

Abstract
Cardiomyocyte apoptosis is initiated by various cellular insults and accumulated cardiomyocyte apoptosis leads to the pathogenesis of heart failure. Excessive reactive oxygen species (ROS) provoke apoptotic cascades. Manganese superoxide dismutase (MnSOD) is an important antioxidant enzyme that converts cellular ROS into harmless products. In this study, we demonstrate that MnSOD is down-regulated upon hydrogen peroxide treatment or ischemia/reperfusion (I/R) injury. Enhanced expression of MnSOD attenuates cardiomyocyte apoptosis and myocardial infarction induced by I/R injury. Further, we show that miR-23a directly regulates the expression of MnSOD. miR-23a regulates cardiomyocyte apoptosis by suppressing the expression of MnSOD. Our study reveals a novel model regulating cardiomyocyte apoptosis which is composed of miR-23a and MnSOD. Our study provides a new method to tackling apoptosis related cardiac diseases.
Keywords: apoptosis, hydrogen peroxide, ischemia/reperfusion, miR-23a, MnSOD
INTRODUCTION
Heart failure is the leading cause of mortality worldwide. Studies have found that apoptotic cell death participates in the pathogenesis of heart failure (Moe and Marin-Garcia, 2016; Olivetti et al., 1997). Elucidating the molecular mechanisms of cardiomyocyte apoptosis would be of great importance for tackling apoptosis related heart disease.
Cardiomyocytes contain a large number of mitochondria which constantly provide energy for the heart (Chen and Zweier, 2014). It is recognized that cellular ROS come from mitochondria (Kalogeris et al., 2014). Physiologically, ROS are considered as signaling molecules that have important biological functions for healthy cells (Lambeth and Neish, 2014; Liochev, 2013; Sena and Chandel, 2012). However, high levels of mitochondrial ROS participate in the regulation of apoptosis (Dixon and Stockwell, 2014). Under pathophysiological conditions, aberrant accumulation of ROS initiates and mediates cardiomyocyte apoptosis (Chen and Zweier, 2014). Superoxide dismutase, catalase and glutathione peroxidase are three major antioxidant enzyme in mammalian cells (Candas and Li, 2014). MnSOD is an important antioxidant located in mitochondrial matrix where it scavenges superoxide and protects cells from oxidative stress (Cramer-Morales et al., 2015).
MicroRNAs (miRNAs) are small noncoding RNAs that regulate target gene expression by repressing target mRNA translation or by promoting the degradation of target mRNA (Bartel, 2004; Filipowicz et al., 2008; Latronico and Condorelli, 2009). It has been demonstrated that miRNAs participate in the regulation of differentiation, proliferation, apoptosis and cell metabolic processes (Barringhaus and Zamore, 2009; Dumortier et al., 2013; Subramanian and Steer, 2010). MiRNAs regulate cardiomyocyte apoptosis by modulating the expression of various targets (Skommer et al., 2014). In addition, dysregulation of miRNAs correlates with the development of heart disease (Duygu et al., 2016; Hata, 2013; Yang et al., 2007).
In this study, we demonstrate that miR-23a participates in the regulation of cardiomyocyte apoptosis in vitro and in vivo. Further studies demonstrate that MnSOD is a target of miR-23a. miR-23a regulates cardiomyocyte apoptosis by inhibiting MnSOD expression. Our study reveals a novel signaling pathway regulating cardiomyocyte apopotosis which is composed of miR-23a and MnSOD.
MATERIAL AND METHODS
Cell culture and treatment
Cardiomyocytes were isolated from male mice (1–2 days) (Long et al., 2013). In brief, the dissected hearts were washed and minced in HEPES-buffered saline solution containing 130 mM NaCl, 3 mM KCl, 1 mM NaH2PO4, 4 mM glucose and 20 mM HEPES (PH adjusted to 7.35 ). Tissues were then dispersed in series of incubations at 37°C in HEPES-buffered saline solution containing 1.2 mg/ml pan-creatin and 0.14 mg/ml collagenase (Worthington). After centrifugation, the cells were re-suspended in Dulbecco’s modified Eagle medium/F-12 (Invitrogen) containing 5% heat-inactivated horse serum, 0.1 mM ascorbate, insulin-transferring-sodium selenite media supplement, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.1 mM bromode-oxyuridine. The dissociated cells were pre-plated at 37°C for 1 h. After that, the cardiomyocytes were diluted to 1 × 106 cells/ml and plated in 10 μg/ml laminin-coated culture dishes according to specific experimental requirement.
Adenovirus construction and infection
The mouse MnSOD open reading frame (ORF) was from Origene. The adenoviruses harboring MnSOD, and β-galactosidase (β-gal) were constructed using the Adeno-X™ expression system (Clontech). The mouse MnSOD RNA interference (siRNA) target sequence was 5′-GCTCTAATCAG GACCCATT-3′. A scramble form was used as a control, 5′-ATCGCTAGATCGTACCTAC-3′. The adenoviruses harboring MnSOD RNAi target sequence and its scramble forms were constructed using the pSilencer™ adeno 1.0-CMV System (Ambion) according to the kit’s instructions. All constructs were amplified in HEK293 cells.
