

Abstract
Aim
Tenapanor (RDX5791/AZD1722), an inhibitor of gastrointestinal Na+/H+ exchanger NHE3, is being evaluated for the treatment of patients with constipation‐predominant irritable bowel syndrome and the treatment of hyperphosphataemia in patients with chronic kidney disease on dialysis. By reducing intestinal H+ secretion, inhibition of NHE3 by tenapanor could indirectly affect H+‐coupled transporter activity, leading to drug–drug interactions. We investigated the effect of tenapanor on the activity of the H+‐coupled peptide transporter PepT1 via assessment of the pharmacokinetics of cefadroxil – a compound transported by PepT1 – in healthy volunteers.
Methods
In this open‐label, two‐period crossover, phase 1 study (NCT02140281), 28 volunteers received in random order: a single dose of cefadroxil 500 mg for 1 day; and tenapanor 15 mg twice daily over 4 days followed by single doses of both cefadroxil 500 mg and tenapanor 15 mg on day 5. There was a 4‐day washout between treatment periods.
Results
Cefadroxil exposure was similar when administered alone or in combination with tenapanor {geometric least‐squares mean ratios [(cefadroxil + tenapanor)/cefadroxil] (90% confidence interval): area under the concentration–time curve 93.3 (90.6–96.0)%; maximum concentration in plasma 95.9 (89.8–103)%}. Tenapanor treatment caused a softening of stool consistency and an increase in stool frequency, consistent with its expected pharmacodynamic effect. No safety concerns were identified and tenapanor was not detected in plasma.
Conclusions
These results suggest that tenapanor 15 mg twice daily does not have a clinically relevant impact on the activity of the H+‐coupled transporter PepT1 in humans. This may guide future research on drug–drug interactions involving NHE3 inhibitors.
Keywords: cefadroxil, drug interactions, PepT1, sodium–hydrogen exchanger 3, tenapanor
What is Already Known about this Subject
Gastrointestinal NHE3 indirectly affects intestinal H+‐coupled transporters.
Tenapanor, a locally‐acting, minimally‐absorbed NHE3 inhibitor, is being investigated for the treatment of patients with IBS‐C and for the treatment of hyperphosphataemia in patients with chronic kidney disease on dialysis.
Cefadroxil, a β‐lactam antibiotic, is transported by the H+‐coupled transporter PepT1.
What this Study Adds
Repeated oral dosing of tenapanor did not have a clinically relevant effect on the pharmacokinetics of the model PepT1‐transported drug cefadroxil in healthy volunteers.
This suggests that pharmacological inhibition of NHE3 by tenapanor is unlikely to have an impact on uptake of drugs mediated by the H+‐coupled transporter PepT1 in humans.
Tables of Links
| LIGANDS |
|---|
| Cefadroxil |
| Tenapanor |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http: //www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2.
Introduction
Sodium/hydrogen (Na+/H+) exchanger isoform 3 (NHE3) 3 plays an important role in sodium transport and fluid homeostasis in the gastrointestinal tract 4, 5. Tenapanor (RDX5791, AZD1722) is a minimally‐absorbed, highly‐selective, small‐molecule inhibitor of gastrointestinal NHE3 that reduces the uptake of dietary sodium and phosphate 3, 6. Tenapanor is currently in development for the treatment of patients with constipation‐predominant irritable bowel syndrome (IBS‐C) (ClinicalTrials.gov identifiers: NCT01923428 7, NCT02621892 8, NCT02686138 9), and for the treatment of hyperphosphataemia in patients with chronic kidney disease (CKD) on dialysis (ClinicalTrials.gov identifiers: NCT02081534 10, NCT02675998 11).
An important step in drug development is to identify any possible drug–drug interactions. As tenapanor has minimal systemic availability, it is unlikely to have effects outside of the gut. In the small intestine, a region of mildly acidic pH (6.1–6.8) adjacent to the luminal surface of the epithelium, known as the acid microclimate, is important for the absorption of molecules from the gut 12. A range of H+‐coupled transporter proteins use the acid microclimate as a driving force for the uptake of molecules across the apical brush‐border membrane 13, 14. This includes transporters of dipeptides and tripeptides, amino acids, vitamins and organic cations 13. NHE3 is present in the apical membrane of gastrointestinal epithelial cells and is expressed more highly in the small intestine than in the colon 15, 16. NHE3 has a major role in the uptake of dietary sodium from the intestinal lumen, in an electroneutral manner, through proton exchange 4. This H+ secretion into the gut lumen plays an important role in generating and maintaining the acid microclimate 12, 17. An NHE3 inhibitor could therefore reduce apical H+ secretion, alter the pH of the microclimate and potentially lead to indirect inhibition of H+‐coupled transporter activity.
