

Abstract
Objective
To assess the differences in ovarian transcriptomes in Shan Ma ducks between their peak and late stages of egg production, and to obtain new transcriptomic data of these egg-producing ducks.
Methods
The Illumina HiSeq 2000 system was used for high throughput sequencing of ovarian transcriptomes from Shan Ma ducks at their peak or late stages of egg production.
Results
Greater than 93% of the sequencing data had a base quality score (Q score) that was not less than 20 (Q20). From ducks at their peak stage of egg production, 42,782,676 reads were obtained, with 4,307,499,083 bp sequenced. From ducks at their late stage of egg production, 45,316,166 reads were obtained, with 4,562,063,363 bp sequenced. A comparison of the two datasets identified 2,002 differentially expressed genes, with 790 upregulated and 1,212 downregulated. Further analysis showed that 1,645 of the 2,002 differentially expressed genes were annotated in the non-redundant (NR) database, with 646 upregulated and 999 downregulated. Among the differentially expressed genes with annotations in the NR database, 696 genes were functionally annotated in the clusters of orthologous groups of proteins database, involving 25 functional categories. One thousand two hundred four of the differentially expressed genes with annotations in the NR database were functionally annotated in the gene ontology (GO) database, and could be divided into three domains and 56 categories. The three domains were cellular component, molecular function, and biological process. Among the genes identified in the GO database, 451 are involved in development and reproduction. Analysis of the differentially expressed genes with annotations in the NR database against the Kyoto encyclopedia of genes and genomes database revealed that 446 of the genes could be assigned to 175 metabolic pathways, of which the peroxisome proliferator-activated receptor signaling pathway, insulin signaling pathway, fructose and mannose metabolic pathways, gonadotropin releasing hormone signaling pathway and transforming growth factor beta signaling pathway were significantly enriched.
Conclusion
The differences in ovarian transcriptomes in Shan Ma ducks between their peak and late stages of egg production were elucidated, which greatly enriched the ovarian transcriptomic information of egg-producing ducks.
Keywords: Transcriptome, Ovary, Differentially Expressed Genes, Shan Ma Ducks
INTRODUCTION
Improving the reproductive performance of poultry is an important goal for genetic selection. Reproductive performance is a trait of low heritability. The reproductive performance of egg-producing ducks has improved progressively by traditional genetic selection, but further improvement is very slow. The egg-producing trait of poultry is closely related to the characteristics of the ovarian tissue. The ovary is the reproductive organ in females, but it is not only an organ that produces and releases eggs (its gametogenic function), but is also an endocrine gland that synthesizes and secretes female hormones (its steroidogenic function). Since ovarian function in poultry exerts a direct impact on egg production [1], poultry breeding scientists usually focus on the ovary to study egg production, including examination of the main genes controlling egg production and also other related differentially expressed genes. The Shan Ma ducks (Anas Platyrhynochos) is an excellent egg-type duck breed in Fujian, as a famous Chinese indigenous duck mentioned in the photograph alblum of china indigenous poultry breeds, with many advantageous traits, including small size, early maturity, great egg production, strong adaptability, and a high feed-conversion rate [2,3]. Its excellent egg-producing performance has therefore brought greater attention from poultry-breeding scientists. Studies of the differences in ovarian transcriptomes of female Shan Ma ducks in different physiologic and reproductive states and the elucidation of the differentially expressed genes involved will have great impacts on our understanding of the molecular genetic basis of egg production and the identification of the associated molecular markers. Recently, with the development of high-throughput sequencing, the transcriptomes of birds, such as goose [4–7], chicken [8], and duck [9,10], have been sequenced and analyzed involving in the ovary, muscle and adipose tissues. Some differentially expressed genes were particularly interesting in ovarian transcriptomic analysis. For example, five of the differentially expressed genes (protein tyrosine kinase 2 beta [PTK2B], phospholipase C [PLCB], cytochrome P450 family 19 subfamily A [CYP19A], hydroxy-delta-5-steroid dehydrogenase, 3 beta [HSD3B], and Insulin-like growth factor-binding protein 3 [IGFBP3]) and were confirmed to be involved in steroid hormone biosynthesis, gonadotropin releasing hormone (GnRH) signaling, prolactin signaling and ovary development. These genes have been extensively studied and are known to be important in mammalian female reproductive organogenesis and reproductive cycle regulation [6]. Luan et al [7] identified several differentially expressed genes in the ovaries from the laying period and ceased period, and these genes play key roles in the regulation of steroid hormone biosynthesis and reproductive functions. However, few reports are available in the literature regarding duck ovarian transcriptomes; and no reports exist on ovarian transcriptomic differences between Shan Ma ducks at peak and late stages of egg production. In our study, we sequenced the ovarian transcriptomes in Shan Ma ducks to assess basic information at different reproductive stages, screen for differentially expressed genes, and provide a foundation for future studies on genes related to reproductive performance and on the regulation of these genes at the molecular level.
MATERIALS AND METHODS
Animals and sample collection
Shan Ma ducks were provided by the Shan Ma Duck Stock Breeding Farm (Longyan, Fujian, China), and kept under the same environmental conditions with free access to feed, water, and commercial corn-soybean-based diets. From hatching to 4 weeks of age, the birds received a starter feed (metabolizable energy [ME], 12.08 MJ/kg; crude protein [CP], 19.0%). From 5 weeks of age to slaughter the birds were fed a grower diet (ME, 11.5 MJ/kg; CP, 16.5%). Six adult female Shan Ma ducks were included in these studies. Samples of ovarian tissue were collected from peak stage of egg production (180 days old) and late stage of egg production (520 days old) (n = 3 each), immediately frozen in liquid nitrogen, and stored at −80°C for later use. Generally, the first egg age of Shan Ma duck was 108 days old, and the peak egg production rate of Shan Ma duck was reached over 95% at 180 days old, and the egg production rate dropped to about 40% to 50% at 520 days old.