Transfection of miR-23a mimic and antagomir
miR-23a mimic, the mimic negative control (mimic-NC), miR-23a antagomir and antagomir negative control (antagomir-NC) were synthesized by GenePhama Co. Ltd. They were transfected at 50 nM. The transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s guideline.
Reporter constructions and luciferase assay
The MnSOD 3′UTR was amplified from mouse genomic DNA by PCR. The forward primer was 5′-GAAGACAGAAGAGCTT GTTGG-3′. The reverse primer was 5′-GGCACACAACCATCT GTAATG-3′. The PCR products were gel-purified and ligated into pGL3 reporter vector (Promega) immediately downstream of the stop codon of the luciferase gene. To generate mutated MnSOD 3′UTR construct, the mutations were introduced by QuickChange II XL site-directed mutagenesis kit (Stratagene).
The luciferase activity assay was performed as described (Long et al., 2013). In brief, cells were cultured in 24 well plates. They were transfected with miR-23a mimic or its negative control, then transfected with plasmid construct of pGL3-MnSOD-3′UTR or pGL3-MnSOD-3′UTR-mut at concentration of 200 ng/well using Lipofectamine 2000 (Invitrogen). The renilla luciferase plasmid was also co-transfected at 2.5 ng/well serving as the internal control. Two days after transfection, cells were lysed and the luciferase activity was detected with Dual Luciferase Reporter Assay kit (Promega).
Quantitative real-time PCR (qRT-PCR)
Stem loop qRT-PCR for mature miR-23a was performed as described (Chen et al., 2005). Total RNA was extracted by TRIzol reagent (Invitrogen). After Dnase I treatment (Takara, Japan), the RNA was reverse transcribed with reverse transcriptase (ReverTra Ace, Toyobo). The samples were run in triplicate in Applied Biosystems ABI 7000 sequence detector system according to the manufacturer’s instructions. The results of qRT-PCR were normalized to that of U6. The primers for U6 were forward, 5′-GCTTCGGCAGCACATATA CTAA-3′; Reverse, 5′-AACGCTTCACGAATTTGCGT-3′. The specificity of the PCR amplification was confirmed by agarose gel electrophoresis.
Immunoblot
Immunoblot was performed as described (Wang et al., 2016). In brief, the cells were lysed at 4°C in a lysis buffer (20 mM Tris, pH 7.5, 2 mM EDTA, 3 mM EGTA, 2 mM dithiothreitol, 250 mM sucrose, 0.1 mM phenylmethylsulfonyl fluoride, 1% Triton X-100, and a protease inhibitor mixture). The samples were subjected to 12% SDS-PAGE and transferred to nitrocellulose membranes. Equal protein loading was controlled by Ponceau red staining of membranes. The blots were probed using the primary antibodies. The anti-MnSOD antibody (1:500, Abcam), anti-Bcl2 antibody (1:500, Abcam), anti-CuZnSOD (1:1000, Abcam), anti-Prohibitin (1:1000, Abcam) and anti-Actin antibody (1:2,000, Abcam) were used in this study.
Antioxidant enzyme activities
Enzyme activities of total SOD and MnSOD were assayed by kits (Beyotime biotechnology, Shanghai, China) according to the manufacturer’s instructions. In general, MnSOD activity of the heart homogenates was tested with the addition of CuZnSOD inhibitors. The CuZnSOD activity was calculated by substracting MnSOD activity from total SOD activities.
Mesurement of hydrogen peroxide in heart tissues
The concentration of hydrogen peroxide was assessed as described (Kopf et al., 2010; Landmesser et al., 2003). Briefly, 25 mg left heart ventricle tissues were incubated with Amplex red and HRP (Molecular Probes, USA) in Krebs/HEPES buffer (130 mM NaCl, 4.7 mM KCl, 1.5 mM CaCl, 1.2 mM MgSO4, 1.2 mM NaH2PO4, 25 mM NaHCO3, 1 mM HEPES, 11.5 mM glucose). The concentration of hydrogen peroxide was determined by fluorescent microplate reader and calculated from a hydrogen peroxide standard curve.
Target protector preparation and transfection
Target protector was designed as described (Choi et al., 2007). Briefly, MnSOD-TPmiR-23a sequence was 5′-AGTTCCAG GACTGCCAGGGATACAC-3′. MnSOD-TPcontrol was 5′-TGACA AATGAGACTCTCTCCTCTCC-3′. Transfection of the target protector was performed using the Endo-Porter kit (Gene Tools) according to the manufacturer′s instructions.
Animal experiment
Adult male C57BL/6 mice (8 weeks old) were purchased from Vital River Laboratory Animal Technology Co. Ltd (Beijing, China). Experiments were performed according to the protocol approved by the Animal Care Committee. The mice received three consecutive days, intravenous injections of miR-23a antagomir, or its control at a dose of 30 mg/kg body weight in a small volume (0.2 ml) per injection.