H+‐coupled peptide transporter 1 (PepT1) is highly expressed throughout the small intestine and is localized to the brush‐border membrane. It is a high‐capacity transporter, with relatively low affinity for a wide range of compounds 13, 18. In addition to transporting dipeptides and tripeptides arising from protein digestion, PepT1 has also been shown to transport a range of hydrophilic oral drugs, including many β‐lactam antibiotics 13, 14, 19. Inhibiting NHE3 activity, by either removing extracellular Na+ or using the NHE3‐selective inhibitor S1611, reduces transepithelial peptide transport by PepT1 in vitro, suggesting that NHE3 contributes to PepT1 transport activity by maintaining the acid microclimate through Na+/H+ exchange 20, 21. Therefore, by pharmacologically inhibiting NHE3, tenapanor has the potential to affect PepT1‐mediated drug uptake.
Based on these theoretical considerations, this phase 1 study was conducted to assess whether tenapanor has a clinically relevant impact on the transport activity of the H+‐coupled transporter PepT1 in humans, by evaluating the pharmacokinetics of the model PepT1‐transported drug cefadroxil 14, 20, 22 when administered alone or in combination with tenapanor.
Methods
The drug/molecular target nomenclature used in this article conforms to the Concise Guide to PHARMACOLOGY 2015/16 2.
Study design
This was a phase 1, randomized, open‐label, two‐period crossover study (ClinicalTrials.gov identifier: NCT02140281) conducted by a contract research organization at a single study centre (Quintiles, Overland Park, KS, USA). The study was conducted in accordance with the Declaration of Helsinki, and International Conference on Harmonisation and Good Clinical Practice guidelines. The study protocol and an amendment were approved by the MidLands Independent Review Board (Overland Park, KS, USA). All study volunteers provided written informed consent before undergoing any study procedure.
Participants
Healthy men and women, aged 18–50 years, with a body mass index of 18–30 kg m–2 and weighing 50–100 kg, were eligible for enrolment in the study. Women of childbearing potential could not be pregnant and had to use effective contraception during the study period. Men were also required to use effective contraception. Volunteers were required to have suitable veins for cannulation or repeated venepuncture.
Key exclusion criteria were: a history or presence of gastrointestinal, hepatic or renal disease, or any other condition known to interfere with the absorption, distribution, metabolism or excretion of drugs; loose stools [Bristol Stool Form Scale (BSFS) score of 6 or 7 23] for 2 or more days in the week before study drug administration; use of medications or supplements known to affect stool consistency and/or gastrointestinal motility, including fibre supplements, antidiarrhoeal agents, prokinetic drugs, enemas, probiotic medications or supplements; or salt or electrolyte supplements containing sodium, potassium, chloride or bicarbonate formulations during the week before randomization.
Study treatments
Following screening, all volunteers underwent two treatments in a 1: 1 randomized sequence (Figure 1): (i) a single oral dose of cefadroxil 500 mg administered in the morning; and (ii) tenapanor 15 mg, administered orally twice daily for 4 days, followed by single doses of both tenapanor 15 mg and cefadroxil 500 mg (administered in the morning) on day 5 (this treatment combination hereafter referred to as cefadroxil + tenapanor). There was a washout period of at least 4 days between the two treatment periods.
Figure 1.
