

Abstract
Gastrointestinal microbiota may play an important role in regulating host mucosal innate immune function. This study was conducted to test the hypothesis that age (non-rumination, transition and rumination) and feeding type [Supplemental feeding (S) vs. Grazing (G)] could alter ruminal microbial diversity and maturation of host mucosal innate immune system in goat kids. MiSeq sequencing was applied to investigate ruminal microbial composition and diversity, and RT-PCR was used to test expression of immune-related genes in ruminal mucosa. Results showed that higher (P < 0.05) relative abundances of Prevotella, Butyrivibrio, Pseudobutyrivibrio, Methanobrevibacter.gottschalkii, Neocallimastix, Anoplodinium–Diplodinium, and Polyplastron, and lower relative abundance of Methanosphaera (P = 0.042) were detected in the rumen of S kids when compared to those in G kids. The expression of genes encoding TLRs, IL1α, IL1β and TICAM2 was down-regulated (P < 0.01), while expression of genes encoding tight junction proteins was up-regulated (P < 0.05) in the ruminal mucosa of S kids when compared to that in G kids. Moreover, irrespective of feeding type, relative abundances of ruminal Prevotella, Fibrobacter, Ruminococcus, Butyrivibrio, Methanobrevibacter, Neocallimastix, and Entodinium increased with age. The expression of most genes encoding TLRs and cytokines increased (P < 0.05) from day 0 to 7, while expression of genes encoding tight junction proteins declined with age (P < 0.05). This study revealed that the composition of each microbial domain changed as animals grew, and these changes might be associated with variations in host mucosal innate immune function. Moreover, supplementing goat kids with concentrate could modulate ruminal microbial composition, enhance barrier function and decrease local inflammation. The findings provide useful information in interpreting microbiota and host interactions, and developing nutritional strategies to improve the productivity and health of rumen during early life.
Keywords: rumen, microbiota, mucosal, innate immune function, animal development process, feeding type
Introduction
Ruminants have a remarkable ability to convert human-indigestible plant biomass into human-digestible food products through rumen microbial fermentation (Jami et al., 2013). Bacteria, fungi and protozoa decompose fibrous plant polymers into short-chain fatty acids, and release hydrogen (Kittelmann and Janssen, 2011; Kittelmann et al., 2012; Jami et al., 2013). Methanogenic archaea can scavenge hydrogen and produce methane (Janssen and Kirs, 2008). During early life, ruminal bacterial populations underwent dynamic changes at both density (Jiao et al., 2015b) and diversity (Jami et al., 2013; Rey et al., 2014) levels as the animals matured. A recent study by Dill-McFarland et al. (2017) revealed succession in colonization of the rumen by the three microbial domains (bacteria, fungi and archaea) at weaning (8 weeks old) and post-weaning (1 and 2 years old) in dairy cows.
The rumen fermentation products are mainly absorbed by the ruminal epithelial wall, and utilized by the host (Graham and Simmons, 2005). In addition to the nutrient uptake, the ruminal epithelium plays a major role in host mucosal innate immune function. The squamous rumen epithelium is comprised of four distinct strata; stratum corneum, granulosum, spinosum and basale layers (Graham and Simmons, 2005). The multicellular structures are connected through junctional complexes, thus forming the primary barrier function (Matter and Balda, 2003). In addition, rumen epithelial cells interact with lymphocytes and dendritic cells in lamina propria to maintain immune homeostasis (Trevisi et al., 2014). In goats, a high-grain diet (65% grain) could cause massive disruption of ruminal epithelial tight junctions when compared to the control diet (0% grain) (Liu et al., 2013). Despite this, a systematic characterization about how rumen mucosal innate immune function developed during early life is still not well-defined.
It is well established that the gastrointestinal commensal bacteria, such as Bacteroides fragilis are capable of activating and modulating immune responses in the host (Troy and Kasper, 2010). For example, Toll like receptors (TLRs) on the host mucosa can recognize conserved microbial domains, and trigger host innate immune responses (Akira et al., 2001; Kawai and Akira, 2010). In the rumen of dairy calves and goats, TLR expression was diet dependent and correlated with bacterial diversity (Chen et al., 2012; Malmuthuge et al., 2013; Liu et al., 2015), suggesting ruminal microbiota may play a role to interact with host mucosa which can mount an immune response. Fungi and protozoa can also encounter with TLR signaling pathway to maintain host–microbe homeostasis (Gazzinelli and Denkers, 2006; Bourgeois and Kuchler, 2012). However, the mechanism by which the ruminal microbiota interact with host mucosal innate immune system is relatively unexplored.
To address this knowledge gap, we hypothesized that age (non-rumination, transition and rumination phases) and feeding type [supplemental feeding, (S) and grazing (G)] could modulate ruminal microbiota, and further manipulate host mucosal innate immune function. To test this hypothesis, we applied a combination of MiSeq sequencing and relative RT-PCR analysis to investigate microbial diversity, as well as expression of genes encoding TLRs, inflammatory cytokines, and toll-IL-1 receptor (TIR)-domain containing adaptors.
Materials and Methods
Animals, Diets and Experimental Design
All management and experimental procedures were approved by the Animal Care Committee, Institute of Subtropical Agriculture, the Chinese Academy of Sciences, Changsha, China.
Thirty-two Liuyang black goat kids were separated from their dams after birth, and maintained within individual pens for the duration of the study. From birth to day 20, kids consumed goat milk and 4 kids were slaughtered at day 0 and 7 (non-rumination phase), respectively. The remaining 24 kids were randomly allocated to two groups based on different feeding types, supplemental feeding (S) and grazing (G). Between day 20 and 40, kids in S group consumed milk, forage plus concentrate, while kids in G group consumed milk and forage. The animals were weaned at day 40. From day 40 to 70, S kids were provided with concentrate and forage, while G kids grazed 8 h on pasture daily, and returned to pens overnight without receiving any extra feed. In both groups, four kids were slaughtered at each of the following ages: day 28, 42 (transition phase) and day 70 (rumination phase). All kids had free access to water. Detailed feeding management, and ingredients of concentrate and forage have been described in our previous parallel study (Jiao et al., 2015b).
Sampling Procedures
After the kids were slaughtered, ruminal digesta samples were collected from five locations (anterior dorsal, anterior ventral, medium ventral, posterior dorsal, and posterior ventral locations) within the rumen (∼10 g each). The samples were composited, and aliquots were stored at -80°C for subsequent DNA extraction and sequencing analysis. Ruminal mucosa samples were collected from the same sites where digesta samples were collected, and rinsed three times with sterile phosphate buffered saline (PBS, pH = 7.4). Afterward, the mucosa samples were separated from the underlying muscular layer, cut into 3 mm × 3 mm fragments, composited, frozen in liquid nitrogen and stored at -80°C for later molecular analysis.
DNA Extraction, PCR Amplification, MiSeq Sequencing and Data Analysis
Genomic DNA was extracted using the QIAamp DNA Stool Mini kit (Qiagen GmbH, Hilden, Germany) and quantified using NanoDrop ND1000 (NanoDrop Technologies, Wilmington, DE, United States). Conventional PCR was conducted to amplify bacterial V2–V3 and archaeal V6–V8 regions of the 16S rRNA genes, as well as fungal V3–V5 and protozoal V4–V5 regions of the 18S rRNA genes using primers detailed in Supplementary Table S1. The PCR conditions were as follows: pre-denaturation at 94°C for 3 min, 35 cycles of denaturation at 94°C for 30 s, annealing at 58°C for bacteria (62°C for archaea, 58°C for fungi and 54°C for protozoa) for 30 s, and elongation at 72°C for 45 s, and following with post-elongation at 72°C for 10 min. The PCR amplification products were purified using Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, WI, United States). Barcoded amplicons were mixed at equal molar rations, and submitted for 300 bp pair-end sequencing on an Illumina MiSeq PE300 instrument (Illumina, San Diego, CA, United States).
Raw data were filtered through Quantitative Insight into Microbial Ecology (QIIME) (Caporaso et al., 2010b) and Mothur (Schloss et al., 2009). Reads were deconvoluted into individual samples based on their barcodes. The pair-end reads were assembled based on their overlapped sequences using COPE (Connecting Overlapped Pair-End) (Liu et al., 2012) and trimmed of primers.