Total RNA extraction
Total RNA from ovarian tissue of the 6 ducks was extracted using Trizol reagent and dissolved in diethyl pyrocarbonate water. The concentrations of the RNA samples were determined using a Nanodrop-2000 UV-Vis Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) for nucleic acids and proteins. Each of the peak egg-production with 10 μg RNAs were pooled and each of the late egg-production with 10 μg RNAs were pooled. We froze the 2 pooled RNA samples at −80°C for later use.
Creating a cDNA library
We used magnetic beads coupled with poly dT to isolate mRNAs from total RNAs in order to obtain mRNA-enriched RNA samples. mRNA-enriched RNAs were then fragmented by sonication. RNA fragments of 200 to 500 bp were reverse-transcribed using random primers to generate first-strand cDNAs. The first-strand cDNAs were used as templates to synthesize the complementary second strand in a reaction system containing buffer, dNTPs, RNase H, and DNA polymerase I. The double-stranded cDNAs were end-repaired, dA-tailed, and ligated to an adaptor for polymerase chain reaction (PCR) amplification. The cDNA library was then ready for sequencing.
Sequencing using the Illumina system
cDNAs were sequenced by Beijing Biomarker Technologies Co., Ltd, using the Illumina HiSeq 2000 system. The original sequence reads were aligned to the reference duck genome (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF_000355885.1_BGI_duck_1.0/GCF_000355885.1_BGI_duck_1.0_genomic.fna.gz) using TopHat2 software [11], allowing up to two base mismatches. The gene expression level was then calculated using the fragments per kilobase of exon per million fragments (FPKM) method by CUFFLINKS V2.1.1 [12]. The expression levels of a gene in different samples were subject to Chi-square test using IDEG6 software (http://telethon.bio.unipd.it/bioinfo/IDEG6). The p values obtained were corrected with the false discovery ratio. The genes with corrected p values less than 0.01 and a ratio of the FPKM of the 2 samples equal or greater than 2 (fold-change≥2) were considered to be significantly differentially expressed.
COG classification, GO classification, and KEGG pathway analysis of the differentially expressed genes
The differentially expressed genes were analyzed in the NR database, and the annotation of the homologous gene in the NR database was used to annotate the differentially expressed genes identified. BLASTX was used to align differentially expressed genes with the orthologous genes from the clusters of orthologous groups of proteins (COG) database to obtain the COG annotation and classification of the genes. BLAST2GO [13] was used to align the differentially expressed genes with genes in the gene ontology (GO) database to obtain the GO annotation of the genes. The obtained annotation was enriched and refined using TopGo (R package version 1.16.2) with the “elim” method and Kolmogorov–Smirnov test. The differentially expressed genes were also aligned in BLASTX with genes in the Kyoto encyclopedia of genes and genomes (KEGG) database to obtain pathway annotation for the genes. KEGG pathways were enriched using the right sided Fisher’s exact test and p value ≤0.05 was taken as a threshold of significance.
Real-time quantitative polymerase chain reaction analysis
To validate the reliability of Illumina analysis, five differentially expressed genes in the significantly enriched pathways were selected and detected by real-time quantitative PCR (qRT-PCR). The relative expression level of each gene was normalized to the control gene β-actin (NM_001310421.1) using the 2−ΔΔCt method [14]. The qRT-PCR primers were designed using Primer Premier 6.0 and Beacon designer 7.8. The primer pairs used for qRT-PCR are listed in Table 1. All primers were synthesized by Invitrogen Biotechnology Co., Ltd (Carlsbad, CA, USA). The qRT-PCR analysis was conducted on the Bio-Rad CFX384 real-time PCR detection System (BIO-RAD, City, CA, USA). The qRT-PCR reaction contained 1 μL of cDNA template, 10.0 μL of Power SYBR Green Master Mix (Applied Biosystem, Foster, CA, USA), 8.0 μL of sterile water, and 0.5 μL of each gene-specific primer. Thermal cycling conditions were 1 cycle at 95°C for 2 min, 40 cycles of 95°C for 15 s and 63°C for 25 s. There were three replicates for each amplification.
Table 1.
Nucleotide sequences of primers used for qRT-PCR amplification
| Target gene | Forward (5′-3′) | Reverse (5′-3′) | Length (bp) |
|---|---|---|---|
| β-actin | TTGTATCTTCCGCCTTAA | GCCTTCATTCACATCTATC | 98 |
| ACSL3 | CGTGCCTAATGGTTACTT | TTCCTGCTGTGTTAATATCA | 136 |
| PPP1R3A | GAATGACACTGGATGACT | TCTGATAAGGAGGCACTA | 120 |
| MTMR7 | AATGAATACGCTTGCTTAG | CTGGTTGAGAGTTGAGTA | 105 |
| FSHβ | TCCTCTACTCTGTTCAAT | GGTCTCTATTCATTCTTACTA | 127 |
| SMURF2 | GTCAGATTCAATAACAAT | CCTCTAACAGTATCATTA | 177 |
qRT-PCR, real-time quantitative polymerase chain reaction; ACSL3, acyl-CoA synthetase long-chain family member 3; PPP1R3A, protein phosphatase 1 regulatory subunit 3A; MTMR7, myotubularin related protein 7; FSHβ, follicle stimulating hormone beta; SMURF2, SMAD specific E3 ubiquitin protein ligase 2.
RESULTS
Quality assessments of RNAs and transcriptomic sequencing data
The quality of RNA preparations used for transcriptome sequencing was examined (Table 2). The optical density 260/optical density 280 (OD260/OD280) ratios were between 1.8 and 2.2, the RNA integrity number values were greater than 8.4, and the 28S/18S ratios were greater than 1. All of these results indicated that the quality of the RNAs used in this study was satisfactory for transcriptome sequencing.