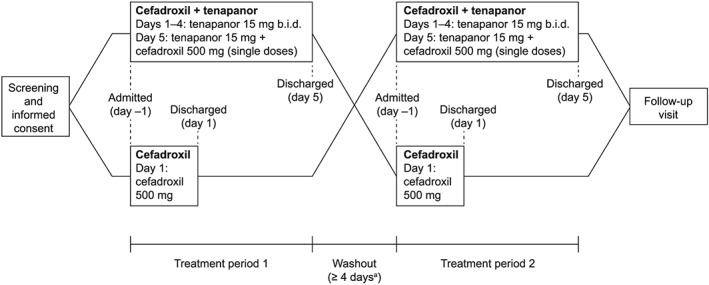
Study design. Participants were admitted to the study centre on the morning of the day before study drug administration (day –1) and discharged on the last day of treatment (day 1 or day 5) following sample collection for pharmacokinetic measurements. aAt least 4 days between the last dose of the first treatment period and the first dose of the second treatment period. b.i.d., twice daily
For each treatment regimen, volunteers were admitted to the study centre on the morning of the day before study drug administration (day –1). Volunteers who received cefadroxil alone were discharged on the day of treatment (day 1), after samples had been collected for pharmacokinetic measurements. Those receiving cefadroxil + tenapanor were resident at the centre until the end of sample collection on day 5. Volunteers returned to the study centre for follow‐up assessments 7–10 days after administration of the final treatment dose.
Volunteers underwent an overnight fast of at least 8 h before study drug administration. Cefadroxil and tenapanor were administered 5–10 min before meals (breakfast and/or dinner), and all volunteers could consume only the same standardized meals and drinks offered during the residential period.
Pharmacokinetic assessments
Blood samples were collected predose and 0.5, 1, 1.5, 2, 2.5, 4, 6, 8, 10 and 12 h postdose on day 1 of the cefadroxil treatment period and at the same time points on day 5 of the cefadroxil + tenapanor treatment period to assess the pharmacokinetics of cefadroxil. Blood samples collected predose and 1, 2 and 4 h postdose on day 5 were also used to assess plasma concentrations of tenapanor.
Samples for determination of drug concentration in plasma were analysed by Covance Laboratories Inc. (Madison, WI, USA). Cefadroxil and its deuterated internal standard were isolated from plasma using protein precipitation and analysed using liquid chromatography followed by tandem mass spectrometric detection. Before being used to analyse cefadroxil levels in the study samples, the method was validated in the range 30.0–30 000 ng ml–1. Tenapanor and its deuterated internal standard were extracted from samples using liquid–liquid extraction. After evaporation under nitrogen, the residue was reconstituted and analysed using liquid chromatography followed by tandem mass spectrometric detection. Before being used to analyse tenapanor concentrations in the samples collected, the method was validated in the range 0.500–100 ng ml–1.
Pharmacokinetic parameters were derived using standard noncompartmental methods with Phoenix WinNonlin Professional version 6.3 (Pharsight Corp., Mountain View, CA, USA). All pharmacokinetic computations were performed using either this software or SAS® version 9.4 (SAS Institute Inc., Cary, NC, USA). The parameters determined were as follows: the maximum concentration in plasma (C max); area under the concentration–time curve in plasma from time zero (predose) to time of last quantifiable concentration (AUC0–t), calculated by linear up/log down trapezoidal summation; area under the concentration–time curve in plasma from time zero (predose) extrapolated to infinite time (AUC), calculated by linear up/log down trapezoidal summation and extrapolated to infinity by addition of the last quantifiable concentration (C t) divided by the apparent elimination rate constant (λz; i.e. AUC = AUC0–t + C t/λz).
Pharmacodynamic assessments
Stool frequency and stool consistency (as measured by the BSFS 23) were assessed daily on day –1 of the first treatment period and on days 1–5 of the cefadroxil + tenapanor treatment period. Assessments were over 24‐h intervals following the morning dosing, except for day 5 of cefadroxil + tenapanor treatment (12‐h interval).
Safety assessments
Safety assessments included vital signs (at screening, on entry to the study clinic on day –1, and at the end of each treatment period), physical examinations (at screening and at the end of the second treatment period), clinical laboratory evaluations (clinical chemistry, haematology and urinalysis: at screening, before dosing on day 1 of each treatment period, and at the end of the second treatment period), electrocardiograms (at screening and at the end of each treatment period) and adverse event (AE) monitoring (throughout the study: from screening until the follow‐up visit).
Statistical analyses
Pharmacokinetic assessments were summarized using descriptive statistics for all plasma measurements of cefadroxil exposure. The pharmacokinetic analysis set consisted of all volunteers who received cefadroxil 500 mg and provided evaluable pharmacokinetic profiles during either treatment period.