For bacteria and archaea, the assembled sequences were assigned to operational taxonomic units (OTUs) at a 97% identity threshold using UPARSE (Edgar, 2013). Chimeric sequences were removed using UCHIME. Taxonomy classifications were assigned against the latest Greengenes database (May 2013 release) for bacteria (DeSantis et al., 2006) and RIM-DB version13_11_13 for archaea (Seedorf et al., 2014), respectively. The taxonomic assignment was performed using Mothur based implementation of the RDP Bayesian classifier with a 0.80 confidence threshold. For fungi and protozoa, sequences were assigned into OTUs using UCLUST at a 97% identity threshold. The OTUs were classified using BLAST against SILVA database for fungi and the reference database constructed by Kittelmann and Janssen (2011) for protozoa. Sequences with identity scores, between 95 and 97% were resolved at the genus level, 90 and 95% at the family level, 85 and 90% at the order level, 80 and 85% at the class level, and 77 and 80% at the phyla level (Hristov et al., 2012). Sequences were aligned by PyNAST (Caporaso et al., 2010a), and phylogenetic trees were constructed using FastTree.
Alpha diversity was examined using the alpha rarefaction pipeline of QIIME. Principle coordinate analysis (PCoA) was performed using unweighted UniFrac distance (jackknifed beta diversity from resampling 100 times at a depth of 75% of least number of sequences). Transformation-based principal component analysis (tb-PCA) was used to ordinate the samples based on OTU abundance data using the Hellinger distance (Legendre and Gallagher, 2001), and only the top 10 OTUs were presented.
RNA Extraction and Gene Expression
Total RNA was extracted from mucosa samples (200 mg) using TRIzol reagent (Invitrogen, Carlsbad, CA, United States). The RNA integrity was evaluated using 1% agarose gel electrophoresis, and its quantity was measured using ND1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, United States). The RNA was reversely transcribed using the First Strand cDNA Synthesis Kit (Toyobo, Osaka, Japan), and the cDNA samples were stored at -20°C until use. The RT-PCR analysis was used to evaluate expression of genes encoding TLRs, inflammatory cytokines, toll-IL-1 receptor (TIR)-domain containing adaptors, and tight junction proteins (TJs), with GADPH and β-actin as the housekeeping genes. The primers are listed in Supplementary Table S2, and procedures are detailed in Supplementary Text 1.
Statistics Analysis
The effect of feeding type was examined from day 28 to 70. Data were analyzed using the MIXED procedures of SAS 8.2 (SAS Institute Inc., Cary, North Carolina, United States). Feeding type, age, and feeding type × age interaction were regarded as fixed effects, animal nested within feeding type × age was regarded as the random effect, and each individual animal was the experimental unit. The following model was used:
where Yijk = dependent variable, μ = overall mean, Ai = fixed effect of age, Fj = fixed effect of feeding type, Anik = random effect of animal nested within feeding type × age, and eijk = the random residual error.
The effect of age on ruminal microbial diversity and expression of genes involved in host mucosal innate immune function was evaluated from day 0 to 70. The MIXED procedure of SAS and Tukey’s test to compare least squares means were used, with age as the fixed effect, animal within age as random effect, and individual animal as the experimental unit. The following model was used:
where Yij = dependent variable, μ = overall mean, Ai = fixed effect of age, Anij = random effect of animal within age, and eij = the random residual error. Orthogonal contrasts were performed to test linear and quadratic effects of age. If feeding type × age interaction was not significant within day 28 to 70, effect of age was assessed using combined data of both groups; otherwise, they were presented separately. All data were presented as least squares means, and significance was declared with P < 0.05.
The PROC CORR procedure of SAS was used to examine the Spearman’s rank correlations between ruminal microbial abundance and expression of mucosal innate immune function related genes. Only those correlation coefficients above 0.50, and P < 0.05 were defined as correlated with each other and plotted using corrplot package in the R studio.
Nucleotide Sequence Accession Numbers
Sequencing data in this study were deposited in the NCBI Sequence Read Archive (SRA) under accession numbers PRJNA362795.
Results
OTU Diversity and Similarity Analysis
As reflected by rarefaction curve, sequencing depth was adequate to cover the community of each microbial domain, with Good’s coverage ranged from 0.980 to 0.999 (Supplementary Figure S1 and Table S3). Alpha diversity of ruminal bacteria, archaea and fungi was more abundant (P < 0.05) in G kids compared to that in S kids. Alpha diversity indices (OTU number, Chao, Ace and Shannon) of ruminal bacteria and archaea in both groups increased with age (P < 0.01). For G kids, alpha diversity indices of ruminal fungi increased (P < 0.01), while those indices of ruminal protozoa decreased quadratically (P < 0.05) with age. PCoA analysis revealed that microbial communities could be clearly discriminated by both age and feeding type (Figure 1). The tb-PCA results revealed the top 10 OTUs within each microbial domain contributing to separation at different ages and for feeding types (Figure 2). Details of these are presented in Supplementary Text 2.
FIGURE 1.
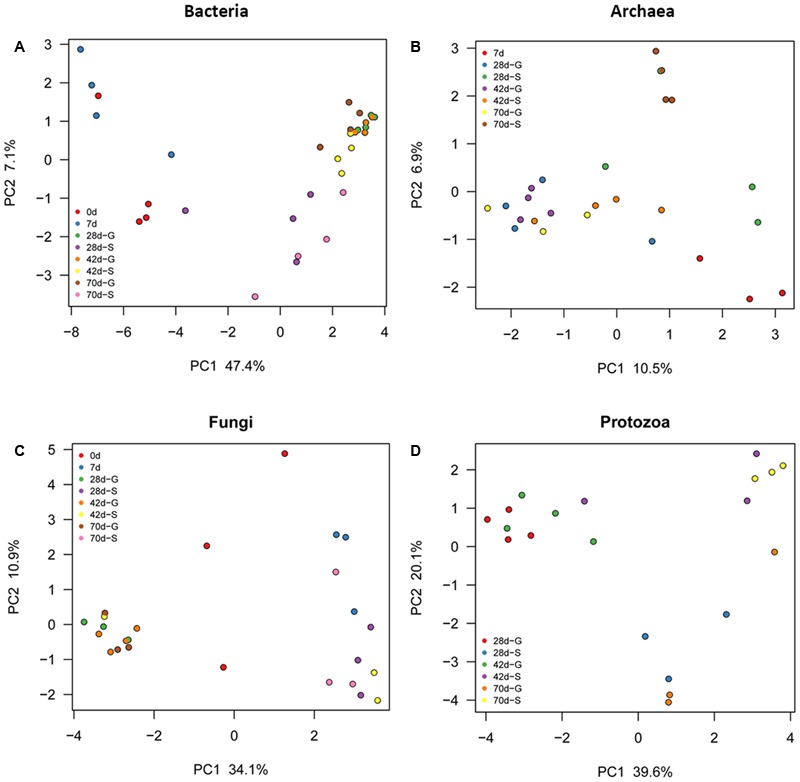
Principal coordinate analysis (PCoA) of each ruminal microbial domain in S and G kids at different ages. (A) Bacteria, (B) Archaea, and (C) Fungi, and (D) Protozoa.
FIGURE 2.
