

Abstract
Cranial neural crest cells are a multipotent cell population that generate all the elements of the pharyngeal cartilage with differentiation into chondrocytes tightly regulated by temporal intracellular and extracellular cues. Here, we demonstrate a novel role for miR-27, a highly enriched microRNA in the pharyngeal arches, as a positive regulator of chondrogenesis. Knock down of miR-27 led to nearly complete loss of pharyngeal cartilage by attenuating proliferation and blocking differentiation of pre-chondrogenic cells. Focal adhesion kinase (FAK) is a key regulator in integrin-mediated extracellular matrix (ECM) adhesion and has been proposed to function as a negative regulator of chondrogenesis. We show that FAK is downregulated in the pharyngeal arches during chondrogenesis and is a direct target of miR-27. Suppressing the accumulation of FAK in miR-27 morphants partially rescued the severe pharyngeal cartilage defects observed upon knock down of miR-27. These data support a crucial role for miR-27 in promoting chondrogenic differentiation in the pharyngeal arches through regulation of FAK.
Keywords: miR-27, Pharyngeal arches, Focal adhesion kinase, Chondrogenesis, Zebrafish
1. Introduction
Craniofacial abnormalities are among the most common human birth defects, cleft lip and palate being among the five most common congenital malformations (Gorlin et al., 1990). Although an increasing number of genetic mutations have been implicated with these malformations, there is limited information about the etiology of congenital craniofacial disorders. In zebrafish, many features that control craniofacial development and pharyngeal skeletal elements are conserved with that of higher vertebrates (Yelick and Schilling, 2002). Most skeletal structures in the skull and the entire pharyngeal skeleton are derived from a unique population of cells, cranial neural crest (CNC) cells (Couly et al., 1993; Lumsden et al., 1991; Schilling and Kimmel, 1994). CNC cells migrate from the dorsal neural tube in three streams to populate the pharyngeal arches. Post-migratory CNC cells go through mesenchymal condensation during which pre-chondrogenic cells (PCCs) aggregate and increase their cell-cell contacts. Coincident with dynamic changes in the extracellular matrix (ECM), PCCs differentiate into chondrocytes surrounded by a type-II collagen and aggrecan rich matrix (Hall and Miyake, 2000; Kozhemyakina et al., 2015).
Vertebrate CNCs are a migratory, multipotent cell population, able to differentiate into cartilage, bone, teeth forming cells, and non-ectomesenchyme derivatives, such as neurons, pigment cells and glia (Baroffio et al., 1991). Chondrogenic differentiation of CNC cells is regulated by various signaling pathways including Tgf-β, Bmp, and Fgf pathways, as well as changes in cell shape (Kozhemyakina et al., 2015). As cell-cell interactions increase during mesenchymal condensation, PCCs become more rounded. Recent studies have shown that restricting cell spreading on synthetic substrates, or by maintaining high-cell density to prevent cell spreading, promotes chondrogenic differentiation of mesenchymal stem cells (Gao et al., 2010; McBride and Knothe Tate, 2008). Interestingly, mechanical forces or changes in the ECM that perturb cell shape lead to the formation of integrin-mediated focal adhesions, which in turn prevents chondrogenesis (Eyckmans et al., 2011; Tang et al., 2013; Yim and Sheetz, 2012). Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase and an essential component of focal adhesions (Parsons, 2003). Apart from its well-established roles in cell adhesion and migration, FAK is also involved in regulating mesenchymal stem cell fates in response to cell shape changes and integrin-β1 activation (Mitra et al., 2005; Pala et al., 2008; Takahashi et al., 2003; Tang et al., 2013). Exactly how FAK is regulated in the pharyngeal arches during chondrogenesis is not known.
miRNAs are small noncoding RNAs that regulate the expression of target mRNAs at the post-transcriptional level. miRNAs bind to the 3′UTR of their targets with imperfect base pairing and induce dead-enylation, translational repression, and degradation of the target mRNA (Huntzinger and Izaurralde, 2011; Krol et al., 2010). Tissue-specific expression of miRNAs allows them to regulate multiple developmental processes in diverse organisms (Flynt et al., 2007; Kloosterman and Plasterk, 2006; Li et al., 2011; Wei et al., 2013; Wienholds, 2005). Previous studies reported that miRNAs are required for skeletal development using mice with conditional deletion of Dicer, an RNaseIII-like enzyme required for miRNA biogenesis, in either NC cells or early chondrocytes in the craniofacial cartilage or growth plate (Kobayashi et al., 2008; Zehir et al., 2010). Global deficiency of all miRNAs in NC cells resulted in the loss of the majority of NC-derived craniofacial cartilage and bone (Zehir et al., 2010). These studies show that miRNA expression is crucial for skeletal development but only a small subset of miRNAs have been characterized as to their targets and control of whole organism cartilage and bone development (miR-92a, miR-140 and miR-452) (Eberhart et al., 2008; Nakamura et al., 2011; Ning et al., 2013; Sheehy et al., 2010).
In this study, we demonstrate a novel role for miR-27, a highly conserved miRNA family, during craniofacial cartilage development. Knock down of miR-27 inhibited pharyngeal arch morphogenesis and caused severe defects in the neurocranium. We show that these craniofacial defects are caused by impaired proliferation and differentiation of chondrogenic progenitors. Knock down of ptk2aa (FAK), a direct target of miR-27, can partially rescue the cartilage defects in miR-27 morphants indicating a novel mechanism whereby miR-27 regulates chondrogenic differentiation in the pharyngeal arches through modulation of FAK levels.
2. Results
2.1. Zebrafish miR-27 is expressed in pharyngeal arches
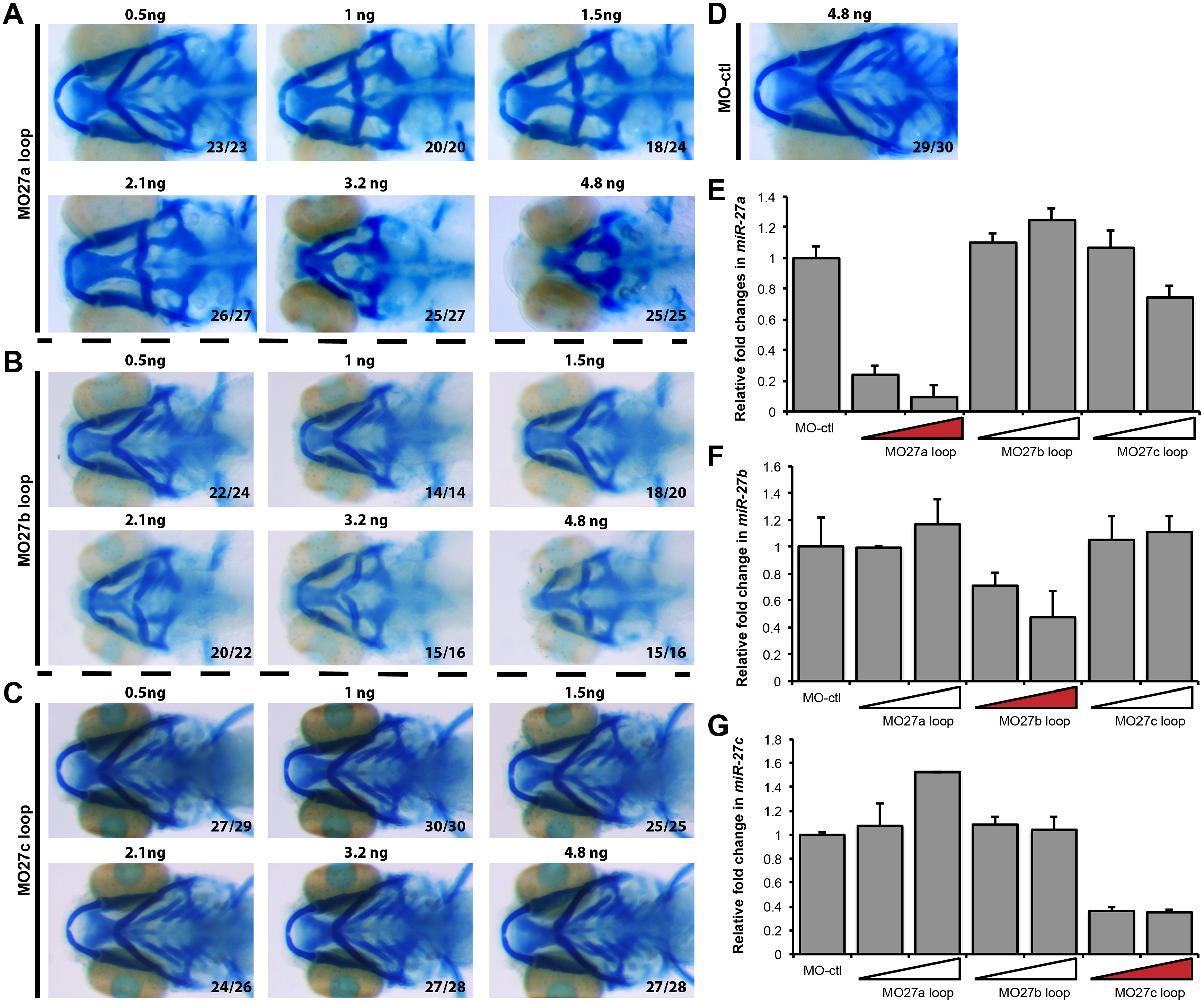
In zebrafish, there are five members of the miR-27 family with nearly identical mature miRNA sequences, but encoded on separate chromosomes (Fig. S1A). miR-27 is a highly conserved miRNA family, the sequences of mature miR-27a among different vertebrate species are identical except for a single nucleotide at the 3′ end (Fig. S1B). The seed sequences are identical among family members suggesting that identical target mRNAs are commonly regulated. Microarray and high-throughput sequencing analyses during early zebrafish development have shown that miR-27 family members are expressed as early as 24 h post fertilization (hpf), but upregulated at later stages (Wei et al., 2012; Wienholds, 2005). To determine the onset of expression of each miR-27 member, we performed qRT-PCR at several key developmental stages starting from the 1-cell stage (Fig. 1A, B). Overall, all miR-27 members have low expression levels between the 2 somite-stage (2ss) and 24hpf, while their expression is upregulated starting at 48hpf, reaching a peak at 72hpf. miR-27c is expressed at significantly higher levels compared to other members, while miR-27d and miR-27e have very low expression levels throughout the stages we analyzed. For this reason, we focused on spatial expression of miR-27a, miR-27b and miR-27c by performing whole-mount in situ hybridization by locked nucleic acid (LNA) probes on zebrafish embryos (Fig. 1C–E, Fig. S1C–F). At 4dpf, miR-27a, b and c are expressed strongly in the pharyngeal arches (Fig. 1C–E, Fig. S1D–F). Earlier during development, miR-27a is detected in the pharyngeal arch primordia that are composed of post-migratory chondrocyte progenitors, as well as in the eye, vasculature, the midbrain-hindbrain boundary (MHB), and the pectoral fins at 24 and 32 hpf (Fig. S1C). At 48 and 72hpf, miR-27a expression is more confined to the pharyngeal cartilage, cartilage joints and pectoral fins along with less expression in the ethmoid plate (EP) and brain. Strong expression of miR-27 in the pharyngeal arches, as well as earlier prechondrogenic mesenchyme, suggest that miR-27 may regulate cartilage development in zebrafish.