Following natural log transformation, the observed C max, AUC and AUC0–t were analysed separately using a mixed effects analysis of variance model, with sequence, period and treatment as fixed effects, and volunteer nested within sequence as a random effect. The point estimate and 90% confidence interval (CI) for the difference between treatments was constructed and exponentially back‐transformed to provide point and CI estimates for the ratio of interest ([cefadroxil + tenapanor]/cefadroxil).
Assuming no effect of tenapanor on the pharmacokinetics of cefadroxil and a standard deviation (SD) of 0.3 or less for the change in log‐transformed pharmacokinetic variables, a sample size of 24 volunteers was expected to provide a 90% probability of the two‐sided 90% CI for the ratio ([cefadroxil + tenapanor]/cefadroxil) being completely contained within 80–125%. The study therefore aimed to include 28 volunteers.
Summary statistics were determined for pharmacodynamic evaluations of stool frequency and stool consistency. The pharmacodynamic (i.e. stool) analysis and safety analysis sets included all volunteers who received at least one dose of tenapanor or cefadroxil and had at least one postdose measurement. All statistical analyses were performed using SAS version 9.4.
Results
Study participants
Twenty‐eight volunteers (18 men) were enrolled in this study. All volunteers completed the study, receiving all treatments according to study protocol, and were included in pharmacokinetic and safety analyses. One participant was excluded from pharmacodynamic (stool) analysis, as only predose data were available. Mean ± SD age of the volunteers was 32 ± 10 years (range 19–49 years) and mean ± SD body mass index was 26.0 ± 2.8 kg m–2 (range 19.4–29.8 kg m–2).
Pharmacokinetics
Cefadroxil plasma concentration–time curves were similar whether cefadroxil was administered alone or in combination with tenapanor (Figure 2). Pharmacokinetic parameters of cefadroxil were also similar when cefadroxil was given alone or in combination with tenapanor [geometric least‐squares mean ratio (90% CI), (cefadroxil + tenapanor)/cefadroxil: AUC, 93.3 (90.6–96.0)%; AUC0–t, 93.4 (90.7–96.1)%; C max, 95.9 (89.8–102.5)%; Table 1]. Median time to maximum plasma concentration was 1.5 h and the geometric mean of the apparent terminal half‐life was approximately 2 h for both cefadroxil and cefadroxil + tenapanor treatments. The plasma concentration of tenapanor was below the lower limit of quantitation (0.5 ng ml–1) in all samples collected and analysed during the study.
Figure 2.
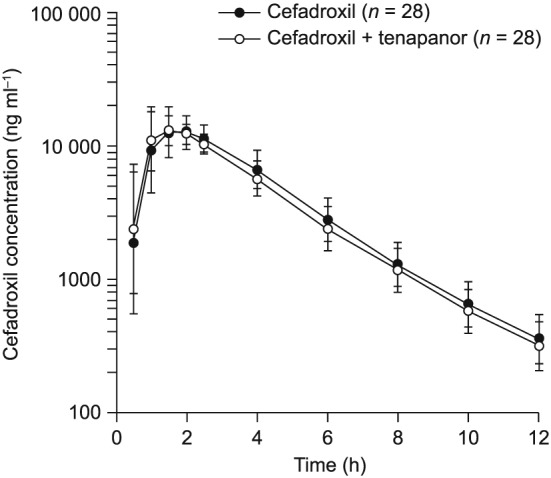
Cefadroxil plasma concentration vs. time following cefadroxil administration alone and in combination with tenapanor. Data shown as geometric mean (± standard deviation). Cefadroxil: a single dose of cefadroxil 500 mg administered on the morning of day 1. Cefadroxil + tenapanor: tenapanor 15 mg twice daily administered from day 1 to day 4, followed by single doses of both tenapanor 15 mg and cefadroxil 500 mg, administered concurrently on the morning of day 5
Table 1.