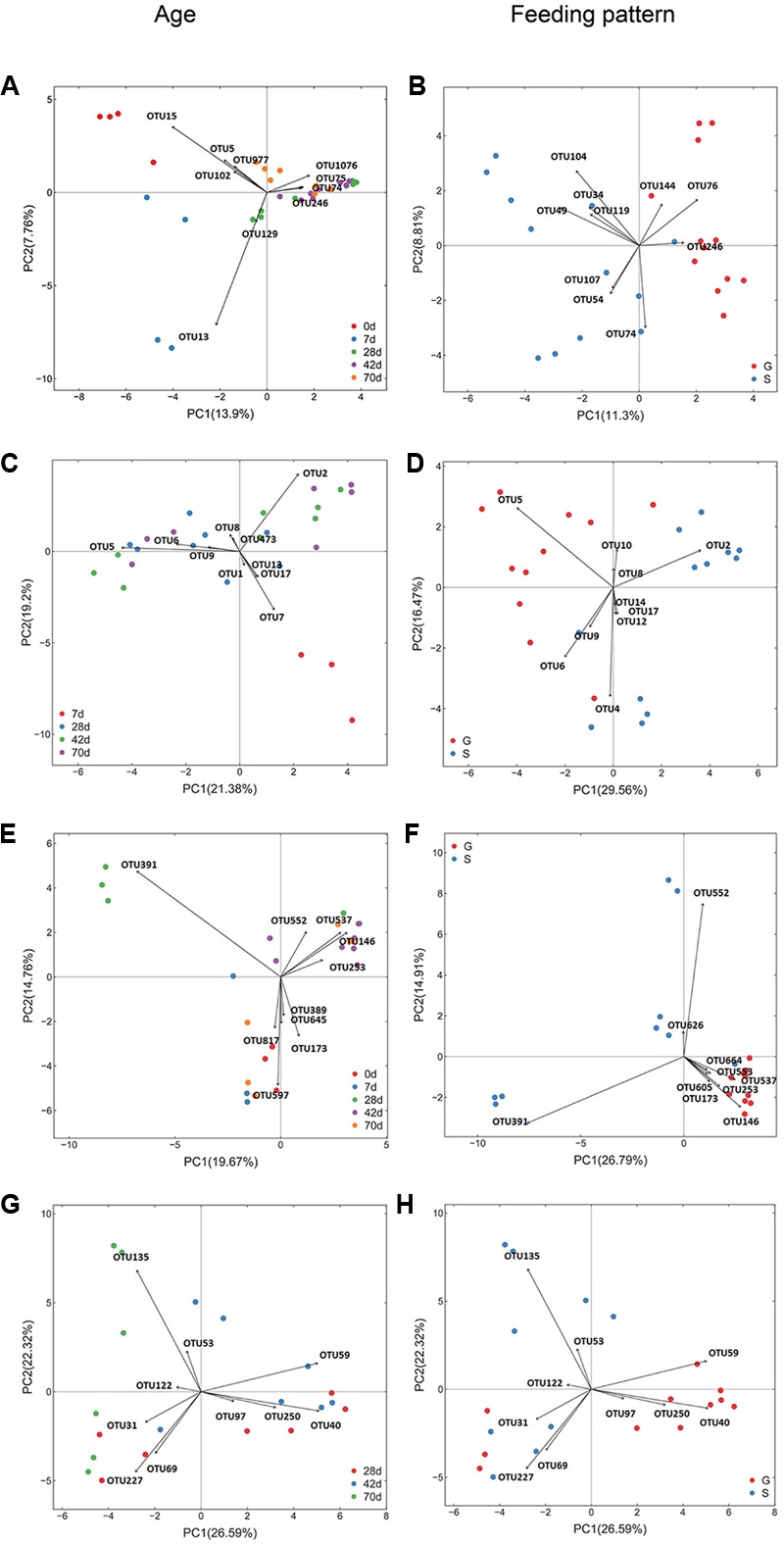
Transform-based PCA of each ruminal microbial domain for separation of different ages and feeding types in kids. The top 10 OTUs contributing to the separation of samples were displayed on the figure (OTU number). (A) Bacterial OTUs for discrimination of different ages, (B) Bacterial OTUs for discrimination of different feeding types. For bacteria (A,B), OTU15, Arcobacter genus; OTU5, Escherichia genus; OUT977, Escherichia genus; OTU102, Pasteurellaceae family; OTU13, Halomonadaceae family; OTU129, Delftia genus; OTU74, Prevotella genus; OTU75, Lachnospiraceae family; OTU1076, Prevotella genus; OTU246, Prevotella genus; OTU76, Prevotella genus; OTU144, Prevotella genus; OTU104, Succinivibrionaceae family; OTU34, Prevotella genus; OTU49, Prevotella genus; OTU119, Bacteroidales order; OTU107, Succinivibrionaceae family; OTU54, Ruminococcaceae family. (C) Archaeal OTUs for discrimination of different ages, (D) Archaeal OTUs for discrimination of different feeding types. For archaea (C,D), OTU5, Methanomassiliicoccaceae family; OTU2, Mbb. gottschalkiiclade; OTU7, Archaea domain; OTU6, Group12_sp._ISO4-H5; OTU9, Group9_sp._ISO4-G1; OTU8, Mbb. Ruminantium clade; OTU4, Methanomassiliicoccaceae family; OTU1, Group10_sp.; OTU10, Mbb. acididurans clade; OTU12, Mbb. mobile clade; OTU13, Euryarchaeota phylum; OTU14, Euryarchaeota phylum; OTU17, Archaea domain; OTU473, Mbb. Ruminantium clade. (E) Fungal OTUs for discrimination of different ages, (F) Fungal OTUs for discrimination of different feeding types. For fungi (E,F), OTU391, Neocallimastix genus; OTU579, Saccharomyces genus; OTU146, Uwebraunia genus; OTU537, Piromyces genus;OTU173, Boeremia genus; OTU817, unclassified fungi; OTU645, Eupenicillium genus; OTU552, Piromyces genus; OTU253, Knufia genus; OTU389, Neurospora genus; OTU553, Neocallimastix genus; OTU664, Microcyclosporella genus; OTU626, Sarocladium genus; OTU605, Neocallimastixgenus. (G) Protozoal OTUs for discrimination of different ages, (H) Protozoal OTUs for discrimination of different feeding types. For protozoa (G,H), OTU135, Polyplastron genus; OTU40, Epidinium genus; OTU59, Entodinium genus; OTU227, Entodinium genus; OTU69, Entodinium genus; OTU50, Eremoplstron-Diploplastron genus; OTU31, Entodinium genus; OTU53, Anoplodinium–Diplodinium; OTU97, Epidinium genus; OTU122, Polyplastron genus.
Rumen Microbial Community Changes-Bacteria
A total of 20 phyla and 48 genera were identified in all samples, with 23 bacterial genera each accounting for >0.5% of all sequences. A great proportion of sequences (31.2–69.2%) remained unclassified at the genus level (Figure 3A). Relative abundances of Prevotella, Asteroleplasma, Coprococcus were greater (P < 0.05), while relative abundances of Butyrivibrio and Pseudobutyrivibrio were lower (P = 0.011) in the rumen of G kids compared to those in S kids. In both groups, Arcobacter (30.1%), Escherichia (13.3%), Porphyromonas (5.7%) and Fusobacterium (2.1%) were predominant ruminal genera at day 0, whist their relative abundances declined to minimal values (<0.2%) afterward. Conversely, Prevotella relative abundance increased drastically (P < 0.01) with age, being the most dominant genus after day 28 (>20%), while Bacteroides relative abundance was high (12.1%) at only day 7. Moreover, relative abundances of ruminal BF311, CF231, Butyrivibrio, Pseudobutyrivibrio, and Fibrobacter in both groups increased quadratically (P < 0.05) with age. After day 28, relative abundances of ruminal Asteroleplasma and Anaeroplasma ranged from 0.3 to 1.2% in G kids, while their ruminal relative abundances were low (<0.2%) in S kids. Relative abundances of ruminal Akkermansia, Corprococcus, Ruminococcus, Selenomonas, Succiniclasticum and Treponema ranged from 0.1 to 3.6% in both groups. And relative abundances of Enterococcus and Lactobacillus were low (<1.0%) in the rumen of both groups.
FIGURE 3.