To perform ischemia/reperfusion surgery, the mice were anesthetized and the chest was opened. A 8-0 silk suture was passed around the left anterior descending coronary artery (LAD) at the inferior border of left auricle. The LAD was occluded by snaring with a vinyl tube through which the ligature had been passed. The coronary artery was occluded by pulling the snare tight and securing it with a hemostat. Forty five minutes later, the ligature was released and the heart was reperfused. The procedure was the same for the sham-operated group except that the snare was left untied.
To perform intracoronary delivery of adenovirus, five days prior to the I/R operation, the mice were anesthetized. The chest was opened and 2 × 1010 moi adenoviruses of MnSOD were injected with a catheter from the apex of the left ventricle into the aortic root while the aorta and pulmonary arteries were cross-clamped. The clamp was maintained for 20 s when the heart pumping against a closed system. The chest was closed and the animal was transferred back to its cage for recovery.
Histology
The harvested hearts were fixed in 10% formalin, embedded in paraffin and sectioned at 6 μm thickness. TUNEL staining was performed according to manufacturer’s instructions (Roche).
Statistical analysis
Data are expressed as mean ± SEM. The statistical comparison among different groups was performed by one-way ANOVA. Paired data were evaluated by Student’s t-test. P < 0.05 was considered statistically significant.
RESULTS
MnSOD inhibits cardiomyocyte apoptosis in vitro and in vivo
To test whether MnSOD participates in cardiomyocyte apoptosis, we employed hydrogen peroxide to treat neonatal cardiomyocyte to induce apoptosis. Hydrogen peroxide treatment led to the reduction of MnSOD in cardiomyocytes in a time-dependent manner (Fig. 1A). Enhanced expression of MnSOD delivered by adenovirus attenuated the percentage of apoptosis in cardiomyocyte induced by hydrogen peroxide as revealed by TUNEL assay (Fig. 1B).
Fig. 1.
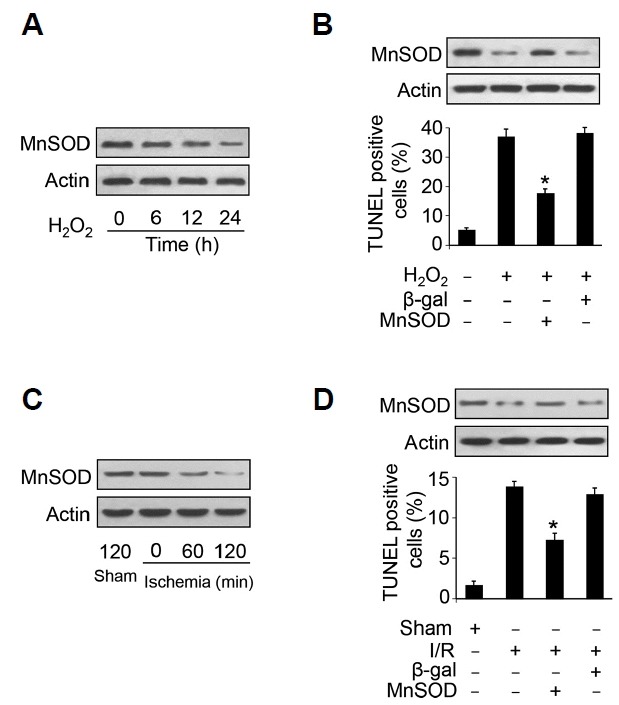
MnSOD suppresses cardiomyocyte apoptosis.
(A) MnSOD is down-regulated upon hydrogen peroxide treatment. Cardiomyocytes were treated with 100 μM hydrogen peroxide, and MnSOD was detected by immunoblot. (B) Overexpression of MnSOD reduces apoptosis induced by hydrogen peroxide. Cardiomyocytes were treated with 100 μM hydrogen peroxide for 24 h, TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labelling) was employed to analyze apoptotic cells. *, P < 0.05 versus hydrogen peroxide alone. (C) MnSOD is downregulated in response to ischemia. Mice were subjected to different period time of ischemia and MnSOD levels were detected by immunoblot. (D) Overexpression of MnSOD attenuates I/R induced apoptosis. Mice were administrated with MnSOD adenovirus and subjected to ischemia for 45 min and reperfusion for 3 h, TUNEL assay was employed to detect apoptotic cells. *, P < 0.05 versus I/R alone.
To test the role of MnSOD in the pathogenesis of myocardial infarction, we subjected the mice to ischemia/reperfusion injury. We found that MnSOD was down-regulated upon ischemia (Fig. 1C). Administration of adenovirus encoding MnSOD elevated the reduced MnSOD expression under I/R insult. Enhanced expression of MnSOD attenuated cardiomyocyte apoptosis upon I/R injury (Fig. 1D). These results suggest that MnSOD inhibits cardiomyocyte apoptosis in cultured cardiomyocyte and I/R injured heart.