Fig. 1.

Knock down of miR-27 leads to craniofacial and pectoral fin defects. (A, B) qRT-PCR for miR-27a-e at the indicated developmental stages normalized to U6 snRNA. Fold changes were calculated using ΔΔC(t) method comparing all miR-27 levels to miR-27c levels at the 3 somite stage (ss). Due to comparably higher levels, miR-27c expression profile is shown separately. (C–E) Expression of miR-27a, miR-27b and miR-27c in 4dpf embryos detected by whole-mount in situ hybridization by locked nucleic acid (LNA) probes. All are ventral views of the head. (F, G) Dorsal view of 3 dpf live embryos injected with either 5 ng standard control morpholino (MO-ctl) or MO-27 at the single-cell stage. Pectoral fins are indicated with arrowheads. (H, I) Morphology of the head in 4 dpf embryos injected with either MO-ctl or MO-27. Lateral views, jaws are indicated with arrowheads. Scale bar, 300 μm. (J) miR-27 and U6 levels in uninjected control and miR-27 morpholino (MO-27) injected embryos at 48hpf detected by Northern blot. (K–P) Head cartilages stained with Alcian blue in 4dpf embryos injected with (K, L) standard control morpholino (MO-ctl), (M, N) 4-mismatch miR-27 morpholino (MO27-4mis) and (O, P) MO-27. Top panels, ventral views; bottom panels, lateral views. The indicated ratio represents the number of embryos with the represented phenotype/total number of observed embryos. Cartilage labels: ep, ethmoid plate; tc, trabeculae cranii; m, Meckel’s cartilage; pq, palatoquadrate; ch, ceratohyal; hs, hyosymplectic; cb, ceratobranchial. Anterior side of the embryos is to the left. (Q, R) Staining of pectoral fin skeleton in 4dpf embryos by Alcian blue. The right side pectoral fin is shown with anterior to the top. The cleithrum (cl) and scapulocoracoid (sc) cartilages and postcoracoid process (pc) of pectoral fins are missing and the endoskeletal disc cartilage (ed) is smaller in miR-27 morphants compared to the controls. Scale bar, 200 μm.
2.2. Knockdown of miR-27 leads to craniofacial and pectoral fin cartilage defects
To determine the function of miR-27, we performed loss-of-function experiments by injecting antisense morpholinos against miR-27. The first morpholino we tested was designed complementary to the mature miR-27 sequence (MO-27) and targets all members of the miR-27 family (Table 1). Injection with MO-27 resulted in approximately 70% loss of miR-27 (Fig. 1J). Knockdown of miR-27 did not lead to any gross morphological changes during early development, but by 3 dpf and even more so by 4 dpf, miR-27 morphants displayed pectoral fin outgrowth defects and severely reduced pharyngeal cartilage, concomitant with smaller heads and eyes (Fig. 1F–I). To analyze the cartilage defects more specifically, we performed Alcian blue staining. In miR-27 morphants, nearly all of the cartilage in the pharyngeal arches, as well as in the pectoral fins, was missing (Fig. 1O–R). In addition, miR-27 knockdown led to an abridged palate in the neurocranium, where the bilateral trabeculae were joined in the midline but the ethmoid plate did not extend properly. Closer examination of the pectoral fins revealed that the cleithrum (cl) and scapulocoracoid (sc) cartilages and postcoracoid process (pc) of pectoral fins were missing and the endoskeletal disc cartilage (ed) was smaller in miR-27 morphants compared to the controls (Fig. 1Q, R).
Table 1.
List of morpholino sequences used in the study.
| MO-standard control (MO-ctl) | 5′-CCTCTTACCTCAGTTACAATTTATA-3′ |
| MO-27-4mis | 5′-GCGCAACTTACCCAGTGTCAACA-3′ |
| MO-27 | 5′-GCGGAACTTAGCCACTGTGAACA-3′ |
| MO-27a loop | 5′-TAGCCACTGTGAACATAGGATATCC-3′ |
| MO-27b loop | 5′-CTTAGCCACTGTGAACAAAGAGTTC-3′ |
| MO-27c loop | 5′-CCACTGTGAACATTGAAGTTCGATC-3′ |
| MO-ptk2 | 5′-TCCAGGAATGCCGTCGCCATGCCTT-3′ |
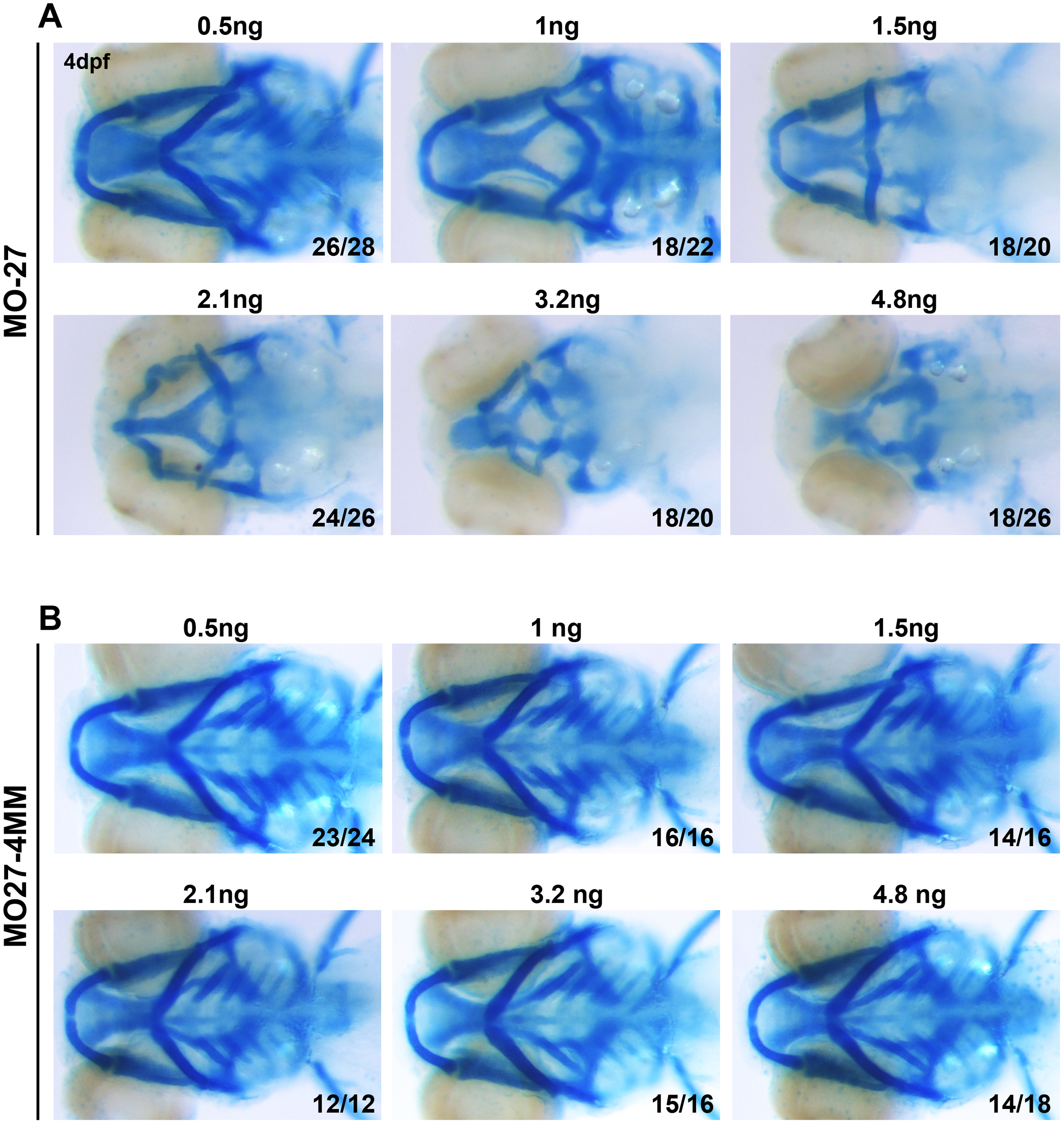
To ensure specificity with the morpholino knockdowns, we per formed a series of control experiments. First, apart from the use of a standard control morpholino (MO-ctl; Gene Tools), we designed a morpholino containing four-base mismatches (MO27-4mis) compared to MO-27 (Fig. 1M, N). MO27-4mis injection did not cause any defects in pharyngeal arch morphogenesis. Second, we designed a third set of morpholinos complementary to the loop sequences of each precursor mir-27 member (Fig. S3A–C, Table 1). Compared to MO-27 which targets all miR-27 family members, loop morpholinos target individual family members by inhibiting the processing of the corresponding precursor miRNA (Kloosterman et al., 2007). Knock down of miR-27a by MO27a loop led to exactly the same phenotype as the MO-27 injections with loss of nearly all the pharyngeal cartilage and a severely reduced neurocranium (Fig. S3A). Knock down of miR-27b using the MO27b loop morpholino led to loss of branchial arches along with severely reduced Meckel’s and ceratohyal cartilage, as well as an abridged neurocranium (Fig. S3B). In contrast, knockdown of miR-27c did not affect the pharyngeal arches (Fig. S3C). These experiments enabled us to determine which miR-27 members are involved in pharyngeal cartilage formation, as well as provide evidence that the defects observed upon loss of all miR-27 family members using the MO-27 morpholino are unlikely to be due to nonspecific effects or toxicity. In addition, the effects of the MO-27 and miR-27 loop morpholinos were dose dependent across an order of magnitude concentration as determined by alcian blue staining on embryos injected with different doses (Fig. S2A–B, Fig. S3A–C). Injection of the mismatch morpholino did not generate any observable phenotypes at any concentration tested (Fig. S2A, B). Knockdown miR-27a and miR-27b separately also showed dose-dependent defects in pharyngeal cartilage formation (Fig. S3A, B). Finally, we tested the knockdown efficiency of each morpholino. By Northern blots, we confirmed approximately 70% loss of miR-27 upon MO-27 injection at the single cell stage (Fig. 1J). To determine the efficacy of the miRNA loop morpholinos, we performed qRT-PCR for miR-27a, miR-27b and miR-27c in 48hpf embryos injected with two different concentrations of MO-27a loop, MO-27b loop and MO27c loop (Fig. S3E–G). All loop morpholinos led to knockdowns in the range of 50–90% of the targeted mature miR-27.
As miR-27 is required for pharyngeal arch development, we next tested whether conditional miR-27 overexpression would induce any craniofacial cartilage defects as well. To conditionally overexpress miR-27, we established two separate transgenic lines, Tg(hsp70l:miR27eGFP) in which pri-miR-27b is expressed under the heat shock promoter (hsp70l), and Tg(sox10:miR-27eGFP) in which pri-miR-27 is expressed under the neural-crest specific sox10 promoter (Fig. S4). We induced the expression of miR-27 in Tg(hsp70l:miR27eGFP) at 24 hpf and confirmed upregulation at 48hpf by qRT-PCR (Fig. S4B). After overexpression, we performed alcian blue staining at 4 dpf and detected no significant differences in the pharyngeal cartilage as well as in the neurocranium in hsp70:miR27eGFP transgenic embryos relative to their non-transgenic siblings (Fig. S4A). When miR-27 was overexpressed in the sox10+ NC cells in Tg(sox10:miR-27beGFP) embryos, we again did not detect any defects in craniofacial cartilage morphogenesis in the transgenic embryos compared to their non-transgenic siblings (Fig. S4C).