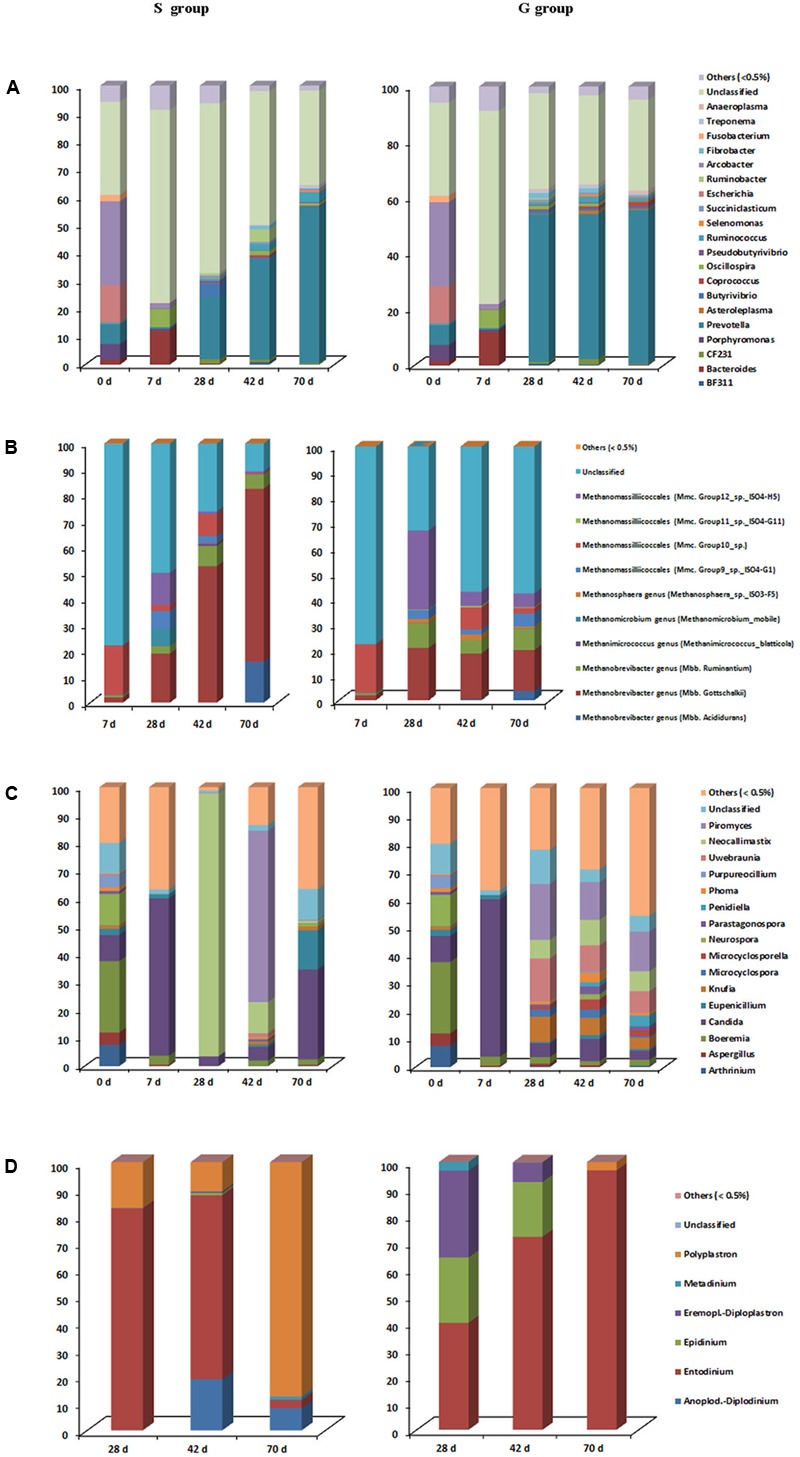
Taxonomic composition of each ruminal microbial domain in S and G kids at different ages. (A) Bacteria, (B) Archaea, (C) Fungi, and (D) Protozoa.
Rumen Microbial Community Changes-Archaea
Archaea were not detectable at day 0, and began to be detected at day 7. The relative abundance of Mbb. gottschalkii within the Methanobrevibacter genus was greater (P = 0.022), while relative abundances of genus Methanosphaera, as well as Group 11 and 12 within the Methanomassilliicoccales order were lower (P < 0.05) in the rumen of S kids compared to those in G kids (Figure 3B). Within the Methanobrevibacter genus, relative abundances of Mbb. gottschalkii (16.1–66.7%) and Mbb. ruminantium (2.9–9.7%) clades increased drastically from day 28, constituting the top two abundant clades. The highest relative abundance of Mbb.acididurans in the rumen of S and G kids was 15.9% (day 70) and 3.7% (day 70), respectively. Relative abundances of Methanimicrococcus and Methanosphaera genera were low (<2.0%) in the rumen of both kids, while Methanomicrobium genus was only detected in the rumen of S kids at day 28 (6.4%). Furthermore, a significant proportion of Methanomassilliicoccales (from 12.1 to 34.8%) was found in the rumen of both groups.
Rumen Microbial Community Changes-Fungi
Two fungal phyla, Ascomycota and Neocallimastigomycota, and 103 fungal genera were identified, with 16 fungal genera each accounting for >0.5% of all sequences (Figure 3C). The relative abundance of Neocallimastix was greater (P < 0.001), while relative abundances of Microcyclosporella, Knufia, Parastagonospora and Uwebraunia were lower (P < 0.05) in the rumen of S kids compared to those in G kids. Ruminal Boeremia (25.6%), Neurospora (11.3%), Arthrinium (7.6%), Aspergillus (4.5%), Purpureocillium (4.3%) were the top five abundant genera at day 0, while their relative abundances declined significantly (P < 0.01) afterward. The highest relative abundance of Candida in the rumen was 56.4% (day 7). The relative abundances of Knufia, Microcyclospora, Microcyclosporella, Parastagonospora, Penidiella, Phoma and Uwebaunia in the rumen of S kids were low (<2%) at all ages, while their relative abundances in the rumen of G kids increased with age (P < 0.05). Relative abundances of ruminal Neocallimastix and Piromyces in S kids increased quadratically with age (P < 0.05), peaking at 94.2% (day 28) and 61.3% (day 42), respectively, while their ruminal relative abundances in G kids increased linearly with age (P < 0.05), peaking at 7.1% (day 70) and 14.3% (day 70), respectively.
Rumen Microbial Community Changes-Protozoa
Protozoa began to colonize after day 28, with >99% sequences classified into six genera (Figure 3D). The relative abundances of Anoplodinium–Diplodinium and Polyplastron were greater (P < 0.05), while Polyplastron relative abundance was lower (P < 0.001) in the rumen of S kids compared to those in G kids. Entodinium was the most abundant genera (39.9–96.8%) in the rumen, with its relative abundance declined linearly (P = 0.001) in S kids, while increased linearly (P = 0.002) in G kids from day 28 to 70. Relative abundances of Anoplodinium–Diplodinium and Polyplastron were high (8.2–87.4%) in the rumen of S kids, but low (<3.0%) in the rumen of G kids. In contrast, the relative abundances of ruminal Epidinium and Eremoplstron-Diploplastron were low (<1.0%) in S kids, but high in G kids (7.3–32.4%).
Expression of Genes Involved in Ruminal Mucosal Innate Immune Function
The expression of genes encoding TLRs (1–10) was down-regulated in the ruminal mucosa of S kids when compared to that in G kids. For S kids, mRNA abundance of TLRs1, 2, 4, 7, and 10 increased slightly (P < 0.05) from day 0 to 28, and then decreased afterward (Figure 4A). The mRNA abundance of other TLRs increased during the first week (P < 0.05), and declined afterward. Conversely, for G kids, expression of TLRs increased from day 0 to 28 (P < 0.01), remaining relatively stable afterward.
FIGURE 4.