miR-23a is responsible for the regulation of MnSOD expression
miRNAs are small noncoding RNAs that can repress the expression of target genes. To elucidate the mechanism by which MnSOD is downregulated upon hydrogen peroxide treatment or I/R injury, we analyzed the 3′ UTR of MnSOD and found that MnSOD is a potential target of miR-23a by using RNA hybrid program (Fig. 2A). We then found that miR-23a is up-regulated upon hydrogen peroxide treatment (Fig. 2B) and the concentration of hydrogen peroxide is significantly elevated upon ischemia in heart tissue (Supplementary Fig. S1B). To test whether miR-23a functionally regulates the expression of MnSOD, we transfected cardiomyocytes with miR-23a mimics and found that miR-23a reduced MnSOD protein levels (Fig. 2C). In addition, knockdown of endogenous miR-23a by antagomir (anta-23a) elevated MnSOD protein levels (Fig. 2D). The expression of Bcl-2 is associated with the expression of MnSOD (Yang et al., 2003). We found that inhibition of endogenous miR-23a abolished the decrease of MnSOD and Bcl-2 protein levels in response to hydrogen peroxide treatment (Figs. 2E and 2H). To confirm the role of miR-23a in cardiomyocyte apoptosis, we performed western blot to detect the expression of Prohibitin which is a known miR-23a target inhibiting apoptosis (Li et al., 2015). The results turned out that Prohibitin levels were restored when miR-23a is inhibited (Fig. 2H). Since SODs have three subtypes and SOD3 is a type of Cu/ZnSOD that exists in the extracellular matrix (Fukai and Ushio-Fukai, 2011), we detected the expression of MnSOD and Cu/ZnSOD in our experiments. We then wonder whether other types of SODs are involved in miR-23a mediated apoptosis in cardiomyocytes. We detected whether the expression of Cu/ZnSOD and activities of SODs are affected by miR-23a. The results showed that inhibition of miR-23a could not restore the expression and activity of Cu/ZnSOD (Fig. 2H and Supplementary Fig. S1A). The activity of MnSOD was elevated when miR-23a was inhibited (Supplementary Fig. S1A). These results suggest that miR-23a regulates the expression and activities of MnSOD rather than other types of SODs in cardiomyocytes.
Fig. 2.
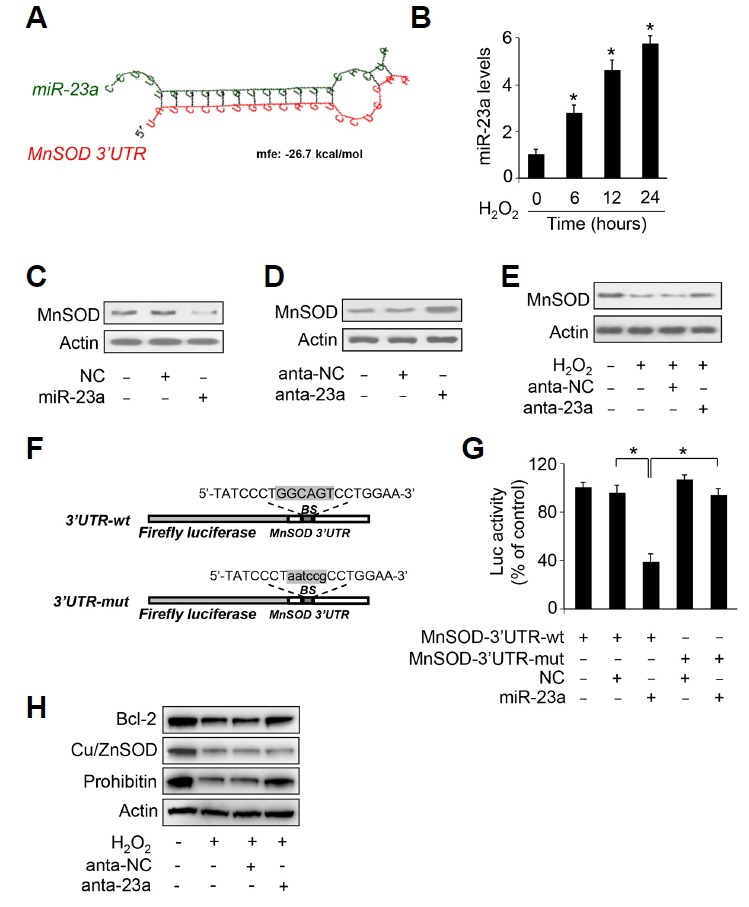
miR-23a regulates the expression of MnSOD by binding with its 3′ UTR.
(A) Analysis of MnSOD 3′ UTR potential binding site of miR-23a. (B) miR-23a is up-regulated upon hydrogen peroxide treatment. Cardiomyocytes were treated with 100 μM hydrogen peroxide, and miR-23a levels were detected by qRT-PCR. (C) miR-23a suppresses the expression of MnSOD. miR-23a mimic or negative control (NC) were used to transfect cardiomyocytes at concentration of 50 nM. Twenty four hours later, the expression of MnSOD was detected by immunoblot. (D) Inhibition of miR-23a elevates MnSOD levels. Cardiomyocytes were transfected with antagomir miR-23a (anta-23a) or antagomir negative control (anta-NC) at the concentration of 50 nM. Forty eight hours later, the expression of MnSOD was detected by immunoblot. (E) Inhibition of miR-23a attenuates hydrogen peroxide induced MnSOD down-regulation. Cardiomyocytes were transfected with antagomir miR-23a (anta-23a) or negative control (NC) at the concentration of 50 nM. Forty eight hours later, the cardiomyocyte were treated with 100 μM hydrogen peroxide for twenty four hours. MnSOD levels were detected by immunoblot. (F) miR-23a binding site was mutated in MnSOD 3′ UTR. (G) miR-23a suppresses the translation of MnSOD. HEK293 cells were transfected with 50 nM of miR-23a mimic or negative control (NC) and with 200 ng per well of pGL3 vector harboring wild type 3′ UTR or mutated 3′ UTR of MnSOD. Forty eight hours later, the cells were harvested, and luciferase activity was measured. *P < 0.05. (H) Cardiomyocytes were transfected with antagomir miR-23a (anta-23a) or negative control (NC) at the concentration of 50 nM. Forty eight hours later, the cardiomyocyte were treated with 100 μM hydrogen peroxide for twenty four hours. Bcl-2, Cu/ZnSOD and Prohibitin levels were detected by immunoblot.