2.3. miR-27 is required in post-migratory CNC cells for proper pharyngeal arch morphogenesis
Because craniofacial cartilage elements are derived from ectomesenchymal CNC cells, we tested whether miR-27 knockdown perturbs early neural crest migration. To determine whether the migration of CNC cells was affected, we analyzed CNC cells during migration into the pharyngeal arches using a transgenic line that expresses another marker of CNC cells, sox10 (Tg(sox10(7.2):mRFP)vu234) (Dutton et al., 2001). The sox10+ arches did not differ in size between embryos at 18 and 24 hpf (Fig. 2A). Complementary in situ hybridization experiments for sox10 and another CNC cell marker, dlx2a, showed the same expression patterns at 16 hpf and 22 hpf in morphants and controls (Fig. 2B, C), suggesting that mir-27 knockdown does not affect CNC cell specification or migration.
Fig. 2.

Early cranial neural crest (CNC) cell specification and migration are not affected in miR-27 morphants. (A) Lateral view of Tg(sox10(7.2):mRFP)vu234 embryos injected either with MO-ctl or MO27 showing cranial neural crest cell streams populating the pharyngeal arches at 18hpf and 24hpf. Anterior is to the left and posterior is to the right. Neural crest streams are numbered from 1 to 3. The indicated ratio represents the number of embryos with the represented phenotype/total number of observed embryos. (B, C) Expression of the neural crest cell marker, sox10, and CNC marker dlx2a in MO-ctl and MO27 embryos at 16 hpf and 22 hpf detected by whole-mount in situ hybridization. Dorsal view of embryos with anterior side to the top.
To determine the earliest time point when the miR-27 morphants show defects in craniofacial development, we analyzed pre-cartilage condensation within the pharyngeal arches using a transgenic line marking the ectomesenchyme lineage of CNC cells (Tg(fli1a:eGFP)y1) (Lawson and Weinstein, 2002; Fig. 3A). In Tg(fli1a:eGFP)y1 embryos at 26 hpf, we did not detect any differences in the size and patterning of the fli:EGFP+ arches. However, at 30 and 36 hpf, the size and fluorescence intensity of the fli1a:eGFP+ arches were significantly perturbed in the miR-27 morphants (Fig. 3A, B). We also detected reduced expression domains for the post-migratory CNC cell marker, dlx2a, at 36 hpf in the posterior pharyngeal arches, while at 30 hpf there was not a detectable reduction in dlx2a expression yet (Fig. 3C, D). It is important to note that by 36 hpf, the patterning of the pharyngeal arches was similar between control embryos and miR-27 morphants, while elongation of the arches along the dorsal-ventral axis was disrupted upon miR-27 knockdown. By 48 hpf, defects in the pharyngeal arch sizes were more severe in the morphants and morphogenesis of the first two arches was not complete (Fig. 3A, B). These data suggest that the onset of the pharyngeal arch morphogenesis defects upon loss of miR-27 is between 30 and 36 hpf.
Fig. 3.

Morphogenesis of the pharyngeal arches is disrupted in miR-27a morphants between 30 and 36hpf. (A) Live images of Tg(fli1a:eGFP)y1 embryos at 26, 30, 36, 48 hpf; lateral views with anterior to the left. Each pharyngeal arch is numbered. Embryos were injected with either 5 ng MO-ctl or MO-27 at the single cell stage and observed with confocal microscopy at the indicated stages. Time-lapse videos captured by spinning-disk confocal microscopy are shown in Supp. Movies 1 and 2. Scale bar, 100 μm. (B) Quantification of normalized and relative fli1a:eGFP fluorescence intensity in the pharyngeal arches of MO-ctl and MO-27 embryos at the indicated time points. The relative fluorescence intensities were normalized to the area of the arches selected and the fluorescence background in each image. Error bars indicate SEM and the number of embryos analyzed is indicated above the bars. For 26 hpf, n=3; for 30 hpf, n=5; for 36 hpf, n=4; for 48 hpf, n=4. Data represent three independent experimental trials. The n.s. non-significant, **p < 0.01, ****p < 0.0001 (Student’s t-test). (C–D) Expression of the CNC marker dlx2a in embryos at 30 and 36 hpf. Embryos were injected with the corresponding morpholinos as described above. Pharyngeal arch expression domains are labeled. The indicated ratio represents the number of embryos with the represented phenotype/total number of observed embryos. Scale bar, 200 μm.
To gain insight into the requirement of miR-27 in pharyngeal arch morphogenetic movements, we performed in vivo time-lapse imaging of pre-chondrogenic crest cells in Tg(fli1a:eGFP)y1 embryos. Timelapse imaging showed that migration of CNC cells into the pharyngeal arches in the anterior-posterior axis was slower in miR-27 morphants compared to the controls. Convergence of the first two arches by 48hpf was not complete in the morphants as the first pouch in between the two arches was still visible, while in control embryos the first two arches did not have any clear boundary at this point (Movies S1 and S2). These findings suggest that miR-27 is required for pharyngeal arch morphogenesis, but not in early patterning of the arches by 30hpf.
Supplementary material related to this article can be found online at: doi:10.1016/j.ydbio.2017.06.013.
2.4. miR-27 is required for the differentiation of pre-chondrogenic crest cells
To identify what stage of cartilage development mir-27 regulates, we analyzed pharyngeal mesenchymal condensation and chondrogenic differentiation of post-migratory PCCs. To assess mesenchymal condensation, we stained with peanut agglutinin (PNA), a lectin that preferentially binds to the cell surface during condensation (Hall and Miyake, 1995). PNA staining of the fli1a:eGFP+ PCCs showed that mesenchymal condensations were equally detectable in miR-27 morphants and control embryos at 48 hpf (data not shown). Next, we analyzed the distribution of the extracellular matrix (ECM) protein fibronectin which is highly expressed during mesenchymal condensation (Hall and Miyake, 2000; Singh and Schwarzbauer, 2014). PCCs in both morphants and control embryos had equal fibronectin matrix distribution at 48 hpf (Fig. S5). To analyze the differentiation of PCCs, we performed in situ hybridization for the chondrogenic differentiation marker sox9a (Bi et al., 1999; Yan et al., 2002) at 55 hpf and for col2a1a, the major collagen in chondrocytes, at 72 hpf (Fig. 4A). Both markers showed reduced expression domains in the ethmoid plate precursor and first two pharyngeal arches, as well as complete loss of expression in the ventral pharyngeal arches of the miR-27 morphants. We could not detect expression of the osteogenic differentiation marker, runx2b (Flores et al., 2004) in the pharyngeal arches of miR-27 morphants at 60hpf.
Fig. 4.

miR-27 is required for differentiation of pre-chondrogenic crest (PCC) cells in pharyngeal arches. (A) Expression of the chondrogenic marker sox9a, the osteogenic marker, runx2b, and the chondrocyte-specific marker col2a1a in MO-ctl or MO-27 (5 ng) injected embryos, detected by whole-mount in situ hybridization. Arrowheads indicate the expression domain of the ceratobranchial arches. All panels except the top panel for col2a1a are lateral views. Top panel for col2a1a expression is a ventral view with the first pharyngeal arch indicated by an arrowhead. (B) Anti-Col2 and anti-GFP immunostaining in the pharyngeal arch region between the eye and otic vesicle (ov) in Tg(fli1a:eGFP)y1 embryos at 61hpf. A single optical section of the confocal stacks is shown in each image, with anterior to the left. Embryos were injected with the corresponding morpholinos as described above. (C) Wheat-germ agglutinin staining and anti-GFP immunostaining in the pharyngeal arch region between the eye and otic vesicle (ov) in Tg(fli1a:eGFP)y1 embryos at 61hpf. The indicated ratio represents the number of embryos with the represented phenotype/total number of observed embryos. Scale bar, 50 μm.
To assess the differentiation of PCCs further, we analyzed mature chondrocytes in the pharyngeal arches by immunostaining with anti-Col2 in fli1a:eGFP embryos at 61hpf. In control embryos we could easily detect fli1a:eGFP+ cells in the first two arch condensations that are secreting Col2 into the ECM, while in miR-27 morphants there were very few to no Col2+ fli1a:eGFP+ cells detected (Fig. 4B). To determine whether miR-27 only perturbs collagen production or completely blocks chondrogenic differentiation, we performed wheat germ agglutinin (WGA) staining to detect mature chondrocytes. WGA staining indicated that in miR-27 morphants, mature chondrocytes are missing, while the control embryos had WGA labeled chondrocytes in fli1a:eGFP+ condensations at 61hpf. These results together suggest that loss of miR-27 does not affect mesenchymal condensation of CNC cells, but completely blocks differentiation of pharyngeal PCCs.
2.5. miR-27 knock down impairs the PCC proliferation and survival
Defects in the extension of the pharyngeal arches in miR-27 morphants suggest that inactivation of miR-27 causes PCC proliferation defects. We analyzed the proliferation of pre-chondrogenic cells at 30 and 36 hpf, the earliest time points where defects can be detected in the pharyngeal arches (Fig. 3A). Immunostaining for phosphorylated histone 3 (pH3) in fli1a: eGFP embryos showed that there was a significant reduction in the number of mitotic PCCs in miR-27 morphants compared to controls at both 30 and 36hpf (Fig. 5A, B). Upon mir-27 knockdown, the impairment in PCC proliferation was more severe at 36hpf compared to 30 hpf (Fig. 5A, B).
Fig. 5.

miR-27 knock-down impairs proliferation and survival of PCC cells. (A) Anti-phospho histone 3 (pH3) and anti-GFP immunostaining in Tg(fli1a:eGFP)y1 embryos at 32 and 36 hpf. Embryos were injected with either 5 ng MO-ctl or MO-27 at the single cell stage. Lateral view with anterior to the left. (B) Quantification of pH3+GFP+ cells normalized to the GFP+ area in each embryo. For 30 hpf, n=7; for 36 hpf, n=8. (C) TUNEL staining and anti-GFP immunostaining in Tg(fli1a:eGFP)y1 embryos at 36 and 54 hpf. Embryos were injected with the corresponding morpholinos and image layouts are as described above. (D) Quantification of TUNEL+GFP+ cells normalized to the GFP+ area in each embryo. Error bars indicate SEM. For 36 hpf, n=9; for 54 hpf, n=5. *p < 0.05, ***p < 0.001 (Student’s t-test). Data are from four independent experiments. Scale bars, 100 μm.
Next, we investigated whether apoptosis in prechondrogenic cells might contribute to the pharyngeal arch extension defects in miR-27 morphants. TUNEL assays in fli:EGFP embryos showed that at 36hpf there was not a significant difference in the number of apoptotic PCCs in miR-27 morphants compared to control embryos. However, at 54hpf, when PCCs are differentiating into mature chondrocytes, there was a significant increase in the number of apoptotic PCCs in the morphants compared to the controls (Fig. 5C, D). These findings suggest that the initial pharyngeal arch growth defects in miR-27 morphants are due to a decrease in PCC proliferation. At later stages, PCCs that were not able to differentiate into chondrocytes undergo apoptosis.