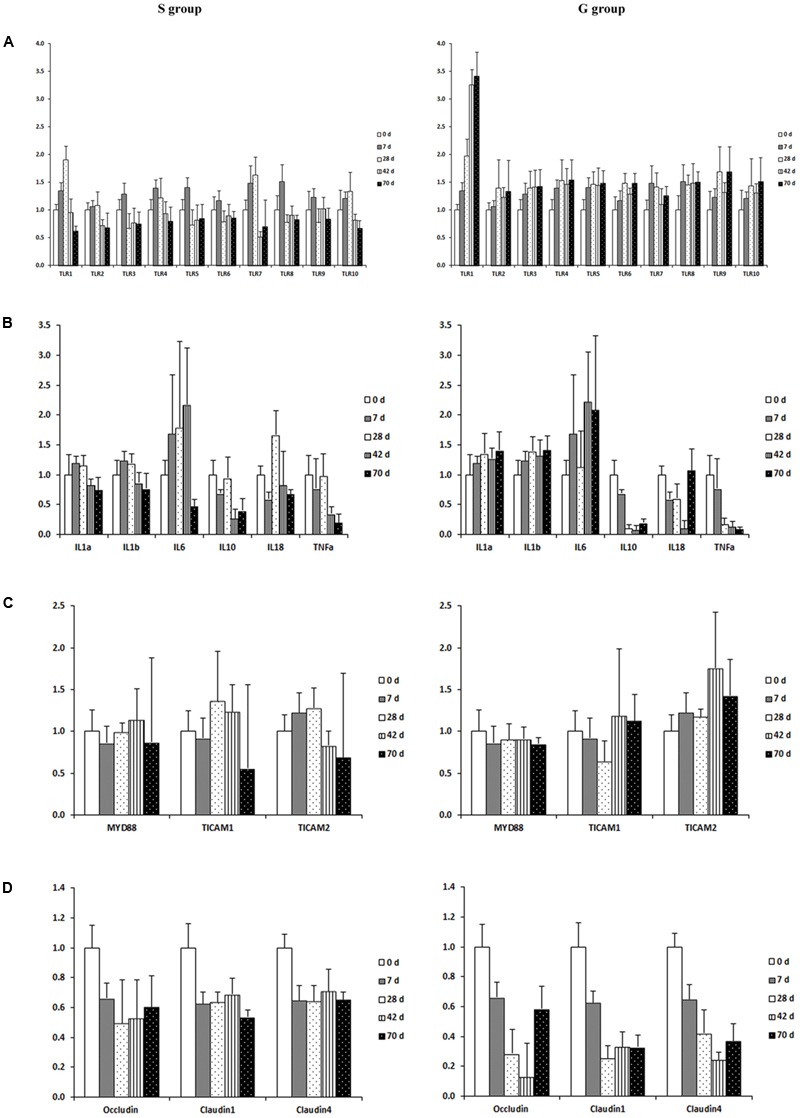
Expression of genes encoding host mucosal innate immune function in the ruminal mucosa of S and G kids at different ages. (A) Toll like receptors, (B) Cytokines; (C) TIR-containing adaptors; (D) Tight junction proteins.
When genes involved in cytokine production were compared, the expression of IL1α and IL1β was up-regulated (P < 0.01), while expression of IL10 and TNFα was down-regulated (P < 0.01) in the ruminal mucosa of G kids when compared to those in S kids (Figure 4B). The mRNA abundance of IL1α, IL1β and IL6 in the ruminal mucosa of S kids increased from day 0 to 28, declined afterward, while their expression in the ruminal mucosa of G kids increased (P < 0.05) from day 0 to d 70. In both groups, mRNA abundance of IL10, IL18 and TNFα in the ruminal mucosa declined with age (P < 0.05).
For genes encoding TIR domain-containing adaptors, MYD88 expression was age- and feeding type-independent (Figure 4C). The expression of TICAM2 (P = 0.003) was up-regulated in the ruminal mucosa of G kids when compared to that in S kids. Age exerted a quadratic effect (P = 0.006) on TICAM1 mRNA abundance in the ruminal mucosa of S kids. TICAM2 mRNA abundance in the ruminal mucosa of S kids increased during the first month and declined afterward, whist its value in G kids increased (P = 0.043) from day 0 to 70.
For genes encoding tight junction proteins (TJs), the expression of Occludin, Claudin1 and Claudin4 was elevated (P < 0.05) in the ruminal mucosa of S kids when compared to those in G kids (Figure 4D). Furthermore, expression of TJs declined (P < 0.05) from day 0 to 70 in the ruminal mucosa of both groups.
Relationship between Microbial Community and Expression of Genes Involved in Ruminal Mucosal Innate Immune Function
As presented in Figure 5, TLR1 mRNA abundance was positively correlated with (R > 0.50) relative abundances of Anaeroplasma, Knufia, Piromyces, Uwebraunia, and Entodinium, while negatively correlated with (R < -0.50) relative abundances of Mbb. gottschalkii and Anoplodinium–Diplodinium. TLR2 mRNA abundance was negatively correlated with (R = -0.52) Anoplodinium–Diplodinium relative abundance, TLR4 mRNA abundance was negatively correlated with (R = -0.50) Mbb. gottschalkii relative abundance, while TLR3 mRNA abundance was positively correlated with (R > 0.50) relative abundances of Knufia and Uwebraunia. TLR6 mRNA abundance was positively correlated with (R > 0.50) relative abundances of Anaeroplasma, Kufia, Neocallimastix, and Uwebraunia). TLR8 mRNA abundance was positively correlated with (R > 0.50) relative abundances of Kufia, Neocallimastix, and Uwebraunia, while negatively correlated with (R = -0.61) Mbb. gottschalkii relative abundance. TLR9 mRNA abundance was positively correlated with (R > 0.50) relative abundances of Fibrobacter, Anaeroplasma, Kufia, Neocallimastix, Uwebraunia, and Epidinium. TLR10 mRNA abundance was positively correlated with (R = 0.51) Neocallimastix relative abundance, while negatively correlated with (R < -0.50) relative abundances of Anoplodinium–Diplodinium and Polyplastron. The mRNA abundance of IL1α and IL1β was negatively correlated with (R < -0.50) relative abundances of Mbb. gottschalkii, Anoplodinium–Diplodinium and Polyplastron. IL6 mRNA abundance was positively correlated with (R = 0.60) Mmc.Group10 relative abundance, while IL18 mRNA abundance was negatively correlated with (R = -0.59) Epidinium relative abundance. The mRNA abundance of IL10 and TNFα was positively correlated with (R > 0.50) Bacteroides relative abundance, but negatively correlated with (R < -0.50) relative abundances of Bacteroides, Prevotella, Coprococcus, Fibrobacter, Anaeroplama, Methanosphaera, Phoma, Piromyces, and Uwebraunia. The mRNA abundance of Occludin, Claudin1 and Claudin4 was negatively correlated with (R < -0.50) relative abundances of most microbes. TICAM1 mRNA abundance was positively correlated with (R = 0.63) Mmc.Group10 relative abundance, while TICAM2 mRNA abundance was negatively correlated with (R < -0.50) relative abundances of Mbb. gottschalkii and Anoplodinium–Diplodinium.
FIGURE 5.
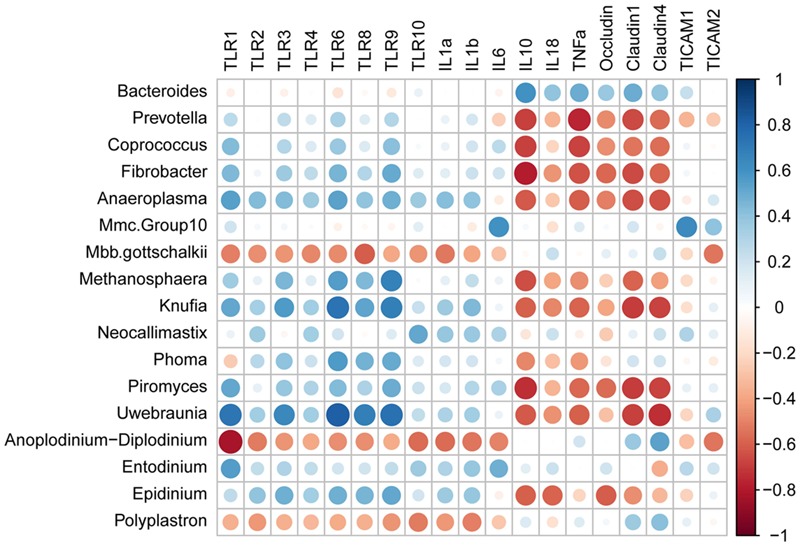
Spearman’s rank correlation coefficients between relative abundances of ruminal microbiota and expression of genes involved in host mucosal innate immune function.
Discussion
The goats underwent a drastic change in nutrient supply from a high-fat milk diet during non-rumination stage to a forage and concentrate based diet during rumination stage (Jiao et al., 2015b). Therefore, the observed shifts in both microbial density (Jiao et al., 2015b) and microbial composition in the rumen could be driven by the interaction between dietary changes and animal growth (ages). Meanwhile, the observed associations between the abundance of certain microbial taxa and expression of genes encoding TLRs and TJ proteins suggest that shifts in microbiota may influence the development of ruminal mucosal innate immune function. Historically, rumen epithelial wall has been considered “immune intact” for its tolerance in highly dynamic and abundant microbiota. However, recent studies have highlighted its innate immune function for the consistently detectable expression of genes encoding TLRs and TJ proteins in dairy and beef cattle (Devant et al., 2016; McCann et al., 2016), and the differences in the expression of these genes have been related to the ruminal microbial population changes (Chen et al., 2012). It is known that the rumen only develops after animals start to consume solid feed, and the undeveloped rumen epithelial structure lacks of distinct corneum, granulosum, spinosum, and basale strata (Graham and Simmons, 2005). This suggests that during early life the ruminal epithelial cells may trigger innate immune responses through molecular recognition pattern mechanism (such as TLRs) similar to the identified mechanism in the intestinal tissues of monogastric animals.