Further, the luciferase assay system was set up to elucidate whether miR-23a interacts with 3′UTR of MnSOD. The luciferase reporter assay showed that construct with wild type MnSOD 3′UTR had significant reduced translational activity in the presence of miR-23a. Introduction of mutations in the miR-23a binding site abolished miR-23a inhibitory effect on MnSOD translational activity (Figs. 2F and 2G). Taken together, these data suggest that MnSOD is a downstream target of miR-23a.
miR-23a participates in the regulation of cardiomyocyte apoptosis
We then study the functional role of miR-23a in cardiomyocyte apoptosis. Enhanced expression of miR-23a induced cardiomyocyte apoptosis (Figs. 3A and 3B). We then treated cardiomyocytes with miR-23a antagomirs that efficiently inhibited upregulation of miR-23a upon hydrogen peroxide treatment (Figs. 3C) and we found that inhibition of miR-23a attenuated cardiomyocyte apoptosis induced by hydrogen peroxide (Figs. 3D). We then sought to elucidate whether miR-23a is involved in the pathogenesis of myocardial infarction. We found that miR-23a is up-regulated upon different period time of ischemia (Figs. 3E). Administration of miR-23a antagomir attenuated apoptosis upon I/R injury (Figs. 3F). These data suggest that miR-23a is an endogenous regulator of cardiomyocyte apoptosis.
Fig. 3.
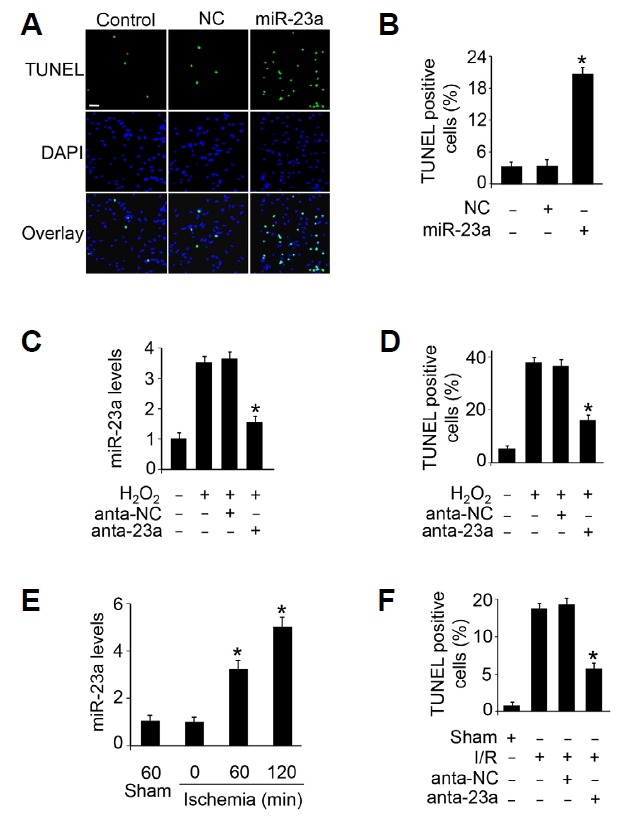
miR-23a regulates apoptosis in vitro and in vivo.
(A, B) miR-23a induced apoptosis in cardiomyocytes. Cardiomyocytes were transfected with 50 nM of miR-23a mimic or negative control (NC). Twenty four hours after transfection, TUNEL was used to analyze apoptotic cardiomyocytes (A). The percentage of apoptotic cardiomyocytes was calculated (B). Scal bar, 20 μm. *P < 0.05 versus NC alone. (C, D) Inhibition of miR-23a attenuates hydrogen peroxide induced apoptosis in cultured cardiomyocytes. Cardiomyocytes were transfected with 50 nM of antagomir miR-23a (anta-23a) or antagomir negative control (anta-NC). Forty eight hours after transfection, the cardiomyocytes were treated with 100 μM hydrogen peroxide for twenty four hours. The expression of miR-23a was detected by qRT-PCR (C). TUNEL was used to analyze apoptotic cardiomyocytes (D). *P < 0.05 versus hydrogen peroxide alone. (E) The expression levels of miR-23a upon myocardial ischemia. Mice were subjected to different period of ischemia and the expression of miR-23a was assayed by qRT-PCR. *P < 0.05 versus sham or 0 min group. (F) Inhibition of miR-23a attenuates apoptosis upon I/R injury. Adult C57BL/6 mice received three consecutive days, intravenous injections of antagomir miR-23a (anta-23a) or antagomir negative control (anta-NC) at a dose of 30 mg/kg body weight. They were then subjected to ischemia for 45 minute and reperfusion for 3 hours. Apoptosis was detected by TUNEL. *P < 0.05 versus I/R alone.