Table 2.
RNA quality analysis
| Sample | Concentration (ng/μL) | OD260/280 | OD260/230 | RIN | 28S/18S |
|---|---|---|---|---|---|
| Peak egg-production stage | 5,737 | 2.02 | 1.90 | 8.4 | 1.1 |
| Late egg-production stage | 6,143 | 1.98 | 2.17 | 8.9 | 1.3 |
OD, optical density; RIN, RNA integrity number.
The quality control results of the ovarian transcriptomic sequencing data from ducks at their peak or late stages of egg production are summarized in Table 3, 4. Table 3 shows that the two samples obtained 42.78 M (million) and 45.32 M reads, respectively, and more than 4.3 Gb of base pairs. The Q20 value was greater than 93% and the Q30 value was greater than 84%. These results indicated that the quality of the transcriptome sequencing data was reliable and could be used for subsequent analysis.
Table 3.
Statistical assessments of the high-throughput transcriptomic sequencing data
| Sample | Total reads | Total nucleotides (bp) | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|
| Peak egg-production stage | 42,782,676 | 4,307,499,083 | 93.12 | 85.00 | 51.86 |
| Late egg-production stage | 45,316, 66 | 4,562,063,363 | 93.11 | 84.94 | 52.93 |
Q20 is the percentage of sequences with base quality score that was no less than 20.
Q30 is the percentage of sequences with base quality score that was no less than 30.
GC is the percentage of bases G and C in the sequences.
Table 4.
Quality assessments of the high-throughput sequencing data of transcriptomes
| Sample1) statistical content | Peak egg-production stage | Late egg-production stage | ||
|---|---|---|---|---|
|
|
|
|||
| Number | Percentage | Number | Percentage | |
| Total reads | 42,782,676 | 100 | 45,316,166 | 100 |
| Mapped reads | 31,101,460 | 72.70 | 32,324,534 | 71.33 |
Total reads, total number of reads. Mapped reads, the number of reads that have matched sequences in the reference genome.
Table 4 shows that the total numbers of reads from sequencing were 42,782,676 and 45,316,166, respectively, for the two samples. Among the total number of reads 31,101,460 and 32,324,534 reads were mapped when compared to the reference genome, accounting for 72.70% and 71.33% of the total reads, respectively. The percentage of mapped reads was high for both samples.
Screening of the differentially expressed genes
The FPKM values of the ovarian tissue from ducks at the peak and late stages of egg production were calculated, as shown in Table 5. At the peak stage, 52.00% of the genes had FPKM values less than 5, and 8.10% of the genes had FPKM values greater than 100. At the late stage, 53.34% of the genes had FPKM values less than 5, and 7.95% of the genes had FPKM values greater than 100. In order to identify the differentially expressed genes, we used IDEG6 software. The genes were evaluated by both fold-change and significance level (p value). Only genes with a fold-change equal to or greater than 2 and a significance level of p< 0.05 were considered differentially expressed. As shown in the volcano plot (Figure 1), there were many differentially expressed genes between ducks at the peak and late stages of their egg-producing performance. We identified 2,002 differentially expressed genes, including 1,212 down-regulated genes and 790 up-regulated genes (Figure 2). These genes were further analyzed in the NR database, and we noted that 1,645 genes were already annotated, among which 999 genes were down-regulated and 646 genes were up-regulated. The top ten up- and down-regulated DEGs from the total of 1,645 DEGs identified between peak and late stage were: homeobox protein Hox-A3 (HOXA3), dihydrofolate reductase (DHFR), cerebellar degeneration related protein 2 like (CDR2L), forkhead box O1 (FOXO1), ER membrane protein complex subunit 3 (EMC3), G protein-coupled receptor 20 (GPR20), stem-loop binding protein (SLBP), acyl-CoA synthetase long-chain family member 3 (ACSL3), cytochrome P450 family 7 subfamily A member 1 (CYP7A1), ATP synthase, alpha subunit 1 (ATP5A1), sphingosine-1-phosphate receptor 3 (S1PR3), and TNF receptor associated protein 1 (TRAP1), transmembrane protein 50B (TMEM50B), nebulin-related anchoring protein (NRAP), glycine receptor, beta (GLRB), zinc finger, CCHC domain containing 24 (ZCCHC24), ribosomal protein L37a (RPL37A), secretion regulating guanine nucleotide exchange factor (SERGEF), solute carrier family 29, member 3 (SLC29A3), THAP domain containing 11 (THAP11), Phosphatidylinositol glycan anchor biosynthesis, class V (PIGV), respectively (Table 6).
Table 5.
The FPKM values of the ovary from ducks at different stages of egg production
| FPKM | 0 to 1 | 1 to 5 | 5 to 10 | 10 to 100 | >100 | Total |
|---|---|---|---|---|---|---|
| Peak egg-production stage | 7,268 (26.91%) | 6,777 (25.09%) | 3,076 (11.39%) | 7,703 (28.51%) | 2,189 (8.10%) | 27,013 |
| Late egg-production stage | 7,873 (29.37%) | 6,427 (23.97%) | 2,913 (10.87%) | 7,464 (27.84%) | 2,131 (7.95%) | 26,808 |
FPKM, fragments per kilobase of exon per million mapped reads.
Figure 1.