Pharmacokinetic parameters of cefadroxil when administered alone or in combination with tenapanor
| Cefadroxil (n = 28) | Cefadroxil + tenapanor (n = 28) | Geometric least‐squares mean ratio,a % (90% CI) | |
|---|---|---|---|
| AUC (ng h ml –1 ) | 53 800 (16.8) | 50 200 (16.6) | 93.3 (90.6–96.0) |
| AUC 0–t (ng h ml –1 ) | 52 700 (16.7) | 49 200 (16.4) | 93.4 (90.7–96.1) |
| C max (ng ml –1 ) | 14 800 (23.2) | 14 200 (21.6) | 95.9 (89.8–102.5) |
| t max (h) b | 1.5 (1.0–4.0) | 1.5 (1.0–4.0) | |
| t ½ (h) | 2.0 (11.1) | 2.1 (11.8) |
Unless otherwise stated, data are shown as geometric mean (GCV, %). AUC, area under the plasma concentration–time curve from time zero to infinity; AUC0–t, area under the plasma concentration–time curve from time zero to the last quantifiable concentration; CI, confidence interval; C max, maximum observed plasma concentration; GCV, geometric coefficient of variation; t max, time to C max; t ½, apparent terminal half‐life.
(cefadroxil + tenapanor)/cefadroxil.
Data are shown as median (range).
Stool frequency and consistency
Stool frequency increased with tenapanor treatment, from a predose mean ± SD of 1.4 ± 0.5 bowel movements per day, measured on the day before study drug administration (day –1), to a mean ± SD of 1.8 ± 0.8 bowel movements per day for the entire 5‐day tenapanor dosing period. Mean ± SD BSFS scores increased from 3.7 ± 1.0 on day –1 to 5.4 ± 1.2 for the overall tenapanor dosing period.
Safety and tolerability
Tenapanor was generally well tolerated and no safety concerns were identified. No serious AEs or discontinuations due to AEs occurred during the study. Overall, 11 volunteers reported AEs, all of which were mild in intensity and resolved. Almost all AEs were gastrointestinal in nature. Ten volunteers reported AEs during administration of tenapanor 15 mg twice daily (days 1–4), while one volunteer reported an AE following cefadroxil + tenapanor administration (day 5). The most common AEs (reported by at least two volunteers) were abdominal distension (n = 4), abdominal pain (n = 4), abnormal gastrointestinal sounds (n = 3), diarrhoea (n = 2) and flatulence (n = 2). No trends or clinically relevant changes in clinical laboratory results, vital signs, electrocardiograms or physical examinations were observed during the study.
Discussion
This phase 1 study aimed to investigate whether gastrointestinal NHE3 inhibition by tenapanor has a clinically relevant impact on the transport activity of the H+‐coupled transporter PepT1 in humans. NHE3 inhibition has been suggested to reduce H+ secretion into the gut, potentially affecting the acid microclimate and thereby reducing H+‐coupled transporter activity 20, 21. PepT1 is involved in the uptake of several drugs 14, 18, 19, and apical dipeptide transport and uptake assays across a range of extracellular pH values suggest that the optimal transport activity of PepT1 occurs at pH 6.5, which is within the physiological pH range of the acid microclimate at the mucosal surface of the intestine (pH 6.1–6.8). To test whether NHE3 inhibition by tenapanor affects PepT1 transport activity, the pharmacokinetics of cefadroxil (a compound transported by PepT1) were compared when cefadroxil was administered alone and in combination with tenapanor in 28 volunteers. Our results suggest that repeated dosing with tenapanor 15 mg twice daily has no clinically relevant effect on PepT1 activity.
Our study was performed in line with regulatory guidance for transporter‐based in vivo drug–drug interaction studies 24, 25. The tenapanor dose of 15 mg twice daily is at the lower end of the range tested so far for the treatment of patients with IBS‐C or the treatment of hyperphosphataemia in patients with CKD on dialysis 7, 10. Additional data may be needed to confirm whether the lack of effect on cefadroxil absorption observed in our study is also seen at higher doses of tenapanor. Tenapanor was administered for 4 days to ensure that the pharmacodynamic effects reached a steady state before administration of a therapeutically relevant dose of cefadroxil. In vitro studies have convincingly shown that cefadroxil has moderate affinity for, and is transported by, PepT1 19. Furthermore, studies in knockout mice indicate that PepT1 plays a key role in the rate and extent of absorption of cefadroxil following oral administration 22. Cefadroxil is therefore a recommended agent for studies examining the pharmaceutical relevance of H+‐coupled peptide transporters 19, 26.