miR-23a regulates cardiomyocyte apoptosis through targeting MnSOD
We explored how miR-23a exerts its effect on cardiomyocyte apoptosis. The results showed that inhibition of miR-23a attenuated the down-regulation of MnSOD (Figs. 4A) and cardiomyocyte apoptosis (Figs. 4B) upon hydrogen peroxide treatment. Knockdown of MnSOD abolished the effect of miR-23a antagomir on MnSOD expression (Figs. 4A) and apoptosis (Figs. 4B) upon hydrogen peroxide treatment. To further figure out the relationship between miR-23a and MnSOD in cardiomyocytes apoptosis, the target protector technology was employed which could disrupt the specific base pairs between miRNA and mRNA. The results demonstrate that down-regulation of MnSOD expression induced by hydrogen peroxide was abolished in the presence of miR-23a target protector (Figs. 4C). The percentage of apoptosis was reduced when the target protector was employed (Figs. 4D). These data suggest that miR-23a regulates cardiomyocyte apoptosis through targeting MnSOD.
Fig. 4.
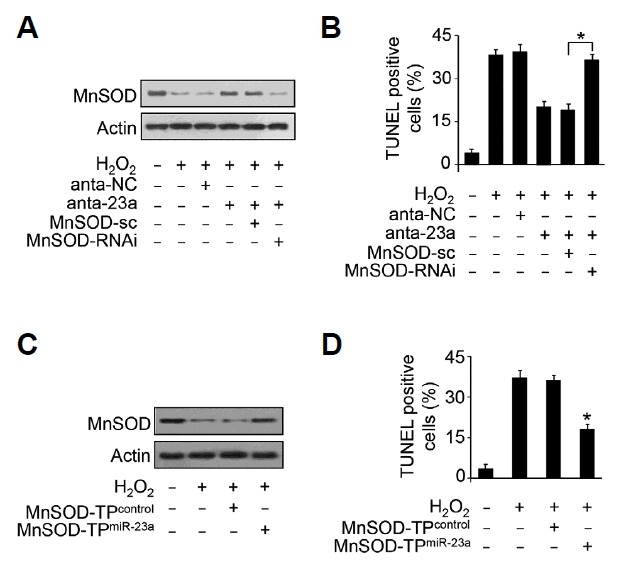
miR-23a regulates cardiomyocyte apoptosis through MnSOD.
(A, B) Knockdown of MnSOD abolished the effect of miR-23a knockdown on MnSOD expression and apoptosis. The cardiomyocytes were infected with MnSOD siRNA adenovirus, and were transfected with 50 nM of antagomir miR-23a (anta-23a) twenty four hours after adenovirus infection. The cells were then treated with 100 μM hydrogen peroxide for twenty four hours. The expression levels of MnSOD were detected by immnoblot (A). Cardiomyocyte apoptosis were detected by TUNEL assay (B). *P <.05. (C, D) The effect of miR-23a on apoptosis is abolished by MnSOD target protector for miR-23a. Cardiomyocytes were transfected with 50 nM of MnSOD target protector for miR-23a or the protector control. Twenty four hours after transfection, the cardiomcyotytes were treated with 100 μM hydrogen peroxide for twenty four hours. The expression of MnSOD was detected by immunoblot (C) and apoptosis (D) were detected by TUNEL. *P < 0.05 versus hydrogen peroxide group alone.
DISCUSSION
Cardiovascular diseases are the leading cause of mortality worldwide. Cardiomyocyte apoptosis is associated with the pathogenesis of myocardial infarction and heart failure. Our present work demonstrates that miR-23a is up-regulated upon hydrogen peroxide treatment or I/R injury. Antagomir mediated inhibition of miR-23a attenuates cardiomyocyte apoptosis in cultured cardiomyocyte and in I/R injured heart. The results suggest that miR-23a inhibits the expression of MnSOD by interacting with its 3′ UTR region. Our results provide evidence that miR-23a and MnSOD constitute a signaling pathway that regulates cardiomyocyte apoptosis.
Recently, studies have found that miRNAs participate in the pathogenesis of heart disease (Barwari et al., 2016; Hata, 2013). miRNAs regulate cardiomyocyte apoptosis by regulating the expression of various targets (Skommer et al., 2014). Modulating the expression of miRNA may be developed to treat myocardial infarction and heart failure (Duygu et al., 2016). Previous study has shown that miR-23a is a pro-hypertrophic miRNA by targeting muscle specific ring finger protein 1 (MuRF1) and its expression is regulated by nuclear factor of activated T cells (NFATc3) (Lin et al., 2009). However, the role of miR-23a in cardiomyocyte apoptosis remains to be characterized. In this study, we demonstrate that miR-23a regulates apoptosis in cultured cardiomyocyte and I/R injured heart. miR-23a may be developed to be a therapeutic target for treating myocardial infarction and heart failure.