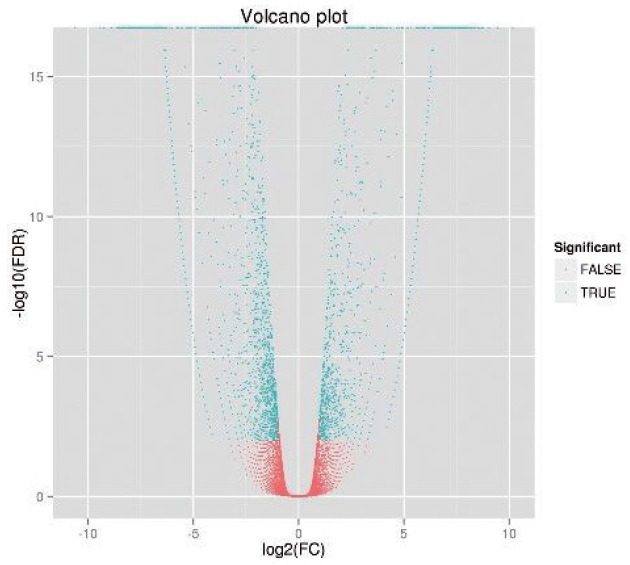
Volcano chart of differentially expressed genes.
Figure 2.
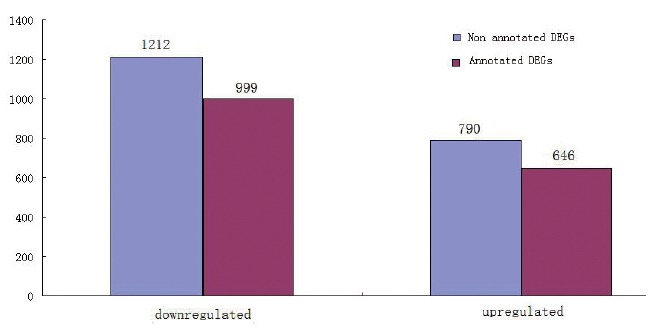
Statistics of differentially expressed genes.
Table 6.
Detailed information on the top ten up- and down-regulated differentially expressed genes
| Gene ID | Gene name | Log2 fold change | p-value | Up/down | Interpro description |
|---|---|---|---|---|---|
| XM_013102012.1 | HOXA3 | 10.24 | 3.92E-11 | Up | Homeobox protein Hox-A3 |
| XM_013099024.1 | DHFR | 9.06 | 1.76E-06 | Up | Dihydrofolate reductase |
| XM_005011990.2 | CDR2L | 9.02 | 0.000203 | Up | Cerebellar degeneration related protein 2 like |
| XM_005014856.2 | FOXO1 | 8.89 | 7.18E-05 | Up | Forkhead box O1 |
| XM_005020890.2 | EMC3 | 8.89 | 2.50E-11 | Up | ER membrane protein complex subunit 3 |
| XM_013096779.1 | GPR20 | 8.50 | 3.08E-06 | Up | G protein-coupled receptor 20 |
| XM_005016138.2 | SLBP | 8.46 | 1.34E-06 | Up | Stem-loop binding protein |
| XM_005026417 | ACSL3 | 8.37 | 2.87E-06 | Up | Acyl-CoA synthetase long-chain family member 3 |
| NM_001310351 | CYP7A1 | 8.28 | 0.001890 | Up | Cytochrome P450 family 7 subfamily A member 1 |
| XM_013100837.1 | ATP5A1 | 8.16 | 1.98E-05 | Up | ATP synthase, alpha subunit 1 |
| XM_005014175.2 | TRAP1 | −10.34 | 4.81E-09 | Down | TNF receptor associated protein 1 |
| XM_013094161.1 | TMEM50B | −9.33 | 1.01E-08 | Down | Transmembrane protein 50B |
| XM_005014451.2 | NRAP | −9.18 | 5.30E-07 | Down | Nebulin-related anchoring protein |
| XM_013096637.1 | GLRB | −8.51 | 0.000161 | Down | Glycine receptor, beta |
| XM_005015787.2 | ZCCHC24 | −8.50 | 0.002266 | Down | Zinc finger, CCHC domain containing 24 |
| XM_005010902.2 | RPL37A | −8.47 | 5.55E-16 | Down | Ribosomal protein L37a |
| XM_013109693.1 | BMP15 | −8.45 | 9.95E-05 | Down | Bone morphogenetic protein 15 |
| XM_005028335.2 | SLC29A3 | −8.38 | 9.91E-13 | Down | Solute carrier family 29, member 3 |
| XM_005029716.2 | THAP11 | −8.30 | 7.33E-05 | Down | THAP domain containing 11 |
| XM_013097508.1 | PIGV | −8.26 | 0.000516 | Down | Phosphatidylinositol glycan anchor biosynthesis, class V |
COG annotation of the differentially expressed genes
The differentially expressed genes annotated in NR database were analyzed in the COG database for functional annotation. The results showed that 696 genes received COG function annotations in 25 functional categories (Figure 3). Among the 25 categories, the one with the greatest number of genes was R (general function prediction only), which had 171 genes; followed by categories K (transcription), L (replication, recombination and repair), and T (signal transduction mechanisms), with 60, 60, and 55 genes each, respectively.
Figure 3.

Cluster of orthologous groups of proteins functional classification of differentially expressed genes.
GO annotation of the differentially expressed genes
The differentially expressed genes annotated in NR database were analyzed using BLAST2GO for functional annotation and genomic dataset analysis in the GO database. GO analysis showed that 1,204 genes received GO functional annotation in 3 domains: cellular component, molecular function, and biological process, and 56 categories (Figure 4). There were 17 categories in the cellular component domain, 17 categories in the molecular function domain, and 22 categories in the biological process domain. In the cellular component domain, the category with the highest number of reads was cell part, which had 804 genes, accounting for the highest percentage—66.78%; the next categories were cell and organelle, with 791 and 610 genes each, accounting for 65.70% and 50.66%, respectively; and the category with the least number of genes was virion, which had only 1 read. In the molecular function domain, the category with the highest number of genes was binding, which had 681 genes, accounting for the highest percentage—56.56%; the next category was catalytic activity, with 430 genes, accounting for 35.71%; and the categories with the fewest genes were translation regulatory activity, chemorepellent activity, nutrient reservoir activity and chemoattractant activity, with only 1 gene each. In the biological process domain, the category with the highest number of genes was cellular process, which had 794 genes, accounting for the highest percentage—65.95%; the next categories were biological regulation and metabolic process, with 587 and 585 genes each, accounting for 48.75% and 48.59%, respectively; and the category with the fewest genes was cell killing, with only 1 gene. In addition, as shown in Figure 5, in GO categories, biological processes related to reproduction and development included reproduction, reproductive process and developmental process, involving 451 corresponding genes.