Indeed, the elevated expression of TLRs was detected in the rumen of kids during the first week of life. It has been reported that the elevated expression of TLRs is associated with cytokine production (Zeytun et al., 2010). Although the production of cytokines was not measured in this study, the enhanced expression of genes encoding pro-inflammatory cytokines, IL1α, IL1β and IL6 may indirectly support the higher cytokine production which could be led by the TLRs. On the other hand, the decreased gene expression of TJ proteins suggests higher mucosal permeability during the first week (Matter and Balda, 2003). Malmuthuge et al. (2013) reported the intake of a calf starter ration decreased expression of TJ genes in the small intestine of 8 weeks calves, and speculated the increased gut permeability could be associated with higher nutrient absorption capacity. Therefore, the potential higher permeability in the rumen could be also related to the nutrient absorption during early life.
In the meantime, our findings showed changes in ubiquitous microbiota, which could directly or indirectly influence ruminal innate immune function during early life. Kids were only exposed to the microbiota of their dams within 12 h of birth, after which they were separated and hand fed goat milk for 20 days which constituted the only nutrient intake. Thereby, we speculate that the microbiota detected at day 0 may be acquired from the mother at birth as well as from the environment and milk during the first day. Firstly, the predominance of Escherichia and Arcobacter at day 0 may play a role in stimulating the expression of TLRs, as several species in these genera, such as E. coli, A. butzleri, A. cryaerophilus, and A. skirrowii have been reported to modulate immune responses in mice (Bambou et al., 2004; Ferreira et al., 2016). Secondly, lactic acid bacteria (LAB), Enterococcus and Lactobacillus were also found in the rumen of new-born goats. Although their relative abundances were lower in the rumen compared to those in the small intestine (Jiao et al., 2016), LAB have been reported to enhance gut immune function, and consequently widely used as probiotics in both humans and animals (Walter, 2008; Franz et al., 2011). Thirdly, mucin degraders Bacteroides and Akkermansia surged in abundance during the first week. And the species, A. muciniphila and B. fragilis are potentially health-promoting bacteria which have anti-inflammatory attributes (Collado et al., 2007; Troy and Kasper, 2010). Finally, several invasive fungal pathogens, such as Aspergillus and Candida are capable of raising host–fungi interactions (Bourgeois and Kuchler, 2012), and can simulate IL1α production (Steele and Fidel, 2002). Their presence during the first week suggests that fungi may also play a role in development of ruminal mucosal innate immune function.
In addition to above findings on the microbial colonization and innate immune function during the first week, we observed that feeding type could impact microbial composition and expression of genes involved in innate immune function. After solid feed offered, differential expression of TLRs and TJs genes was detected between two feeding types, with lower expression of genes involved in inflammation and higher expression of genes encoding barrier function in the ruminal mucosa of S kids (detailed later). In intensive farming, supplementation of concentrate to young ruminants at a relative early age is a common practice which is considered to enhance the development of rumen (Jiao et al., 2015b). Therefore, the following discussion will mainly focus on S kids.
The expression profile for all TLRs was not consistent, which could reflect varied responses to the changes in the microbiota driven by the diet through the growth of S-kids. Indeed, relative abundances of bacterial members of Prevotella and Fibrobacter, archaeal members of Mbb. gottschallkii and Mbb. ruminantium, fungal members of Neocallimastix and Priomyces, protozoal members of Entodinium and Epidinium increased markedly after solid feed offered. Such changes are similar to a recent study reported by Dill-McFarland et al. (2017) who tracked ruminal microbiota from weaning (8 week) to first lactation (2 year) in the same heifers. These microbes are common members of gastrointestinal microbiota in ruminants, and are well recognized as efficient in degrading plant cellulosic fibers (Kittelmann et al., 2012; Henderson et al., 2015). Although the correlation relationship does not necessarily indicate a direct causal effect, the observed multiple significant correlations between relative abundances of these microbial taxa and expression of TLRs provide some insights on potential host–microbiota interaction in the rumen. Considering ruminal digesta and epithelium harbor more microbes in elder than younger ruminants (Jiao et al., 2015a,b), the down-regulated TLR expression after d 20 suggests a mechanism to minimize uperfluous inflammatory responses to commensal flora as proposed previously (Malmuthuge et al., 2012). Our observed down-regulated expression of genes encoding pro-inflammatory cytokines and stable expression of genes encoding TJ proteins confirm this speculation. Two possible reasons might contribute to the minimized immune responses. Firstly, relative abundances of butyrate-producing bacteria in ruminal digesta (Butyrivibrio and Pseudobutyrivirio) and epithelium (Butyrivibrio and Campylobacter) (Segain et al., 2000; Jiao et al., 2015a) increased, and then butyrate production was elevated (Jiao et al., 2015b). It is well established that butyrate can inhibit inflammatory responses via inhibition of NF kappa B activation (Segain et al., 2000). Secondly, relative abundances of mucin-degraders, such as Ruminococcus increased, and the enhanced mucus production could partly promote barrier function and alleviate inflammatory responses (Crost et al., 2013).
As described above, supplementing kids with concentrate in early life altered ruminal microbial diversity and increased expression of genes involved in barrier function. Compared to G kids, down-regulation of TLR signaling and pro-inflammatory cytokines revealed a potential to resist local inflammation in the ruminal mucosa of S kids (Liu et al., 2013). In general, TLRs can recognize microbial conserved molecular patterns, through TIR-domain containing adaptors such as MYD88, TICAM1, TICAM2 and TIRAF (Akira et al., 2001). The lower TICAM2 expression in the ruminal mucosa of S kids indicates that this molecule protein may play an essential role in differentiating immune responses to these two feeding types. Another explanation is the reduced permeability of the maturing rumen mucosa, as indicated by elevated expression of genes encoding TJ proteins in the rumen of S kids, preventing luminal microbiota entering the mucosa (Groschwitz and Hogan, 2009). The third important factor can be attributed to greater butyrate production observed in the same S kids (Jiao et al., 2015b), which is known to decrease production of pro-inflammatory cytokines (Segain et al., 2000).
On the other hand, the altered expression of genes in the ruminal mucosa of S kids could be due to the suppressed microbial diversity when compared to that in G kids. For example, increased relative abundances of Pseudobutyrivibrio and Butyrivibrio in the rumen of S kids could be associated with enhanced anti-inflammatory effects (Kopecny et al., 2003). Secondly, some species of Methanosphera, which have been reported to activate host dendritic cells, and induce release of inflammatory cytokines TNFα and IL1β (Bang et al., 2014), were more abundant in the rumen of G kids due to the pectins in the forages (Janssen and Kirs, 2008). Finally, Microcyclosporella and Parastagonospora are well-known fungal plant–pathogens (Frank et al., 2010; John et al., 2016), and their greater relative abundances in the rumen of G kids suggest the notion that fungal composition is diet-driven (Denman et al., 2008; Kittelmann et al., 2012), which provokes further consideration of their roles in modulating host immune function.
To our knowledge, limited studies research on microbiota and host innate immune function cross-talk in the rumen, and how such interaction can be impacted by dietary interventions. Based on the findings from current study, we proposed a working model for interactions between ruminal microbiota and mucosal innate immune function during animal development in S kids (Figure 6A) and the effect of feeding type intervention (Figure 6B). During the first week of life, milk nutrients facilitate colonization of mucin-degraders and fungi (Candida), which activate TLR signaling and provoke potential local inflammation. The starch and fiber alters microbial composition and diversity in ruminal digesta and mucosa, leading to enhanced SCFAs, especially butyrate production. These partly contribute to decreased inflammation by lower TLR signaling, and enhanced barrier function through stable TJs signaling, suggesting a mechanism to minimize superfluous inflammatory responses to commensal microbiota in S kids (Figure 6A). Furthermore, supplementing kids with concentrate alters ruminal microbial composition, diversity and products, giving rise to lower inflammation and enhanced barrier function (Figure 6B). Although the role of commensal microbiota on host mucosal immune function in the rumen is far from clear, our results provide some cues on the communication between ruminal microbiota and ruminal mucosal immune function that can be manipulated by dietary changes and developmental ages.