Pharmacological activity of tenapanor was evident owing to changes in stool consistency and frequency, consistent with NHE3 inhibition and previous findings in healthy volunteers 27. In mouse models, deletion of the NHE3 gene results in severe impairment of sodium–fluid volume homeostasis with subsequent chronic diarrhoea 4, and a decrease in calcium absorption 28. However, our study showed that pharmacological inhibition of NHE3 in humans resulted in a phenotype nowhere near as extreme as that observed in NHE3 knockout mouse models. The ratios of plasma cefadroxil AUC, AUC0–t and C max for cefadroxil alone and cefadroxil in combination with tenapanor were within the bioequivalence range (80–125%), indicating that there were no clinically significant differences in cefadroxil uptake between the two treatments.
Other clinical studies that have investigated the effect of changes in the acid microclimate on PepT1 transport activity have shown inconsistent results 29, 30. Amiloride, an inhibitor of Na+/H+ exchange, was found to decrease the gastrointestinal uptake of the β‐lactam antibiotic amoxicillin in healthy volunteers 29. By contrast, in another study, jejunal perfusion with amiloride and amoxicillin in healthy volunteers did not affect amoxicillin absorption in the gut 30. Our study showed no evidence of an indirect effect of tenapanor on PepT1 transport activity. This lack of effect suggests that inhibition of gastrointestinal NHE3 by tenapanor may not be sufficient to alter the pH of the acid microclimate to an extent that could significantly affect PepT1 function. Although the reported pH of the acid microclimate varies across studies and in different regions of the intestine, values for the proximal jejunum typically range from pH 6.1 to 6.8 31, 32, 33, 34, 35. In vitro studies have shown that, although PepT1 activity is reduced as the pH increases, PepT1 is still active at pH 7.0 20. It therefore seems unlikely that any increase in the pH of the acid microclimate as a consequence of tenapanor inhibition of NHE3 would be sufficient to cause complete loss of PepT1 activity. Furthermore, most H+‐coupled transporters are spread over a large area of the intestine and are high‐capacity and low‐affinity transporters, making it difficult to inhibit the transport of any one particular compound effectively and hence difficult to create a relevant drug–drug interaction. PepT1 is considerably more abundant in the small intestine than in the colon 36; however, as this expression pattern is similar to that of NHE3 15, 16, it seems unlikely that differences in the relative regional expression of gastrointestinal PepT1 and NHE3 account for the lack of drug–drug interaction seen here. Throughout our phase 3 programme for tenapanor we are investigating any data trends that may reflect drug–drug interactions; none have been identified to date.
In conclusion, our study found that repeated dosing of tenapanor 15 mg twice daily had no clinically relevant effect on cefadroxil exposure in humans. This suggests that pharmacological inhibition of NHE3 by tenapanor is unlikely to have any clinically relevant impact on the activity of the H+‐coupled transporter PepT1 in the gut. This may guide future research on drug–drug interactions during the development of NHE3 inhibitors.
Competing Interests
S.J. and C.H. are employees of, and have ownership interest in, AstraZeneca. J.P., B.S. and M.K. are employees of AstraZeneca. D.P.R. is an employee of, and has ownership interest in, Ardelyx Inc. E.L. is an employee of Quintiles. This study was funded by AstraZeneca.
We thank all volunteers and investigators. Medical writing support was provided by Steven Inglis (PhD) and Sarah Graham (PhD) from PharmaGenesis London, London, UK, and was funded by AstraZeneca Gothenburg, Mölndal, Sweden and Ardelyx Inc., Fremont, CA, USA.
Contributors
S.J., J.P. and C.H. designed the research. E.L. was the principal investigator of the study. M.K. was responsible for the statistical analyses. All authors contributed to the interpretation of the results and the writing of the manuscript.