Figure 4.

Gene ontology functional classification of differentially expressed genes.
Figure 5.

Quantitative expression of five gene were detected by real-time quantitative polymerase chain reaction.
KEGG enrichment analysis of the differentially expressed genes
The differentially expressed genes annotated in the NR database were analyzed in the KEGG database. The result showed that 446 genes were annotated into 175 KEGG pathways, in which 5 were significantly enriched pathways (p<0.05). As shown in Table 7, significantly enriched pathways are mainly involved in peroxisome proliferator-activated receptor (PPAR) signaling pathway, insulin signaling pathway, fructose and mannose metabolic pathways, GnRH signaling pathway, and transforming growth factor beta (TGF-β) signaling pathway.
Table 7.
Differentially expressed genes enriched in KEGG pathways
| Number | Metabolic pathway | Number of DEGs | Genes | Pathway ID | p-value |
|---|---|---|---|---|---|
| 1 | PPAR signaling pathway | 12 | CYP8B1, APOA1, AQP7, SLC27A6, CYP7A1, ACSL3, ACOX3, FABP2, PEPCK, FABP7, PDK1, CPT1 | Ko03320 | 0.0034 |
| 2 | Insulin signaling pathway | 14 | SOCS2, LIPE, FOXO1A, PIK3CG, PRKAR1A, PPP1R3B, PHKG1, HK2, PPP1R3A, PIK3CA, PCK2, SHC3, PDPK1, AKT2 | Ko04910 | 0.0067 |
| 3 | Fructose and mannose metabolism | 4 | TPI1, MTMR7, PFKM, HK2 | Ko00051 | 0.0175 |
| 4 | GnRH signaling pathway | 5 | FSHβ, PLA2G2E, GNAQ, MAP2K4, CALM3B | Ko04912 | 0.0297 |
| 5 | TGF-beta signaling pathway | 4 | EP300, ppp2r1a, BMPR1A, SMURF2 | Ko04350 | 0.0383 |
KEGG, Kyoto encyclopedia of genes and genomes; DEGs, differentially expressed genes; PPAR, peroxisome proliferator-activated receptor; GnRH, gonadotropin releasing hormone; TGF-β, transforming growth factor beta.
Real-time quantitative PCR analysis
To validate the reliability of Illumina analysis, we further used qRT-PCR to examine expression levels of five DEGs (acyl-CoA synthetase long-chain family member 3 [ACSL3], protein phosphatase 1 regulatory subunit 3A [PPP1R3A], myotubularin related protein 7 [MTMR7], follicle stimulating hormone beta [FSHβ], and SMAD specific E3 ubiquitin protein ligase 2 [SMURF2]) and in the significantly enriched pathways from peak and late stages of ovary. As shown in Table 8 and Figure 5, expression of these five genes can be detected, and the patterns of differential expression are consistent with the results from Illumina sequencing. This result suggested that the Illumina sequencing data was credible.
Table 8.
Comparison of the Illumina sequencing expression data and qRT-PCR result for five selected genes
| Gene | Illumina sequencing | Real-time PCR | ||
|---|---|---|---|---|
|
|
|
|||
| Fold change1) | q-value2) | Fold change3) | p-value4) | |
| ACSL3 | 1.22 | 0.88 | 1.56 | 0.37 |
| PPP1R3A | 26.7 | 0.006 | 184.17 | 0.03 |
| MTMR7 | 1.17 | 0.99 | 1.42 | 0.30 |
| FSHβ | 36.41 | 0.008 | 162.75 | 0.02 |
| SMURF2 | 16.83 | 0.03 | 198.49 | 0.02 |
qRT-PCR, real-time quantitative polymerase chain reaction; ACSL3, acyl-CoA synthetase long-chain family member 3; PPP1R3A, protein phosphatase 1 regulatory subunit 3A; MTMR7, myotubularin related protein 7; FSHβ, follicle stimulating hormone beta; SMURF2, SMAD specific E3 ubiquitin protein ligase 2; FPKM, fragments per kilobase of exon per million mapped reads.
Ratio of FPKM values between peak stage and late stage.
False discovery rate, q-value <0.05 was considered as significant.
Relative expression of genes was normalized to the control gene β-actin using 2−ΔΔCt method.
Student’s t-test p-values between peak stage and late stage, p-value <0.05 was considered as significant.
DISCUSSION
Creating the cDNA library
High-throughput transcriptomic sequencing technology has in recent years played an important role in revealing gene expression and regulation in animals and plants. It attracted considerable attention in studies focusing on gene expression and differential expression in particular aspects of animal development [15–17]. In this study, we used an Illumina HiSeq 2000 system for high-throughput sequencing of the ovarian transcriptomes from Shan Ma ducks at their peak and late stages of egg production, and obtained a great deal of transcriptional information. High-throughput sequencing results showed that sample quality was good and high-quality sequencing data Q20 accounted for 93% of the data. Transcriptomic sequencing data from ducks at their peak and late stages of egg production received 42,782,676 and 45,316,166 reads, respectively. The percentages of aligned reads to the reference genome were high, which were 72.70% and 71.33%, respectively. This suggests that the sequencing data have high coverage and the cDNA library was created successfully.