FIGURE 6.
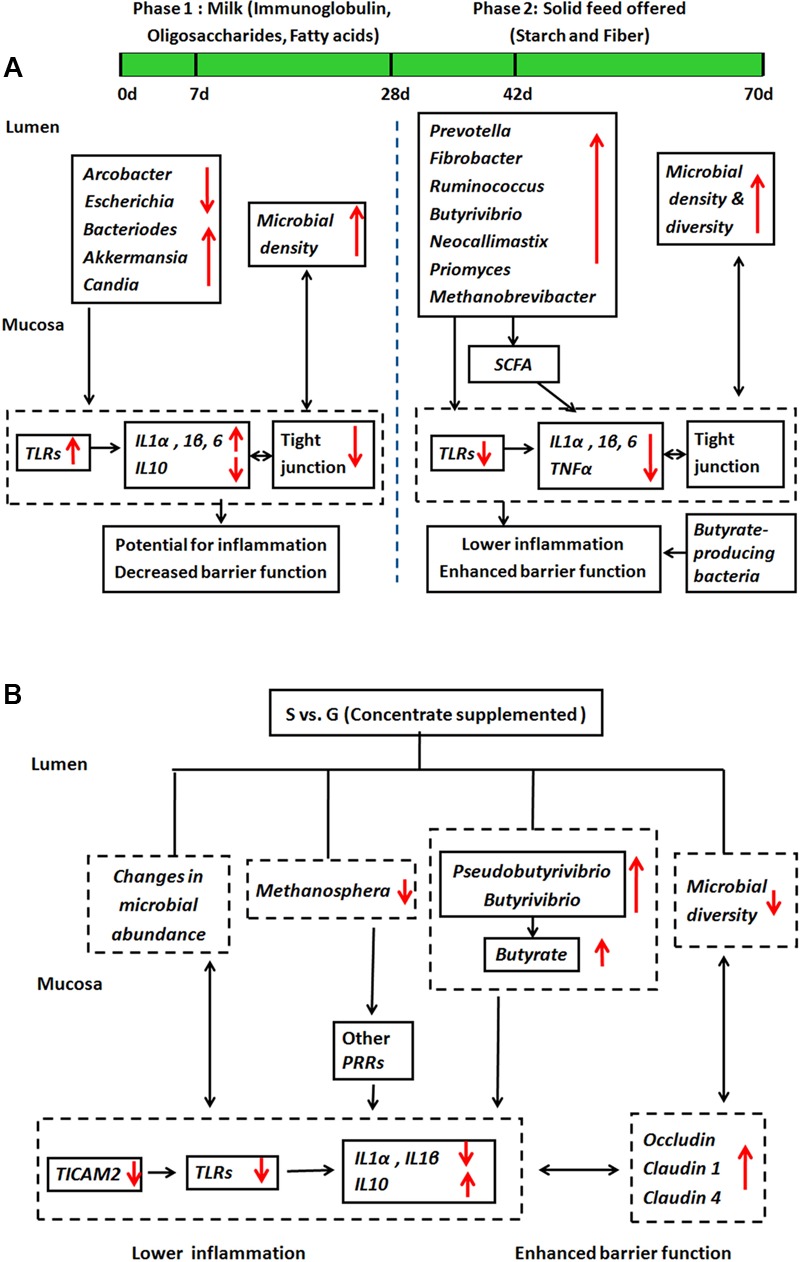
A working hypothesis of how ruminal microbiota affect host mucosal innate immune function. (A) During animal development in S kids, and (B) the effect of feeding type.
Conclusion
Ruminal microbial diversity (bacteria, archaea, fungi and protozoa) and expression of genes involved in ruminal mucosal innate immune function was age-related in both supplemental feeding and grazing goat kids. Concentrate supplementation during the early stages of life had the potential to modulate ruminal microbial composition, enhance barrier function and decrease local inflammation. Our results provide a model for integrating information on ruminal microbial diversity and function with measures of host mucosal immunity in the maturing ruminants. Future studies are warranted to investigate the actual contribution of specific microbial species in modulating host mucosal innate immune maturation.
Author Contributions
JJ and ZT designed the research; JJ and CZ conducted the research; ST and MW were involved in the animal experiments; JJ, LG, and CM performed the statistical analysis and contributed to the interpretation of results. JJ and LG wrote the draft of the manuscript. All authors read, approved the final manuscript, and have no competing interests.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors appreciated the joint financial support from National Natural Science Foundation of China (grants 31601967, 31320103917, and 31561143009). And the authors thank Graeme T. Attwood from Ruminant Nutrition and Animal Health, AgResearch for his critical revision of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01655/full#supplementary-material
Rarefaction curves of each ruminal microbial domain in S and G kids at different ages. (A) Bacteria, (B) Archaea, (C) Fungi, and (D) Protozoa.
References
- Akira S., Takeda K., Kaisho T. (2001). Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2 675–680. 10.1038/90609 [DOI] [PubMed] [Google Scholar]
- Bambou J. C., Giraud A., Menard S., Begue B., Rakotobe S., Heyman M., et al. (2004). In vitro and ex vivo activation of the TLR5 signaling pathway in intestinal epithelial cells by a commensal Escherichia coli strain. J. Biol. Chem. 279 42984–42992. 10.1074/jbc.M405410200 [DOI] [PubMed] [Google Scholar]
- Bang C., Weidenbach K., Gutsmann T., Heine H., Schmitz R. A. (2014). The intestinal archaea Methanosphaera stadtmaniae and Methanobrevibacter smithii activate human dendritic cells. PLoS ONE 9:e99411 10.1371/journal.pone.0099411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois C., Kuchler K. (2012). Fungal pathogens-a sweet and sour treat for toll-like receptors. Front. Cell. Infect. Microbiol. 2:142 10.3389/fcimb.2012.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G., Bittinger K., Bushman F. D., DeSantis T. Z., Andersen G. L., Knight R. (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 26 266–267. 10.1093/bioinformatics/btp636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Oba M., Guan L. L. (2012). Variation of bacterial communities and expression of Toll-like receptor genes in the rumen of steers differing in susceptibility to subacute ruminal acidosis. Vet Microbiol. 159 451–459. 10.1016/j.vetmic.2012.04.032 [DOI] [PubMed] [Google Scholar]
- Collado M. C., Derrien M., Isolauri E., de Vos W. M., Salminen S. (2007). Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl. Environ. Microbiol. 73 7767–7770. 10.1128/AEM.01477-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crost E. H., Tailford L. E., Le Gall G., Fons M., Henrissat B., Juge N. (2013). Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS ONE 8:e76341 10.1371/journal.pone.0076341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denman S. E., Nicholson M. J., Brookman J. L., Theodorou M. K., McSweeney C. S. (2008). Detection and monitoring of anaerobic rumen fungi using an ARISA method. Lett. Appl. Microbiol. 47 492–499. 10.1111/j.1472-765X.2008.02449.x [DOI] [PubMed] [Google Scholar]
- DeSantis T. Z., Hugenholtz P., Larsen N., Rojas M., Brodie E. L., Keller K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72 5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devant M., Penner G., Marti S., Quintana B., Fábregas F., Bach A., et al. (2016). Behavior and inflammation of the rumen and cecum in Holstein bulls fed high-concentrate diets with different concentrate presentation forms with or without straw supplementation. J. Anim. Sci. 94 3902–3917. 10.2527/jas.2016-0594 [DOI] [PubMed] [Google Scholar]
- Dill-McFarland K. A., Breaker J. D., Suen G. (2017). Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci. Rep. 7:40864 10.1038/srep40864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- Ferreira S., Queiroz J. A., Oleastro M., Domingues F. C. (2016). Insights in the pathogenesis and resistance of Arcobacter: a review. Crit. Rev. Microbiol. 42 364–383. 10.3109/1040841x.2014.954523 [DOI] [PubMed] [Google Scholar]
- Frank J., Crous P. W., Groenewald J. Z., Oertel B., Hyde K. D., Phengsintham P., et al. (2010). Microcyclospora and Microcyclosporella: novel genera accommodating epiphytic fungi causing sooty blotch on apple. Persoonia 24 93–105. 10.3767/003158510X510560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz C. M., Huch M., Abriouel H., Holzapfel W., Galvez A. (2011). Enterococci as probiotics and their implications in food safety. Int. J. Food Microbiol. 151 125–140. 10.1016/j.ijfoodmicro.2011.08.014 [DOI] [PubMed] [Google Scholar]
- Gazzinelli R. T., Denkers E. Y. (2006). Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat. Rev. Immunol. 6 895–906. 10.1038/nri1978 [DOI] [PubMed] [Google Scholar]
- Graham C., Simmons N. L. (2005). Functional organization of the bovine rumen epithelium. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288 R173–R181. 10.1152/ajpregu.00425.2004 [DOI] [PubMed] [Google Scholar]
- Groschwitz K. R., Hogan S. P. (2009). Intestinal barrier function: molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 124 3–22. 10.1016/j.jaci.2009.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson G., Cox F., Ganesh S., Jonker A., Young W., Janssen P. H. (2015). Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 5:14567 10.1038/srep14567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristov A. N., Callaway T. R., Lee C., Dowd S. E. (2012). Rumen bacterial, archaeal, and fungal diversity of dairy cows in response to ingestion of lauric or myristic acid. J. Anim. Sci. 90 4449–4457. 10.2527/jas.2011-4624 [DOI] [PubMed] [Google Scholar]
