zhang X, Hering BJ, Miller SD, Luo X. Transient B-cell depletion combined with apoptotic donor splenocytes induces xeno-specific T-and B-cell tolerance to islet xenografts. Diabetes. 2013;62:3143–3150. doi: 10.2337/db12-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Song J, Huang J, Chen X, Teng X, Song Z, Xing Y, Wang M, Chen K, Wang Z, Yang P. Donor-derived exosomes induce specific regulatory T cells to suppress immune inflammation in the allograft heart. Sci Rep. 2016;7 doi: 10.1038/srep20077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li X, Li JJ, Yang JY, Wang DS, Zhao W, Song WJ, Li WM, Wang JF, Han W, Zhang ZC. Tolerance induction by exosomes from immature dendritic cells and rapamycin in a mouse cardiac allograft model. PLoS ONE. 2012;7:e44045. doi: 10.1371/journal.pone.0044045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, Broady R, Levings MK. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. 2016;126:1413–1424. doi: 10.1172/JCI82771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, Zahorchak AF, Logar AJ, Wang Z, Watkins SC. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. 2004;104:3257–3266. doi: 10.1182/blood-2004-03-0824. [DOI] [PubMed] [Google Scholar]
- 109.Larche M, Akdis CA, Valenta R. Immunological mechanisms of allergen-specific immunotherapy. Nat Rev Immunol. 2006;6:761–771. doi: 10.1038/nri1934. [DOI] [PubMed] [Google Scholar]
- 110.Cox L, Nelson H, Lockey R, Calabria C, Chacko T, Finegold I, Nelson M, Weber R, Bernstein DI, Blessing-Moore J, Khan DA, Lang DM, Nicklas RA, Oppenheimer J, Portnoy JM, Randolph C, Schuller DE, Spector SL, Tilles S, Wallace D. Allergen immunotherapy: a practice parameter third update. J Allergy Clin Immunol. 2011;127:S1–55. doi: 10.1016/j.jaci.2010.09.034. [DOI] [PubMed] [Google Scholar]
- 111.Senti G, Prinz Vavricka BM, Erdmann I, Diaz MI, Markus R, McCormack SJ, Simard JJ, Wüthrich B, Crameri R, Graf N, Johansen P, Kündig TM. Intralymphatic allergen administration renders specific immunotherapy faster and safer: A randomized controlled trial. Proc Natl Acad Sci U S A. 2008;105:17908–17912. doi: 10.1073/pnas.0803725105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hylander T, Latif L, Petersson-Westin U, Cardell LO. Intralymphatic allergen-specific immunotherapy: an effective and safe alternative treatment route for pollen-induced allergic rhinitis. J Allergy Clin Immunol. 2013;131:412–420. doi: 10.1016/j.jaci.2012.10.056. [DOI] [PubMed] [Google Scholar]
- 113.Smarr CB, Bryce PJ, Miller SD. Antigen-specific tolerance in immunotherapy of Th2-associated allergic diseases. Crit Rev Immunol. 2013;33:389–414. doi: 10.1615/critrevimmunol.2013007046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fujita H, Soyka MB, Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. Clin Transl Allergy. 2012;2:2. doi: 10.1186/2045-7022-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. 2014;14:653–666. doi: 10.1038/nri3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Martin AJ, McCarthy D, Waltenbaugh C, Goings G, Luo X, Miller SD. Ethylenecarbodiimide-Treated Splenocytes Carrying Male CD4 Epitopes Confer Histocompatability Y Chromosome Antigen Transplant Protection by Inhibiting CD154 Upregulation. J Immunol. 2010;185:3326–3336. doi: 10.4049/jimmunol.1000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lorentz KM, Kontos S, Diaceri G, Henry H, Hubbell JA. Engineered binding to erythrocytes induces immunological tolerance to E. coli asparaginase. Sci Adv. 2015;1:e1500112. doi: 10.1126/sciadv.1500112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kishimoto TK, Ferrari JD, LaMothe RA, Kolte PN, Griset AP, O'Neil C, Chan V, Browning E, Chalishazar A, Kuhlman W, Fu FN, Viseux N, Altreuter DH, Johnston L, R AM. Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat Nanotechnol. 2016;11:890–899. doi: 10.1038/nnano.2016.135. [DOI] [PubMed] [Google Scholar]