The role of miR-23a in apoptosis was found to be distinct in different cell types and tissues. A previous study found that miR-23a inhibited apoptosis in nero-2a cells and decreased cerebral infarction volume following middle cerebral artery occlusion. In addition, miR-23a increased the expression of MnSOD (Zhao et al., 2014). These findings are opposite to our results and this may be attributed to miR-23a playing different roles in different cells and tissues. To strengthen our findings, we detected the expression of Bcl-2 protein which is associated with the expression of MnSOD (Yang et al., 2003). We found that miR-23a inhibits the expression of MnSOD and Bcl-2. Also, we detected the expression of Prohibitin which is a known apoptosis-related target of miR-23a and the results suggest that miR-23a inhibits Prohibitin expression which is consistent with previously published results (Li et al., 2015). Taken together, our results demonstrate that miR-23a promotes apoptosis by inhibiting the expression of MnSOD in cardiomyocytes.
Cellular ROS physiologically function as messenger molecules regulating various biological processes. However, uncontrolled ROS accumulation causes damage to the cell (Murphy et al., 2011). ROS are considered as hazardous molecules causing oxidative stress under pathological conditions. Oxidative stress is an important factor that contributes to the cardiomyocyte apoptosis. It is conceived that ROS comes from mitochondrial respiratory chain (Murphy, 2009). Superoxide dismutase (SOD) is one of the major antioxidant enzymes. Three different SODs exist in mammalian cells. SOD1 encodes cytosolic copper-zinc SOD. SOD2 encodes MnSOD which is distributed in mitochondrial matrix. Whereas SOD3 is anchored to the extracellular matrix (Sena and Chandel, 2012). All of the SODs catalyze the dismutation of superoxide radicals and prevent cellular damage caused by oxidative stress. Mice homozygous for loss of MnSOD die from dilated cardiomyopathy shortly after birth (Li et al., 1995). We have conducted an experiment to detect the expression and activities of SODs, the results show that the levels and activities of MnSOD and CuZnSOD are decreased upon hydrogen peroxide treatment. However, only the expression and activities of MnSOD recovered when miR-23a is inhibited. These results suggest that miR-23a regulates the expression and activity of MnSOD rather than CuZnSOD in cardiomyocytes. Since a miRNA may functions by targeting different genes, whether miR-23a directly regulate other genes in cardiomyocyte apoptosis need to be further clarified in our future research. In addition, as a gene may be directly regulated by many miRNAs, whether other miRNAs are involved in the regulation of MnSOD needs further elucidation.
Our results suggest that the expression of MnSOD is inhibited by hydrogen peroxide. Previous experiments showed that the expression of MnSOD was decreased when retinal ganglion cells were treated with hydrogen peroxide (Khan et al., 2012). However, in primary hepatocytes and human macrophages, the expression of MnSOD was increased when the cells were treated with hydrogen peroxide (Kinscherf et al., 1998; Takami et al., 2010). Also, no significant change of MnSOD was observed when pancreatic beta cells were treated with hydrogen peroxide (Kimoto et al., 2003). Different expression of MnSOD may depend on the concentration, cell type and duration of hydrogen peroxide treatment. In summary, our data demonstrate that miR-23a regulate cardiomyocyte apoptosis in vitro and in vivo. Inhibition of miR-23a attenuates hydrogen peroxide or I/R induced cardiomyocte apoptosis. We further found that miR-23a regulates cardiomyocyte apoptosis through MnSOD. Our results provide evidence that miR-23a may become a therapeutic target for treating apoptosis-related heart disease.