2.6. Ptk2aa (FAK) is a direct target of miR-27 in vivo
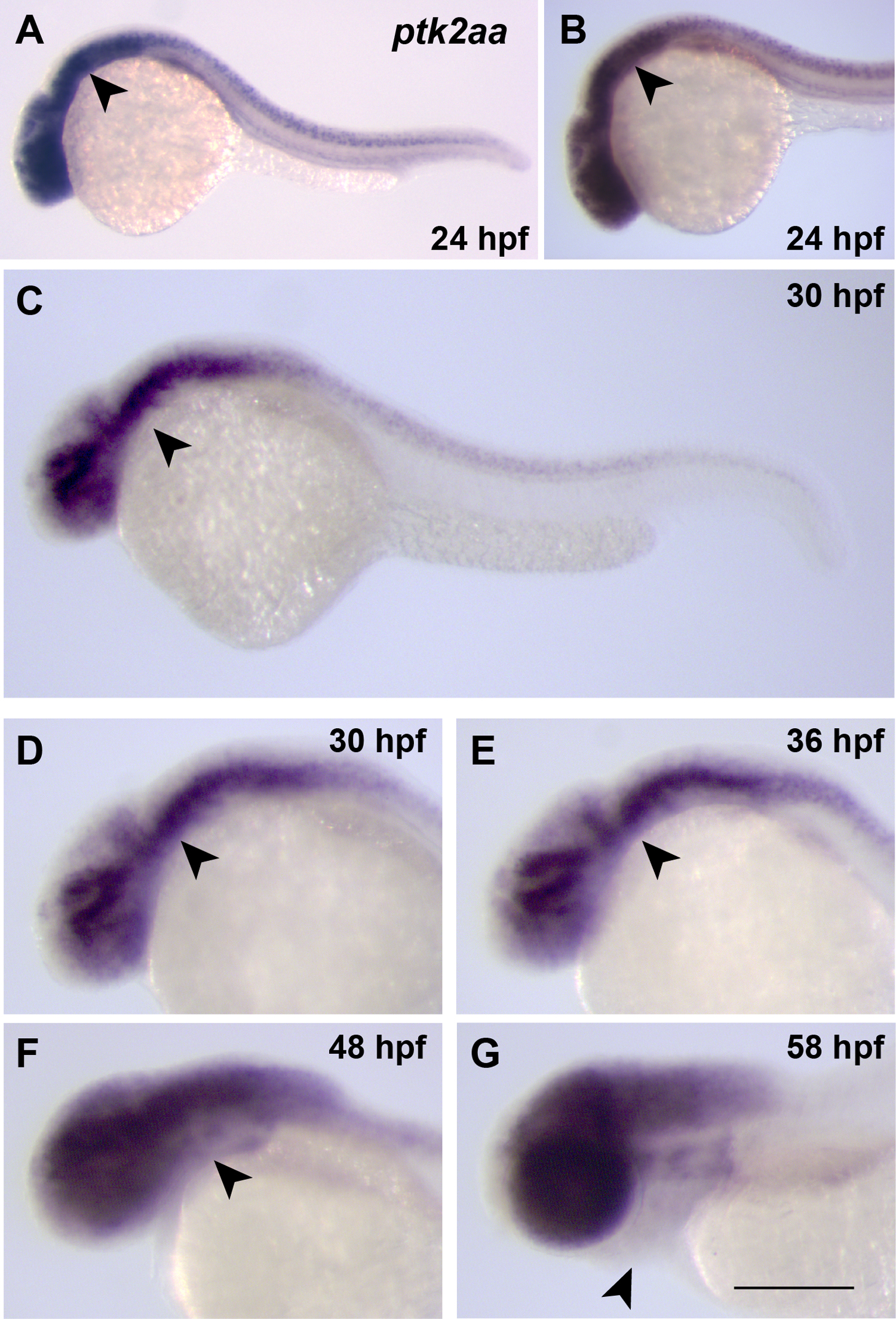
To investigate the molecular mechanism through which miR-27 regulates pharyngeal cartilage development, we tested candidate mRNA targets of miR-27 using the online target prediction algorithm TargetScanFish (Lewis et al., 2005). We focused on a subset of potential target mRNAs based on their spatiotemporal expression patterns at different developmental stages. After initial testing of multiple candidate mRNAs, ptk2aa was selected for further functional analyses. Ptk2aa is a zebrafish focal adhesion kinase (FAK) gene whose expression is ubiquitous at the shield stage and accumulates in the head, axial region and tail at the 10-somite stage (Crawford et al., 2003). At 24 hpf, there is strong expression detected in the head and dorsal axis (Fig. S6A, B). Since we detected miR-27 expression in the pharyngeal arch region starting at 24 hpf (Fig. S1C), and because pharyngeal arch defects in miR-27 morphants are first observed around 30–36 hpf (Fig. 3A), we decided to determine the expression pattern of ptk2aa at these crucial time points. Early ptk2aa expression was detected strongly in the head, as previously reported, and between 30 and 48 hpf, was detected in the midbrain, MHB, and paraxial mesoderm axis, with low expression in the pharyngeal region (Fig. S6C–F; Crawford et al., 2003). At 58 hpf, ptk2aa expression in the jaw cartilage was not detectable (Fig. S6G). Consistent with the in situ data, active FAK protein (FAK-pY397) showed little to no co-localization with the arches as marked by fli1a:eGFP at 36 and 48hpf (Fig. S7A, B). The reduction of ptk2aa expression in the pharyngeal arch region compared to the higher expression in the head at 24hpf is consistent with the prediction that ptk2aa might be targeted by miR-27.
Ptk2aa has two miRNA binding elements (MREs) for miR-27 in the 3′UTR (Fig. 6A). To test whether miR-27 can target ptk2aa, we performed GFP reporter assays. As shown in Fig. 6B, injection of GFP-ptk2aa 3′UTR mRNA into one-cell stage embryos caused strong GFP expression at 24hpf, while co-injection of the mRNA and mature miR-27 mimic RNAs decreased GFP fluorescence levels. Western blots of pooled protein lysates from these embryos confirmed that co-injection of miR-27 significantly decreased GFP protein levels (Fig. 6D, E). To demonstrate specificity of targeting, we created mutations that disrupted the miR-27 seed regions within both MREs in the ptk2aa 3′UTR. Co-injection of the mutated transcripts with miR-27 did not decrease the GFP fluorescence compared to injection of the reporters alone (Fig. 6C). These results support the hypothesis that miR-27 can regulate ptk2aa through the two MREs in its 3′UTR.
Fig. 6.

miR-27 targets FAK and regulates FAK levels in vivo. (A) Schematic of the reporter mRNA consisting of the coding sequence of GFP fused to the ptk2aa 3′UTR. Two predicted miRNA recognition elements (MREs) are indicated. Predicted base-pairing between MREs (shown in green) and the miR-27a sequence (shown in red). (B) Embryos injected with either GFP-ptk2aa 3′UTR reporter mRNA alone or co-injected with miR-27a imaged at 24hpf. (C) Reporter assays with GFP-ptk2aa 3′UTR mRNA containing mutations in the miR-27 seed sites. (D) Western blots with anti-GFP and anti-tubulin antibodies using the lysates from embryos injected with the GFP-ptk2aa 3′UTR reporter mRNA alone or co-injected with miR-27a. (E) Quantification of GFP levels by Western blots normalized to the levels of tubulin. (F) Western blots with anti-FAK and anti-GAPDH antibodies using lysates from 24hpf embryos either uninjected or injected with 100 or 200 pg miR-27a. (G) Western blots with anti-FAK and anti-GAPDH antibodies using lysates from 48hpf embryos injected either with MO-ctl or MO-27. (H) Quantification of FAK levels from Western blots shown in (G) normalized to the levels of tubulin. At least 20 embryos were pooled for protein lysates. Error bars indicate SEM. **p < 0.01 (Student’s t-test).
Next, we tested whether miR-27 regulates endogenous ptk2aa. We injected mature miR-27 mimic RNAs in two different concentrations into single cell embryos and then prepared protein lysates from injected embryos at 24 hpf followed by western blots with antibodies against FAK. Increasing doses of miR-27 led to a 50–70% decrease in endogenous FAK protein levels (Fig. 6F). As a complementary experiment, we analyzed FAK protein levels when miR-27 was knocked down. We injected either miR-27 morpholinos or control morpholinos into single cell embryos at the same concentration as we used in the functional analyses described above. Western blot analyses showed that FAK levels were more than 2-fold upregulated in the miR-27 morphants compared to control embryos at 2dpf (Fig. 6G, H). Collectively, these results indicate that miR-27 regulates FAK in vivo.
2.7. Knock down of ptk2aa (FAK) partially rescues pharyngeal cartilage defects in miR-27 morphants
Next, we investigated whether the pharyngeal cartilage defects in miR-27 morphants are caused by misregulation of ptk2aa (FAK). Given that FAK is upregulated in miR-27 morphants, we hypothesized that knocking down FAK in the morphants would rescue the pharyngeal cartilage defects. To knock down FAK, we designed antisense morpholinos that block the translation start site of both ptk2aa and ptk2ab. We titrated the ptk2 morpholinos and determined the highest concentration (3 ng) that did not disrupt normal morphological development at 1 dpf (data not shown). At this concentration, Ptk2 morpholinos (MO-ptk2) efficiently blocked translation of a ptk2aa reporter construct (Fig. S8). We then injected morpholinos against miR-27 in two different concentrations, co-injected with either control or ptk2 morpholinos, and examined whether the craniofacial defects were suppressed upon knockdown of miR-27. We categorized the pharyngeal cartilage phenotypes observed at 4 dpf into three groups (CH1, CH2 and CH3) based on the ceratohyal position and size (Fig. 7A). CH1 had the least severe cartilage phenotype with Meckel’s cartilage not as anteriorly extended as compared to wild type embryos, along with three missing posterior branchial arches. In CH2, the sizes of all visible pharyngeal cartilage elements were shorter in comparison to CH1, the ceratohyal cartilage was not able to extend anteriorly, and all branchial arches were missing. CH3 had the most severe cartilage phenotype with Meckel’s cartilage and the branchial arch phenotypes similar to CH2, but the ceratohyal cartilage had a more severe phenotype by shifting posteriorly. Co-injection of ptk2 morpholinos and miR-27 morpholinos both at low (2 ng) and high (4 ng) concentrations increased the frequency of the CH1 phenotype, while significantly decreasing the frequency of the more severe phenotypes (CH2, CH3) (Fig. 7B). The ability to suppress the cartilage defects in miR-27 morphants by coincident knockdown of ptk2aa is consistent with regulation of FAK by miR-27.
Fig. 7.