- 119.Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, Paulson JC. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J Clin Invest. 2013;123:3074–3083. doi: 10.1172/JCI69187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sestak JO, Sullivan BP, Thati S, Northrup L, Hartwell B, Antunez L, Forrest ML, Vines CM, Siahaan TJ, Berkland C. Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis. Molecular Therapy - Methods & Clinical Development. 2014;1:14008. doi: 10.1038/mtm.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, Mucida D, Merad M, Steinman RM. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest. 2013;123:844–854. doi: 10.1172/JCI65260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 123.Nimmerjahn F, Ravetch JV. FcgammaRs in health and disease. Current Topics in Microbiology and Immunology. 2011;350:105–125. doi: 10.1007/82_2010_86. [DOI] [PubMed] [Google Scholar]
- 124.Krishnamoorthy S, Liu T, Drager D, Patarroyo-White S, Chhabra ES, Peters R, Josephson N, Lillicrap D, Blumberg RS, Pierce GF, Jiang H. Recombinant factor VIII Fc (rFVIIIFc) fusion protein reduces immunogenicity and induces tolerance in hemophilia A mice. Cell Immunol. 2016;301:30–39. doi: 10.1016/j.cellimm.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stern JN, Keskin DB, Kato Z, Waldner H, Schallenberg S, Anderson A, von Boehmer H, Kretschmer K, Strominger JL. Promoting tolerance to proteolipid protein-induced experimental autoimmune encephalomyelitis through targeting dendritic cells. Proc Natl Acad Sci U S A. 2010;107:17280–17285. doi: 10.1073/pnas.1010263107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mukhopadhaya A, Hanafusa T, Jarchum I, Chen YG, Iwai Y, Serreze DV, Steinman RM, Tarbell KV, DiLorenzo TP. Selective delivery of beta cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8+ T cells in NOD mice. Proc Natl Acad Sci U S A. 2008;105:6374–6379. doi: 10.1073/pnas.0802644105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Spiering R, Margry B, Keijzer C, Petzold C, Hoek A, Wagenaar-Hilbers J, van der Zee R, van Eden W, Kretschmer K, Broere F. DEC205+ Dendritic Cell-Targeted Tolerogenic Vaccination Promotes Immune Tolerance in Experimental Autoimmune Arthritis. J Immunol. 2015;194:4804–4813. doi: 10.4049/jimmunol.1400986. [DOI] [PubMed] [Google Scholar]
- 128.Chittasupho C, Sestak J, Shannon L, Siahaan TJ, Vines CM, Berkland C. Hyaluronic acid graft polymers displaying peptide antigen modulate dendritic cell response in vitro. Mol Pharm. 2014;11:367–373. doi: 10.1021/mp4003909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 130.Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol. 2007;7:585–598. doi: 10.1038/nri2138. [DOI] [PubMed] [Google Scholar]
- 131.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353:179–184. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther. 2014;22:1018–1028. doi: 10.1038/mt.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C, Agrawal S, Keough MB, Yong VW, James E, Moore A, Yang Y, Stratmann T, Serra P, Santamaria P. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. 2016;530:434–440. doi: 10.1038/nature16962. [DOI] [PubMed] [Google Scholar]
- 134.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, Rudensky AY. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. 2015;162:1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, Jiang S, Kuchroo VK, Mathis D, Roncarolo MG, Rudensky A, Sakaguchi S, Shevach EM, Vignali DA, Ziegler SF. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. 2013;14:307–308. doi: 10.1038/ni.2554. [DOI] [PubMed] [Google Scholar]
- 137.Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. 2014;259:88–102. doi: 10.1111/imr.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Davidson TS, Shevach EM. Polyclonal Treg cells modulate T effector cell trafficking. Eur J Immunol. 2011;41:2862–2870. doi: 10.1002/eji.201141503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol. 2004;172:3983–3988. doi: 10.4049/jimmunol.172.7.3983. [DOI] [PubMed] [Google Scholar]
- 140.Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–462. doi: 10.1038/ni1455. [DOI] [PubMed] [Google Scholar]