- Jami E., Israel A., Kotser A., Mizrahi I. (2013). Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 7 1069–1079. 10.1038/ismej.2013.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen P. H., Kirs M. (2008). Structure of the archaeal community of the rumen. Appl. Environ. Microbiol. 74 3619–3625. 10.1128/aem.02812-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J., Huang J., Zhou C., Tan Z. (2015a). Taxonomic identification of ruminal epithelial bacterial diversity during rumen development in goats. Appl. Environ. Microbiol. 81 3502–3509. 10.1128/AEM.00203-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J., Li X., Beauchemin K. A., Tan Z., Tang S., Zhou C. (2015b). Rumen development process in goats as affected by supplemental feeding v. grazing: age-related anatomic development, functional achievement and microbial colonisation. Br. J. Nutr. 113 888–900. 10.1017/S0007114514004413 [DOI] [PubMed] [Google Scholar]
- Jiao J., Wu J., Zhou C., Tang S., Wang M., Tan Z. (2016). Composition of ileal bacterial community in grazing goats varies across non-rumination, transition and rumination stages of life. Front. Microbiol. 7:1364 10.3389/fmicb.2016.01364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John E., Lopez-Ruiz F., Rybak K., Mousley C. J., Oliver R. P., Tan K. C. (2016). Dissecting the role of histidine kinase and HOG1 mitogen-activated protein kinase signalling in stress tolerance and pathogenicity of Parastagonospora nodorum on wheat. Microbiology 162 1023–1036. 10.1099/mic.0.000280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Akira S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11 373–384. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- Kittelmann S., Janssen P. H. (2011). Characterization of rumen ciliate community composition in domestic sheep, deer, and cattle, feeding on varying diets, by means of PCR-DGGE and clone libraries. FEMS Microbiol. Ecol. 75 468–481. 10.1111/j.1574-6941.2010.01022.x [DOI] [PubMed] [Google Scholar]
- Kittelmann S., Naylor G. E., Koolaard J. P., Janssen P. H. (2012). A proposed taxonomy of anaerobic fungi (class neocallimastigomycetes) suitable for large-scale sequence-based community structure analysis. PLoS ONE 7:e36866 10.1371/journal.pone.0036866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecny J., Zorec M., Mrazek J., Kobayashi Y., Marinsek-Logar R. (2003). Butyrivibrio hungatei sp. nov. and Pseudobutyrivibrio xylanivorans sp. nov., butyrate-producing bacteria from the rumen. Int. J. Syst. Evol. Microbiol. 53 201–209. 10.1099/ijs.0.02345-0 [DOI] [PubMed] [Google Scholar]
- Legendre P., Gallagher E. (2001). Ecologically meaningful transformations for ordination of species data. Oecologia 129 271–280. 10.1007/s004420100716 [DOI] [PubMed] [Google Scholar]
- Liu B., Yuan J., Yiu S. M., Li Z., Xie Y., Chen Y., et al. (2012). COPE: an accurate k-mer-based pair-end reads connection tool to facilitate genome assembly. Bioinformatics 28 2870–2874. 10.1093/bioinformatics/bts563 [DOI] [PubMed] [Google Scholar]
- Liu J. H., Bian G. R., Zhu W. Y., Mao S. Y. (2015). High-grain feeding causes strong shifts in ruminal epithelial bacterial community and expression of Toll-like receptor genes in goats. Front. Microbiol. 6:167 10.3389/fmicb.2015.00167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. H., Xu T. T., Liu Y. J., Zhu W. Y., Mao S. Y. (2013). A high-grain diet causes massive disruption of ruminal epithelial tight junctions in goats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305 R232–R241. 10.1152/ajpregu.00068.2013 [DOI] [PubMed] [Google Scholar]
- Malmuthuge N., Li M. J., Fries P., Griebel P. J., Guan L. L. (2012). Regional and age dependent changes in gene expression of Toll-like receptors and key antimicrobial defence molecules throughout the gastrointestinal tract of dairy calves. Vet. Immunol. Immunopathol. 146 18–26. 10.1016/j.vetimm.2012.01.010 [DOI] [PubMed] [Google Scholar]
- Malmuthuge N., Li M., Goonewardene L. A., Oba M., Guan L. L. (2013). Effect of calf starter feeding on gut microbial diversity and expression of genes involved in host immune responses and tight junctions in dairy calves during weaning transition. J. Dairy Sci. 96 3189–3200. 10.3168/jds.2012-6200 [DOI] [PubMed] [Google Scholar]
- Matter K., Balda M. S. (2003). Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 4 225–236. 10.1038/nrm1055 [DOI] [PubMed] [Google Scholar]
- McCann J. C., Luan S., Cardoso F. C., Derakhshani H., Khafipour E., Loor J. J. (2016). Induction of subacute ruminal acidosis affects the ruminal microbiome and epithelium. Front. Microbiol. 7:701 10.3389/fmicb.2016.00701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey M., Enjalbert F., Combes S., Cauquil L., Bouchez O., Monteils V. (2014). Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J. Appl. Microbiol. 116 245–257. 10.1111/jam.12405 [DOI] [PubMed] [Google Scholar]
- Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75 7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seedorf H., Kittelmann S., Henderson G., Janssen P. H. (2014). RIM-DB: a taxonomic framework for community structure analysis of methanogenic archaea from the rumen and other intestinal environments. PeerJ 2:e494 10.7717/peerj.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segain J. P., de la Bletiere D. R., Bourreille A., Leray V., Gervois N., Rosales C., et al. (2000). Butyrate inhibits inflammatory responses through NF kappa B inhibition: implications for Crohn’s disease. Gut 47 397–403. 10.1136/gut.47.3.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele C., Fidel P. L. (2002). Cytokine and chemokine production by human oral and vaginal epithelial cells in response to Candida albicans. Infec Immun. 70 577–583. 10.1128/iai.70.2.577-583.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisi E., Amadori M., Riva F., Bertoni G., Bani P. (2014). Evaluation of innate immune responses in bovine forestomachs. Res. Vet. Sci. 96 69–78. 10.1016/j.rvsc.2013.11.011 [DOI] [PubMed] [Google Scholar]
- Troy E. B., Kasper D. L. (2010). Beneficial effects of Bacteroides fragilis polysaccharides on the immune system. Front. Biosci. 15 25–34. 10.2741/3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J. (2008). Ecological role of lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Appl. Environ. Microbiol. 74 4985–4996. 10.1128/aem.00753-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeytun A., Chaudhary A., Pardington P., Cary R. B., Gupta G. (2010). Induction of cytokines and chemokines by Toll-like receptor signaling: strategies for control of inflammation. Crit. Rev. Immunol. 30 53–67. 10.1615/CritRevImmunol.v30.i1.40 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rarefaction curves of each ruminal microbial domain in S and G kids at different ages. (A) Bacteria, (B) Archaea, (C) Fungi, and (D) Protozoa.