Suppressing FAK in miR-27 morphants partially rescues cartilage defects. (A) Three categories of representative head cartilage phenotypes in 4dpf old embryos injected with MO-27, along with either standard control morpholino MO-ctl or translation blocking MO-ptk2 at the single-cell stage. Ventral views of head cartilage stained with alcian blue. Ceratohyal cartilage indicated by an arrowhead and Meckel’s cartilage indicated with an arrow. (B) Embryos were injected with 2 ng or 4 ng MO-27 either co-injected with 3 ng MO-ctl or MO-ptk2. Percentage of embryos with the corresponding head cartilage phenotypes (CH1-CH3) at 4dpf. The distribution of phenotypes in MO-ptk2 and MO-27 injected embryos are compared to those injected with MO-ctl and MO-27. ***p < 0.0001 (Chi-squared goodness of fit test), n=40–60 embryos. Data are from three independent experiments. (C) Analysis of cartilage positions. Ventral view of head cartilages in wild-type embryos stained with alcian blue. A represents the distance between the palataquadrate (pq) and ceratohyal (ch) cartilage joints. B represents the distance from the anterior joint of the two ceratohyals to the baseline shown by a dashed line. C represents the distance from the anterior end of Meckel’s cartilage (m) to the baseline. (D) (C-B)/A ratio was calculated for quantitative analysis of anterior-posterior extension of the ceratohyal cartilage. This ratio increases as the ceratohyal position extends posteriorly instead of anteriorly. Percentage increase of the (C-B)/A ratio in MO-27 and MO-ctl injected embryos or embryos co-injected with both MO-27 and MO-ptk2 compared to the ratio in wild-type embryos. Error bars represent SEM. n=40–60 embryos, **p < 0.01 (student’s t-test).
Next, we quantitatively evaluated the level of rescue in extension of the ceratohyal cartilage in the anterior-posterior axis. We measured the distance between Meckel’s cartilage and the ceratohyal cartilage to a reference point where the palatoquadrate cartilage and hyosymplectic cartilage joint (Fig. 7C). To normalize these lengths to the width of the pharyngeal skeleton, we measured the distance between the palatoquadrate cartilage and the hyosymplectic cartilage joins, as previously described (Wu et al., 2015). The ratio (C/A), which represents the normalized length of Meckel’s cartilage, did not show any significant difference in MO-ptk2 and MO-27 (2 ng) co-injected embryos compared to injection of only MO-27 (2 ng) (data not shown). The ratio (C-B)/A represents the positional relationship between Meckel’s cartilage and the ceratohyal cartilage. As the phenotypes get more severe, the ceratohyal cartilage shifts more posteriorly and the (C-B)/A ratio increased in miR-27 morphants compared to controls (Fig. 7D). However, when we knocked down ptk2 along with miR-27, the ratio was significantly smaller than in miR-27 morphants, consistent with defects observed when we used higher miR-27 morpholino concentrations (data not shown). This indicates that ptk2 knockdown partially suppresses the severe ceratohyal positioning in the mild miR-27 morphants. Overall, these results demonstrate that miR-27 knock down leads to craniofacial cartilage defects due to misregulation of ptk2.
3. Discussion
3.1. miR-27 is essential for chondrogenic differentiation
We show that miR-27 is required for cartilage development both in the pharyngeal arches and the pectoral fins, which are derived from CNC cells and the paraxial mesoderm, respectively (Le Douarin et al., 2004; Lee et al., 2013). Using loss-of-function studies, we show that miR-27 knockdown does not affect neural crest specification and migration of the CNC cell streams, but is instead required for the proliferation and differentiation of PCCs. This suggests that miR-27 promotes a chondrogenic differentiation program regardless of the origin of the pre-chondrogenic progenitor cells. Biallelic knockout clones for the miR-23a~27a~24-2 and miR-23b~27b~24-1 clusters in ESCs demonstrated that these clusters are indispensable for ESC differentiation in vitro and in vivo (Ma et al., 2014). Our study shows that either miR-27a or miR-27b knockdown leads to severe craniofacial defects (Fig. S3), consistent with the requirement for both isoforms in ESC differentiation and supporting the idea that both family members cooperate to control chondrogenic differentiation.
Although the role of miR-27 in chondrogenesis has not been reported before, the miR-23a~27a~24-2 cluster was implicated in skeletal development by negatively regulating in vitro osteogenic differentiation (Hassan et al., 2010). Interestingly, one of the most crucial bone-specific transcription factors, Runx2, was reported to suppress the transcription of the miR-23a~27a~24-2 cluster early in osteogenesis, while in the final osteocyte stage of differentiation this cluster is upregulated and functions to attenuate continuing bone formation. These findings, along with our results demonstrating that miR-27 is a positive regulator of chondrogenesis, suggest that miR-27 plays a major role in the cell fate commitment program of skeletal stem cells.
3.2. miRNA knockdown and knockout experiments
Loss-of-function studies using morpholinos must be interpreted carefully with multiple controls to avoid possible off-target effects, as well as a reliable confirmation of morpholino efficacy. We have performed multiple control experiments following published guidelines such as the use of mismatch control morpholinos, targeting the same gene with two morpholinos, testing dose dependent effects, and ensuring the efficacy of knock downs (Eisen and Smith, 2008). In addition, the TUNEL assays showed that there was no increase in apoptosis in pre-chondrogenic cells at the stage when pharyngeal arch defects were detected suggesting that the morphant phenotype is not due to p53-induced apoptosis (Robu et al., 2007). Due to the fact that MO-27 targets the mature full length miR-27 RNA, we do not have a miR-27 isoform that is immune to the MO which could be used to rescue the morphant phenotype. However, and most importantly, we were able to suppress the effects of miR-27 knockdown by co-injection of morpholinos against the miR-27 target ptk2aa.
An ideal strategy to confirm morpholino associated phenotypes is to analyze mutant alleles of the same gene. There have been concerns about the lack of concordance between mutant and morphant phenotypes of the same genes in zebrafish (Kok et al., 2015; Stainier et al., 2015). To address these concerns, we generated a miR-27a mutant line (miR-27aΔ/Δ) that carried a 6 bp deletion in the miRNA seed region using the CRISPR/Cas9 system (Fig. S9A, B). Analysis of F2 and F3 embryos of heterozygous mutant parents either did not show any detectable craniofacial defects or the phenotypes similar to the miR27 morphants were observed with low penetrance in the homozygous mutants. There could be multiple reasons for the discrepancy between the miR-27 morphants and the mutants. One possibility is genetic compensation upon CRISPR-mediated mutation of the miR-27a gene, as has been reported recently, after other CRISPR and TALEN mediated mutations (Blum et al., 2015; Rossi et al., 2015). The candidate genes most likely contributing to the genetic compensation would be the other members of miR-27 family. Since we already showed that miR-27b is also required for pharyngeal cartilage morphogenesis (Fig. S3B), we hypothesized that miR-27b expression might compensate for the loss of miR-27a in miR-27aΔ/Δ embryos. Injection of MO27b-loop at low concentration normally results in no detectable defects in the pharyngeal cartilage (Fig. S3B, Fig. S9C). However, we found that exclusively homozygous miR-27aΔ/Δ embryos are sensitized to limited loss of miR-27b knockdown compared to miR-27a+/Δ and WT siblings (Fig. S9C–F). miR-27b knockdown with low doses of MO injection resulted in shorter pharyngeal elements, as shown by significant decreases in Meckel’s extension compared to miR-27a+/Δ heterozygotes and WT siblings (Fig. S9E–F). This indicates that miR-27b can contribute to compensation of the miR-27a loss in miR-27aΔ/Δ embryos. However, there could also be other genes that compensate for the loss of miR-27a, including protein coding genes as well as other miRNAs. Most miRNAs target multiple mRNAs, it is unlikely that ptk2aa (FAK) is only targeted by miR-27 family members, consistent with the finding that we were only able to partially suppress the severe ceratohyal positioning defect in the miR-27 morphants by coincident ptk2 knockdown. TargetScan predicts 7 miRNAs with significant seed matches that could target ptk2.2 and could conceivably compensate for the loss of miR-27 and retain proper regulation of ptk2.2. It remains to be determined whether any of these miRNAs are temporally and spatially expressed in a manner consistent with regulation of pharyngeal arch development.
While generation of 5 independent lines and crosses to generate a line lacking expression of all miR-27 family members is beyond the scope of this report, we did attempt to utilize multiplex CRISPR/Cas9 using guide RNAs that target all miR-27 family members in early embryos (Narayanan et al., 2016). Unfortunately, we did not observe any resulting phenotypes at 4dpf, potentially due to the fact that the level of miR-27 knockdown in early embryos was not sufficient, averaging less than 50%.
3.3. miR-27 regulates pharyngeal cartilage development through targeting FAK
miRNAs are well-known for their role in preventing translation and accelerating decay of target mRNAs, thereby providing a precise mechanism for spatiotemporal control of developmental processes (Krol et al., 2010; Zhao and Srivastava, 2007). Given that miR-27 knockdown prevents chondrogenic differentiation, the target mRNA of miR-27 would be hypothesized to be a negative regulator of PCC proliferation and differentiation. Here, we identify ptk2aa (FAK) as a novel in vivo target of miR-27. Ptk2aa and ptk2ab are paralogs of the zebrafish focal adhesion kinase (FAK) gene (Crawford et al., 2003; Henry et al., 2001). ptk2ab also carries two MREs for miR-27 in its 3′UTR but our GFP reporter assays showed no detectable regulation of ptk2ab 3′UTR by miR-27. Importantly, we show that craniofacial cartilage defects can be partially rescued by ptk2a (FAK) knockdown (Fig. 7). This suggests that FAK accumulation is required for proper chondrogenesis.
3.4. Potential roles of FAK during chondrogenic differentiation
Loss of miR-27 does not prevent mesenchymal condensation of the PCCs (Fig. S3). However, PCC proliferation was significantly impaired in miR-27 morphants during the condensation stage (Fig. 5A, B). We hypothesize that FAK accumulation due to miR-27 knockdown does not affect condensation, but rather proliferation and differentiation of PCCs. A remaining question is whether in miR-27 morphants, the effect of FAK accumulation during chondrogenesis is cell-autonomous. Previous studies have shown a cell-autonomous role of FAK on mesenchymal stem cells by performing in vitro chondrogenesis assays (Bursell et al., 2007; DeLise et al., 2000; Pala et al., 2008; Tang et al., 2013). Conditional deletion of the membrane-anchored metalloproteinase MT1-MMP in mesenchymal progenitors caused a loss of β1 integrin/FAK signaling activation and thereby promoted differentiation towards the chondrogenic versus adipogenic or osteogenic lineages (Tang et al., 2013). Similarly, using a micromass model of chondrogenesis with FAK+/+ and FAK−/− embryonic fibroblasts, FAK signaling was reported as a negative regulator of chondrogenesis (Pala et al., 2008). Loss of FAK in embryonic mesenchymal cells resulted in a significant increase in chondrogenic differentiation along with a higher expression of chondrogenic genes compared to wild-type cells (Pala et al., 2008). Induction of chondrogenic differentiation observed in FAK null cells is strikingly similar to effects reported upon inhibition of other components in the FAK signaling pathway. For example, inhibition of the FAK interacting kinase Src and the RhoA/ROCK pathway downstream of FAK/Src complex resulted in cell rounding and loss of stress fibers along with upregulation of chrondrogenic markers, Sox9, collagen type II and aggrecan during in vitro chondrogenesis (Bursell et al., 2007; Woods et al., 2005; Woods and Beier, 2006). All these findings demonstrate that FAK signaling suppresses chondrogenic differentiation with likely involvement of downstream RhoA/Rock signaling
An important role for FAK in focal adhesion complexes is to control mechanotransduction in cells that are subject to external mechanical forces or ECM remodeling (Eyckmans et al., 2011; Yim and Sheetz, 2012). Interestingly, mechanical stretching or ECM-mediated cell shape changes activate β1-integrin/FAK signaling and consequently inhibit chondrogenesis of mesenchymal stem cells (Onodera et al., 2005; Takahashi et al., 2003; Tang et al., 2013; Woods et al., 2007). These studies support our hypothesis that miR-27 regulates chondrogenesis in the pharyngeal arches by maintaining low levels of FAK.