- 141.Yadav M, Bluestone J, Stephan S. Peripherally Induced Tregs – Role in Immune Homeostasis and Autoimmunity. Front Immunol. 2013;4 doi: 10.3389/fimmu.2013.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kasagi S, Zhang P, Che L, Abbatiello B, Maruyama T, Nakatsukasa H, Zanvit P, Jin W, Konkel JE, Chen W. In vivo-generated antigen-specific regulatory T cells treat autoimmunity without compromising antibacterial immune response. Sci Transl Med. 2014;6:241ra278. doi: 10.1126/scitranslmed.3008895. [DOI] [PubMed] [Google Scholar]
- 143.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86:10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Joly P, Mouquet H, Roujeau JC, D'Incan M, Gilbert D, Jacquot S, Gougeon ML, Bedane C, Muller R, Dreno B. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545–552. doi: 10.1056/NEJMoa067752. [DOI] [PubMed] [Google Scholar]
- 148.Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, Harris RA, Magnusson PU, Brittebo E, Loskog AS. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. 2012;9:1. doi: 10.1186/1742-2094-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, Santamaria P. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity. 2010;32:568–580. doi: 10.1016/j.immuni.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 150.Rios PD, Zhang X, Luo X, Shea LD. Mold-casted non-degradable, islet macro-encapsulating hydrogel devices for restoration of normoglycemia in diabetic mice. Biotechnol Bioeng. 2016;113:2485–2495. doi: 10.1002/bit.26005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Stein-Streilein J, Caspi RR. Immune privilege and the philosophy of immunology. Front Immunol. 2014;5 doi: 10.3389/fimmu.2014.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Desai T, Shea LD. Advances in islet encapsulation technologies. Nat Rev Drug Discov. 2016 doi: 10.1038/nrd.2016.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Graham JG, Zhang X, Goodman A, Pothoven K, Houlihan J, Wang S, Gower RM, Luo X, Shea LD. PLG scaffold delivered antigen-specific regulatory T cells induce systemic tolerance in autoimmune diabetes. Tissue Eng Part A. 2013;19:1465–1475. doi: 10.1089/ten.tea.2012.0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kuo R, Saito E, Miller SD, Shea LD. Peptide-conjugated nanoparticles reduce positive co-stimulatory expression and T cell activity to induce tolerance. Mol Ther. 2017 doi: 10.1016/j.ymthe.2017.03.032. 10.1016/j.ymthe.2017.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kim WU, Lee WK, Ryoo JW, Kim SH, Kim J, Youn J, Min SY, Bae EY, Hwang SY, Park SH, Cho CS, Park JS, Kim HY. Suppression of collagen-induced arthritis by single administration of poly(lactic-co-glycolic acid) nanoparticles entrapping type II collagen: a novel treatment strategy for induction of oral tolerance. Arthritis Rheum. 2002;46:1109–1120. doi: 10.1002/art.10198. [DOI] [PubMed] [Google Scholar]
- 156.Fessler MB, Keijzer C, Slütter B, van der Zee R, Jiskoot W, van Eden W, Broere F. PLGA, PLGA-TMC and TMC-TPP Nanoparticles Differentially Modulate the Outcome of Nasal Vaccination by Inducing Tolerance or Enhancing Humoral Immunity. PLoS ONE. 2011;6:e26684. doi: 10.1371/journal.pone.0026684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schöll I, Weissenböck A, Förster-Waldl E, Untersmayr E, Walter F, Willheim M, Boltz-Nitulescu G, Scheiner O, Gabor F, Jensen-Jarolim E. Allergen-loaded biodegradable poly(d,l-lactic-co-glycolic) acid nanoparticles down-regulate an ongoing Th2 response in the BALB/c mouse model. Clin Exp Allergy. 2004;34:315–321. doi: 10.1111/j.1365-2222.2004.01884.x. [DOI] [PubMed] [Google Scholar]
- 158.Marazuela EG, Prado N, Moro E, Fernandez-Garcia H, Villalba M, Rodriguez R, Batanero E. Intranasal vaccination with poly(lactide-co-glycolide) microparticles containing a peptide T of Ole e 1 prevents mice against sensitization. Clin Exp Allergy. 2008;38:520–528. doi: 10.1111/j.1365-2222.2007.02922.x. [DOI] [PubMed] [Google Scholar]