Johansson, S. , Rosenbaum, D. P. , Palm, J. , Stefansson, B. , Knutsson, M. , Lisbon, E. A. , and Hilgendorf, C. (2017) Tenapanor administration and the activity of the H+‐coupled transporter PepT1 in healthy volunteers. Br J Clin Pharmacol, 83: 2008–2014. doi: 10.1111/bcp.13313.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1D68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 2015; 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med 2014; 6: 227ra36. [DOI] [PubMed] [Google Scholar]
- 4. Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet 1998; 19: 282–285. [DOI] [PubMed] [Google Scholar]
- 5. Tse CM, Brant SR, Walker MS, Pouyssegur J, Donowitz M. Cloning and sequencing of a rabbit cDNA encoding an intestinal and kidney‐specific Na+/H+ exchanger isoform (NHE‐3). J Biol Chem 1992; 267: 9340–9346. [PubMed] [Google Scholar]
- 6. Labonte ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo‐McCoy S, et al. Gastrointestinal inhibition of sodium‐hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol 2015; 26: 1138–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chey WD, Lembo AJ, Rosenbaum DP. Tenapanor treatment of patients with constipation‐predominant irritable bowel syndrome: a phase 2, randomized, placebo‐controlled efficacy and safety trial. Am J Gastroenterol 2017; 112: 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. ClinicalTrials.gov. A 12‐week study with a 4‐week randomized withdrawal period to evaluate the efficacy and safety of tenapanor for the treatment of IBS‐C (T3MPO‐1). Available at: https://clinicaltrials.gov/ct2/show/NCT02621892. (last accessed 11 July 2016).
- 9. ClinicalTrials.gov. A 26‐week study to evaluate the efficacy and safety of tenapanor in IBS‐C (T3MPO‐2). Available at: https://clinicaltrials.gov/ct2/show/NCT02686138. (last accessed 1 June 2016).
- 10. Block GA, Rosenbaum DP, Leonsson‐Zachrisson M, Åstrand M, Johansson S, Knutsson M, et al. Effect of tenapanor on serum phosphate in patients receiving hemodialysis. J Am Soc Nephrol 2017. https://doi.org/10.1681/ASN.2016080855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. ClinicalTrials.Gov. An 8‐week study to evaluate tenapanor in the treatment of hyperphosphatemia in end‐stage renal disease patients on hemodialysis (ESRD‐HD). Available at: https://clinicaltrials.gov/ct2/show/NCT02675998. (last accessed 28 June 2016).
- 12. Thwaites DT, Anderson CM. H+‐coupled nutrient, micronutrient and drug transporters in the mammalian small intestine. Exp Physiol 2007; 92: 603–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anderson CM, Thwaites DT. Hijacking solute carriers for proton‐coupled drug transport. Physiology (Bethesda) 2010; 25: 364–377. [DOI] [PubMed] [Google Scholar]
- 14. Estudante M, Morais JG, Soveral G, Benet LZ. Intestinal drug transporters: an overview. Adv Drug Deliv Rev 2013; 65: 1340–1356. [DOI] [PubMed] [Google Scholar]
- 15. Brant SR, Yun CH, Donowitz M, Tse CM. Cloning, tissue distribution, and functional analysis of the human Na+/N+ exchanger isoform, NHE3. Am J Physiol—Cell Physiol 1995; 269: C198–C206. [DOI] [PubMed] [Google Scholar]
- 16. Dudeja PK, Rao DD, Syed I, Joshi V, Dahdal RY, Gardner C, et al. Intestinal distribution of human Na+/H+ exchanger isoforms NHE‐1, NHE‐2, and NHE‐3 mRNA. Am J Physiol—Gastrointest Liver Physiol 1996; 271: G483–GG93. [DOI] [PubMed] [Google Scholar]
- 17. Watanabe C, Kato Y, Ito S, Kubo Y, Sai Y, Tsuji A. Na+/H+ exchanger 3 affects transport property of H+/oligopeptide transporter 1. Drug Metab Pharmacokinet 2005; 20: 443–451. [DOI] [PubMed] [Google Scholar]
- 18. Brandsch M, Knutter I, Bosse‐Doenecke E. Pharmaceutical and pharmacological importance of peptide transporters. J Pharm Pharmacol 2008; 60: 543–585. [DOI] [PubMed] [Google Scholar]
- 19. Brandsch M. Transport of drugs by proton‐coupled peptide transporters: pearls and pitfalls. Expert Opin Drug Metab Toxicol 2009; 5: 887–905. [DOI] [PubMed] [Google Scholar]
- 20. Kennedy DJ, Leibach FH, Ganapathy V, Thwaites DT. Optimal absorptive transport of the dipeptide glycylsarcosine is dependent on functional Na+/H+ exchange activity. Pflugers Arch 2002; 445: 139–146. [DOI] [PubMed] [Google Scholar]
- 21. Thwaites DT, Kennedy DJ, Raldua D, Anderson CM, Mendoza ME, Bladen CL, et al. H/dipeptide absorption across the human intestinal epithelium is controlled indirectly via a functional Na/H exchanger. Gastroenterology 2002; 122: 1322–1333. [DOI] [PubMed] [Google Scholar]
- 22. Posada MM, Smith DE. In vivo absorption and disposition of cefadroxil after escalating oral doses in wild‐type and PepT1 knockout mice. Pharm Res 2013; 30: 2931–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol 1997; 32: 920–924. [DOI] [PubMed] [Google Scholar]
- 24. European Medicines Agency . Guideline on the investigation of drug interactions. CPMP/EWP/560/95/Rev. 1 Corr. 2012. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf. (last accessed 16 April 2015).