3.5. miR-27 regulates differentiation programs in other tissue types
The miR-23~27~24 cluster has been implicated as a positive regulator for mesoderm differentiation of embryonic stem cells (ESC) by directly targeting pluripotency-maintenance factors (Ma et al., 2014). We show that miR-27 is also essential for development of specific mesoderm and neural crest-derived cell/tissue types in vivo. Previous reports showed that miR-27 is required for angiogenesis, adipogenesis, and development of the embryonic vasculature consistent with phenotypes we observe upon knockdown of miR-27 (Biyashev et al., 2012; Kang et al., 2013; Urbich et al., 2012; Zhou et al., 2011; Fig. S10). Together, miR-27 emerges as a key factor that negatively regulates the stemness of progenitor cells promoting the differentiation of multiple tissue types.
4. Materials and methods
4.1. Zebrafish husbandry and lines
Wild-type (AB) (Walker, 1999), Tg(fli1a:eGFP)y1 (Lawson and Weinstein, 2002), Tg(sox10(7.2):mRFP)vu234 (Kirby et al., 2006) lines were maintained at 28.5 °C on a 14:10 h light:dark cycle. Embryos were raised in egg water (0.03% Instant Ocean) at 28.5°C, staged by morphology (Kimmel et al., 1995) and hours post fertilization (hpf). For whole-mount immunohistochemistry and in situ hybridization analyses, embryos were raised in egg water supplemented with 0.003% N-phenylthiourea (PTU; Sigma-Aldrich) to prevent melanin formation. Zebrafish maintenance, embryo collection, and analyses were performed with the approval of the Vanderbilt University Institutional Animal Care and Use Committee (M/09/398).
4.2. Constructs
The ptk2aa (NM_198819.1) mRNA 3′UTR was amplified by RT-PCR using forward (5′-GGCGAATTCGACCTCCACACTGGCTGGAT CATC-3′) and reverse (5′-CGGCTCGACCTGAGCATTCGGTACACA CTTTCTGTATTA-3′) primers. ptk2ab (NM_131796.1) mRNA 3′UTR was amplified by RT-PCR using forward (5′-CGGACTAGTCTACTC ACCCACCCTCACGTTAAGC-3′) and reverse (5′-CGGCTCGAGTGCCTT GCTGTTAAACATCATTTGG-3′) primers. Each 3′UTR was cloned downstream of the GFP coding sequence in the PCS2+ vector. miRNA recognition elements (MREs) within the ptk2aa 3′UTR were deleted using the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene). The first MRE was deleted using sense (5′-GAATAATAATACTGAAGCTGACGGAGGGCTGAGGTA -3′) and anti-sense (5′-TACCTCAGCCCTCCGTCAGCTTCAGTATTATTATTC -3′) primers. The second MRE was deleted using sense (5′-CAAAAT CAGTTCTATGGTGAAGGGGCGGGATTAAACAA -3′) and anti-sense (5′-TTGTTTAATCCCGCCCCTTCACCATAGAACTGATTTTG -3′) primers. For the ptk2aa in situ probe, a 1.1 kb region was amplified by RT-PCR using forward (5′-GTAGTAGGATCCTCAGAAACAGAC GACTACGCA-3′) and reverse (5′-GTAGTACTCGAGTGGTTC CAGCTCTCAAGCG-3′) primers containing BamHI and XhoI sites for cloning into PCS2+. The ptk2aa coding region without the 3′UTR was amplified by RT-PCR using forward (5′-GTAGTAGAATTCCCTA GCGTACGGTAAAGGCA-3′) and reverse (5′-GTAGTACTCGAGAAG GGTGATGTTCCTCCGTG-3′) primers and cloned into PCS2+. For the reporter construct for MO-ptk2, GFP was amplified with a forward primer carrying the MO binding site overlapping the ptk2aa translation start site (5′-GTAGTAGGATCCAAGGCATGGCG ACGGCATTCCTGGACATGGTGAGCAAGGGCGAGG-3′) and reverse (5′-GTAGTAGAATTCGCCTTCTAGAGCTCGTCCA -3′) primer and cloned into pCS2+.
512 pME-miR was a gift from Nathan Lawson (Addgene plasmid # 26032). Around 300 bp fragment spanning the miR-27b gene was amplified using genomic DNA from 24 hpf old embryos and cloned into 512pME-miR using EcoRI restriction digestion site. Hsp70l:miR27b-eGFP and sox10:miR27b-eGFP plasmids were generated using pDestTol2CG2 vector, p5E-hsp70l (Kwan et al., 2007), p5E-sox10 (Das and Crump, 2012), p3E-eGFPpA (Kwan et al., 2007) and assembled using the Multisite Gateway cloning system (Invitrogen).
4.3. Transgenesis
To generate the Tg(hsp70l:miR27b-eGFP) and Tg(sox10:miR-27b-eGFP) lines, the mixture of Tol2 mRNA (25 pg) and plasmid (20 pg) was injected into the single-cell stage embryos. Injected embryos were prescreened for GFP fluorescence and raised in egg water at 28°C for five days before transferring them Aquatic Habitats system. Transgenic F1 lines were established by crossing founder and wild-type adults.
4.4. RNA synthesis
mRNAs were in vitro synthesized from linearized constructs using mMESSAGE mMACHINE® SP6 Transcription Kit (Life Technologies). Digoxigenin-UTP-labeled anti-sense RNA probes were in vitro synthesized from linearized constructs using either T7 or T3 RNA polymerases and DIG RNA labeling mix (Roche Applied Sciences). In vitro transcribed RNA was purified by NucAway™ Spin Columns (Life Technologies).
4.5. Morpholinos, microinjections
Morpholinos were purchased from Gene Tools and their sequences are listed in Table 1. MO-27 was designed complementary to mature miR-27a and the MO-27a loop targets the Dicer cleavage site and the loop of the miR-27a precursor. MO-ptk2 was designed against the translation start site of ptk2aa. All injections were performed in fertilized 1-cell stage embryos. For reporter assays, GFP-ptk2aa3′UTR mRNA was injected at 150 pg/embryo concentration either alone or with a synthetic miR-27a duplex (Dharmacon) at 75 pg/embryo. Double stranded mature miR-27a was synthesized with 3′-UU overhangs for the following target sequence: 5′-UUCACAGUGGCUAAGUUCCGCU-3′. MO-27 morpholinos were injected at 5 ng/embryo concentration unless specified.
4.6. Alcian Blue Staining
Embryos were fixed with 4% phosphate-buffered paraformaldehyde (PFA) for 1 h at room temperature. Fixed embryos were rinsed in PBS with 0.1% Tween-20 two times and rinsed in 50% EtOH for 10 min on a rocker. Embryos were then stained in 0.2% Alcian blue, 30 mM MgCl2 in 75% EtOH overnight on a rocker and bleached with 1.5% H2O2 and 1% KOH for 20 min.
4.7. Generation of miRNA mutants by CRIPSR/Cas9
sgRNAs were designed using CRISPRscan (http://www.crisprscan.org). sgRNA and Cas9 mRNAs were prepared as described previously (Yin et al., 2015). The sgRNA target region for miR-27a gene was 5′-GGATATCCTATGTTCACAG-3′. WT embryos were injected at the one-cell stage with 300 ng/μL Cas9 mRNA and 50 ng/μL sgRNA. Mutagenesis efficiency was confirmed by heteroduplex mobility assay analysis of the PCR amplicon of the CRISPR targeted genomic region. F1 heterozygous mutant embryos were obtained by crossing founder adults carrying germline miR-27a mutations with WT adults. Indels in the miR-27a gene in the F1 adults were identified and a 6 bp deletion in the miRNA seed region was selected for further breeding.
4.8. qRT-PCR
Taqman small RNA assays (Life Technologies) were used to perform qRT-PCR of the indicated miRNAs. 5 ng of total RNA isolated from 50 pooled embryos at the indicated stages were used per RT reaction and 1.33 μL of 1:2 diluted resultant cDNA was used in 10 μL qPCR reaction in technical triplicates. qPCR reactions were conducted in either 96-well plates using Bio-Rad CFX96 Real-time system or in 384-well plates using Bio-Rad CFX384 Real-time System. All quantifications were normalized to an endogenous U6 snRNA control. Fold changes were calculated using the ΔΔC(t) method, where Δ = C(t) miRNA – C(t)U6 snRNA, and ΔΔC(t) = ΔC(t)condition1 – ΔC(t) condition2, and FC = 2–ΔΔC(t). Taqman probe #: U6 snRNA: 001973; dre-miR-27a-3p: 007138_mat; dre-miR-27b: 008075_mat; dre-miR-27c: 006826_mat; dre-miR-27d: 003373_mat; dre-miR-27e: 007922_mat.
4.9. Immunoblotting and Northern Blots
Embryos were deyolked at the indicated time points and placed in RIPA buffer with Complete Protease Inhibitor Cocktail (Roche 04693159001), followed by homogenization with a pestle. Separation of total proteins and transfer was performed as described (Olena et al., 2015). The following antibodies were used for western blots: rabbit anti-GFP (1:1000, Torrey Pines), rabbit anti-α-tubulin (1:1000, Abcam), mouse anti-FAK (1:300, H-1 Santa Cruz Biotechnology), mouse anti-GAPDH (1:20,000, Ambion), anti-rabbit and anti-mouse HRP-conjugated secondary antibodies (1:5000, GE Healthcare). Quantification of band intensities was performed in ImageJ and intensities for each protein of interest were normalized to the loading control levels (either α-tubulin or gapdh). Data was represented as the mean for normalized band intensities from at least 3 independent pools of protein extract. Northern blots were performed as described (Flynt et al., 2007; Wei et al., 2013). Quantification of the band intensities was performed in ImageJ and intensities for miR-27 were normalized to the loading control levels (U6).
4.10. In situ hybridization
Whole-mount in situ hybridization was performed as described (Thisse and Thisse, 2008). Embryos were hybridized to digoxygenin-UTP labeled RNA probes for foxd3 (Kelsh et al., 2000), sox10 (Dutton et al., 2001), dlx2a (Akimenko et al., 1994), sox9a (Chiang et al., 2001) and col2a1 (Yan et al., 1995) at 70 °C in a hybridization solution containing 50% formamide. Hybridized probes were detected using anti-digoxigenin alkaline-phosphatase conjugated antibodies, followed by incubation with nitro blue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl-phosphate (NBT/BCIP) solution (Roche Applied Sciences). Whole-mount miRNA in situ hybridization was performed as described (Olena et al., 2015) using miRCURY 5′-and 3′-DIG labeled LNA (locked nucleic acid) probes (Exiqon). LNA probe #: dre-miR-27a: 613249-360; dre-miR-27b: 613734-360; dre-miR-27c-3p: 613613-360.
4.11. Immunofluorescent staining
To visualize mature chondrocytes, anti-type II collagen (anti-Col2) antibodies and wheat germ agglutinin (WGA) were used for staining of sections. Tg(fli1a:eGFP)y1 embryos at 61 h post fertilization (hpf) were fixed in 4%PFA at 4°C overnight, washed with PBT twice and permeabilized with proteinase K (10 μg/ml) for 30 min. Embryos were then incubated in blocking buffer (2 mg/ml BSA, 2% donkey serum, 4% DMSO, 0.1% Triton-X in PBS) for 2 h and stained with anti-GFP (1:500, A-11120 Invitrogen), anti-collagen type II (Col2) (1:200, Rockland) or WGA–Alexa-Fluor-555 conjugate (1:200, Molecular Probes) overnight at 4°C, followed by Cy3-conjugated and Alexa Fluor 488-conjugated secondary antibody (1:100 and 1:200, Jackson Immuno) staining for 2 h.