Analysis of the differentially expressed genes between peak stage and late stage
Analysis for differentially expressed genes between ovarian transcriptomes of ducks at their peak or late stages of egg production showed that there were 2,002 differentially expressed genes, with 790 genes upregulated and 1,212 genes downregulated. Further analysis of these genes in the NR database received 1,645 annotated genes, with 646 genes upregulated and 999 genes downregulated. There were 357 differentially expressed genes that did not have annotations. Genes that are highly expressed in reproductive tissues or cells may also indicate that the gene itself is activated, as shown in previous studies [18–21]. Therefore, in our study, we identified the top 10 up- and down-regulated DEGs in the ovaries of peak and late stage, respectively (Table 6). Some of them may be involved in specific reproductive processes. For example, previous research has shown that bone morphogenetic protein 15 gene is a member of TGF-β superfamily, and its function is to regulate the ovary development and to promote the proliferation and differentiation of follicular cells [22,23]. ATP5A1 gene involves in oxidative phosphorylation and ATP synthesis with an important role in aerobic respiration [24], and the study indicated that the expression level of ATP5A1 in the ovarian tissue during egg laying period was significantly higher than that in the early laying [25]. In oocytes, some related factors are coupled to the SLBP, which contains the only RNA binding region and interaction with stem-loop structure [26]. The pervious study showed SLBP played an important role in oocyte maturation, involving in translation regulation, processing and transport of histone mRNA [27,28]. The other most DEGs may play other important roles in biological processes. These DEGs may play an important role in regulating the function of the ovary and regulation of the egg production. So it should be further investigated the function of these genes.
From our GO analysis, the 1,645 genes with annotation in the NR database were analyzed in the GO database for functional classification. We observed that 1,204 of the genes received GO annotations and 441 did not. From our KEGG analysis, the 1,645 genes with annotations in the NR database were also analyzed in the KEGG database for pathway analysis. The 466 genes were annotated to KEGG pathways and 1,199 were not. We hypothesize that the lack of annotations of some genes was because studies on the duck transcriptome were still at early stages, and because there were not enough annotated genes available. Various types of biological information databases are constantly being updated and improved, and as a result, some of the sequencing data do not currently have corresponding annotations.
COG analysis results showed that 696 of the differentially expressed genes received COG functional annotations in 25 functional categories. This suggests that COG annotation is more comprehensive and covers the majority of life activities. GO analysis results showed that the differentially expressed genes involved 17 categories in the cellular component domain, of which cell-related (including cell and organelle) categories accounted for the majority, and membrane-related categories also accounted for a significant proportion. This result implies that gene products involved in the physiologic activities of the Shan Ma ovary are not only in the cells, but also widely present on membranes, which is consistent with studies on the ovarian transcriptomes of yaks [29], and suggests that the cell membrane plays an important role in the physiologic and reproductive activities of the ovary. GO analysis results showed that the differentially expressed genes also involved 17 categories in the molecular function domain, of which the binding category accounted for the greatest number. We further confirmed that binding proteins accounted for a high percentage in proteins expressed in oocytes. Other researchers have also shown that a high percentage of the proteins expressed in oocytes are binding proteins [30,31]. The fact that we confirmed that binding proteins were abundantly expressed in the ovary suggests that binding proteins play important roles in the physiologic and reproductive activities of the ovary. GO analysis results showed that the differentially expressed genes also involved 22 categories in the biological process domain, of which the cellular process category accounted for the largest percentage, followed by the biological regulation and metabolic process categories. In addition to the above biological processes, there were genes in the categories related to development and reproduction, including reproduction, reproductive process, and developmental process categories, involving 451 genes. This shows that these 451 reproduction- and development-related genes play different roles in Shan Ma ducks at their peak and late stages of egg-production.
In vivo, different genes are coordinated to perform biological functions. Pathway enrichment analysis will contribute to further comprehension and better annotation of differentially expressed genes, and can help to identify the main metabolic and signal-transduction pathways in which these genes are involved. In the present study, we performed KEGG pathway enrichment analysis, and our results showed that the differentially expressed genes identified involved 175 pathways, of which the PPAR signaling pathway, the insulin signaling pathway, fructose and mannose metabolism, the gonadotropin releasing signaling pathway, and the TGF-β signaling pathway are the 5 enriched pathways. Insulin is a polypeptide with multiple functions, and its binding to the insulin receptor can cause a series of cascading amplifications of its signal. It has been confirmed that insulin is an important factor in promoting the proliferation of ovarian granulosa cells and facilitating their physiologic functions, and its signaling pathway is involved in regulating granulosa cell proliferation, cellular metabolism and steroid hormone biosynthesis [32,33]. Previous study has showed that insulin signaling plays important roles in the physiologic and reproductive activities within the yak ovary; and is involved in cellular differentiation, development, and apoptosis, and in the integration of energy metabolism and reproductive activity in the ovary [29]. In our study, 14 of the differentially expressed genes we identified (suppressor of cytokine signaling 2 [SOCS2], lipase E [LIPE], forkhead box O1 a [FOXO1A], phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma [PIK3CG], protein kinase cAMP-dependent type I regulatory subunit alpha [PRKAR1A], protein phosphatase 1 regulatory subunit 3B [PPP1R3B], phosphorylase kinase gamma 1 [PHKG1], hexokinase 2 [HK2], protein phosphatase 1 regulatory subunit 3A [PPP1R3A], phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha [PIK3CA], phosphoenolpyruvate carboxykinase 2 [PCK2], SHC adaptor protein 3 [SHC3], 3-phosphoinositide dependent protein kinase 1 [PDPK1], and AKT serine/threonine kinase 2 [AKT2]), are involved in insulin signaling. GnRH is a neurohormone secreted by the hypothalamus that promotes the synthesis and release of gonadotropins by the pituitary (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]), which in turn play important roles in animal reproduction [34], including sex hormone biosynthesis [35,36]. In this study, 5 of the differentially expressed genes we identified (follicle stimulating hormone beta subunit [FSHβ], phospholipase A2 group IIE [PLA2G2E], G protein subunit alpha q [GNAQ], mitogen-activated protein kinase kinase [MAP2K4], and calmodulin 3b [CALM3B]), are involved in GnRH signaling. TGF-β signaling is one of the most important signal-transduction pathways, and plays important roles in the development of various organs [37], and in regulating animal reproduction, especially ovarian functions [38,39]. We identified 4 differentially expressed genes (E1A binding protein p300 [EP300], protein phosphatase 2 scaffold subunit Aalpha [ppp2r1a], bone morphogenetic protein receptor, type 1A [BMPR1A], and SMAD specific E3 ubiquitin protein ligase 2 [SMURF2]) as being involved in TGF-β signaling. Collectively, we have confirmed that insulin-, GnRH-, and TGF-β-signaling pathways are important for physiologic and reproductive activities of Shan Ma ducks. In addition, other pathways such as the PPAR-signaling pathway and the fructose and mannose metabolic pathways were also enriched in our study. The functions of these two latter pathways in physiologic and reproductive activities within the ovary are not clear and require further elucidation.