- 25. Food and Drug Administration . Guidance for industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations 2012. Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf. (last accessed 16 April 2015).
- 26. Brandsch M. Drug transport via the intestinal peptide transporter PepT1. Curr Opin Pharmacol 2013; 13: 881–887. [DOI] [PubMed] [Google Scholar]
- 27. Johansson S, Rosenbaum DP, Knutsson M, Leonsson‐Zachrisson M. A phase 1 study of the safety, tolerability, pharmacodynamics, and pharmacokinetics of tenapanor in healthy Japanese volunteers. Clin Exp Nephrol 2016. https://doi.org/10.1007/s10157‐016‐1302‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pan W, Borovac J, Spicer Z, Hoenderop JG, Bindels RJ, Shull GE, et al. The epithelial sodium/proton exchanger, NHE3, is necessary for renal and intestinal calcium (re)absorption. Am J Physiol Renal Physiol 2012; 302: F943–F956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Westphal JF, Jehl F, Brogard JM, Carbon C. Amoxicillin intestinal absorption reduction by amiloride: possible role of the Na+‐H+ exchanger. Clin Pharmacol Ther 1995; 57: 257–264. [DOI] [PubMed] [Google Scholar]
- 30. Lennernas H, Knutson L, Knutson T, Hussain A, Lesko L, Salmonson T, et al. The effect of amiloride on the in vivo effective permeability of amoxicillin in human jejunum: experience from a regional perfusion technique. Eur J Pharm Sci 2002; 15: 271–277. [DOI] [PubMed] [Google Scholar]
- 31. Legen I, Kristl A. Factors affecting the microclimate pH of the rat jejunum in ringer bicarbonate buffer. Biol Pharm Bull 2003; 26: 886–889. [DOI] [PubMed] [Google Scholar]
- 32. Shimada T. Factors affecting the microclimate pH in rat jejunum. J Physiol 1987; 392: 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iwatsubo T, Miyamoto Y, Sugiyama Y, Yuasa H, Iga T, Hanano M. Effects of potential damaging agents on the microclimate‐pH in the rat jejunum. J Pharm Sci 1986; 75: 1162–1165. [DOI] [PubMed] [Google Scholar]
- 34. Ikuma M, Hanai H, Kaneko E, Hayashi H, Hoshi T. Effects of aging on the microclimate pH of the rat jejunum. Biochim Biophys Acta 1996; 1280: 19–26. [DOI] [PubMed] [Google Scholar]
- 35. Shimada T, Hoshi T. Na+–dependent elevation of the acidic cell surface pH (microclimate pH) of rat jejunal villus cells induced by cyclic nucleotides and phorbol ester: possible mediators of the regulation of the Na+/H+ antiporter. Biochim Biophys Acta 1988; 937: 328–334. [DOI] [PubMed] [Google Scholar]
- 36. Drozdzik M, Gröer C, Penski J, Lapczuk J, Ostrowski M, Lai Y, et al. Protein abundance of clinically relevant multidrug transporters along the entire length of the human intestine. Mol Pharm 2014; 11: 3547–3555. [DOI] [PubMed] [Google Scholar]