Supplementary Information
ACKNOWLEDGMENTS
This work was supported by National Natural Science Foundation of China (81600252), Beijing Natural Science Foundation (7174335).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Barringhaus KG, Zamore PD. MicroRNAs: regulating a change of heart. Circulation. 2009;119:2217–2224. doi: 10.1161/CIRCULATIONAHA.107.715839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Barwari T, Joshi A, Mayr M. MicroRNAs in cardiovascular disease. J Am Coll Cardiol. 2016;68:2577–2584. doi: 10.1016/j.jacc.2016.09.945. [DOI] [PubMed] [Google Scholar]
- Candas D, Li JJ. MnSOD in oxidative stress responsepotential regulation via mitochondrial protein influx. Antioxid Redox Signal. 2014;20:1599–1617. doi: 10.1089/ars.2013.5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524–537. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WY, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- Cramer-Morales K, Heer CD, Mapuskar KA, Domann FE. SOD2 targeted gene editing by CRISPR/Cas9 yields Human cells devoid of MnSOD. Free Radic Biol Med. 2015;89:379–386. doi: 10.1016/j.freeradbiomed.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- Dumortier O, Hinault C, Van Obberghen E. MicroRNAs and metabolism crosstalk in energy homeostasis. Cell Metabol. 2013;18:312–324. doi: 10.1016/j.cmet.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Duygu B, de Windt LJ, da Costa Martins PA. Targeting microRNAs in heart failure. Trends Cardiovasc Med. 2016;26:99–110. doi: 10.1016/j.tcm.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A. Functions of microRNAs in cardiovascular biology and disease. Ann Rev Physiol. 2013;75:69–93. doi: 10.1146/annurev-physiol-030212-183737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014;2:702–714. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan RS, Fonseca-Kelly Z, Callinan C, Zuo L, Sachdeva MM, Shindler KS. SIRT1 activating compounds reduce oxidative stress and prevent cell death in neuronal cells. Front Cell Neurosci. 2012;6:63. doi: 10.3389/fncel.2012.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimoto K, Suzuki K, Kizaki T, Hitomi Y, Ishida H, Katsuta H, Itoh E, Ookawara T, Honke K, Ohno H. Gliclazide protects pancreatic beta-cells from damage by hydrogen peroxide. Biochem Biophy Res Commun. 2003;303:112–119. doi: 10.1016/s0006-291x(03)00310-3. [DOI] [PubMed] [Google Scholar]
- Kinscherf R, Claus R, Wagner M, Gehrke C, Kamencic H, Hou D, Nauen O, Schmiedt W, Kovacs G, Pill J, et al. Apoptosis caused by oxidized LDL is manganese superoxide dismutase and p53 dependent. FASEB J. 1998;12:461–467. doi: 10.1096/fasebj.12.6.461. [DOI] [PubMed] [Google Scholar]
- Kopf PG, Scott JA, Agbor LN, Boberg JR, Elased KM, Huwe JK, Walker MK. Cytochrome P4501A1 is required for vascular dysfunction and hypertension induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci. 2010;117:537–546. doi: 10.1093/toxsci/kfq218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD, Neish AS. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Ann Rev Pathol. 2014;9:119–145. doi: 10.1146/annurev-pathol-012513-104651. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol. 2009;6:419–429. doi: 10.1038/nrcardio.2009.56. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Li J, Aung LH, Long B, Qin D, An S, Li P. miR-23a binds to p53 and enhances its association with miR-128 promoter. Sci Rep. 2015;5:16422. doi: 10.1038/srep16422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Rad Biol Med. 2013;60:1–4. doi: 10.1016/j.freeradbiomed.2013.02.011. [DOI] [PubMed] [Google Scholar]
- Long B, Wang K, Li N, Murtaza I, Xiao JY, Fan YY, Liu CY, Li WH, Cheng Z, Li P. miR-761 regulates the mitochondrial network by targeting mitochondrial fission factor. Free Rad Biol Med. 2013;65:371–379. doi: 10.1016/j.freeradbiomed.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe GW, Marin-Garcia J. Role of cell death in the progression of heart failure. Heart Fail Rev. 2016;21:157–167. doi: 10.1007/s10741-016-9532-0. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, Rhee SG, Thornalley PJ, Partridge L, Gems D, et al. Unraveling the biological roles of reactive oxygen species. Cell Metabol. 2011;3:361–366. doi: 10.1016/j.cmet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skommer J, Rana I, Marques FZ, Zhu W, Du Z, Charchar FJ. Small molecules, big effects: the role of microRNAs in regulation of cardiomyocyte death. Cell Death Dis. 2014;5:e1325. doi: 10.1038/cddis.2014.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Steer CJ. MicroRNAs as gatekeepers of apoptosis. J Cell Physiol. 2010;223:289–298. doi: 10.1002/jcp.22066. [DOI] [PubMed] [Google Scholar]
- Takami Y, Uto H, Tamai T, Sato Y, Ishida Y, Morinaga H, Sakakibara Y, Moriuchi A, Oketani M, Ido A, et al. Identification of a novel biomarker for oxidative stress induced by hydrogen peroxide in primary human hepatocytes using the 2-nitrobenzenesulfenyl chloride isotope labeling method. Hepatol Res. 2010;40:438–445. doi: 10.1111/j.1872-034X.2009.00615.x. [DOI] [PubMed] [Google Scholar]
- Wang K, Long B, Liu F, Wang JX, Liu CY, Zhao B, Zhou LY, Sun T, Wang M, Yu T, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016;37:2602–2611. doi: 10.1093/eurheartj/ehv713. [DOI] [PubMed] [Google Scholar]
- Yang J, Marden JJ, Fan C, Sanlioglu S, Weiss RM, Ritchie TC, Davisson RL, Engelhardt JF. Genetic redox preconditioning differentially modulates AP-1 and NF kappa B responses following cardiac ischemia/reperfusion injury and protects against necrosis and apoptosis. Mol Therapy. 2003;7:341–353. doi: 10.1016/s1525-0016(02)00061-8. [DOI] [PubMed] [Google Scholar]
- Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- Zhao H, Tao Z, Wang R, Liu P, Yan F, Li J, Zhang C, Ji X, Luo Y. MicroRNA-23a-3p attenuates oxidative stress injury in a mouse model of focal cerebral ischemia-reperfusion. Brain Res. 2014;1592:65–72. doi: 10.1016/j.brainres.2014.09.055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.