For phospho-histone 3 (pH3) staining, embryos were fixed, permeabilized as above, and incubated in blocking buffer (10 mg/ml BSA, 2% donkey serum, 1%DMSO, 0.1% Triton-X in PBS). Embryos were then stained with anti-pH3 (1:200, 06–570 Millipore) in blocking buffer. For anti-fibronectin immunostainings, Tg(fli1a:eGFP)y1 embryos were fixed, cryopreserved, and mounted in Cryomatrix (Thermo). 12 μm thick cryosections were incubated in blocking buffer (1 mg/ml BSA, 1%DMSO, 1% Triton-X in PBS) and then stained with anti-fibronectin (1:100, Sigma F-3648), Alexa 568 conjugate (Life Technologies, L32458) along with anti-GFP (1:500, Life Technologies, A11120) in blocking buffer.
To detect apoptosis, whole-mount TUNEL labeling was performed using an in situ Cell Death Detection Kit, TMR red (Roche), followed by anti-GFP (1:500, Torrey Pines Biolabs) staining.
Phospho-FAK pTyr397 (pFAK) staining was performed as described (Crawford et al., 2003; Koshida et al., 2005) using anti-pFAK [pY397] antibody (Invitrogen, 44–624 G, originally from BioSource). This antibody was validated in zebrafish embryos previously by Western blotting and immunohistochemistry (Crawford et al., 2003; Henry et al., 2001). We also validated the specificity of the anti-pFAK [pY397] antibody by loss of pFAK signal in in MO-ptk2 embryos (Fig. S7C). 12 μm thick cryosections of Tg(fli1a:eGFP)y1 embryos were incubated with anti-pFAK at 1:300 and anti-GFP (A-11120 Invitrogen) at 1:500, then stained with Cy3-conjugated and Alexa Fluor 488-conjugated secondary antibodies (1:100 and 1:200, Jackson Immuno) for 2 h. For pFAK staining in wild-type embryos shown in Supplemental Fig. 7C, embryos were stained with Alexa Fluor 488-conjugated phalloidin (1:100, Molecular Probes) and TO-PRO-3 (1:1000, Molecular Probes) along with the Cy3-conjugated secondary antibody for pFAK.
4.12. Imaging and image processing
For time-lapse imaging, Tg(fli1a:eGFP)y1 embryos were anesthetized at the indicated time points and mounted laterally in 0.6% agarose. Confocal stacks of the pharyngeal arch region were taken at 15 min intervals using a PerkinElmer spinning disk confocal microscope with a heating unit (PerkinElmer, 20× objective). For imaging of immunofluorescent stainings, either a PerkinElmer spinning disk confocal microscope or a META Zeiss LSM 510 Meta confocal microscope were used. Images were processed using either Volocity software (Improvision/PerkinElmer) or ImageJ software.
4.13. Cell counts and statistical analyses
For quantifying the pH3+ and TUNEL+ cells in fli1a:eGFP+ pharyngeal arches, cells positive for both GFP and marker of interest were counted, normalized to the whole area for fli1a:eGFP+ arch region using ImageJ software. Data were represented as a mean for normalized counts for the marker of interest and statistical analyses was performed using a two-tailed Student’s t-test.
Fluorescence intensity measurements are done using ImageJ software as described (Gavet and Pines, 2010). For each image, “integrated density”, “area” and “mean gray value” of the fli1a:eGFP+ region, as well as background were measured. Corrected fluorescence intensity of the selected region was calculated according to the formula: “Corrected fluorescence intensity= Integrated Density - (Area of selected region * mean fluorescence of background)”. Data were represented as a mean of corrected fluorescence intensity for each experimental condition and statistical analyses were performed using a two-tailed Student’s t-test.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to thank Patton lab members for helpful discussions, Dr. Stefania Nicoli and Emma Ristori for providing assistance in multiplex CRISPR/Cas9 mutagenesis, Drs. Ela Knapik, Josh Gamse and Seok-Hyung Kim for providing plasmids for RNA probes and transgenic zebrafish lines, Qiang Guan for zebrafish maintenance. This study was supported by National Eye Institute–National Institutes of Health (R21 EY19759) to JGP, the Stevenson Family Endowment to Vanderbilt University, and the Gisela Mosig Fund in the Department of Biological Sciences.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at: doi:10.1016/j.ydbio.2017.06.013.
Footnotes
Author contributions
N.K., C.W. and J.G.P. conceived and designed the experiments. N.K., C.W. and A.C. performed the experiments and analyzed the data. N.K. and J.G.P. wrote the manuscript.
References
- Akimenko MA, Ekker M, Wegner J, Lin W, Westerfield M. Combinatorial expression of three zebrafish genes related to distal-less: part of a homeobox gene code for the head. J Neurosci. 1994;14:3475–3486. doi: 10.1523/JNEUROSCI.14-06-03475.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroffio A, Dupin E, Le Douarin NM. Common precursors for neural and mesectodermal derivatives in the cephalic neural crest. Development. 1991;112:301–305. doi: 10.1242/dev.112.1.301. [DOI] [PubMed] [Google Scholar]
- Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- Biyashev D, Veliceasa D, Topczewski J, Topczewska JM, Mizgirev I, Vinokour E, Reddi AL, Licht JD, Revskoy SY, Volpert OV. miR-27b controls venous specification and tip cell fate. Blood. 2012;119:2679–2687. doi: 10.1182/blood-2011-07-370635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum M, De Robertis EM, Wallingford JB, Niehrs C. Morpholinos: antisense and Sensibility. Dev Cell. 2015;35:145–149. doi: 10.1016/j.devcel.2015.09.017. [DOI] [PubMed] [Google Scholar]
- Bursell L, Woods A, James CG, Pala D, Leask A, Beier F. Src kinase inhibition promotes the chondrocyte phenotype. Arthritis Res Ther. 2007;9:R105. doi: 10.1186/ar2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang EF, Pai CI, Wyatt M, Yan YL, Postlethwait J, Chung B. Two sox9 genes on duplicated zebrafish chromosomes: expression of similar transcription activators in distinct sites. Dev Biol. 2001;231:149–163. doi: 10.1006/dbio.2000.0129. [DOI] [PubMed] [Google Scholar]
- Couly GF, Coltey PM, Le Douarin NM. The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development. 1993;117:409–429. doi: 10.1242/dev.117.2.409. [DOI] [PubMed] [Google Scholar]
- Crawford BD, Henry CA, Clason TA, Becker AL, Hille MB. Activity and distribution of paxillin, focal adhesion kinase, and cadherin indicate cooperative roles during zebrafish morphogenesis. Mol Biol Cell. 2003;14:3065–3081. doi: 10.1091/mbc.E02-08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Crump JG. Bmps and Id2a act upstream of twist1 to restrict ectomesenchyme potential of the cranial neural crest. PLoS Genet. 2012:8. doi: 10.1371/journal.pgen.1002710. [DOI] [PMC free article] [PubMed]
- DeLise AM, Stringa E, Woodward WA, Mello MA, Tuan RS. Embryonic limb mesenchyme micromass culture as an in vitro model for chondrogenesis and cartilage maturation. Methods Mol Biol. 2000;137:359–375. doi: 10.1385/1-59259-066-7:359. [DOI] [PubMed] [Google Scholar]
- Dutton KA, Pauliny A, Lopes SS, Elworthy S, Carney TJ, Rauch J, Geisler R, Haffter P, Kelsh RN. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development. 2001;128:4113–4125. doi: 10.1242/dev.128.21.4113. [DOI] [PubMed] [Google Scholar]
- Eberhart JK, He X, Swartz ME, Yan YL, Song H, Boling TC, Kunerth AK, Walker MB, Kimmel CB, Postlethwait JH. MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat Genet. 2008;40:290–298. doi: 10.1038/ng.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JS, Smith JC. Controlling morpholino experiments: don’t stop making antisense. Development. 2008;135:1735–1743. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- Eyckmans J, Boudou T, Yu X, Chen CS. A Hitchhiker’s guide to mechanobiology. Dev Cell. 2011 doi: 10.1016/j.devcel.2011.06.015. [DOI] [PMC free article] [PubMed]
- Flores MV, Tsang VWK, Hu W, Kalev-Zylinska M, Postlethwait J, Crosier P, Crosier K, Fisher S. Duplicate zebrafish runx2 orthologues are expressed in developing skeletal elements. Gene Expr Patterns. 2004;4:573–581. doi: 10.1016/j.modgep.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Flynt AS, Li N, Thatcher EJ, Solnica-Krezel L, Patton JG. Zebrafish miR-214 modulates Hedgehog signaling to specify muscle cell fate. Nat Genet. 2007;39:259–263. doi: 10.1038/ng1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, McBeath R, Chen CS. Stem cell shape regulates a chondrogenic versus myogenic fate through rac1 and N-cadherin. Stem Cells. 2010;28:564–572. doi: 10.1002/stem.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJ, Stefan LL, Cohen MM. Syndromes of the Head and Neck. 3. Oxford University Press; New York: 1990. [DOI] [Google Scholar]
- Hall BK, Miyake T. Divide, accumulate, differentiate: cell condensation in skeletal development revisited. Int J Dev Biol. 1995;39:881–893. [PubMed] [Google Scholar]
- Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. BioEssays. 2000;22:138–147. doi: 10.1002/(SICI)1521-1878(200002)22:2<138::AID-BIES5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Hassan MQ, Gordon JaR, Beloti MM, Croce CM, van Wijnen AJ, Stein JL, Stein GS, Lian JB. A network connecting Runx2, SATB2, and the miR-23a~27a~24-2 cluster regulates the osteoblast differentiation program. Proc Natl Acad Sci USA. 2010;107:19879–19884. doi: 10.1073/pnas.1007698107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry Ca, Crawford BD, Yan YL, Postlethwait J, Cooper MS, Hille MB. Roles for zebrafish focal adhesion kinase in notochord and somite morphogenesis. Dev Biol. 2001;240:474–487. doi: 10.1006/dbio.2001.0467. [DOI] [PubMed] [Google Scholar]
- Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- Kang T, Lu W, Xu W, Anderson L, Bacanamwo M, Thompson W, Chen YE, Liu D. MicroRNA-27 (miR-27) targets prohibitin and impairs adipocyte differentiation and mitochondrial function in human adipose-derived stem cells. J Biol Chem. 2013;288:34394–34402. doi: 10.1074/jbc.M113.514372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsh RN, Dutton K, Medlin J, Eisen JS. Expression of zebrafish fkd6 in neural crest-derived glia. Mech Dev. 2000;93:161–164. doi: 10.1016/S0925-4773(00)00250-1. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kirby BB, Takada N, Latimer AJ, Shin J, Carney TJ, Kelsh RN, Appel B. In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat Neurosci. 2006;9:1506–1511. doi: 10.1038/nn1803. [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Plasterk RHA. The diverse functions of MicroRNAs in animal development and disease. Dev Cell. 2006 doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed]