CONCLUSION
In the present study, high-throughput sequencing technology was used to study the ovarian transcriptomes of Shan Ma ducks at their peak and late stages of egg production. Information related to transcriptomes of Shan Ma ducks at different stages of egg production was explored, and the differentially expressed genes in ovaries of Shan Ma ducks in different physiologic and reproductive states were elucidated. Our study greatly enriched the transcriptomic information regarding egg-producing ducks and laid the foundation for further studies on the genes responsible for reproductive traits in egg-producing ducks, and the molecular regulation of these genes.
ACKNOWLEDGMENTS
This study was supported by the major projects in Fujian province in China (2012NZ0003). This work was also supported by seed industry innovation project in Fujian province in China (2014S 1477-13) and Fujian province public class special research institutes (2014R1023-2).
Footnotes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
REFERENCES
- 1.Dong CY, Kang B, Jia XJ, Yang HM. Construction of the full-length cDNA libarary and analysis in part of ESTs in Zi goose ovary. J Agric Biotechnol. 2010;18:389–93. [Google Scholar]
- 2.Xu GF, Chen KW. Photograh album of China indigeneous poultry breeds. Beijing, China: China Agricultural Press; 2003. [Google Scholar]
- 3.Lin RL, Chen HP, Rourvier R, Marie-Etancelin C. Genetic parameters of body weight, egg production, and shell quality traits in the Shan Ma laying duck (Anas platyrhynchos) Poult Sci. 2016;95:2514–9. doi: 10.3382/ps/pew222. [DOI] [PubMed] [Google Scholar]
- 4.Tariq M, Chen R, Yuan HY, et al. De novo transcriptomic analysis of peripheral blood lymphocytes from the Chinese goose: gene discovery and immune system pathway description. PLOS ONE. 2015;10:e0121015. doi: 10.1371/journal.pone.0121015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao GL, Zhao XZ, Li Q, Su J, Wang QG. Gene expression profiles in the pituitary glands of sichuan white geese during prelaying and laying periods. Genet Mol Res. 2015;14:12636–45. doi: 10.4238/2015.October.19.7. [DOI] [PubMed] [Google Scholar]
- 6.Ding N, Han Q, Zhao XZ, et al. Differential gene expression in pre-laying and laying period ovaries of Sichuan white geese (Anser cygnoides) Genet Mol Res. 2015;14:6773–85. doi: 10.4238/2015.June.18.20. [DOI] [PubMed] [Google Scholar]
- 7.Luan XH, Liu DW, Gao ZZ, et al. Transcriptome profiling identifies differentially expressed genes in huoyan goose ovaries between the laying period and ceased period. PLOS ONE. 2014;9:e113211. doi: 10.1371/journal.pone.0113211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yi B, Chen L, Sa RN, et al. Transcriptome profile analysis of breast muscle tissues from high or low levels of atmospheric ammonia exposed broilers (gallus gallus) PLOS ONE. 2016;11:e0162631. doi: 10.1371/journal.pone.0162631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu TS, Gu LH, Schachtschneider KM, et al. Identification of differentially expressed genes in breast muscle and skin fat of postnatal pekin duck. PLOS ONE. 2014;9:e107574. doi: 10.1371/journal.pone.0107574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Luo J, Li JX, et al. Transcriptome analysis of adiposity in domestic ducks by transcriptomic comparison with their wild counterparts. Anim Genet. 2015;46:299–307. doi: 10.1111/age.12294. [DOI] [PubMed] [Google Scholar]
- 11.Trapnell C, Pachter L, Saizberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;9:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conesa A, Gotz S, Garcir-Gomez JM, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 14.Livaka KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Colonello-Frattini NA, Hartfelder K. Differential gene expression profiling in mucus glands of honey bee (Apis mellifera) drones during sexual maturation. Apidologie. 2009;40:481–95. [Google Scholar]
- 16.Du C, Fu SY, Gao HY, et al. Transcriptome analysis of intramuscular preadipocytes and matureadipocyte in cashmere goats. Acta Vet Zoot Sin. 2014;45:714–21. [Google Scholar]
- 17.Zeng T, Zhang LP, Li JJ, et al. De novo assembly and characterization of Muscovy duck liver transcriptome and analysis of differentially regulated genes in response to heat stress. Cell Stress Chaperones. 2015;20:483–93. doi: 10.1007/s12192-015-0573-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang XD, Huang L, Wu T, et al. Transcriptomic analysis of ovaries from pigs with high and low litter size. PLOS ONE. 2015;10:e0139514. doi: 10.1371/journal.pone.0139514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gan L, Xie L, Zuo F, Xiang Z, He N. Transcriptomic analysis of Rongchang pig brains and livers. Gene. 2015;560:96–106. doi: 10.1016/j.gene.2015.01.051. [DOI] [PubMed] [Google Scholar]