- Kloosterman WP, Lagendijk AK, Ketting RF, Moulton JD, Plasterk RHA. Targeted inhibition of miRNA maturation with morpholinos reveals a role for miR-375 in pancreatic Islet development. PLoS Biol. 2007;5:e203. doi: 10.1371/journal.pbio.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Lu J, Cobb BS, Rodda SJ, McMahon AP, Schipani E, Merkenschlager M, Kronenberg HM. Dicer-dependent pathways regulate chondrocyte proliferation and differentiation. Proc Natl Acad Sci USA. 2008;105:1949–1954. doi: 10.1073/pnas.0707900105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok FO, Shin M, Ni CW, Gupta A, Grosse AS, van Impel A, Kirchmaier BC, Peterson-Maduro J, Kourkoulis G, Male I, DeSantis DF, Sheppard-Tindell S, Ebarasi L, Betsholtz C, Schulte-Merker S, Wolfe SA, Lawson ND. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in Zebrafish. Dev Cell. 2015;32:97–108. doi: 10.1016/j.devcel.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshida S, Kishimoto Y, Ustumi H, Shimizu T, Furutani-Seiki M, Kondoh H, Takada S. Integrinα5-dependent fibronectin accumulation for maintenance of somite boundaries in zebrafish embryos. Dev Cell. 2005;8:587–598. doi: 10.1016/j.devcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Kozhemyakina E, Lassar aB, Zelzer E. A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development. 2015;142:817–831. doi: 10.1242/dev.105536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. The Tol2kit: a multisite gateway-based construction kit forTol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248:307–318. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Creuzet S, Couly G, Dupin E. Neural crest cell plasticity and its limits. Development. 2004;131:4637–4650. doi: 10.1242/dev.01350. [DOI] [PubMed] [Google Scholar]
- Lee RTH, Knapik EW, Thiery JP, Carney TJ. An exclusively mesodermal origin of fin mesenchyme demonstrates that zebrafish trunk neural crest does not generate ectomesenchyme. Development. 2013;140:2923–2932. doi: 10.1242/dev.093534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Li N, Wei C, Olena AF, Patton JG. Regulation of endoderm formation and left-right asymmetry by miR-92 during early zebrafish development. Development. 2011;138:1817–1826. doi: 10.1242/dev.056697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden A, Sprawson N, Graham A. Segmental origin and migration of neural crest cells in the hindbrain region of the chick embryo. Development. 1991;113:1281–1291. doi: 10.1242/dev.113.4.1281. [DOI] [PubMed] [Google Scholar]
- Ma Y, Yao N, Liu G, Dong L, Liu Y, Zhang M, Wang F, Wang B, Wei X, Dong H, Wang L, Ji S, Zhang J, Wang Y, Huang Y, Yu J. Functional screen reveals essential roles of miR- 27 a/24 in differentiation of embryonic stem cells. 2014:1–19. doi: 10.15252/embj.201489957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride SH, Knothe Tate ML. Modulation of stem cell shape and fate A: the role of density and seeding protocol on nucleus shape and gene expression. Tissue Eng Part A. 2008;14:1561–1572. doi: 10.1089/ten.tea.2008.0112. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Inloes JB, Katagiri T, Kobayashi T. Chondrocyte-specific microRNA-140 regulates endochondral bone development and targets Dnpep to modulate bone morphogenetic protein signaling. Mol Cell Biol. 2011;31:3019–3028. doi: 10.1128/MCB.05178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan A, Hill-Teran G, Moro A, Ristori E, Kasper DM, Roden A, Lu C, Nicoli SJ. In vivo mutagenesis of miRNA gene families using a scalable multiplexed CRISPR/Cas9 nuclease system. Sci Rep. 2016;6:32386. doi: 10.1038/srep32386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning G, Liu X, Dai M, Meng A, Wang Q. MicroRNA-92a upholds Bmp signaling by targeting noggin3 during pharyngeal cartilage formation. Dev Cell. 2013;24:283–295. doi: 10.1016/j.devcel.2012.12.016. [DOI] [PubMed] [Google Scholar]
- Olena AF, Rao MB, Thatcher EJ, Wu SY, Patton JG. miR-216a regulates snx5, a novel notch signaling pathway component, during zebrafish retinal development. Dev Biol. 2015;400:72–81. doi: 10.1016/j.ydbio.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera K, Takahashi I, Sasano Y, Bae JW, Mitani H, Kagayama M, Mitani H. Stepwise mechanical stretching inhibits chondrogenesis through cell-matrix adhesion mediated by integrins in embryonic rat limb-bud mesenchymal cells. Eur J Cell Biol. 2005;84:45–58. doi: 10.1016/j.ejcb.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Pala D, Kapoor M, Woods A, Kennedy L, Liu S, Chen S, Bursell L, Lyons KM, Carter DE, Beier F, Leask A. Focal adhesion kinase/Src suppresses early chondrogenesis: central role of CCN2. J Biol Chem. 2008;283:9239–9247. doi: 10.1074/jbc.M705175200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Robu ME, Larson JD, Nasevicius A, Beiraghi S, Brenner C, Farber SA, Ekker SC. p53 activation by knockdown technologies. PLoS Genet. 2007;3:787–801. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M, Stainier DYR. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–233. doi: 10.1038/nature14580. [DOI] [PubMed] [Google Scholar]
- Schilling TF, Kimmel CB. Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development. 1994;120:483–494. doi: 10.1242/dev.120.3.483. [DOI] [PubMed] [Google Scholar]
- Sheehy NT, Cordes KR, White MP, Ivey KN, Srivastava D. The neural crest-enriched microRNA miR-452 regulates epithelial-mesenchymal signaling in the first pharyngeal arch. Development. 2010;137:4307–4316. doi: 10.1242/dev.052647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Schwarzbauer JE. Fibronectin matrix assembly is essential for cell condensation during chondrogenesis. 2014:4420–4428. doi: 10.1242/jcs.150276. <. >. [DOI] [PMC free article] [PubMed]
- Stainier DYR, Kontarakis Z, Rossi A. Making sense of anti-sense data. Dev Cell. 2015;32:7–8. doi: 10.1016/j.devcel.2014.12.012. [DOI] [PubMed] [Google Scholar]
- Takahashi I, Onodera K, Sasano Y, Mizoguchi I, Bae JW, Mitani H, Kagayama M, Mitani H. Effect of stretching on gene expression of beta1 integrin and focal adhesion kinase and on chondrogenesis through cell-extracellular matrix interactions. Eur J Cell Biol. 2003;82:182–192. doi: 10.1078/0171-9335-00307. [DOI] [PubMed] [Google Scholar]
- Tang Y, Rowe RG, Botvinick E, Kurup A, Putnam A, Seiki M, Weaver V, Keller E, Goldstein S, Dai J, Begun D, Saunders T, Weiss SJ. MT1-MMP-dependent control of skeletal stem cell commitment via a β1-Integrin/YAP/TAZ signaling axis. Dev Cell. 2013;25:402–416. doi: 10.1016/j.devcel.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Urbich C, Kaluza D, Frömel T, Knau A, Bennewitz K, Boon Ra, Bonauer A, Doebele C, Boeckel J-N, Hergenreider E, Zeiher AM, Kroll J, Fleming I, Dimmeler S. MicroRNA-27a/b controls endothelial cell repulsion and angiogenesis by targeting semaphorin 6A. Blood. 2012;119:1607–1616. doi: 10.1182/blood-2011-08-373886. [DOI] [PubMed] [Google Scholar]
- Walker C. Haploid screens and gamma-ray mutagenesis. Methods Cell Biol. 1999;60:43–70. doi: 10.1016/s0091-679x(08)61893-2. [DOI] [PubMed] [Google Scholar]
- Wei C, Salichos L, Wittgrove CM, Rokas A, Patton JG. Transcriptome-wide analysis of small RNA expression in early zebrafish development. RNA. 2012 doi: 10.1261/rna.029090.111. [DOI] [PMC free article] [PubMed]
- Wei C, Thatcher EJ, Olena AF, Cha DJ, Perdigoto AL, Marshall AF, Carter BD, Broadie K, Patton JG. miR-153 regulates SNAP-25, synaptic transmission, and neuronal development. PLoS One. 2013:8. doi: 10.1371/journal.pone.0057080. [DOI] [PMC free article] [PubMed]
- Wienholds E. MicroRNA expression in Zebrafish embryonic development. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- Woods A, Beier F. RhoA/ROCK signaling regulates chondrogenesis in a context-dependent manner. J Biol Chem. 2006;281:13134–13140. doi: 10.1074/jbc.M509433200. [DOI] [PubMed] [Google Scholar]
- Woods A, Wang G, Beier F. RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J Biol Chem. 2005;280:11626–11634. doi: 10.1074/jbc.M409158200. [DOI] [PubMed] [Google Scholar]
- Woods A, Wang G, Beier F. Regulation of chondrocyte differentiation by the act in cytoskeleton and adhesive interactions. J Cell Physiol. 2007 doi: 10.1002/jcp.21110. [DOI] [PubMed]
- Wu BT, Wen SH, Hwang SPL, Huang CJ, Kuan YS. Control of Wnt5b secretion by Wntless modulates chondrogenic cell proliferation through fine-tuning fgf3 expression. J Cell Sci. 2015;128:2328–2339. doi: 10.1242/jcs.167403. [DOI] [PubMed] [Google Scholar]
- Yan YL, Hatta K, Riggleman B, Postlethwait JH. Expression of a type II collagen gene in the zebrafish embryonic axis. Dev Dyn. 1995;203:363–376. doi: 10.1002/aja.1002030308. [DOI] [PubMed] [Google Scholar]
- Yan YL, Miller CT, Nissen RM, Singer A, Liu D, Kirn A, Draper B, Willoughby J, Morcos Pa, Amsterdam A, Chung BC, Westerfield M, Haffter P, Hopkins N, Kimmel C, Postlethwait JH. A zebrafish sox9 gene required for cartilage morphogenesis. Development. 2002;129:5065–5079. doi: 10.1242/dev.129.21.5065. [DOI] [PubMed] [Google Scholar]
- Yelick PC, Schilling TF. Molecular dissection of craniofacial development using Zebrafish. Crit Rev Oral Biol Med. 2002;13:308–322. doi: 10.1177/154411130201300402. [DOI] [PubMed] [Google Scholar]
- Yim EK, Sheetz MP. Force-dependent cell signaling in stem cell differentiation. Stem Cell Res Ther. 2012;3:41. doi: 10.1186/scrt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Jao LE, Chen W. Generation of targeted mutations in Zebrafish using the CRISPR/Cas system. Methods Mol Biol. 2015;1332:205–217. doi: 10.1007/978-1-4939-2917-7_16. [DOI] [PubMed] [Google Scholar]
- Zehir A, Hua LL, Maska EL, Morikawa Y, Cserjesi P. Dicer is required for survival of differentiating neural crest cells. Dev Biol. 2010;340:459–467. doi: 10.1016/j.ydbio.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Srivastava D. A developmental view of microRNA function. Trends Biochem Sci. 2007 doi: 10.1016/j.tibs.2007.02.006. [DOI] [PubMed]
- Zhou Q, Gallagher R, Ufret-Vincenty R, Li X, Olson EN, Wang S. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23~27~24 clusters. Proc Natl Acad Sci USA. 2011;108:8287–8292. doi: 10.1073/pnas.1105254108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.