- 20.Xia JH, Yuan J, Xin LL, et al. Transcriptome analysis on the inflammatory cell infiltration of nonalcoholic steatohepatitis in bama minipigs induced by along-term high-fat, high-sucrose diet. PLOS ONE. 2014;9:e113724. doi: 10.1371/journal.pone.0113724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang XL, Zhou GX, Xu XC, et al. Transcriptome profile analysis of adipose tissues from fat and short-tailed sheep. Gene. 2014;549:252–7. doi: 10.1016/j.gene.2014.07.072. [DOI] [PubMed] [Google Scholar]
- 22.Pierre A, Pisselet C, Dupont J, et al. Molecular basis of bone Morphogenetic protein-4 inhibitory action on progesterone secretion by ovine granulose cells. J Mol Endocrinol. 2005;33:805–17. doi: 10.1677/jme.1.01545. [DOI] [PubMed] [Google Scholar]
- 23.Dube JL, Wang P, Elvin J, et al. The bone morphogenetic protein 15 Gene is X-linked and expressed in oocytes. Mol Endocrinol. 1999;12:1809–17. doi: 10.1210/mend.12.12.0206. [DOI] [PubMed] [Google Scholar]
- 24.Ba6ran A, Silverman KA, Zeskand J, et al. The modifier of min2 (mom2) locus: embryonic lethality of a mutation in the apt5a1 gene suggests a novel mechanism of polyp suppression. Genome Res. 2007;17:566–77. doi: 10.1101/gr.6089707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang B, Guo JR, Yang HM, et al. Differential expression profiling of ovarian genes in prelaying and laying geese. Poult Sci. 2009;88:1975–83. doi: 10.3382/ps.2008-00519. [DOI] [PubMed] [Google Scholar]
- 26.Wang ZF, Whitfield ML, Ingledue TC, Dominski Z, Marzluff WF. The protein that binds the 3V end of histone mRNA: a novel RNA-binding protein required for histone pre-mRNA processing. Genes Dev. 1996;10:3028–40. doi: 10.1101/gad.10.23.3028. [DOI] [PubMed] [Google Scholar]
- 27.Dominski Z, Zheng LX, Sanchez R, Marzluff WF. The stem-loop binding protein facilitates 3′end formation by stabilizing U7 snRNP binding to the histone pre-mRNA. Mol Cell Biol. 1999;19:3561–70. doi: 10.1128/mcb.19.5.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patrick A, Yang Q, Marzluff WF, Clarke HJ. The stem-loop binding protein regulates translation of histone mRNA during mammalian oogenesis. Dev Biol. 2005;286:195–206. doi: 10.1016/j.ydbio.2005.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lan DL, Xiong XR, Wei YL, et al. RNA-Seq analysis of yak ovary: improving yak gene structure information and mining reproduction-related genes. Sci China Life Sci. 2014;44:307–17. doi: 10.1007/s11427-014-4678-2. [DOI] [PubMed] [Google Scholar]
- 30.Regassa A, Rings F, Hoelker M, et al. Transcriptome dynamics and molecular cross-talk between bovine oocyte and its companion cumulus cells. BMC Genomics. 2011;12:57. doi: 10.1186/1471-2164-12-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mamo S, Carter F, Lonergan P, et al. Sequential analysis of global gene expression profiles in immature and in vitro matured bovine oocytes: potential molecular markers of oocyte maturation. BMC Genomics. 2011;12:151. doi: 10.1186/1471-2164-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seto-Young D, Zajac J, Liu HC, Rosenwaks Z, Poretsky L. The role of mitogen-activated protein kinase in insulin and insulin-like growth factor I (IGF-I) signaling cascades for progesterone and IGF-binding protein-1 production in human granulosa cells. J Clin Endocrinol Metab. 2003;88:3385–91. doi: 10.1210/jc.2002-021965. [DOI] [PubMed] [Google Scholar]
- 33.Richardson MC, Cameron IT, Simonis CD, et al. Insulin and human chorionic gonadotropin cause a shift in the balance of sterol regulatory element-binding protein (SREBP) isoforms toward the SREBP-1c isoform in cultures of human granulosa cells. J Clin Endocrinol Metab. 2005;90:3738–46. doi: 10.1210/jc.2004-2057. [DOI] [PubMed] [Google Scholar]
- 34.Clarke IJ, Smith JT, Caraty A, Goodman R, Lehman MN. Kisspeptin and seasonality in sheep. Peptides. 2009;30:154–63. doi: 10.1016/j.peptides.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casan EM, Raga F, Bonilla-Musoles F, Polan M. Human oviductal gonadotropin-releasing hormone: Possible implications in fertilization, early embryonic development and implantation. J Clin Endocrinol Metab. 2000;85:1377–81. doi: 10.1210/jcem.85.4.6503. [DOI] [PubMed] [Google Scholar]
- 36.Lee VH, Lee LT, Chow BK. Gonadotropin-releasing hormone: regulation of the GnRH gene. FEBS J. 2008;275:5458–78. doi: 10.1111/j.1742-4658.2008.06676.x. [DOI] [PubMed] [Google Scholar]
- 37.Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;13:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 38.Drummond AE. TGFβ signaling in the development of ovary function. Cell Tissue Res. 2005;322:107–15. doi: 10.1007/s00441-005-1153-1. [DOI] [PubMed] [Google Scholar]
- 39.Konrad L, Keilani M, Laible L, Nottelmann U, Hofmann R. Effects of TGF-betas and a specific antagonist on apoptosis of immature rat male germ cells in vitro. Apoptosis. 2006;11:739–45. doi: 10.1007/s10495-006-5542-z. [DOI] [PubMed] [Google Scholar]